

Abstract
Fatty acid synthase (FAS) is necessary for growth and survival of tumor cells and is a promising drug target for oncology. Here, we report on the syntheses and activity of novel inhibitors of the thioesterase domain of FAS. Using the structure of orlistat as a starting point, which contains a β-lactone as the central pharmacophore, 28 novel congeners were synthesized and examined. Structural features such as the length of the α- and β-alkyl chains, their chemical composition, and amino ester substitutions were altered and the resulting compounds explored for inhibitory activity toward the thioesterase domain of FAS. Nineteen congeners show improved potency for FAS in biochemical assays relative to orlistat. Three of that subset, including the natural product valilactone, also display an increased potency in inducing tumor cell death and improved solubility compared to orlistat. These findings support the idea that an orlistat congener can be optimized for use in a preclinical drug design and for clinical drug development.
Introduction
Most human carcinomas, including those of the breast and prostate, overexpress fatty acid synthase (FASa), the sole enzyme responsible for de novo biosynthesis of fatty acids.1–6 In the vast majority of cases, FAS is required for tumor cell survival and it also seems to play a role in conferring chemoresistance.7,8 In contrast, most normal cells utilize dietary fats and therefore FAS is not required for survival. Consequently, FAS is a promising drug target for the treatment of human carcinomas.
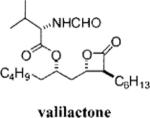
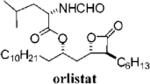
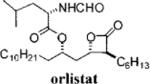
Orlistat is a pancreatic lipase inhibitor that is currently marketed for the treatment of obesity. In the gut, orlistat forms a covalent, but reversible, bond with the active site serine residue of pancreatic lipases, rendering them unable to hydrolyze dietary fat into free fatty acids and thereby reducing the absorption of dietary fat.9 Previously, we showed that orlistat is also a potent inhibitor of the thioesterase activity of FAS and that it has antitumor activity in vitro and in vivo.10 The three-dimensional structure of orlistat bound to FAS shows that the compound forms a covalent adduct with the enzyme's active site serine, the same mechanism by which it inhibits pancreatic lipase.11
Despite its ability to inhibit FAS and elicit tumor cell death, there are a number of challenges that prevent the deployment of orlistat as an antitumor drug: it has poor solubility and bioavailability and it lacks sufficient potency. Here we sought to take the first step toward the synthesis of an orlistat derivative suitable for use as an antitumor drug. The specific objectives of the present study were to (1) synthesize derivatives of orlistat with increased solubility, (2) determine the structural alterations that can be made to orlist at without loss of activity toward FAS, and (3) identify orlistat derivatives with increased potency toward FAS and increased cytotoxicity toward tumor cells. Twenty-eight novel congeners of orlistat were synthesized, most having increased solubility and inhibitory activity compared to orlistat. The α- and β-side chains extending from the β-lactone were shown to be amenable to optimization, and alkenyl bonds can be incorporated into their structure without loss of activity. The amino ester can be changed without substantial loss of activity toward FAS. Reversal of chirality at Cα and Cδ from S to R is tolerated, but compounds with R-Cβ chirality had substantially reduced activity. The results of the study reveal several new FAS inhibitors that are more suitable for preclinical drug development and also provide the foundation for a more detailed structure-based drug design centered on the β-lactone pharmacophore.
Results and Discussion
Synthesis of Orlistat Congeners
Twenty-eight congeners of orlistat were synthesized to identify the structural components important for activity toward FAS (Table 1). Four structural features of orlistat were altered in the test set: the α- and β-side chains, the amino ester, and chirality (Figure 1). The described β-lactones were synthesized employing the tandem Mukaiyama aldol-lactonization (TMAL) process previously developed in our group as the key step.12 Previously we reported general strategies for the synthesis of orlistat derivatives employing the TMAL process in conjunction with coupling reactions that enable facile modification of both the α- and δ-side chain including cuprate addition, simple alkylation of ethylacetoacetate for subsequent Noyori reduction, and an olefin metathesis/hydrogenation sequence.13 These methods were also employed to prepare the derivatives studied herein.
Table 1.
Oiiistat Derivatives Prepared by the TMAL Process Followed by Either Inversion or Retention of C6 Stereochemistry during Amino Ester side Chain Introductiona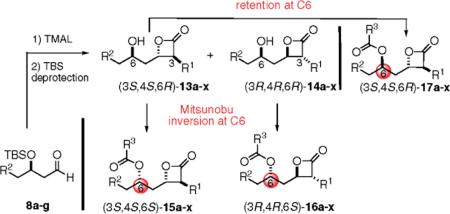
| entry | R1 | R2 | TMAL % yieldb | TMAL ratio (13:14)a | % yield Mitsunobu/ acylanon | Orlistat derivatives 15–17 |
|---|---|---|---|---|---|---|
| 1 | C6H13 | C12H25 | 58 | 13a: 14a= 6:1 | 80 (15a) |
|
| 73 (15b) | ||||||
| 80 (15c) | ||||||
| 17 (15d) | ||||||
| 17 (16a) | ||||||
|
| ||||||
| 2 | C6H13 | C10H21 | 58 | 13b: 14b = 8:1 | 87 (15g) |
|
| 97 (17c) | ||||||
|
| ||||||
| 3 | C4H9 | C10H21 | 49 | 13c: 14c = 7.7:1 | 52 (17d) |
|
|
| ||||||
| 4 | C2H5 | C10H21 | 58 | 13d: 14d = 6:1 | 25 (15f) |
|
| 99 (17a) | ||||||
|
| ||||||
| 5 | Me E/Z | C10H21 | 20 | 13e:14e = 1.5:1 | 38 (15e) |
|
| 24 (16b) | ||||||
|
| ||||||
| 6 | C8H17 | C8H17 | 42 | 13f:14f = 8 4:1 | ref. 22 |
|
|
| ||||||
| 7 | C6H13 | ally1 | 60 | 13g:14g = ND | 68 (15j) |
|
| 46 (15k) | ||||||
| 55 (17f) | ||||||
|
| ||||||
| 8 | C6H13 | trans-croty l | 62 | 13h:14b = 9:1 | 45 (15h) |
|
| ND (15i) | ||||||
| 57 (17g) | ||||||
| ND (17h) | ||||||
|
| ||||||
| 9 | C6H13 | H | 42 | 13i:14i = 7.1:1 | 52 (17d) |
|
|
| ||||||
| 10 | C6H13 | TsO | 42 | 13j:14j = 7.7:1 | 58 (17e) |
|
|
| ||||||
| 11 | C6H13 | C10H21 | NDa | 13j:14j= 3:1 | 51 (17i) |
|
ND = not determined.
Includes yield for TBS deprotection step.
Figure 1.
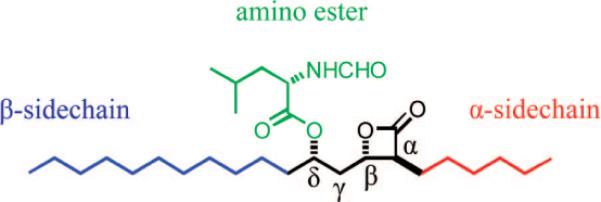
Molecular structure of tetrahydrolipstatin (orlistat) and the structural features modified in this study. Modifications were made to reduce the overall hydrophobicity of the compounds and then tested for retention of activity. Several compounds with variation at one or a combination of these features were shown to have increased potency toward FASTE in a fluorogenic biochemical assay.
The TMAL process employs readily available thiopyridyl ketene acetals 3 and optically active aldehydes 8 and 12. A series of thiopyridyl ketene acetals 3 were obtained according to previously reported methods involving generation of thiopyridyl esters 2 from the corresponding acid chlorides 1 and an enolization/silylation sequence (Scheme 1).
Scheme 1.

Syntheses of the Thiopryridyl Ketene Acetals
One route studied for the preparation of optically active β-hydroxy aldehydes 8 made use of the chiral pool and involved cuprate addition to a common tosylate 6 derived from malic acid to install the δ-chain, which would enable its facile variation by appropriate choice of cuprate reagent (Scheme 2). Thus, (S)-malic acid was converted to tosylate ester 6 in a four-step sequence by following literature procedures (the synthesis of aldehyde 8a was described previously; see supporting documentation in ref 13). After some experimentation, suitable cuprate conditions were identified and the addition of the octyl Gilman reagent gave ester 7 in 78% yield. However, this process was not readily extended to other cuprates, so an alternative route to optically active aldehydes was studied (vide infra, Scheme 3). Following ester half-reduction, aldehyde 8a could be obtained in 86% yield.
Scheme 2.

Synthesis of the Chiral Aldehyde 8a from Malic Acid Involving Cuprate Addition
Scheme 3.

Syntheses of Chiral Aldehydes 8b–g via Noyori Reduction
In an alternative route to optically active aldehydes 8, simple alkylation of ethylacetoacetate was employed to provide several β-ketoesters 10 and subsequent Noyori reduction, alcohol protection, and ester half-reduction provided aldehydes 8b–g. This enabled variation of the δ-chain of orlistat derivatives in a lengthier but high-yielding route (Scheme 3).
Treatment of various aldehydes 8 in a dichloromethane slurry of freshly fused ZnCl2 and thioketeneacetals 3 delivered the desired β-lactones as mixtures of anti/syn diastereomers (7–9:1, dr) with complete selectivity for the trans-β-lactone. The β-lactones were directly desilylated to afford the more readily separable γ-hydroxy β-lactones 13 and 14 (Scheme 4). The trans relative stereochemistry of the β-lactone core was verified by analysis of coupling constants (JHa,Hb ~ 4.0 Hz) as described previously, whereas the relative stereochemistry of the major diastereomers with respect to the γ-stereocenter (C5) is consistent with Evans' model for additions to β-silyloxy aldehydes as previously observed for these TMAL reactions.14 Further verification came following completion of the synthesis of orlistat and valilactone and comparison to literature data (vide infra). To obtain a cis-β-lactone to determine the effect of β-lactone relative stereochemistry on FAS inhibition, we employed our stereocomplimentary approach to β-lactones, which employs SnCl4 as Lewis acid for the aldol-lactonization process.15 Reaction of aldehyde 8f and ketene acetal 3d under these conditions gave a mixture of diastereomeric cis-β-lactones from which the major β-lactone cis-13 could be isolated.
Scheme 4.

Tandem Mukaiyama Aldol-Lactonization Process and Deprotection Delivering β-Lactones 13/14
To obtain the required syn relative stereochemistry with respect to the β- and γ-stereocenters as found in orlistat, the major diastereomeric hydroxy-β-lactones 13, possessing the 6R configuration, were subjected to Mitsunobu conditions to invert this stereocenter (Scheme 5). This was accomplished with N-formyl amino acids and proceeded with the expected inversion of configuration, providing the 3S,4S,6S-diastereomers 15. To probe the effect of stereochemistry on FASTE inhibition, the minor 3R,4R-diastereomers 14 obtained in the TMAL process were also converted to the 3R,4R,6R-diastereomers 16 via the Mitsunobu reaction. In addition, retention of stereochemistry was possible by simple acylation of β-lactones 13 with N-formyl amino acids to provide 3S,4S,6R-diastereomers 17 in high yield.
Scheme 5.
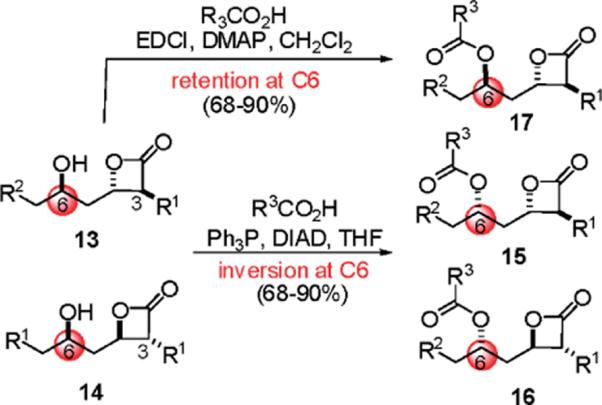
Introduction of the α-Amino Ester Moiety via Mitsunobu Reaction (with Inversion) or Acylation (with Retention) at C6, Providing Orlistat Derivatives 15–17
Finally, we also studied olefin cross metathesis as a possible tactic for modification of the δ-side chain from olefin precursors 18 involving two routes that differ only in reaction order, olefin metathesis, and introduction of the α-amino ester moiety (methods A and B, Scheme 6). In one example, cross-metathesis with excess n-nonene in the presence of the first generation Grubb's catalyst gave low conversion (20% yield, unoptimized) of intermediate 20a (~4:1 E/Z).16 Subsequent hydrogenation of the derived olefinic derivatives under standard conditions afforded saturated δ-alkyl substituted derivatives 22 in nearly quantitative yield. This overall process provides an expedient route to modify the δ-side chain of orlistat. However, while this strategy successfully provided olefinic orlistat derivatives 21a–c (~4:1 E/Z with exception of 21c, which gave exclusively E), generally, this route suffered from low conversion in the olefin metathesis step.
Scheme 6.

Alternative Strategy for Modification of the δ-Side Chain Involving Olefin Cross-Metathesis and Hydrogenation
Effect of Novel β-Lactones on the Thioesterase Activity of FAS
The thioesterase domain of FAS cleaves palmitate from the acyl carrier protein once its synthesis is complete.17 Compounds were compared for their ability to inhibit the activity of the recombinant thioesterase using methods we have previously described.18,19 Four structural components of the orlistat molecule were modified to test for effect on potency: the α- and β-side chains, the amino ester, and both relative and absolute stereochemistry (Figure 1). The results of this analysis are summarized below:
(i) Modifications to the α-Chain
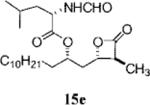
In general, the inhibitory activity toward FAS is tolerant to changes in the length of the α-chain. An α-chain of just two carbons appears to be sufficient for good potency. Compounds 17a (Table 2, entry 2), 15f, and 17b (Table 3, entries 14 and 18, respectively) all have α-chains less than the six carbons found in orlistat, and each has an IC50 equal to or less than that of orlistat. In fact, 17a, with an ethyl α-chain, has an IC50 of 0.23 μM, which is greater than 6-fold more potent than orlistat (IC50) 1.35 μM). The reduction of the α-chain to a methyl group, however, generally resulted in reduced potency. Compound 15e (Table 4, entry 7), which has the same structure as orlistat with the exception of having a methyl group in the α-chain position, is approximately 8-fold less potent than orlistat.
Table 2.
Compounds that Displayed Enhanced Inhibitory Activity in the Biochemical Assay as Well as Better Selectivity for Lipogenic Cells Relative to Orlistata
| Entry | Structure | FASTE1 IC50 (μM) | MDA-MB 2312 IC50 (μM) | Hs58.Fs2 IC50 (μM) | Ratio3 | cLogP4 |
|---|---|---|---|---|---|---|
| 1 |
|
1.35±0.34 | 16.8±0.5 | 70.2±15.9 | 4.2 | 8.609 |
|
| ||||||
| 2 |
|
0.23±0.02 | 1.3±0.1 | 10.0±2.4 | 7.7 | 4.727 |
| 3 |
|
0.28±0.04 | 3.3±0.6 | 29.6±2.3 | 9.0 | 4.422 |
| 4 |
|
0.30±0.07 | 10.5±2.5 | >100 | >9.5 | 4.906 |
(1)Results are presented as mean and 95% CI. (2) Results are presented as the mean ± SD of at least two independent experiments. (3) Ratio of IC50 values (Hs58.Fs/MDA-MB-231). (4) cLogP values were calculated with ChemDraw Ultra 10.0 software (CambridgeSoft).
Table 3.
Compounds that Displayed Enhanced Inhibitory Activity Relative to Orlistat but Less Cellular Selectivitya
| Entry | Structure | FASTE1IC50 (μM) | MDA-MB-2312 IC50 (μM) | Hs58.Fs2 IC50 (μM) | Ratio3 | cLogP4 | Entry | Structure | FASTE1 IC50 (μM) | MDA-MB-2312 IC50 (μM) | Hs58.Fs2 IC 50 (μM) | Ratio3 | cLogP4 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
|
1.35±0.34 | 16.8±0.5 | 70.2±15.9 | 4.2 | 8.609 | 10 |
|
0.50=0.12 | 14.1 | 32.9 | 2.3 | 6.163 |
| 2 |
|
0.10 ± 0.03 | 48.0 ± 5.7 | 52.2 ± 3.9 | 1.1 | 3.140 | 11 |
|
0.50±0.18 | 54.0±9.4 | >100 | >1.9 | 2.656 |
| 3 |
|
0.12±0.29 | >100 | 43.9±13.9 | <0.4 | 8.125 | 12 |
|
0.52±0.14 | >1OO | >1OO | ND | 1.553 |
| 4 |
|
0.18±0.01 | >100 | >100 | ND | 7.901 | 13 |
|
0.68±0.12 | 56.9±19.0 | >100 | >l.8 | 4.422 |
| 5 |
|
0.20+0.04 | 26.9+12.2 | 37.3−0.3 | 1.4 | 7.901 | 14 |
|
0.79±0.12 | 88.4±7.3 | >100 | >1.1 | 4.727 |
| 6 |
|
0.29+0.04 | >100 | >100 | ND | 9.628 | 15 |
|
0.90±0.10 | 24.5±0.8 | 73.6±2.0 | 3.0 | 4.383 |
| 7 |
|
0.37±0.06 | >100 | >100 | ND | 2.508 | 16 |
|
1.02±0.38 | 4.8=0.5 | 19.9±4.4 | 4.2 | 6.843 |
| 8 |
|
0.40±0.05 | >100 | 24.9=0.1 | <0.3 | 8.086 | 17 |
|
1.04+0.33 | 38.3+0.9 | 61.2+3.0 | 1.6 | 4.912 |
| 9 |
|
0.45±0.10 | 37.1 ±11.8 | 15.8±3.4 | 0.4 | 7.227 | 18 |
|
1.35+0.07 | 35.7+1.3 | 31.1+3.2 | 0.9 | 5.785 |
(1) Results are presented as mean and 95% CI. (2) Results are presented as the mean ± SD of at least two independent experiments. (3) Ratio of IC50 values (Hs58.Fs/MDA-MB-231). (4) cLogP values were calculated with ChemDraw Ultra 10.0 software (CambridgeSoft). ND = not determined.
Table 4.
Compounds with Structural Changes that Proved Deleterious to the Biochemical Inhibition of PASTEa
| Entry | Structure | FASTE1 IC50 (μM) | MDA-MB-2312 IC50 (μM) | Hs58.Fs2 IC50 (μM) | Ratio3 | cLogP4 |
|---|---|---|---|---|---|---|
| 1 |
|
l.35±0.34 | 16.5±0.5 | 70.2±15.9 | 4.2 | 8.609 |
|
| ||||||
| 2 |
|
1.72±0.09 | 11.7±0.9 | 46.8±0.3 | 4.0 | 6.843 |
| 3 |
|
3.00±2.21 | >100 | 87.2±10.5 | <1.0 | 8.609 |
| 4 |
|
3.67±0.89 | >100 | >100 | ND | 9.667 |
| 5 |
|
6.24±0.46 | ND | ND | ND | 3.185 |
| 6 |
|
9.74±6.04 | ND | ND | ND | 9.667 |
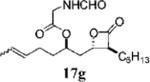
| 7 |
|
10.81 + 1.66 | ND | ND | ND | 5.964 |
| 8 |
|
13.54±2.54 | ND | ND | ND | 4.422 |
| 9 |
|
45.1 1±11.39 | ND | ND | ND | 5.964 |
Compounds with an IC50 > 5 μM in the FASTE biochemical assay were not tested in cells. (1) Results are presented as mean and 95% CI. (2) Results are presented as the mean ± SD of at least two independent experiments. (3) Ratio of IC50 values (Hs58.Fs/MDA-MB-231). (4) cLogP values were calculated with ChemDraw Ultra 10.0 software (Cambridge-Soft). ND = not determined.
We have also observed that ebelactone B, which contains an ethyl α-chain, is roughly 10 times more potent than ebelactone A, which contains a methyl α-chain20 (data not shown). Altogether these data establish that an α-chain of just two carbons is sufficient for retention of good potency toward FAS. This observation is consistent with the recent finding from the crystal structure of orlistat bound to the FAS thioesterase which shows the α-chain packs against the histidine residue of the catalytic triad.11 On the basis of this structure, an ethyl moiety is expected to maintain such packing where a methyl group would be insufficient to extend to this histidine.
(ii) Modifications to the β-Chain
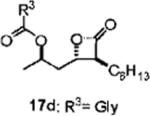
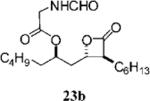
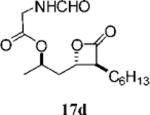
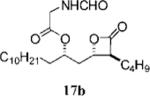
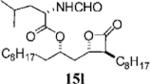
Optimization of the β-chain would seem to present a greater challenge because it binds within the putative palmitate binding pocket of the FAS thioesterase, helping to properly position the β-lactone in the vicinity of the catalytic triad.11,21 Therefore, truncation or other alteration of the β-chain could lead to decreased potency if the conjugate bond cannot be formed. In a direct head-to-head comparison, the reduction of the β-chain from 11 carbons (17c, Table 4, entry 2) to just four carbons (23b, Table 3, entry 2) resulted in a 15-fold increase in potency against FASTE in the biochemical assay, suggesting that the full length β-chain is not necessary to position the β-lactone at the catalytic triad. Even though they have additional structural changes, compounds 17d and 15f (Table 3, entries 12 and 14, respectively) also have β-chains of reduced length and have increased potency compared to orlistat, providing further support that the full length chain is not necessary for activity.
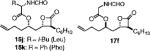
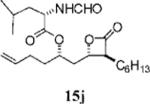
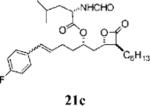
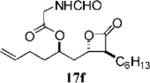
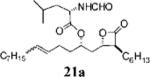
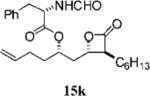
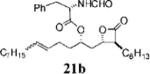
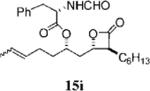
Other alterations to the β-chain were also well-tolerated. The incorporation of a single alkenyl bond into the β-chain (21a, Table 3, entry 3) increased the potency of the compound 10-fold compared to orlistat. Several other olefinic compounds showed good potency toward FASTE, i.e., 15j (Table 2, entry 3), 21a–c (Table 3, entries 3, 8, and 10, respectively), and 15k (Table 3, entry 15) even though they were not part of direct head-to-head comparisons. Even the placement of an alkene at the terminal position of the β-chain is tolerated as in compounds 15i (Table 3, entry 17) and 17f (Table 3, entry 11).
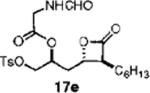
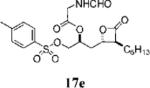
A small set of compounds were synthesized with β-chains other than simple hydrocarbons. Two of these compounds, 17e and 21c (Table 3, entries 7 and 10, respectively), were at least as active as orlistat in the biochemical assay, providing additional support for the idea that significant structural variability within the β-chain can be accommodated by FASTE.
(iii) Modifications to the Amino Ester Side Chain
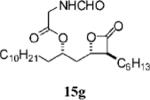
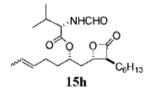
Orlistat contains an N-formyl-l-leucine moiety branching from the δ carbon (C6) of the lactone. Interactions between the enzyme and the amino ester may contribute to positioning orlistat at the active site.11 Interestingly, the substitution of glycine for leucine at this position (15g, Table 3, entry 16) had little effect on potency toward the FAS thioesterase. Results from other head-to-head comparisons of compounds with long β-chains, i.e., 15b (Table 3, entry 5) versus 15c (Table 3, entry 6), were similar, indicating such substitutions are tolerated but provide no significant enhancement in potency by themselves. Importantly, though, in compounds with much shorter β-chains that contain an alkenyl bond, substitution of the amino ester yielded a greater than 10-fold increase in potency as with 15i (Table 3, entry 17) versus 15h (Table 4, entry 8).
Overall, 12 of the congeners we present here contain an N-formyl-glycine moiety that lends greater conformational flexibility than the N-formyl-leucine moiety and significantly decreases the compounds' overall hydrophobicity. Nine of these compounds had significantly improved inhibitory activity toward FASTE compared to orlistat. A small set of congeners (8) containing valine, phenylalanine, and histidine at the amino ester position were also synthesized and tested. These compounds also exhibited good potency toward the enzyme, suggesting a wide range of substitutions at this position are accommodated by the enzyme.
(iv) Modifications in Chirality
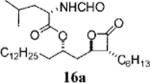
Several compounds (11) with differing relative and absolute stereochemistry at Cα, Cβ, and/or Cδ were also synthesized and tested. While no head-to-head comparisons were made, initial indications suggest that stereochemical changes at Cβ (R versus S) abrogates inhibitory activity toward the FASTE such as with compounds 16a (Table 4, entry 6; IC50) 9.74 μM) and 16b (Table 4, entry 9; IC50) 45.11 μM). However, changes to chirality at Cα appear to be tolerated, i.e., 17i (Table 3, entry 4; IC50) 0.18 μM), as long as the S-configuration is retained at the β-chain. Somewhat surprisingly, inverted stereochemistry at Cδ, the carbon from which the amino ester side chain branches off the main β-chain, was also tolerated. In fact, compound 17a (Table 2, entry 2), with increased potency both toward FASTE and in cell-based assays, has inverted stereochemistry at Cδ. The vast majority of compounds with this inverted stereochemistry contained an N-formyl-glycine moiety that allows for greater conformational flexibility; however, we acknowledge the possibility of an alternate binding mode to the enzyme.
Summary of All Orlistat Derivatives Synthesized
In Table 1, we summarize all orlistat derivatives prepared in this study with yields and diastereoselectivity of the key TMAL reaction process as well as yields for subsequent amino ester introduction via Mitsunobu or acylation chemistry.
Effect of Compounds on Tumor Cell Apoptosis
Each compound was also tested in MDA-MB-231 breast carcinoma cells and Hs58.Fs foreskin fibroblasts to distinguish mechanism-based cell death elicited by inhibition of FAS from general cytotoxicity. To establish the degree to which each cell type depends on FAS for survival, we treated them with siRNA targeting FAS or with 25 μM orlistat (Figure 2). Both treatments induced death in the cancer cells but had only minimal effect on the fibroblasts. Importantly, the absolute level of apoptosis was essentially equivalent in both cell lines when they were treated with staurosporine, a compound known to induce apoptosis via an unrelated cAMP-mediated mechanism22 (data not shown). These results establish that the cancer cells are dependent on FAS for survival but the fibroblasts are not.
Figure 2.
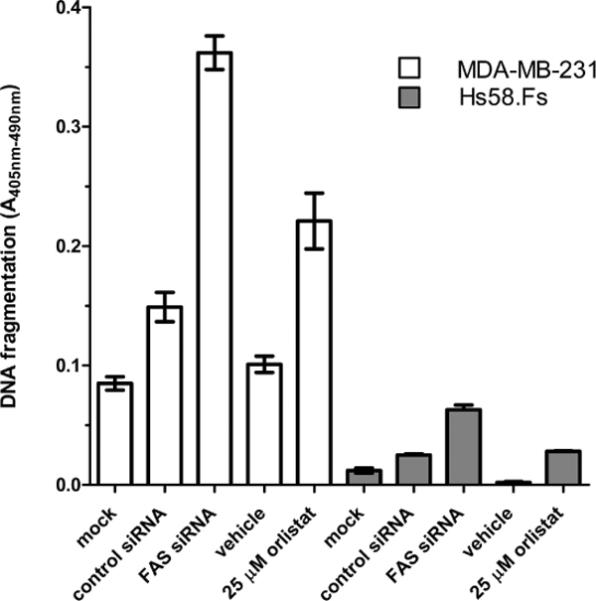
Differential cytotoxicity in MDA-MB-231 and Hs58.Fs cells are a model platform for screening FASTE inhibitors, as shown by the pharmacological and siRNA inhibition of FAS. MDA-MB-231 tumor cells and primary cultures of Hs58.5s fibroblasts were exposed to 100 nM FAS siRNA or nonsilencing control siRNA and 25 μM orlistat or vehicle control for 72 h. Cellular cytotoxicity was monitored by measuring DNA fragmentation. Values are means ± SE of four samples per treatment group.
Three compounds (17a, 15j, and valilactone) were identified that had increased potency toward the thioesterase of FAS and in cell-based assays and that showed selective toxicity to the cancer cells compared to fibroblasts (Table 2). The enhanced potency of this subset of compounds toward cells could be due to their increased affinity for the thioesterase domain, their enhanced aqueous solubility (cLogP values of less than 5 vs 8.6 for orlistat), or some combination of both. One other compound, 15g (Table 3, entry 16) had better potency than orlistat in the cell-based assays, even though its performance against the recombinant thioesterase was not significantly different from orlistat. The increase in cellular potency of this compound is probably due to a single structural variation, the substitution of a glycine at the amino ester position, which makes the compound less hydrophobic and reduces its cLogP.
The remainder of the compounds either had a reduction in potency toward tumor cells or lost selectivity for tumor cells over the fibroblasts. Most of the compounds having good potency toward the recombinant thioesterase exhibited an IC50 in tumor cells greater than that of orlistat. There are two possibilities to explain this observation. First, it is conceivable that the FAS holoenzyme, in the context of the cellular environment, displays a thioesterase with a catalytic cleft that is structurally distinct from that of the recombinant thioesterase. Second, and probably more likely, is the possibility that these compounds simply have reduced ability to traverse the plasma membrane. Some of the compounds also lost selectivity for tumor cells over fibroblasts, which may be indicative of the reactivity of these compounds for other enzymes, especially other serine hydrolases that may be important for survival of all cells.
Conclusion
Synthesis of 28 orlistat congeners led to the identification of molecular features that can be optimized to generate β-lactones with improved potency toward the thioesterase domain of FAS. Three of these compounds had an improved affinity for the thioesterase, an increased potency toward tumor cells, and greater selectivity for tumor cells over fibroblasts. Interestingly, these three compounds show considerable structural diversity. Each includes a unique amino ester substitution and a unique β-chain. Additionally, compound 17a (Table 2, entry 2) has a truncated α-chain and opposite chirality (R versus S) at the δ carbon. These results provide an important foundation for structure-based design of β-lactones with optimal cytotoxicity profiles and the heterogeneity of the most promising compounds suggests a larger, more diverse study is warranted. The data presented here also underpin the idea that an orlistat congener can be optimized for use in preclinical drug design and for clinical drug development for the treatment of carcinomas.
Materials and Methods
Chemical Synthesis and Characterization
All reactions were carried out under nitrogen atmosphere in flame-dried glassware. Dichloromethane, acetonitrile, methanol, tetrahydrofuran, and ethyl ether were purified by passage through activated molecular sieves. Hünig's base and triethylamine were distilled from potassium hydroxide prior to use. All other commercially obtained reagents were used as received. 1H NMR chemical shifts are reported as δ values in ppm relative to CDCl3 (7.27 ppm) and coupling constants (J) are reported in hertz (Hz). Unless indicated otherwise, deutero-chloroform (CDCl3) served as an internal standard (77.23 ppm) for all 13C spectra. Flash column chromatography was performed using 60 Å silica gel (Silicycle, 230–400 mesh) as stationary phase. Mass spectra were obtained at the Mass Spectrometry Application and Collaboration Facility (Texas A&M University). Thin layer chromatography (TLC) was performed using glass-backed silica gel 60F254 (Silicycle, 250 μM thickness). 1H and 13C NMR spectra were acquired using Varian spectrometers at the frequency indicated. Solvents are indicated for each compound. Chemical shifts are expressed in ppm referenced to the residual nondeuterated solvent as an internal standard. 1H NMR coupling constants (J) are reported in hertz (Hz) and multiplicities are abbreviated as follows: app = apparent, s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, br = broadband signal. For representative procedures for synthesis of ketene acetals 3a–d and aldehydes 8a–g, please see ref 22.
Representative Procedure for Synthesis of β-Lactones 13/14 via the TMAL Reaction/Desilylation, as Described for (3S,4S)-3-Hexyl-4-((R)-2-hydroxytridecyl)-oxetan-2-one (13) [reproduced from ref 22]. TMAL Reaction
ZnCl2 (398 mg, 2.92 mmol) was fused under vacuum and then allowed to cool to room temperature under a flow of nitrogen. Then 7.5 mL of dry CH2Cl2 was then added via syringe, followed by a solution of aldehyde 8f (500 mg, 1.46 mmol) in 2.5 mL of CH2Cl2 and ketene acetal 3d (719 mg, 2.04 mmol). The reaction mixture was stirred for 60 h at ambient temperature. Then 5 mL of pH 7 buffer were added, and the mixture was stirred vigorously for 30 min, filtered through a pad of Celite, and washed with CH2Cl2. The organic filtrate was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography on SiO2 (10:1, hexanes:EtOAc) to provide the mixture of silyloxy-β-lactone diastereomers as a pale-yellow oil. 1H NMR (500 MHz) analysis of the mixture of diastereomers indicated a diastereomeric ratio of 8:1. Without further purification, the mixture was used directly in the next step.
Desilylation
To a stirred solution of the mixture of silyloxy-β-lactones in 24 mL of CH3CN cooled to 0 °C, 2.4 mL of HF (48%) was added dropwise. The mixture was stirred at 0 °C for 2 h, allowed to warm to ambient temperature, and stirred for an additional 5 h. The reaction mixture was diluted with 100 mL of Et2O, quenched carefully with cold saturated NaHCO3, and washed with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by flash chromatography on SiO2 (10:1, hexanes:EtOAc) to provide the hydroxy-β-lactone 13 (190 mg, 44%) and a mixture of the two diastereomers (112 mg, 14%) as white solids (58% overall, 2 steps). Spectroscopic data are reported for the major diastereomeric β-lactone 13: Rf = 0.54 (20% EtOAc/hexanes); [α]23d −30.9 (c 0.7, CHCl3). IR (thin film) 1819 cm−1. 1H NMR (500 MHz, CDCl3) δ 4.51 (dt, J = 4.3, 8.5 Hz, 1H), 3.79–3.86 (m, 1H), 3.27 (ddd, J = 4.0, 7.0, 8.5 Hz, 1H), 1.93 (ddd, J = 3.0, 8.5, 14.5 Hz, 1H), 1.72–1.87 (m, 3H), 1.24–1.56 (m, 28H), 0.891 (t, J = 6.0 Hz, 3H), 0.889 (t, J = 6.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 171.8, 75.8, 68.7, 56.8, 42.0, 38.3, 32.1, 31.7, 29.9, 29.83, 29.77 (2), 29.7, 29.6, 29.2, 27.9, 27.0, 25.6, 22.9, 22.7, 14.3, 14.2. LRMS (ESI) calcd for C22H42O3 [M + Li], 361; found, 361.
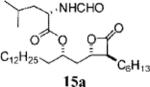
Representative Mitsunobu Reaction Procedure as Described for (S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3S,4S)-3-Hexyl-4-oxo-oxetan-2-ylmethyl)-tetradecyl Ester (15a)
To a solution of β-lactone 13a (30.0 mg, 0.0784 mmol) in THF (1.0 mL) at 0 °C was added triphenylphosphine (66.6 mg, 0.254 mmol), N-formyl-l-leucin (47.3 mg, 0.296 mmol), and DIAD (49 μL, 0.25 mmol). The reaction was monitored by TLC. After the reaction was complete, solvent was removed in vacuo and purified by flash chromatography, eluting with Hexanes/EtOAc (4:1), to give β-lactone 15a (33 mg, 80%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.22 (s, 1H), 5.97 (d, J = 8.1 Hz, 1H), 4.99–5.07 (m, 1H), 4.69 (dt, J = 5.1, 9.0 Hz, 1H), 4.30 (quint, J = 4.5 Hz, 1H), 3.22 (dt, J = 4.2, 7.8 Hz, 1H), 2.17 (dt, J = 7.8, 14.7 Hz, 1H), 2.00 (dt, J = 4.8, 14.7 Hz, 1H), 1.25–1.82 (m, 37H), 0.86–0.98 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 172.2, 171.0, 160.8, 75.0, 73.0, 57.2, 49.8, 41.8, 38.9, 34.3, 32.1, 31.7, 29.89, 29.86(3), 29.7, 29.64, 29.56, 29.5, 29.2, 27.8, 26.9, 25.3, 25.1, 23.1, 22.9, 22.7, 22.0, 14.3, 14.2. LRMS HRMS (ESI) calcd for C31H57NO5 [M + Li], 530.4397; found, 530.4282.
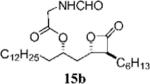
(2S,3S,4S)-Formylamino-acetic Acid 1-(3-Hexyl-4-oxo-oxetan-2-ylmethyl)-tetradecyl Ester (15b)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13a (20.0 mg, 0.0523 mmol), triphenylphosphine (44.4 mg, 0.169 mmol), N-formylglycine (20.4 mg, 0.197 mmol), and DIAD (33 μL, 0.169 mmol) in 0.7 mL of THF. Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 15b (17.8 mg, 73%). 1H NMR (300 MHz, C6D6) δ 7.72 (s, 1H), 5.16 (br, 1H), 4.94–5.01 (m, 1H), 4.06 (dt, J = 4.8, 7.5 Hz, 1H), 3.78 (dd, J = 6.0, 18.0 Hz, 1H), 3.69 (dd, J = 5.7, 18.0 Hz, 1H), 2.74 (dt, J = 4.2, 7.5 Hz, 1H), 1.72 (dt, J = 8.1, 15.0 Hz, 1H), 1.15–1.47 (m, 35H), 0.88–0.94 (m, 6H). 13C NMR (75 MHz, C6D6) δ 170.7, 169.6, 160.9, 74.8, 72.8, 57.6, 40.5, 39.2, 34.8, 32.7, 32.2, 30.53 (2), 30.51, 30.5, 30.4, 30.3, 30.2, 30.1, 29.7, 28.1, 27.3, 25.9, 23.5, 23.3, 14.7, 14.6.
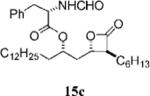
(S)-2-Formylamino-3-phenyl-propionic Acid 1-((2S,3S,4S)-3-Hexyl-4-oxo-oxetan-2-ylmethyl)-tetradecyl Ester (15c)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13a (20.0 mg, 0.0523 mmol), triphenylphosphine (44.4 mg, 0.169 mmol), N-formyl-l-leucin (47.3 mg, 0.296 mmol), and DIAD (49 μL, 0.25 mmol) in 1.0 mL of THF. Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 15c (33 mg, 80%). 1H NMR (300 MHz, C6D6) δ 7.57 (s, 1H), 6.96–7.12 (m, 5H), 5.08 (d, J = 8.1 Hz, 1H), 4.92–4.98 (m, 1H), 4.87 (q, J = 7.5 Hz, 1H), 3.93–3.99 (m, 1H), 3.01 (dd, J = 6.3, 14.1 Hz, 1H), 2.84 (dd, J = 7.2, 14.1 Hz, 1H), 2.70 (dt, J = 4.2, 7.8 Hz, 1H), 1.73 (dt, J = 7.5, 15.0 Hz, 1H), 1.14–1.54 (m, 35H), 0.88–0.95 (m, 6H). HRMS (ESI) calcd for C34H55NO5 [M + Li], 564.4240; found, 564.4166.
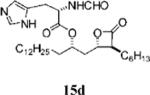
(S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3S,4S)-3-Methyl-4-oxo-oxetan-2-ylmethyl)-dodecyl Ester (15d)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13a (40.0 mg, 0.105 mmol), triphenylphosphine (88.9 mg, 0.339 mmol), N-formyl-l-histidine (72.3 mg, 0.395 mmol), and DIAD (66 μL, 0.34 mmol) in 3 mL of THF. Purification by flash chromatography on SiO2 (2:1, hexanes:EtOAc) gave β-lactone 15d (10 mg, 17%). 1H NMR (300 MHz, C6D6) δ 7.91 (s, 1H), 7.78 (s, 1H), 6.97 (s, 1H), 6.82 (d, J = 7.5 Hz, 1H), 4.93–4.99 (m, 2H), 4.82 (quint, J = 6.6 Hz, 1H), 4.22 (dt, J = 3.9, 6.3 Hz, 1H), 2.99 (dd, J = 5.1, 14.1 Hz, 1H), 2.86 (dd, J = 5.4, 15.0 Hz, 1H), 2.78–2.82 (m, 1H), 1.88 (dt, J = 6.9, 14.7 Hz, 1H), 1.16–1.63 (m, 31H), 0.87–0.96 (m, 10H).
(S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3S,4S)-3-Methyl-4-oxo-oxetan-2-ylmethyl)-dodecyl Ester (15e)
Prepared according to the representative Mitsunobu reaction procedure using the mixture of β-lactones 13e and 14e (11.9 mg, 0.0418 mmol), triphenylphosphine (32.8 mg, 0.125 mmol), N-formylleucine (23.2 mg, 0.146 mmol), and DIAD (25 μL, 0.13 mmol) in 1.0 mL of THF. Purification by flash chromatography on SiO2 (3:2, hexanes: EtOAc) followed by a second purification by flash chromatography on SiO2 (0.4:1:3 THF:CHCl3:hexanes) gave the desired β-lactone as a mixture of diastereomers. The two diastereomers could be separated by MPLC (65:35, hexanes: EtOAc), affording β-lactone 15e (6.8 mg, 38%) and diastereomeric β-lactone 16b (4.4 mg, 24%). Major diastereomer (15e): 1H NMR (300 MHz, CDCl3) δ 8.23 (s, 1H), 5.92 (d, J = 8.7 Hz, 1H), 4.98–5.10 (m, 1H), 4.69 (td, J = 4.8, 8.4, 1H), 4.23 (ddd, J = 8.4, 4.2, 4.2 Hz, 1H), 3.28 (dq, J = 4.2, 7.8 Hz, 1H), 1.96–2.26 (m, 2H), 1.51–1.75 (m, 5H), 1.40 (d, J = 7.8 Hz, 3H), 1.26 (br. s, 18H), 0.98 (d, J = 6.0 Hz, 6H), 0.89 (t, J = 6.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 172.2, 171.4, 160.8, 76.5, 72.8, 51.8, 49.9, 41.8, 38.7, 34.4, 32.1, 29.92, 29.83, 29.75, 29.65, 29.56, 29.51, 25.3, 25.1, 23.1, 22.9, 22.0, 14.3, 12.6.
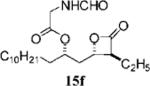
(S)-((R)-1-((2S,3S)-3-Ethyl-4-oxooxetan-2-yl)tridecan-2-yl) 2-Methanamido-4-methylpentanoate (15f)
To a mixture of hydroxy β-lactone 13d (17.7 mg, 0.0594 mmol), N-formylleucine (33.4 mg, 0.209 mmol), and triphenylphosphine (53.4 mg, 0.203 mmol) in a 10 mL round-bottom flask was added 0.5 mL of xylene, and the mixture was azeotroped in vacuo. After 1.0 h, THF (1.4 mL) was added under N2 and the solution was cooled to 0 °C. Next, DIAD (37.9 μL, 0.1947 mmol) was added and the reaction mixture was stirred at 0 °C for 10 min. The reaction was allowed to warm to room temperature and stirred overnight. The mixture was concentrated in vacuo and purified twice by chromatography (SiO2, 10 → 30% EtOAc:hexanes) to afford β-lactone 15f (6 mg, 25%). Rf = 0.26 (30% EtOAc/hexanes); [α]22d = −3.68 (c 0.4, CHCl3). IR (thin film) 2926, 2856, 1828, 1740, 1671 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.23 (br s, 1H), 5.90 (d, J = 8.5 Hz, 1H), 5.04 (app quint, J = 5.5 Hz, 1H), 4.69 (ddd, J = 5.0, 8.5, 13.0 Hz, 1H), 4.30–4.33 (m, 1H), 3.18–3.22 (m, 1H), 2.19 (app quint, J = 7.0 Hz, 1H), 2.00 (dt, J = 4.5, 19.0 Hz, 1H), 1.64–1.70 (m, 2H), 1.54–1.60 (m, 19H) 1.22–1.32 (m, 6H), 1.042 (dppt, J = 7.5 Hz, 3H) 0.97 (dd, J = 1.5, 4.5 Hz, 4H), 0.90 (appt, J = 7.5 Hz, 3H). 13C NMR (125 HMz, CDCl3) δ 172.2, 169.5, 161.2, 74.7, 73.3, 58.6, 40.4, 39.1, 34.4, 32.2, 30.0, 29.9, 29.8, 29.7, 29.6, 29.5, 25.4, 22.9, 21.1, 14.4, 11.3. HRMS (ESI) calcd for 38C21H45NO5 [M + H], 384.2744; found, 384.2786.
(2S,3S,4S)-Formylamino-acetic Acid 1-(3-Hexyl-4-oxo-oxetan-2-ylmethyl)-dodecyl Ester (15g)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13b (40.0 mg, 0.113 mmol), triphenylphosphine (74.1 mg, 0.283 mmol), N-formylglycine (40.7 mg, 0.395 mmol), and DIAD (55 μL, 0.28 mmol) in 2 mL of THF. Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 15g (43 mg, 87%). 1H NMR (300 MHz, C6D6) δ 7.79 (s, 1H), 5.45 (br, 1H), 4.95–5.02 (m, 1H), 4.08 (dt, J = 5.1, 7.2 Hz, 1H), 3.81(dd, J = 5.7, 18.0 Hz, 1H), 3.73 (dd, J = 5.7, 18.3 Hz, 1H), 2.78 (dt, J = 4.2, 7.8 Hz, 1H), 1.75 (dt, J = 8.1, 15.0 Hz, 1H), 1.17–1.52 (m, 31H), 0.87–0.94 (m, 6H). 13C NMR (75 MHz, C6D6) δ 170.8, 169.7, 161.1, 74.9, 72.8, 57.6, 40.6, 39.2, 34.8, 32.7, 32.2, 30.47, 30.45, 30.40, 30.3, 30.2, 30.1, 29.7, 28.2, 27.4, 25.9, 23.5, 23.3, 14.7, 14.6.
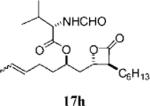
2-Formylamino-3-methyl-butyric Acid (2>S,2R,3S,4S)-1-(3-Hexyl-4-oxo-oxetan-2-ylmethyl)-hex-4-enyl Ester (15h)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13h (20.0 mg, 0.0745 mmol), triphenylphosphine (27.4 mg, 0.104 mmol), N-formyl-l-valine (21.6 mg, 0.149 mmol), DIAD (20 μL, 0.10 mmol) in 1 mL of THF. Purification by flash chromatography on SiO2 (9:1, hexanes:EtOAc) gave β-lactone 15h (13.3 mg, 45%) as a colorless oil as a E/Z-mixture, only peaks for major olefin isomer are assigned. Rf = 0.24 (30% EtOAc/hexanes); [α]22d −7.95 (0.9, CHCl3). IR (thin film) 1823, 1737, 1670 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.28 (s, 1H), 6.05 (d, J = 8.0 Hz, 1H), 5.32–5.50 (m, 2H), 5.03–5.08 (m, 1H), 4.64 (ddd, J = 4.5, 7.5, 16.5 Hz, 1H), 4.30 (dt, J = 4.0, 8.0 Hz, 1H), 3.23 (dt, J = 4.5, 7.0 Hz, 1H), 2.06–2.26 (m, 3H), 2.02 (dt, J = 5.5, 14.5 Hz, 2H), 1.64–1.85 (m, 6H), 1.28–1.48 (m, 9H), 1.01 (d, J = 6.5 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H), 0.89 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 171.2, 170.9, 161.0, 129.4, 126.6, 74.8, 72.5, 57.3, 56.1, 38.8, 33.9, 31.7, 31.2, 29.2, 28.4, 27.9, 26.9, 22.7, 19.5, 18.11, 17.7, 14.2. HRMS (ESI) calcd for C22H37NO5 [M + H], 396.2744; found, 396.2833.
(S)-2-Formylamino-3-phenyl-propionic Acid 1-((2S,3S,4S)-3-Hexyl-4-oxo-oxetan-2-ylmethyl)-hex-4-enyl Ester (15i)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13h (20.0 mg, 0.0745 mmol), triphenylphosphine (27.4 mg, 0.104 mmol), N-formyl-l-phenylalanine (28.8 mg, 0.149 mmol), and DIAD (20 μL, 0.10 mmol) in 2 mL of THF. Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 15i (yield not determined) as a E/Z-mixture; only peaks for major olefin isomer are assigned. 1H NMR (300 MHz, CDCl3) δ 8.18 (s, 1H), 7.17–7.32 (m, 5H), 5.99 (d, J = 7.8 Hz, 1H) 5.30–5.47 (m, 2H), 4.97–5.05 (m, 1H), 4.91 (q, J = 6.9 Hz, 1H), 4.28 (dt, J = 5.1, 7.8 Hz, 1H), 3.06–3.24 (m, 3H), 1.26–2.10 (m, 19H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 171.0, 170.9, 160.6, 135.7, 129.5, 129.4, 128.9, 127.6, 126.5, 74.7, 72.7, 57.3, 52.3, 38.8, 38.0, 33.9, 31.7, 29.2, 28.3, 27.8, 26.9, 22.7, 18.1, 14.3. HRMS (ESI) calcd for C26H37NO5 [M + H], 444.2744; found, 444.2730.
(S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3S,4S)-3-Hexyl-4-oxo-oxetan-2-ylmethyl)-pent-4-enyl Ester (15j)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13g (80.0 mg, 0.315 mmol), triphenylphosphine (116 mg, 0.441 mmol), N-formylleucin (126 mg, 0.788 mmol), and DIAD (85 μL, 0.44 mmol) in 5.0 mL of THF, purified by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 15j (85 mg, 68%). 1H NMR (300 MHz, C6D6) δ 7.99 (s, 1H), 5.94 (d, J = 8.1 Hz, 1H), 5.64–5.78 (m, 1H), 4.94–5.09 (m, 3H), 4.75 (dt, J = 5.1, 9.6 Hz, 1H), 4.10 (dt, J = 4.8, 8.1 Hz, 1H), 2.79 (dt, J = 3.9, 7.5 Hz, 1H), 1.96–2.05 (m, 1H), 1.17–1.83 (m, 18H), 1.05 (d, J = 6.0 Hz, 3H), 0.86–0.91 (m, 6H). 13C NMR (75 MHz, C6D6) δ 172.6, 170.8, 161.3, 137.9, 116.0, 74.8, 72.2, 57.6, 50.3, 41.5, 39.2, 33.9, 32.2, 30.0, 29.7, 28.1, 27.3, 25.4, 23.35, 23.27, 21.9, 14.6. HRMS (ESI) calcd for C22H37NO5 [M + Li], 402.2832; found, 402.2830.
(S)-2-Formylamino-3-phenyl-propionic Acid 1-((2S,3S,4S)-3-Hexyl-4-oxo-oxetan-2-ylmethyl)-pent-4-enyl Ester (15k)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 13g (40.0 mg, 0.157 mmol), triphenylphosphine (57.6 mg, 0.220 mmol), N-formyl-l-phenylalanine (70.0 mg, 0.362 mmol), and DIAD (43 μL, 0.220 mmol) in 3.0 mL of THF. Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 15k (31 mg, 46%). 1H NMR (300 MHz, C6D6) δ 7.56 (s, 1H), 6.94–7.12 (m, 5H), 5.60–5.75 (m, 1H), 4.82–5.06 (m, 4H), 3.90–3.92 (m, 1H), 2.99 (dd, J = 6.3, 14.4 Hz, 1H), 2.82 (dd, J = 6.9, 14.4 Hz, 1H), 2.67 (dt, J = 3.9, 7.5 Hz, 1H), 1.91 (q, J = 6.9 Hz, 1H), 1.22–1.68 (m, 16H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 170.9 (2), 160.7, 137.1, 135.7, 129.4, 128.9, 127.6, 115.9, 74.6, 72.6, 57.3, 52.3, 38.9, 38.0, 33.2, 31.7, 29.5, 29.2, 27.8, 26.9, 22.7, 14.3.
(S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3S,4S)-3-Octyl-4-oxo-oxetan-2-ylmethyl)-decyl Ester (15l)
Prepared as previously reported.23
(S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3R,4R)-3-Hexyl-4-oxo-oxetan-2-ylmethyl)-tetradecyl Ester (16a)
Prepared according to the representative Mitsunobu reaction procedure using β-lactone 14a (20.0 mg, 0.052 mmol), triphenylphosphine (44.4 mg, 0.169 mmol), N-formylluecin (31.6 mg, 0.197 mmol), and DIAD (33 μL, 0.17 mmol) in 1.0 mL of THF. Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 16a (10 mg, 17%). 1H NMR (300 MHz, C6D6) δ 7.80 (s, 1H), 5.05–5.13 (m, 2H), 4.79 (dt, J = 4.2, 8.7 Hz, 1H), 4.05 (quint, J = 4.5 Hz, 1H), 2.81 (ddd, J = 4.2, 4.9, 11.1 Hz, 1H), 1.12–1.71 (m, 39H), 0.83–0.92 (m, 12H). 13C NMR (75 MHz, C6D6) δ 172.6, 170.4, 160.7, 74.2, 72.6, 57.2, 50.1, 41.9, 39.4, 34.9, 32.7, 32.2, 30.54 (2), 30.52, 30.5, 30.4, 30.3, 30.2, 30.1, 29.7, 28.2, 27.4, 25.8, 25.3, 23.5, 23.31, 23.27, 22.0, 14.7, 14.6.
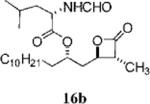
(S)-2-Formylamino-4-methyl-pentanoic Acid 1-((2S,3R,4R)-3-Methyl-4-oxo-oxetan-2-ylmethyl)-dodecyl Ester (16b)
Prepared according to the representative Mitsunobu reaction procedure using the mixture of β-lactones 13e and 14e (11.9 mg, 0. 042 mmol), triphenylphosphine (32.8 mg, 0.125 mmol), N-formylleucine (23.2 mg, 0.146 mmol), and DIAD (25 μL, 0.13 mmol) in 1.0 mL of THF. Purification by flash chromatography on SiO2 (3:2, hexanes: EtOAc) followed by a second purification by flash chromatography on SiO2 (0.4:1:3 THF:CHCl3:hexanes) gave the desired β-lactone as a mixture of diastereomers. The two diastereomers could be separated by MPLC (65:35, hexanes: EtOAc), affording the β-lactone diastereomer 16b (4.4 mg, 24%). 1H NMR minor diastereomer (16b) (300 MHz, CDCl3) δ 8.23 (s, 1H), δ 5.92 (d, J = 8.7 Hz, 1H), δ 5.08 – 4.98 (m, 1H), δ 4.71 (td, J = 8.4, 4.8, 1H), δ 4.27 – 4.19 (m, 1H), δ 3.29 (qd, J = 7.5, 3.9 Hz, 1H), δ 2.12 – 2.05 (m, 2H), δ 1.75 – 1.50 (m, 5H), δ 1.41 (d, J = 7.5 Hz, 3H), δ 1.26 (br s, 18H), δ 0.98 (dd, J = 6.3, 2.4 Hz, 6H), δ 0.89 (t, J = 6.6 Hz, 3H). HRMS (ESI) calcd for C24H43NO5 [M + Li], 432.3301; found, 432.3309.
Representative Procedure for Acylation as Described for (R)-1-((2S,3S)-3-Ethyl-4-oxooxetan-2-yl)tridecan-2-yl-2-methanamidoethanoate (17a)
A solution of hydroxy β-lactone 13d (11.0 mg, 0.0369 mmol), 1-ethyl-3-(3dimethylaminopropyl)-carbodiimide (9.20 mg, 0.048 mmol), and 4-dimethylaminopyridine (5.45 mg, 0.044 mmol) in 0.5 mL of xylene was azeotroped in vacuo for 1.0 h. The residue was dissolved in CH2Cl2 (1.0 mL), and N-formyl glycine (5.71 mg, 0.055 mmol) was added. After 12 h, the mixture was extracted with water (3 × 1 mL) and dichloromethane. The organic extract was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by chromatography (SiO2, 20% EtOAc:hexanes) to afford β-lactone 17a (17.4 mg, 99%). Rf = 0.22 (50% EtOAc/hexanes); [α]22d =−9.4 (c 0.4, CHCl3). IR (thin film) 2926, 2855, 1823, 1738, 1651. 1H NMR (500 MHz, CDCl3) δ 8.25 (br s, 1H), 6.22 (br s, 1H), 5.05–5.09 (m, 1H), 4.30–4.27 (m, 1H), 4.08 (d, J = 5.5 Hz, 2H), 3.21 (dq, J = 1.0, 4.5 Hz, 1H), 2.05–2.09 (m, 2H), 1.75–1.87 (m, 2H), 1.59–1.68 (m, 2H), 1.25–1.33 (m, 19H), 1.04 (app t, J = 7.0 Hz, 3H), 0.88 (app t, J = 6.5 Hz, 3H). 13C (125 MHz, CDCl3) δ 171.0, 169.4, 161.3, 74.1, 73.2, 58.3, 40.4, 39.1, 34.5, 32.2, 30.0 (2), 29.9, 29.8, 29.7, 29.6, 25.4, 23.0, 21.2 (2), 14.5, 11.4.
(R)-1-((2S,3S)-3-Butyl-4-oxooxetan-2-yl)tridecan-2-yl-2-methanamidoethanoate (17b)
Prepared according to the representative acylation procedure, hydroxy β-lactone 13c (8.5 mg, 0.026 mmol), 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide (6.5 mg, 0.034 mmol), 4-dimethylaminopyridine (3.8 mg, 0.031 mmol), and N-formyl glycine (4.0 mg, 0.039 mmol), purified twice by chromatography (SiO2, 40% EtOAc:hexanes) to afford β-lactone 17b (7.4 mg, 69%). Rf = 0.24 (40% EtOAc/hexanes); [α]22d = −19.5 (c 0.8, CHCl3). IR (thin film) 3386, 2926, 2855, 1825, 1745, 1681, 1519 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.27 (br s, 1H), 6.09 (br s 1H), 5.09 (app quint, J = 6.5 Hz, 1H), 4.26–4.30 (m, 1H), 4.13 (dd, J = 1.0, 7.0 Hz, 1H), 4.10 (d, J = 5.0 Hz, 2H), 3.22–3.26 (m, 1H), 2.07 (app t, J = 6.5 Hz, 1H), 2.05 (d, J = 1.00 Hz, 1H), 1.55–1.87 (m, 7H), 1.26–1.38 (m, 20H), 0.93 (app t, J = 7.5 Hz, 2H), 0.89 (app t, J = 7.5 Hz, 2H). 13C (125 MHz, CDCl3) δ 171.0, 169.3, 161.145, 74.5, 73.1, 56.9, 40.3, 39.0, 34.4, 32.1, 29.84, 29.83, 29.75, 29.67, 29.7, 29.53 29.2, 27.6, 25.3, 22.9, 22.6, 14.4, 14.0. HRMS (ESI) calcd for C23H41NO5 [M + Li], 418.3145; found, 418.3324.
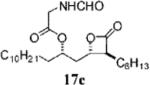
(2R,3S,4S)-Formylamino-acetic Acid 1-(3-Hexyl-4-oxo-oxetan-2-ylmethyl)-dodecyl Ester (17c)
Prepared according to the representative acylation procedure using β-lactone 13b (15.0 mg, 0.0423 mmol), N-formylglycin (4.8 mg, 0.047 mmol), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (10.5 mg, 0.0550 mmol), and 4-dimethylaminopyridine (5.2 mg, 0.042 mmol). Purification by flash chromatography on SiO2 (4:1, hexanes:EtOAc) gave β-lactone 17c (18 mg, 97%). 1H NMR (500 MHz, CDCl3) δ 8.28 (s, 1H), 6.15 (br, 1H), 5.06–5.11 (m, 1H), 4.27–4.30 (m, 1H), 4.03–4.14 (m, 2H), 3.22–3.26 (m, 1H), 2.07 (t, J = 3.9 Hz, 2H), 1.26–1.85 (m, 30H), 0.87–0.91 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 170.9, 169.3, 161.1, 74.4, 73.0, 56.9, 40.3, 39.0, 34.4, 32.1, 31.7, 29.8 (2), 29.7, 29.6, 29.54, 29.52, 29.1, 27.9, 27.0, 25.3, 22.9, 22.7, 14.3, 14.2. LRMS (ESI) calcd for C25H45NO5 [M + H], 440; found, 440.
(R)-1-((2S,3S)-3-Hexyl-4-oxooxetan-2-yl)propan-2-yl-2-methanamidoethanoate (17d)
Prepared according to the representative acylation procedure using hydroxy β-lactone 13i (10.9 mg, 0.051 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (12.6 mg, 0.066 mmol), 4-dimethylaminopyridine (7.5 mg, 0.061 mmol), and N-formyl glycine (7.9 mg, 0.077 mmol). Purification by chromatography (SiO2, 10 → 40% EtOAc:hexanes) afforded β-lactone 17d (7.9 mg, 52%). Rf = 0.11 (40% EtOAc/hexanes); [α]22D = −30.9 (c 0.60, CHCl3). IR (thin film) 3376, 2931, 1820, 1747, 1685 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.23 (br s, 1H), 6.07 (brs, 1H), 5.10–5.17 (m, 1H), 4.30 (app quint, J = 4.0 Hz, 1H), 4.09 (dd, J = 0.5, 5.0 Hz, 1H), 3.25 (ddd, J = 1.5, 4.0, 6.5 Hz, 1H), 2.10–2.15 (m, 1H), 2.01–2.06 (m, 1H), 1.80–1.87 (m, 1H), 1.70–1.77 (m, 1H), 1.30–1.47 (m, 14H), 0.89 (app t, J = 3.5 Hz, 3H). 13C (125 MHz, CDCl3) δ 170.9, 169.1, 161.2, 74.3, 59.8, 57.0, 40.8, 40.3, 31.7, 29.1, 27.9, 27.0 22.7, 20.5, 14.2. HRMS (ESI) calcd for C16H27NO5 [M + Li], 306.1893; found, 306.1987.
(S)-1-((2S,3S)-3-Hexyl-4-oxooxetan-2-yl)-3-(tosyloxy)propan-2-yl-2-methanamidoethanoate (17e)
Prepared according to the representative acylation procedure using hydroxy β-lactone 13j (13.0 mg, 0.034 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (8.50 mg, 0.044 mmol), 4-dimethylaminopyridine (5.0 mg, 0.041 mmol), and N-formyl glycine (5.26 mg, 0.051 mmol). Purification by chromatography (SiO2, 20% EtOAc:hexanes) afforded β-lactone 17e (9.2 mg, 58%). Rf = 0.22 (40% EtOAc/hexanes); [α]D22=−6.5 (c 0.4, CHCl3). IR (thin film) 2955, 2925, 2855, 1738, 1607, 1516, 1466. 1H NMR (125 MHz, CDCl3) δ 8.27 (br s, 1H), 7.77 (d, J = 7 Hz, 2H), 7.38 (d, J = 8 Hz, 2H), 6.07 (br s, 1H), 5.14–5.19 (m, 1H), 4.29 (app t, J = 12 Hz, 1H), 4.05–4.16 (m, 4H), 3.23–3.27 (m, 1H), 2.47 (s, 3H), 2.26–2.31 (m, 2H), 1.99–2.05 (m, 2H), 1.72–1.86 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 170.3, 168.3, 161.2, 145.8, 132.7, 130.3 (2), 128.12 (2), 73.7, 69.7, 57.2, 40.1, 35.6, 31.6, 29.1, 27.8, 27.0, 22.7, 21.9, 14.3. HRMS (ESI) calcd for C22H31NO8S, 476.1930; found, 476.1955.
(R)-1-((2S,3S)-3-Hexyl-4-oxooxetan-2-yl)hex-5-en-2-yl-2-methanamidoethanoate (17f)
Prepared according to the representative acylation procedure using hydroxy β-lactone 13g (15.0 mg, 0.059 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (14.8 mg, 0.077 mmol), 4-dimethylaminopyridine (8.71 mg, 0.071 mmol), and N-formyl glycine (9.12 mg, 0.089 mmol). Purification by chromatography (SiO2,10 → 40% EtOAc:hexanes) afforded β-lactone 17f (10.8 mg, 55%). Rf = 0.33 (40% EtOAc/hexanes); [α]22D = −5.1 (c 1.8, CHCl3). IR (thin film) 3418, 2925, 2854, 1819, 1740, 1667 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.27 (br s, 1H), 6.10 (br s, 1H), 5.73–5.81 (m, 1H), 5.07–5.14 (m, 1H), 5.02 (dt, J = 1.5, 10.5 Hz, 2H), 4.27–4.30 (m, 1H), 4.09 (dt, J = 0.5, 5.0 Hz, 2H), 3.21–3.25 (m, 1H), 2.09 (app t, J = 7.0 Hz, 4H), 1.64–1.85 (m, 4H), 1.24–1.45 (m, 8H), 0.89 (app t, J = 7.0 Hz, 3H). 13C (125 MHz, CDCl3) δ 170.9, 169.3, 161.1, 137.0, 116.0, 74.4, 72.5, 57.0, 40.3, 39.1, 33.5, 31.7, 29.5, 29.2, 27.9, 27.0, 22.7, 14.3. HRMS (ESI) calcd for C18H29NO5 [M + Li], 346.2206; found, 346.2276.
(R)-1-((2S,3S)-3-Hexyl-4-oxooxetan-2-yl)hept-5-en-2-yl-2-methanamidoethanoate (17g)
Prepared according to the representative acylation procedure using hydroxy β-lactone 13h (15.6 mg, 0.058 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (14.4 mg, 0.075 mmol), 4-dimethylaminopyridine (8.7 mg, 0.070 mmol), and N-formyl glycine (9.0 mg, 0.087 mmol). Purification by chromatography (SiO2, 10→40% EtOAc:hexanes) afforded β-lactone 17g (11.7 mg, 57%). Rf = 0.38 (40% EtOAc/hexanes); [α]22D =−22 (c 1.1, CHCl3). IR (thin film) 2926, 2855, 1823, 1712 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.27 (br s, 1H), 6.11 (br s, 1H), 5.42–5.48 (m, 1H), 5.34–5.39 (m, 1H), 5.10 (app quint, J = 5.0 Hz, 1H), 4.26–4.29 (m, 1H), 4.09 (d, J = 5.5 Hz, 2H), 3.22–3.60 (m, 1H), 1.99–2.10 (m, 4H), 1.64–1.85 (m, 4H), 1.25–1.45 (m, 8H), 0.89 (app t, J = 9.0 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 170.9, 169.3, 161.1, 129.5, 126.6, 74.4, 72.5, 56.9, 40.3, 39.0, 34.2, 31.7, 29.2, 28.4, 27.0, 22.7, 18.1, 14.3. HRMS (ESI) calcd for C19H31NO5 [M + Li], 360.2362; found, 360.2367.
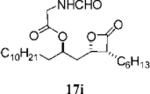
2-Formylamino-3-methyl-butyric Acid (2S,2R,3S,4S)-1-(3-Hexyl-4-oxo-oxetan-2-ylmethyl)-hex-4-enyl Ester (17h)
Prepared according to the representative acylation procedure using β-lactone 13h (20.0 mg, 0.0745 mmol), N-formyl-l-valine (11.9 mg, 0.0820 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (18.6 mg, 0.0969 mmol), and 4-dimethylaminopyridine (9.2 mg, 0.0745 mmol). Purification by flash chromatography on SiO2 (0.4:1:3 THF/CHCl3/hexane) gave β-lactone 17h as a E/Z-mixture; only peaks for major olefin isomer are assigned. 1H NMR (300 MHz, CDCl3) δ 8.28 (s, 1H), 6.08 (br, 1H), 5.34–5.50 (m, 2H), 5.02–5.10 (m, 1H), 4.63–4.68 (m, 1H), 4.28 (dt, J = 3.9, 8.1 Hz, 1H), 3.20–3.28 (m, 1H), 2.18–2.26 (m, 1H), 1.30–2.08 (m, 19H), 1.01 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 171.3 (2), 161.1, 129.4, 126.6, 74.4, 72.3, 57.0, 55.9, 39.2, 34.3, 31.7, 31.4, 29.1, 28.3, 27.8, 27.0, 22.7, 19.4, 18.1, 17.6, 14.2.
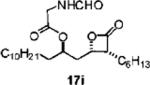
Mukaiyama Aldol-Lactonization Process using SnCl4 as Described for (R)-1-((2S,3R)-3-Hexyl-4-oxooxetan-2-yl)tridecan-2-yl-2-methanamidoethanoate (17i)
To a solution of aldehyde 8f (0.189 g, 0.55 mmol) and thiopyridyl ketene acetal 3d (0.21 g, 1.10 mmol) in CH2Cl2 (4 mL) at −78 °C was added SnCl4 (0.66 mL, 0.66 mmol, 1.0 M in CH2Cl2) via syringe pump over 2 h. After 10 h, the reaction was warmed to −50 °C for additional 6 h. Next, the reaction was quenched with pH 7 buffer (10 mL) at −50 °C and warmed to 23 °C with vigorous stirring for 20 min, and the resulting mixture was filtered through a small pad of celite. After the filtrate was dried over Na2SO4 and filtered, the volume of CH2Cl2 was adjusted to a final concentration of 0.15 M. CuBr2 (0.26 g, 1.10 mmol) was added, and the mixture was stirred for 2 h. The resulting suspension was filtered through celite and washed with 10% aqueous K2CO3 (2 × 10 mL) solution and brine (2 × 20 mL). The organic phase was separated and dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by chromatography (SiO2, 30% EtOAc in hexanes) to afford a mixture of cis-β-lactone diastereomers, which was taken on directly to desilylation (dr = 3:1).
Representatitve Procedure for Desilylation of Crude Silyloxy-β-Lactones from the Aldol-Lactonization Process
To a solution of the diastereoisomeric mixture of silyloxy-β-lactones (obtained from the TMAL process) in dry CH3CN (20 mL) at 0 °C was added 48% aqueous HF (0.43 mL). The reaction was stirred at 0 °C for 2 h and warmed to room temperature. After stirring for additional 2 h, the reaction mixture was diluted with 15 mL of ether. The organic layer was separated, washed with a saturated solution of NaHCO3 (2 × 20 mL) with caution, followed with brine (2 × 20 mL). The residue was dried over Na2SO4, filtered, and concentrated in vacuo. The mixture was purified by flash chromatography (2–10% EtOAc in hexanes). The diastereomers were partially separated via MPLC on SiO2 (10% EtOAc in hexanes). Only 12 mg of the major diastereoisomer cis-13j (dr >19:1) was separated completely.
β-Lactone 17i
Acylation was performed according to the representative procedure using hydroxy β-lactone cis-13 (12.0 mg, 0.032 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (7.8 mg, 0.041 mmol), 4-dimethylamino-pyridine (54.7 mg, 0.038 mmol), and N-formyl glycine (5.0 mg, 0.048 mmol). Purification by chromatography (SiO2, 20% EtOAc:hexanes) to afford β-lactone 17i (7.2 mg, 51%). Rf = 0.32 (40% EtOAc/hexanes); [α]22D = −8.8 (c 0.45, CHCl3). IR (thin film) 2925, 2854, 1823, 1748, 1824, 1748 1654 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.27 (br s, 1H), 6.09 (br s, 1H), 5.11–5.15 (m, 1H), 4.58–4.67 (m, 1H), 4.12–4.13 (m, 2H), 3.67 (app d, J = 7.0 Hz, 1H), 1.90–2.05 (m, 2H), 1.70–1.80 (m, 8H), 1.20–1.40 (m, 25H), 0.99 (m, 7H). 13C NMR (125 MHz, CDCl3) δ171.6, 169.3, 161.2, 73.1, 71.8, 53.2, 40.2, 34.9, 34.5, 32.1, 31.7, 29.9, 29.85 (2), 29.83, 29.77, 29.7, 29.57, 29.56, 29.2, 27.6, 25.3, 24.3, 22.9, 22.8, 14.4, 14.3. LRMS (ESI) calcd for C27H49NO5 [M + Li], 446.68; found, 446.35.
Representative Cross-Metathesis Procedure as Described for 2-Formylamino-4-methyl-pentanoic Acid 1-(3-Hexyl-4-oxooxetan-2-ylmethyl)-dodec-4-enyl Ester (21a)
To a solution of β-lactone 15j (23.5 mg, 0.0594 mmol) and n-nonene (51 μL, 0.297 mmol) in CH2Cl2 (0.2 mL) at rt was added a solution of Grubb's catalyst (4.9 mg, 0.00594 mmol) in CH2Cl2 (0.1 mL). The mixture was heated to 45 °C for 24 h and directly purified using flash chromatography (4:1, hexanes:EtOAc) gave β-lactone 21a (yield not determined). 1H NMR (500 MHz, CDCl3) δ 8.23 (s, 1H), 5.90 (d, J = 8.5 Hz, 1H), 5.31–5.47 (m, 2H), 5.04–5.08 (m, 1H), 4.70 (dt, J = 4.0, 8.5 Hz, 1H), 4.30 (quint, J = 4.5 Hz, 1H), 3.20–3.25 (m, 1H), 1.95–2.20 (m, 6H), 1.57–1.82 (m, 8H), 1.25–1.46 (m, 17H), 0.87–0.99 (m, 12H). HRMS (ESI) calcd for C29H51NO5 [M + Li], 500.3927; found, 500.3734.
2-Formylamino-3-phenyl-propionic Acid 1-(3-Hexyl-4-oxooxetan-2-ylmethyl)-dodec-4-enyl Ester (21b)
Prepared according to the representative cross-metathesis procedure using β-lactone 15i (10 mg, 0.023 mmol), n-nonene (20 μL, 0.12 mmol), and Grubb's catalyst (3.8 mg, 0.0047 mmol). Purification by flash chromatography (10:1, hexanes:EtOAc) gave desired β-lactone 21b (5.5 mg, 45%) (3.8 mg, 38%) as a E/Z-mixture; only peaks for major olefin isomer are assigned. 1H NMR (300 MHz, CDCl3) δ 8.20 (s, 1H), 7.20–7.30 (m, 5H), 6.02 (d, J = 7.2 Hz, 1H), 5.30–5.50 (m, 2H), 5.00–5.08 (m, 1H), 4.94 (q, J = 6.9 Hz, 1H), 4.17–4.23 (m, 1H), 3.13–3.21 (m, 4H), 1.30–2.12 (m, 27H), 0.89–0.93 (m, 6H). HRMS (ESI) calcd for C32H49NO5 [M + Li], 534.3771; found, 534.3659.
2-Formylamino-4-methyl-pentanoic Acid 5-(4-Fluoro-phenyl)-1-(3-hexyl-4-oxo-oxetan-2-ylmethyl)-pent-4-enyl Ester (21c)
Prepared according to the cross-metathesis procedure using β-lactone 15j (15.0 mg, 0.0379 mmol), Grubb's catalyst (3.1 mg, 0.0038 mmol), and 4-fluorostyrene (18 μL, 0.15 mmol). Purification by flash chromatography (4:1, hexanes:EtOAc) gave β-lactone 21c (yield not determined). 1H NMR (300 MHz, CDCl3) δ 8.15 (s, 1H), 7.30–7.34 (m, 2H), 6.98–17.04 (m, 2H), 6.40 (d, J = 15.6 Hz, 1H), 6.10 (dt, J = 6.6, 15.6 Hz, 1H), 5.90 (d, J = 8.1 Hz, 1H), 5.10–5.20 (m, 1H), 4.72 (dt, J = 5.1, 9.3 Hz, 1H), 4.30–4.36 (m, 1H), 3.22–3.29 (m, 1H), 1.25–2.70 (m, 19H), 0.89–1.01 (m, 9H). HRMS (ESI) calcd for C23H40FNO5 [M + Na], 512.2788; found, 512.2828.
(R)-1-((2S,3S)-3-Hexyl-4-oxooxetan-2-yl)hexan-2-yl-2-methanamidoethanoate (23b)
β-lactone 17f (6.2 mg, 0.018 mmol) and 5 wt % palladium on carbon (5 mg) in 2 mL of CH2Cl2 was stirred at ambient temperature for 12 h under H2 atmosphere. Filtration and chromatography (SiO2, 40% EtOAc:hexanes) gave the desired β-lactone 23b (5.8 mg, 95%). Rf = 0.24 (40% EtOH/hexanes); [α]22D = −28.9 (c 0.8, CHCl3). IR (thin film) 2927, 2858, 1819, 1745, 1675 cm−1. 1H NMR (500 MHz, CDCl3) δ 8.27 (br s, 1H), 6.09 (br s, 1H), 5.06–5.12 (m, 1H), 4.27–4.30 (m, 1H), 4.10 (dd, J = 2.0, 5.0 Hz, 2H), 3.22–3.27 (m, 1H), 2.07 (dt, J = 1.5, 6.5 Hz, 2H), 1.53–1.87 (m, 8H), 1.23–1.47 (m, 10H), 0.90 (dd, J = 7.0, 15.0 Hz, 4H). 13C NMR (125 MHz, CDCl3) δ 171.0, 169.3, 161.2, 74.5, 73.0, 56.9, 40.3, 39.0, 34.1, 31.7, 29.2, 27.9, 27.4, 27.0, 22.7, 22.6, 14.3, 14.1. HRMS (ESI) calcd for C18H31NO5 [M + Li], 348. 2362; found, 348.2311.
Fluorogenic Assay for Detection of FASTE Inhibition
The synthetic fluorogenic substrate, 4-methylumbelliferyl heptanoate (4-MUH), was purchased from Sigma (St. Louis, MO). The reaction mixture consisted of 500 nM FASTE in buffer (100 mM Tris-HCl, 50 mM NaCl at pH 7.4), which was preincubated with 2.5 μL of test compounds dissolved in DMSO at final concentrations of 0.32–100 μM and/or 0.08–10 μM at 37 °C for 30 min. The reaction was initiated by addition of 5 μL of 1.25 mM 4-MUH in 1:1 DMSO:buffer. The resulting fluorescence from liberated 4-methylumbelliferone was measured every 5 min at 350/450 nm for 40–60 min. 4-MUH incubated without enzyme served as a background control. Results are the average of triplicate data points.
Cell Viability Assays for Determination of Test Compound Potencies
MB-MDA-231 or human fibroblast cells were plated in 96-well plates in appropriate media and incubated overnight at 37 °C and 5% CO2. Cells were treated with test compounds (0.1–100 μM) or vehicle in triplicate, with a final percentage of DMSO not exceeding 1% (v/v). At 48 h, the medium was aspirated and replaced with complete MEM, containing 333 μg/mL [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and 25 μM phenazine methosulfate (PMS), using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega). Plates were incubated for 2 h and absorbance was assayed at 490 nm. Background levels of formazan formation were measured in medium alone. IC50 values were derived from dose–response curves.
Cell Lines
The MDA-MB-231 cell line and Hs 58.Fs cells were purchased from American Type Culture Collection (Manassas, VA). MDA-MB-231 tumor cells were maintained in minimum essential medium Eagle (MEM) with Earl's Salts (Mediatech, Inc., Herndon, VA) supplemented with 10% fetal bovine serum (FBS; Irvine Scientific, Santa Ana, CA), 2 mM L-glutamine (Invitrogen Life Technologies, Inc., Carlsbad, CA), MEM vitamins (Invitrogen Life Technologies, Inc.), nonessential amino acids (Mediatech, Inc.), and antibiotics (Omega Scientific, Inc., Tarzana, CA). Human fibroblast (Hs 58.Fs) cells were maintained in Dulbecco's modified Eagle's medium (Irvine Scientific) supplemented with 10% FBS and antibiotics. All cells were grown at 37 °C under a humidified, 5% CO2 atmosphere.
RNA Interference
siRNA was purchased from Dharmacon (Lafayette, CO). Cells were plated in 96-well plates and grown for 48 h prior to transfection with 100 nM FAS SMARTpool (M-003954-02) or nonsilencing control (D-001206-13) siRNA using LipofectAMINE 2000 reagent (Invitrogen) as previously described.24
Inhibition of Fatty Acid Synthase with Orlistat
Orlistat (Xenical) was extracted from capsules (Roche Applied Science, Indianapolis, IN) as previously described.24 Cells were plated in 96-well plates for 48 h, followed by treatment with 25 μM orlistat or vehicle control for 72 h.
DNA Fragmentation Assay
Apoptosis was monitored by measuring the level of DNA fragments present following 72 h exposure to FAS siRNA or orlistat using the Cell Death Detection ELISAplus kit (Roche) according to manufacturer instructions.
Supplementary Material
Acknowledgment
We thank the NIH (NCI CA 10658, D.R. and J.W.S.; CA81713, J.W.S.) and the Welch Foundation (D.R.) for support of these investigations. We thank Dr. Wei Zhang for assistance with manuscript preparation and Ms. Li Yang for technical assistance with purity chromatograms. Helpful conversations with Dr. Adam Richardson and Dr. Russell Dahl are gratefully acknowledged.
Footnotes
Supporting Information Available: Purity data for synthesized compounds is available online. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations FAS, fatty acid synthase; TE, thioesterase; MUH, methylumbelliferyl heptanoate; TMAL, tandem Mukaiyama aldol-lactonization.
References
- (1).Alo PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996;77:474–482. doi: 10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- (2).Nakamura I, Kimijima I, Zhang GJ, Onogi H, Endo Y, Suzuki S, Tuchiya A, Takenoshita S, Kusakabe T, Suzuki T. Fatty acid synthase expression in Japanese breast carcinoma patients. Int. J. Mol. Med. 1999;4:381–387. doi: 10.3892/ijmm.4.4.381. [DOI] [PubMed] [Google Scholar]
- (3).Wang YY, Kuhajda FP, Li J, Finch TT, Cheng P, Koh C, Li T, Sokoll LJ, Chan DW. Fatty acid synthase as a tumor marker: Its extracellular expression in human breast cancer. J. Exp. Ther. Oncol. 2004;4:101–110. [PubMed] [Google Scholar]
- (4).Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, Heyns W, Verhoeven G. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int. J. Cancer. 2002;98:19–22. doi: 10.1002/ijc.10127. [DOI] [PubMed] [Google Scholar]
- (5).Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S, Loda M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol. Cancer. Res. 2003;1:707–715. [PubMed] [Google Scholar]
- (6).Pizer ES, Wood FD, Heine HS, Romantsev FE, Pasternack GR, Kuhajda FP. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res. 1996;56:1189–93. [PubMed] [Google Scholar]
- (7).Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer. 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- (8).Kridel SJ, Lowther WT, Pemble C. W. t. Fatty acid synthase inhibitors: new directions for oncology. Expert Opin. Invest. Drugs. 2007;16:1817–29. doi: 10.1517/13543784.16.11.1817. [DOI] [PubMed] [Google Scholar]
- (9).Guerciolini R. Mode of action of orlistat. Int. J. Obes. Relat. Metab. Disord. 1997;21(Suppl 3):S12–23. [PubMed] [Google Scholar]
- (10).Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64:2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- (11).Pemble C. W. t., Johnson LC, Kridel SJ, Lowther WT. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat. Struct. Mol. Biol. 2007;14:704–9. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- (12).Yang HW, Romo D. A Highly Diastereoselective, Tandem Mukaiyama Aldol-Lactonization Route to beta-Lactones: Application to a Concise Synthesis of the Potent Pancreatic Lipase Inhibitor, (−)-Panclicin D. J. Org. Chem. 1997;62:4–5. doi: 10.1021/jo9619488. [DOI] [PubMed] [Google Scholar]
- (13).Yang HW, Zhao CX, Romo D. Studies of the Tandem Mukaiyama Aldollactonization (TMAL) Reaction: A Concise and Highly Diastereoselective Route to B-Lactones Applied to the Total Synthesis of the Potent Pancreatic Lipase Inhibitor, (−)-Panclicin D. Tetrahedron. 1997;53:16471–16488. [Google Scholar]
- (14).Evans DA, Dart MJ, Duffy JL, Yang MG. A Stereochemical Model for Merged 1,2- and 1,3-Asymmetric Induction in Diastereoselective Mukaiyama Aldol Addition Reactions and Related Processes. J. Am. Chem. Soc. 1996;118:4322–4343. [Google Scholar]
- (15).Wang Y, Zhao C, Romo D. A Stereocomplementary Approach to β-Lactones: Highly Diastereoselective Synthesis of cis- β-Lactones, a β-Chloroacid, and a Tetrahydrofuran. Org. Lett. 1999;1:1197–1199. [Google Scholar]
- (16).Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. A general model for selectivity in olefin cross metathesis. J. Am. Chem. Soc. 2003;125:11360–70. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- (17).Chirala SS, Wakil SJ. Structure and function of animal fatty acid synthase. Lipids. 2004;39:1045–1053. doi: 10.1007/s11745-004-1329-9. [DOI] [PubMed] [Google Scholar]
- (18).Purohit VC, Richardson RD, Smith JW, Romo D. Practical, catalytic, asymmetric synthesis of beta-lactones via a sequential ketene dimerization/hydrogenation process: inhibitors of the thioesterase domain of fatty acid synthase. J. Org. Chem. 2006;71:4549–58. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- (19).Richardson RD, Smith JW. Novel antagonists of the thioesterase domain of human fatty acid synthase. Mol. Cancer Ther. 2007;6:2120–6. doi: 10.1158/1535-7163.MCT-07-0187. [DOI] [PubMed] [Google Scholar]
- (20).Umezawa H, Aoyagi T, Uotani K, Hamada M, Takeuchi T, Takahashi S. Ebelactone, an inhibitor of esterase, produced by actinomycetes. J. Antibiot. (Tokyo) 1980;33:1594–6. doi: 10.7164/antibiotics.33.1594. [DOI] [PubMed] [Google Scholar]
- (21).Chakravarty B, Gu Z, Chirala SS, Wakil SJ, Quiocho FA. Human fatty acid synthase: structure and substrate selectivity of the thioesterase domain. Proc. Natl. Acad. Sci. U.S.A. 2004;101:15567–15572. doi: 10.1073/pnas.0406901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bertrand R, Solary E, O'Connor P, Kohn KW, Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp. Cell Res. 1994;211:314–21. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- (23).Ma G, Zancanella M, Oyola Y, Richardson RD, Smith JW, Romo D. Total synthesis and comparative analysis of orlistat, valilactone, and a transposed orlistat derivative: Inhibitors of fatty acid synthase. Org. Lett. 2006;8:4497–500. doi: 10.1021/ol061651o. [DOI] [PubMed] [Google Scholar]
- (24).Knowles LM, Axelrod F, Browne CD, Smith JW. A fatty acid synthase blockade induces tumor cell-cycle arrest by down-regulating Skp2. J. Biol. Chem. 2004;279:30540–30545. doi: 10.1074/jbc.M405061200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.