

Abstract
Combinatorial regulation of transcription implies flexible yet precise assembly of multiprotein regulatory complexes in response to signals. Biochemical and crystallographic analyses revealed that hormone binding leads to the formation of a hydrophobic groove within the ligand binding domain (LBD) of the thyroid hormone receptor that interacts with an LxxLL motif-containing α-helix from GRIP1, a coactivator. Residues immediately adjacent to the motif modulate the affinity of the interaction; the motif and the adjacent sequences are employed to different extents in binding to different receptors. Such interactions of amphipathic α-helices with hydrophobic grooves define protein interfaces in other regulatory complexes as well. We suggest that these common structural elements impart flexibility to combinatorial regulation, whereas side chains at the interface impart specificity.
Keywords: Nuclear receptors, coactivators, GRIP1, specificity, interaction site
Transcriptional regulatory factors integrate input from multiple signals, producing distinct regulatory patterns in different signaling contexts. Regulators typically function in multiprotein regulatory complexes, and the signals evoke structural changes that facilitate or preclude particular protein–protein interactions. By this view, functional regulatory complexes would be structurally dynamic, their composition governed by the specific DNA-binding sites arrayed within a given genomic response element, by the particular mixture of regulatory factors produced in a given cell type, and by the physiologic status of that cell as represented by the activities of signaling networks that influence regulatory factor activities (Yamamoto et al. 1992). Such a ‘mixed assembly’ model for combinatorial regulation implies that DNA-binding regulatory factors and their various coactivators and corepressors must interact rather flexibly to enable assembly into multiple final complexes yet also quite specifically to ensure precise assembly into the appropriate complex. How are these conditions satisfied at the molecular level? We have approached this question by studying nuclear receptors (NRs), a class of regulatory factors that binds to signals, to response elements, and to protein cofactors.
The ligand binding domains (LBDs) of NRs are signal responsive regulatory modules, adopting distinct conformations as aporeceptors, agonist-bound or antagonist-bound species, that support or preclude interactions with proteins such as chaperones, corepressors, or coactivators (Moras and Gronemeyer 1998). Within their normal context of intact receptors, LBDs can control virtually all receptor activities, including nuclear localization and DNA binding (Yamamoto et al. 1988), and even as isolated domains, LBDs remain functional as compact ‘molecular switches.’ Structural analyses of the LBDs from receptors for retinoids (RAR and RXR) (Bourguet et al. 1995; Renaud et al. 1995), thyroid hormone (TR) (Wagner et al. 1995), estrogen (ER) (Brzozowski et al. 1997), and progesterone (PR) (Williams and Sigler 1998) revealed a common overall fold for the domain, despite substantial sequence divergence (Wurtz et al. 1996). That ligand binding induces conformational changes, affecting especially the position of an α-helix at or near the LBD carboxyl terminus, can be inferred from comparisons of structures of the unliganded RXR with liganded RAR and TR LBDs, and of the agonist- and antagonist-bound ER LBD. This helix, usually denoted helix 12, is an essential component of a ligand-dependent transcriptional activation function, AF-2, within the LBD (Danielian et al. 1992; Barettino et al. 1994; Durand et al. 1994; Tone et al. 1994; Jurutka et al. 1997).
Agonist-bound LBDs can bind coactivators from the p160 family, which includes at least three distinct members, SRC-1 [Oñate et al. 1995; also NcoA-1 (Kamei et al. 1996)], p/CIP [Torchia et al. 1997; also AIB1 (Anzick et al. 1997), TRAM-1 (Takeshita et al. 1997), RAC3 (Li et al. 1997), ACTR (Chen et al. 1997)], and TIF2 [Voegel et al. 1996; also GRIP1 (Hong et al. 1996, 1997), NcoA-2 (Torchia et al. 1997)] (for review, see Moras and Gronemeyer 1998; Glass et al. 1997). These ∼160-kD proteins include amino-terminal bHLH and PAS domains, domains for interaction with nuclear receptors (NR interaction domain, NID) and CREB-binding protein (CBP), and carboxy-terminal activation domains (Fig. 1a) (Ding et al. 1998; Kalkhoven et al. 1998; Voegel et al. 1998). In p160 coactivators, and in unrelated coactivators such as TIF1 or RIP140, interaction with NRs is mediated by multiple LxxLL motifs, each residing within distinct patches of conserved sequence termed NR boxes (Fig. 1b,c) (Le Douarin et al. 1996; Heery et al. 1997; Torchia et al. 1997; Ding et al. 1998; Voegel et al. 1998). Particular NRs show overlapping but distinct preferences for individual NR boxes (Torchia et al. 1997; Ding et al. 1998; Voegel et al. 1998).
Figure 1.
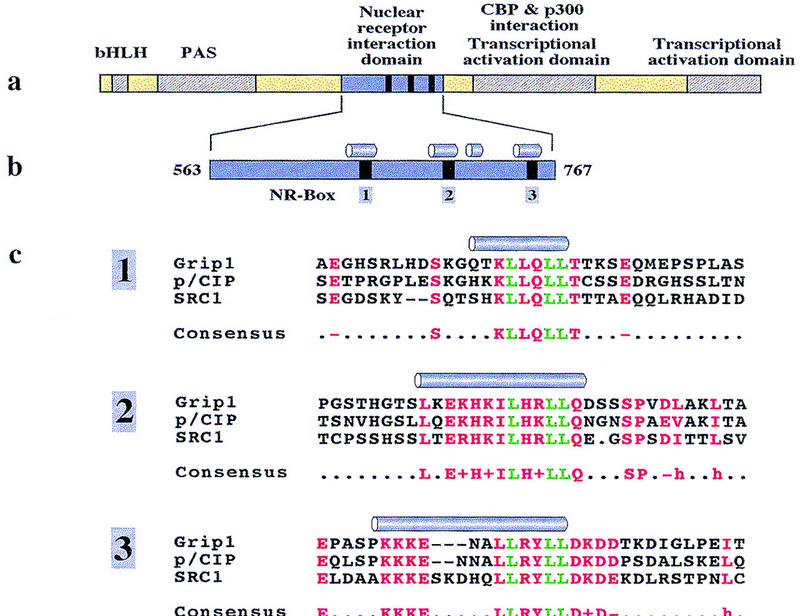
(a) Functional domains of p160 family coactivators (Ding et al. 1998; Voegel et al. 1998). (b) The NID of GRIP1 (563–767) contains four predicted α-helices (Rost and Sander 1994); three of them include the conserved LxxLL motifs of NR-boxes 1, 2, and 3. (c) Sequence alignment of LxxLL motifs in members of the p160 coactivator family. Motif leucine residues are green; box-specific conserved residues are red. GRIP1 (Hong et al. 1996, 1997), pCIP (Torchia et al. 1997), and SRC-1 (Oñate et al. 1995) were taken as representatives for the three distinct classes of coactivators in the p160 family. Predicted α-helices are shown for each box.
We sought to define the structural and molecular basis for the interaction of NRs with the LxxLL motif of p160 coactivators, and to identify the determinants of specificity of these interactions. To this end we studied the interaction of the p160 coactivator GRIP1 with TR or with the glucocorticoid receptor (GR) in vitro, and initiated a structural analysis of the TRβ LBD in complex with a peptide containing NR-box 2 of GRIP1.
Results
Affinity of the GRIP1–TRβ interaction
We purified a 205-amino acid fragment of GRIP1 (amino acids 563–767), denoted NID, that contained three NR boxes (1–3) (Fig. 1b) and was readily soluble. Secondary structure prediction (Rost and Sander 1994) placed these NR boxes within separate α-helical regions, in each case close to the carboxyl terminus of the helix.
To measure the interaction between TRβ and GRIP1 in vitro, fusions bearing glutathione S-transferase (GST) and NID or NID derivatives bearing mutant NR boxes were used to monitor interactions with 35S-labeled TRβ or TRβ LBD by retention on glutathione agarose; full-length TRβ or TRβ LBD bound the NID of GRIP1 with equal affinity and specificity (data not shown). Purification of the GST–NID fusions to near homogeneity enabled quantitative comparisons of interactions between different GRIP1 derivatives and receptors; each assay was performed with known concentrations of the GST–NID and receptor derivatives.
The TRβ LBD:NID interaction was strongly hormone-dependent (Fig. 2a). In a qualitative assay carried out under standard conditions (see legend to Fig. 2; Materials and Methods), substitution of the bulky hydrophobic residues of NR-box 3 by alanine (NID3−) did not reduce binding of TRβ LBD significantly, whereas similar substitutions in NR-box 2 (NID2−) resulted in a 50% loss of TRβ LBD binding. In the absence of functional NR-boxes 2 and 3 (NID2−3−), no ligand-dependent binding to TRβ LBD was detected under our experimental conditions, suggesting that multiple binding sites are required or that NR-box 1 is not a binding site for TRβ LBD. The latter possibility was strongly supported by subsequent peptide competition assays (see below). The observed preference of TRβ LBD for NR-box 2 is consistent with previous results from yeast two-hybrid and mammalian transfection assays (Ding et al. 1998). Thus, our in vitro assay reflects the selectivity of TR:GRIP1 interactions observed in vivo.
Figure 2.

(a) GRIP1 NR-boxes 1, 2, and 3 interact differentially with TRβ LBD. (GST) An isolated GST domain; (NID) GST fusion of the GRIP1 fragment (563–767)His6 that contains NR-boxes 1, 2, and 3 (shaded bars). In NID2− (open bar) or NID3− (solid bar) the bulky hydrophobic residues of NR-box 2 (ILHRLL) or NR-box 3 (LLRYLL) were replaced by alanine yielding AAHRAA and AARAAA, respectively. NID2−3− (hatched bar) contains replacement of both NR-boxes 2 and 3. Assays were carried out using 10 nm labeled TRβ LBD and 1.6 μm purified, glutathione–agarose bound GST–NID proteins either in the absence (−) or presence (+) of 10 μm T3. The yield of bound receptor is given as percentage relative to the input. The data show the average of more than three independent experiments together with the standard deviation. (b) NR-box 2 interacts with TRβ LBD with a fourfold higher affinity than NR-box 3. Labeled TRβ LBD (10 nm) was incubated in the presence of 10 μm T3 and various concentrations of purified and glutathione–agarose-bound GST–NID (shaded diamond) and GST–NID2− (open square) or GST–NID3− (solid square) lacking either a functional NR-box 2 or 3, respectively. As in a, the amount of bound receptor is relative to the receptor input. The data represent the average and range of at least two independent experiments. (c) Peptides containing NR boxes compete the interaction of TRβ LBD with the NID. Labeled TRβ LBD (10 nm) was incubated with 1.6 μm glutathione–agarose-bound GST–NID3− in the presence of 10 μm T3 and increasing concentrations of NR-box 2 peptides EKHKILHRLLQDS (solid circle) or TSLKEKHKILHRLLQDSS (solid triangle), or NR-box 3 peptides LLRYLL (open square), KENALLRYLLDKDD (open circle) or PKKKENALLRYLLDKDDTKD (open triangle). Bound receptor is shown relative to the amount retained in the absence of peptide. The data and calculated IC50 values represent the average and standard deviation of three independent experiments.
To measure the affinities of the interactions of NID, NID2−, and NID3− with the TRβ LBD and to test for cooperativity between NR-box 2 and 3, we titrated the levels of NID (NR-box 2 and 3 functional), NID3− (NR-box 2 functional), and NID2− (NR-box 3 functional) (Fig. 2b). The results demonstrated that the binding of the TRβ LBD to NR-box 2 of NID3− (EC50 = 0.9 ± 0.2 μm) was three- to fourfold stronger than to NR-box 3 of NID2− (EC50 = 3.2 ± 0.9 μm) and that binding to NR-box 2 and 3 was noncooperative.
Peptide competition of NR-box TRβ LBD interaction
To begin to define the source of the specificity of the interactions seen, we compared the ability of peptides containing individual NR boxes and different lengths of adjacent sequences to compete the interaction of NID3− with TRβ LBD. A peptide containing NR-box 1 competed the interaction of GRIP1 with TRβ LBD only at very high concentrations (EC50 = 130 μm; data not shown), consistent with our failure to observe an interaction between TRβ LBD and NR-box 1 in the context of the NID in the qualitative assay above (Fig. 2a). The central hexapeptide of NR-box 3, LLRYLL, competed the interaction of NID3− with TRβ LBD with very low efficiency, demonstrating that an isolated motif is not sufficient for binding to the receptor (Fig. 2c). Extending this hexapeptide to the 14-mer KENALLRYLLDKDD decreased the IC50 for competition by >60-fold, from >600 μm to 9.0 ± 2.0 μm; further extension of the peptide to PKKKENALLRYLLDKDDTKD decreased the IC50 only threefold farther, to 2.9 ± 1.0 μm. Similarly, the two NR-box 2 peptides EKHKILHRLLQDS and TSLKEKHKILHRLLQDSS were potent competitors of the TRβ LBD:NID3− interaction (IC50 = 2.1 ± 0.5 μm or 0.4 ± 0.1 μm, respectively), demonstrating that 13 and 14 amino acid peptides encompassing NR-boxes 2 and 3, respectively, are sufficient to interact with TRβ LBD. Competition of the TRβ LBD:NID interaction by the NR-box 2 peptide EKHKILHRLLQDS and NR-box 3 peptide KENALLRYLLDKDD yielded similar results (IC50 = 2.2 ± 0.7 μm or IC50 = 6.0 ± 2.7 μm, respectively) (data not shown).
Based on the equation of Cheng and Prussoff (1973), the apparent affinities of TRβ LBD for the NR-box 2 peptide EKHKILHRLLQDS (Kdapp = 0.8 ± 0.3 μm) and for NR-box 3 peptide KENALLRYLLDKDD (Kdapp = 3.2 ± 1.2 μm) were indistinguishable from affinities for the full domain derivatives NID3− (functional NR-box 2) or NID2− (functional NR-box 3), respectively. We conclude that the interaction of these NR boxes with TRβ LBD is highly localized to the LxxLL motif and sequences immediately adjacent, and that the preference of TRβ LBD for particular NR boxes depends on the sequence rather than the structural context of the NR boxes within the NID.
Structure of the TRβ LBD:NR-box 2 peptide complex
Having demonstrated that peptides containing NR boxes mimic the interaction of the TRβ LBD with the GRIP1 NID domain, we cocrystallized the 13-amino-acid NR-box 2 peptide KHKILHRLLQDSS with TRβ LBD; under our crystallization conditions, the peptide or TRβ LBD alone precipitated. These crystals diffracted to a limiting resolution of 3.6 Å resolution; persistent efforts to improve the resolution of the crystals or identify alternate crystal forms were unsuccessful. The structure of the NR-box 2 peptide complex was determined by molecular replacement using a structure of the human TRβ LBD (R.L. Wagner, unpubl.), and refined to an R-factor of 25% (Table 1).
Table 1.
Data collection, phasing, and refinement statistics
 |
The asymmetric unit of the crystal contained two monomers of the TRβ LBD and two NR-box 2 peptides (Fig. 3a). Difference density for the peptide was present in the initial map, calculated from the molecular replacement solution; the density was further improved by refinement of the TRβ LBD against a maximum-likelihood target, to reduce bias toward the search model. Despite the low resolution of the data, the density for the NR-box 2 peptide was of high quality, as the structure of the TRβ LBD, determined independently to 2.4 Å resolution, dominates the maximum-likelihood weighted phases.
Figure 3.
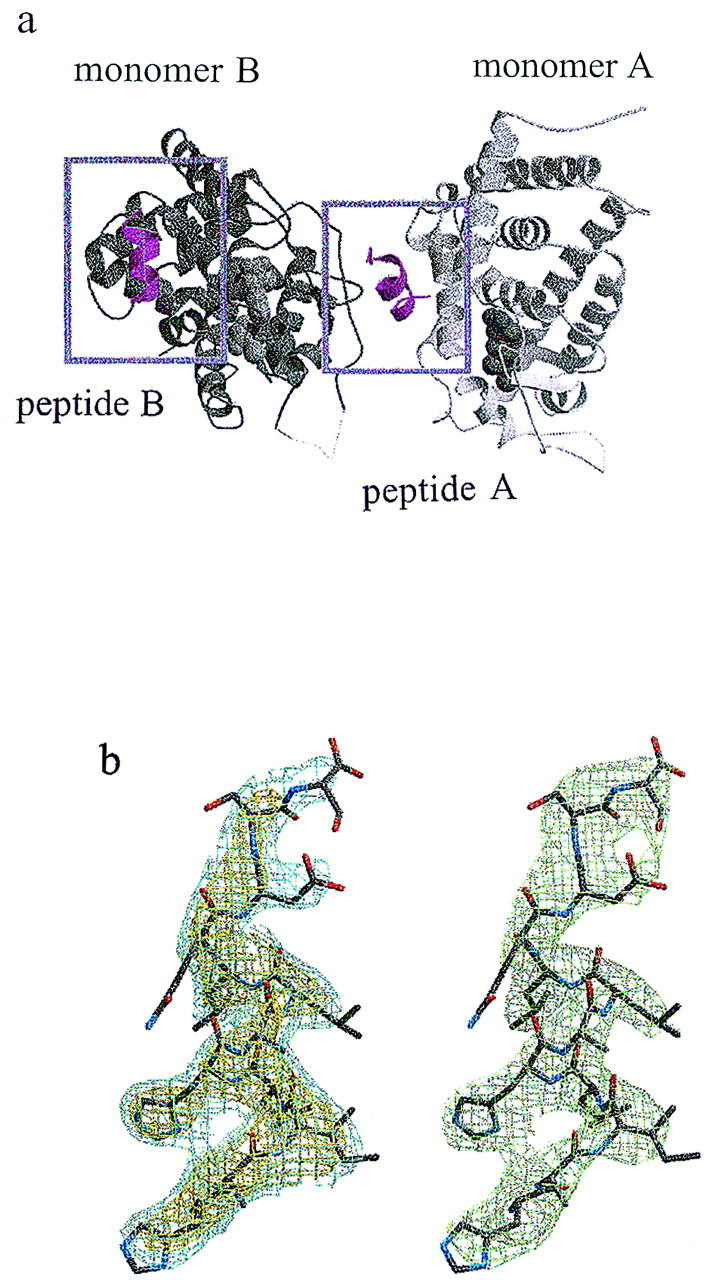
(a) The contents of the asymmetric unit of the crystallized TRβ LBD:NR-box 2 complex. The repeating unit of the crystal lattice contains a 2:2 complex of TRβ LBD and NR-box 2 peptide. The two monomers of the TRβ LBD are shown as a ribbon drawing, with monomer A in light gray, and monomer B in dark gray. The positions of the two NR-box 2 peptides, depicted as a magenta secondary structure ribbon, are boxed. The interaction of peptide A with monomer A and of peptide B with monomer B observe the same noncrystallographic symmetry relation as the two TRβ LBD monomers. The ligand T3 is shown in a space-filling representation. (b) Electron density for NR-box 2 peptide A and stereo representation of the peptide model. (Left) Initial Fo–Fc electron density map for the NR-box 2 peptide A, using phases calculated from the TRβ LBD model (without peptide), following one cycle of positional refinement against a maximum likelihood target in crystallography NMR systems (CNS) (A.T. Brunger, pers. comm.), with strict noncrystallographic symmetry enforced. The electron density is contoured at 3.0σ (yellow) and 2.0σ (cyan). The peptide appears as a stick representation, using the final refined atomic model. The same map shows strong, corroborating difference density for the central seven residues of peptide B. (Right) Final 2F0–Fc electron density map for the NR-box 2 peptide A (green).
The structure of the TRβ LBD consisted of 12 α-helices and 4 β-strands organized in three layers, as observed for the rTRα LBD (Wagner et al. 1995), and was not significantly different from the search model (R.L. Wagner, unpubl.). The periodicity of the side chain density of the NR-box 2 peptides clearly defines an amphipathic α-helix of nearly three turns for the central residues, K688–Q694, of the NR-box 2 peptide, with residues H687 and D695–S698 on either end in extended coil conformation (peptide A) or disordered (peptide B); the registration of the residues in the peptide was fixed by the side chain density for residues H687, I689, and H691. In the absence of the TRβ LBD, the far UV–CD spectrum of the NR-box 2 peptide indicated a random coil conformation (data not shown); thus, helix formation appears to be induced by complex formation. Figure 3b shows a comparison of the electron density for the peptide before modeling the peptide and after completion of the refinement.
One of the NR-box 2 peptides, peptide A, bridged the two TR monomers: the hydrophobic face of the α-helix contacted TRβ LBD monomer A at H3, H4, H5, and H12, whereas the hydrophilic face of peptide A contacted monomer B at the hairpin turn preceding β-strand S3. In contrast, the interaction of peptide B with TRβ LBD was restricted to monomer B, also contacting H3, H4, H5, and H12. The interface common to both TRβ LBD:NR-box 2 peptide complexes buried the LxxLL motif (GRIP1 residues L690, L693, and L694) against a hydrophobic surface of the receptor LBD (Fig. 4a,b). The interactions displayed by both peptides in the asymmetric unit are likely specific for the interaction of the peptide with the TRβ LBD and not driven by crystallization. These conclusions provide a structural rationale for functional studies that identified residues in the LBDs of various NRs that are important for ligand-dependent transcriptional activation and coactivator binding (Tone et al. 1994; Renaud et al. 1995; Collingwood et al. 1997; Henttu et al. 1997; Jurutka et al. 1997; Masuyama et al. 1997; Saatcioglu et al. 1997; Feng et al. 1998): All map to the common interface of the TRβ LBD with the NR-box 2 peptides A and B. In contrast, the interaction of the hydrophilic face of peptide A with monomer B likely represents a gratuitous crystal contact.
Figure 4.
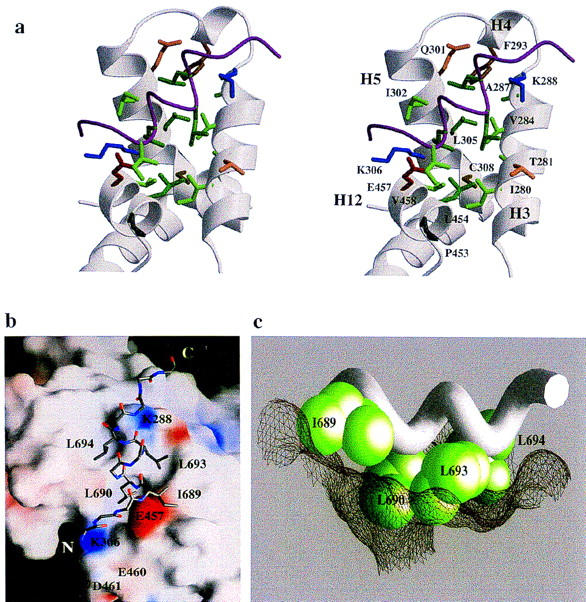
(a) Interface between the NR-box 2 peptide and the TRβ LBD. The side chains of those residues of the TRβ LBD within 4.5 Å of the NR-box 2 peptide are labeled. Acidic residues are red, basic residue are blue, aliphatic residues are green, aromatic residues are brown, and polar residues are orange. The peptide is depicted as a Cα trace, with the side chains of ILxxLL motif shown explicitly. (b) Surface of the TRβ LBD. The side chains of the leucines residues from the NR-box 2 peptide fit within a hydrophobic groove on the surface of the TRβ LBD formed from helices H3, H4, H5, and H12, whereas the side chain of the nonconserved isoleucine residue packs against the outside edge of the groove. The remainder of the peptide is shown as main chain. Areas of positive electrostatic potential are shown in blue; areas of negative electrostatic potential are shown in red. Individual charged residues of the TRβ LBD at the NR-box interface are labeled. Electrostatic and surface calculations used GRASP (Nicholls et al. 1991). (c) The TRβ LBD:LxxLL interface. The side chains of the NR-box 2 ILxxLL motif are shown in a CPK representation, with the main chain of the peptide drawn as a Cα worm. The three leucine residues fit into pockets on the molecular surface of the TRβ LBD, depicted as mesh, whereas the nonconserved isoleucine residue rests on the edge of the surface cleft.
The TRβ LBD:LxxLL interface
The surface of TRβ LBD that interacted with the NR-box 2 peptide was formed by 16 residues from four helices, H3, H4, H5, and H12, that packed against one another to create a shallow, hydrophobic groove (Fig. 4). The residues that formed the groove are I280, T281, V283, V284, A287, and K288 from H3; F293 from H4; Q301, I302, L305, K306, and C308 from H5; and L454, E457, V458, and F459 from H12 (Fig. 4a). Hydrophobic residues formed the floor of the groove, with charged residues lining the rim (Fig. 4b).
NR-box 2 peptide A bound at the junction of H3 and H12 of LBD monomer A (Fig. 4a). The hydrophobic residues I689, L690, L693, and L694 were modeled as the best-fit rotamers; the presence or absence of individual van der Waals contacts are unambiguous. L690 was inserted into a shallow depression at the base of the groove, making van der Waals contact with L454 and V458 of H12, whereas I689 packed against L454 of H12 outside the edge of the groove; L454 interdigitated between the two peptide residues. One further turn carboxy-terminal along the peptide A α-helix, L693 and L694, packed into complementary pockets within the groove. L693 formed a van der Waals contact with V284 of H3, whereas L694, bound more deeply in the groove, contacted F293 and L305 of H4 and H5. The TRβ LBD:NR-box 2 interface buried 980 Å2 of surface area, most of which is hydrophobic. As noted above, similar hydrophobic interactions were observed between peptide B and monomer B.
The crucial role of the hydrophobic residues within the LxxLL motif for the formation of a complex with the LBD of NRs is consistent with and accounts for several mutational studies in vitro and in vivo, showing that substitution of multiple leucine residues of the motif by alanines abrogates physical and functional interactions between p160 family coactivators and NRs (Le Douarin 1996; Heery et al. 1997; Torchia et al. 1997; Ding et al. 1998; Voegel et al. 1998). To investigate the role of the individual hydrophobic residues in NR-box 2, we replaced single residues of the ILHRLL motif by alanine in the background of NID3−. Replacement of any one of the three leucines severely compromised binding to TRβ LBD (Fig. 5a), whereas replacement of the isoleucine specific to NR-box 2, which is not as deeply buried as the conserved leucine residues, reduced binding to a lesser degree.
Figure 5.

(a) Individual leucine residues of the LxxLL motif are crucial for binding of GRIP1 NID to TRβ LBD. Shown are 1.6 μm (solid bars) or 4.0 μm (hatched bars) of glutathione–agarose-bound GST–NID3− or variants containing alanine substitutions of individual hydrophobic residues of the NR-box 2 ILxxLL motif (ALxxLL: I689A; IAxxLL: L690A; ILxxAL: L693A; ILxxLA: L694A; IAxxLA: L690A + L694A) were incubated with labeled TRβ LBD in the presence of 10 μm T3 (mutations are in boldface type). The amount of bound receptor is relative to the receptor input. These are the results of a representative experiment. (b,c) Pairwise or single conservative substitutions of the ILxxLL leucine residues drastically reduce the affinity of NR-box 2 peptides for TRβ LBD. Interaction of 1.6 μm glutathione–agarose-bound NID3− with 10 nm labeled TRβ LBD + 10 μm T3 was competed with increasing concentrations of variants of the NR-box 2 peptide KHKILHRLLQDSS, containing either pairwise alanine substitutions KHKAAHRLLQDSS, KHKILAALLQDSS, KHKILHRAAQDSS (b), or single phenylalanine substitutions of conserved residues of the hLxxLL motif (KHKFLHRLLQDSS, KHKIFHRLLQDSS, KHKILHRFLQDSS, KHKILHRLFQDSS) (c) (mutations are in boldface type). The amount of bound receptor is relative to the amount of retained receptor in the absence of peptide. The data represent the average and standard deviation of three independent experiments. (d,e) Sequences adjacent to the LxxLL motif affect the affinity of the TRβ LBD:NR-box 2 interaction. Labeled TRβ LBD (10 nm) was incubated with 1.6 μm glutathione–agarose-bound GST–NID3− in the presence of 10 μm T3 and increasing concentrations of peptides containing NR-box 2 (solid triangle), NR-box 3 (open triangle), and the LLRYLL motif of NR-box 3 in the context of flanking sequences from NR-box 2 (shaded diamond) (d), or NR-box 2 (solid circle), VP16 (solid square), and the ILHRLL motif of NR-box 2 in the context of the adjacent sequences from the VP16 peptide (shaded diamond) (e). The amount of bound receptor is relative to the amount of retained receptor in the absence of peptide. The data and IC50 values represent the average and standard deviation of three independent experiments. (f) Labeled TRβ LBD (solid line) or TRβ LBD ED–TN (broken line) (10 nm) was incubated with 1.6 μm glutathione–agarose-bound GST–NID3− in the presence of 10 μm T3 and increasing concentrations of peptides containing NR-box 2 (solid circle), or NR-box 3 (open circle). The amount of bound receptor is relative to the amount of retained receptor in the absence of peptide. The data represent the average and standard deviation of three independent experiments.
Parallel results were obtained by competing the interaction of TRβ LBD with NID3− using peptides in which either IL, HR or LL of the NR-box 2 motif were replaced pairwise by alanines (Fig. 5b). Whereas the peptides containing the IL or LL replacements failed to compete for TRβ LBD even at very high concentrations, replacement of the ‘HR spacer’ by alanines had little effect on competition, consistent with the α-helical structure of the motif. In the TRβ:GRIP1 complex, the NR-box 2 residues I689 and L690 packed against L454 and V458 of H12, whereas one helical turn further in the motif, L693 and L694, contacted V284, F293, and L305 from H3, H4, and H5, respectively, of the TRβ LBD (Fig. 4a). The strong dependence of the interaction on the integrity of the entire motif suggests that these interactions are not independent and drive the interaction through cooperative rather than additive contributions.
Replacement of single leucine residues of NR-box 2 by phenylalanine reduced competition by the mutant peptides for TRβ LBD by 60- to 100-fold relative to wild-type peptide, whereas replacement of the isoleucine resulted in an ∼10-fold reduction (Fig. 5c). Therefore, efficient interaction of TRβ with GRIP1 relies not simply on the hydrophobicity of the LxxLL motif, but rather on stereochemical properties of the leucine side chains, consistent with the observed structural complementarity of the interacting surfaces (Fig. 4c). The lesser effect on affinity by the replacement of I689 by phenylalanine may be explained by the location of I689 on the rim of the hydrophobic groove, where steric changes may be accommodated more easily. The stronger effect of the replacement of L690 correlates well with position of this residue deep within the hydrophobic groove. It is notable, however, that mutant peptides bearing bulky hydrophobic motifs other than LxxLL retain some affinity, albeit substantially weaker, for the TRβ LBD.
The role of the adjacent sequences
Our analyses demonstrated that the conserved LxxLL motif of NR-box 2 is necessary for strong interaction of GRIP1 with TRβ LBD. However, the lower affinity of the NR-box 3 hexapeptide LLRYLL for TRβ LBD (Fig. 2c) implied that an isolated LxxLL motif is not sufficient. Sequence identity among all the NR boxes of the p160 coactivator family is limited to the conserved leucine residues of the LxxLL motif, but for each NR box, the sequence conservation extends into adjacent residues (Fig. 1b).
NR-box 2 interacted with the TRβ LBD with a fourfold higher affinity than NR-box 3. To determine whether this affinity differential reflects differences in the respective hydrophobic motifs (NR-box 2, ILHRLL; NR-box 3, LLRYLL) or their adjacent sequences, we constructed a chimeric peptide containing the NR-box 3 motif in the context of adjacent sequences from NR-box 2. This peptide competed the interaction of NID3− with TRβ LBD with similar potency as the intact NR-box 2 peptide, demonstrating that the higher affinity of TRβ LBD for GRIP1 NR-box 2 is determined by the sequences adjacent of the LxxLL motif (Fig. 5d).
In principle, adjacent sequences might also decrease the affinity of a motif-driven interaction. We uncovered an example of such an effect when we constructed a chimeric peptide bearing the NR-box 2 LxxLL motif flanked by sequences normally adjacent to a VP16 Fxxhh motif, which interacts with TAFII31 in a complex (Uesugi et al. 1997) that is structurally similar to the TRβ LBD:NR box complex. The intact VP16 peptide EFEQMFTDALGIDE did not interact with TRβ LBD even at very high concentrations (Fig. 5e), demonstrating that the hydrophobicity of peptides is not sufficient to mediate interaction with TRβ. The chimeric peptide EFEQILHRLLGIDE competed the interaction of TRβ LBD with NID but with an ∼100-fold lower potency than the NR-box 2 peptide EKHKILHRLLQDS. We conclude that the affinity of the interaction between the NR boxes and TRβ LBD depends on both the hydrophobic motif and the adjacent sequences. As single substitutions of the conserved leucine residues of the LxxLL motif by alanines compromise interaction of the GRIP1 NID with TRβ LBD, the contributions of the adjacent sequences appear to depend on the interaction of the hydrophobic motif with TRβ LBD and are not sufficient to mediate interaction by themselves. However, the affinity of the LxxLL motif for its interaction with the hydrophobic cleft of the TRβ LBD is dependent on the flanking sequences.
The discrimination of sequences flanking the LxxLL motif by the TRβ LBD likely reflects the interaction of specific NR-box residues with the TRβ LBD interaction surface. In the TRβ LBD:peptide A complex, the positively charged amino terminus of the NR-box 2 peptide KHKILHRLLQDSS followed a shallow groove between H5 and H12 (Fig. 4b). Interaction of the negatively charged residues at the carboxyl terminus of the TRβ LBD (E460, D461) with the positively charged amino terminus of the NR-box 2 peptide may provide a favorable contact, which would be missing in the case of the NR-box 3 peptide ENALLRYLLDKDD or repulsive in the case of the NR-box 2/VP16 chimeric peptide EFEQILHRLLGIDE.
To test this hypothesis, we analyzed the interaction of peptides containing NR-box 2 or 3 with a mutant TRβ LBD, in which the residues E460 and D461 were substituted by threonine and asparagine, respectively, which are the homologous residues in GR (Fig. 6a). The affinity for the NR-box 2 peptide of this mutant was decreased about fourfold compared to wild-type TRβ LBD, whereas the affinity for NR-box 3 was marginally increased (Fig. 5f). We conclude that the higher affinity of NR-box 2 for TRβ LBD reflects a favorable interaction between positively charged residues amino-terminal to the LxxLL motif and negatively charged residues at the carboxyl terminus of the TRβ LBD.
Figure 6.
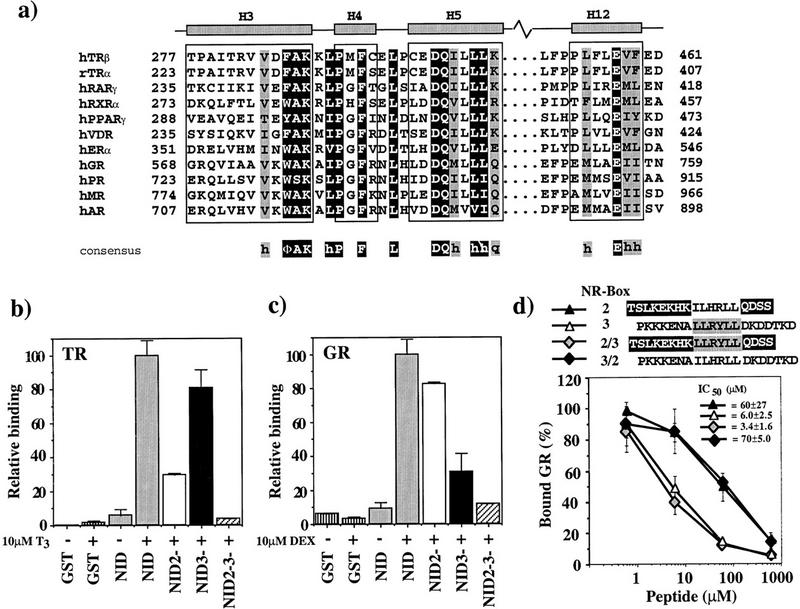
(a) Sequence alignment of LBD helices 3, 4, 5, and 12. NR sequences (Wurtz et al. 1996) are shown for LBD regions that interact with GRIP1. Residues representing the NR signature sequence, ΦAKxhPxFxxLxxxDQxxhh, are on dark background. Conserved residues, always hydrophobic (h) or strong polar (q), are shaded. The borders for helices H3, H4, H5, and H12 are those for the TRβ LBD. (Φ) Bulky hydrophobic residue. (b,c) GR and TR interact preferentially with different GRIP1 NR boxes. Labeled TRβ (b) or rat GR (c) (10 nm) was incubated with 1.6 μm glutathione–agarose-bound GST (vertically striped), GST–NID (shaded) or the NID variants NID2− (open), NID3− (solid), and NID2−3− (hatched) in the absence (−) or presence (+) of 10 μm T3 (TR) or 10 μm dexamethasone (DEX) (GR). The amount of bound receptor is normalized to the fraction bound to GST–NID (100%). The data represent the average and standard deviation of about three independent experiments. (d) Preferential interaction of GR with NR-box 3 is not specified by sequences adjacent to the LLRYLL motif. Labeled GR (10 nm) was incubated with 1.6 μm glutathione–agarose-bound GST–NID3− in the presence of 10 μm DEX and increasing concentrations of NR-box 2 peptide (solid triangle), NR-box 3 peptide (open triangle), and either a chimeric peptide bearing the LLRYLL motif of NR-box 3 with the adjacent sequences from NR-box 2 (shaded diamond), or a chimeric peptide bearing the ILHRLL motif of NR-box 2 with the adjacent sequences from NR-box 3 (solid diamond). The amount of bound receptor is relative to the amount of retained receptor in the absence of peptide. The data and IC50 values represent the average and standard deviation of three independent experiments.
A role for helix stabilization?
One explanation for poor competition by the NR-box 3 hexapeptide LLRYLL for TRβ LBD (Fig. 2c) may be the high energy of α-helix formation by short peptides. In fact, in the TRβ LBD:NR-box 2 complex, the peptide helix appears to be stabilized by two helix-capping interactions: one between the side chain of TRβ K288, which interacts with the free carbonyl groups of L694 of peptide A; and one between the side chain of TRβ E457 in H12, which interacts with the backbone amides of I689 and L690 in both peptide complexes. The TRβ residues K288 and E457 are highly conserved among NRs, and mutations in these or homologous residues eliminate the interaction of TRβ or the ER with p160 coactivators, perhaps implying that capping interactions are functionally important in coactivator binding, by restricting the length of the interacting α-helix (Collingwood et al. 1997; Tone et al. 1994; Henttu et al. 1997; Saatcioglu et al. 1997; Feng et al. 1998).
Interaction specificity
The features of the interface in the TRβ LBD that interacts with the NR-box 2 LxxLL motif are highly conserved within the NR family (Fig. 6a) (Bourguet et al. 1995; Renaud et al. 1995; Wagner et al. 1995; Wurtz et al. 1996; Brzozowski et al. 1997; Williams and Sigler 1998): The hydrophobic groove is present in all known structures of receptor LBDs, and many residues within and surrounding the groove were identical or, as demonstrated by mutational analysis, functionally conserved across the family (Tone et al. 1994; Renaud et al. 1995; Collingwood et al. 1997; Henttu et al. 1997; Jurutka et al. 1997; Masuyama et al. 1997; Saatcioglu et al. 1997; Feng et al. 1998). The conservation of the interacting surfaces may explain the broad specificity of members of the p160 coactivator family for NRs.
However, it is apparent that receptors interact selectively with particular p160 family members, and with individual NR boxes within a given coactivator. In fact, functional and biochemical studies revealed NR-selective interactions with various p160 coactivators (Ding et al. 1998; Voegel et al. 1998). To investigate the determinants of selectivity, we compared the interaction of GRIP1 with TRβ and the GR, another NR family.
Similar to TRβ and TRβ LBD, GR bound the GRIP1 NID in a ligand-dependent manner and failed to bind to NR-box 1; in contrast to TRβ, however, GR preferred NR-box 3 over NR-box 2 (Fig. 6b,c). These in vitro results parallel those obtained in yeast two-hybrid assays in vivo, using mutants of NR-boxes 2 or 3 in the context of full-length GRIP1 (Ding et al. 1998). Consistent with this observation, a peptide containing NR-box 3 was more efficient than a peptide containing NR-box 2 in competing the GR NID interaction (Fig. 6d). Surprisingly, NR-box 3 peptide PKKKENALLRYLLDKDDTKD displayed similar efficiency for inhibition of the interactions of the NID with GR (IC50 = 5 ± 3.8 μm) and TRβ (IC50 = 2.9 ± 1.0 μm), whereas NR-box 2 peptide TSLKEKHKILHRLLQDSS was 100-fold weaker in competing the GR:NID interaction (IC50 = 64 ± 29 μm) than the TRβ:NID interaction (IC50 = 0.4 ± 0.1 μm) [cf. Fig. 2c, competition of the interactions between TRβ or TRβ LBD and NID or NID3− by these peptides was comparable (data not shown)]. Unlike the TRβ, for GR there was no difference in the efficiency of competition between the NR-box 3 peptides PKKKENALLRYLLDKDDTKD and KENALLRYLLDKDD (data not shown).
In the case of TRβ LBD, preference of NR-box 2 over 3 was dependent on the adjacent sequences of NR-box 2, as demonstrated by a chimeric peptide containing the hydrophobic LLRYLL motif of NR-box 3 in the context of the adjacent sequences of the NR-box 2 motif (Fig. 5d). In contrast, the chimeric peptide and the NR-box 3 peptide competed the interaction of the NID with GR comparably (Fig. 6d), whereas chimeric peptides containing the ILHRLL motif of NR-box 2 in the context of either the adjacent sequences of NR-box 3 (PKKENAILHRLLDKDDTKD) (Fig. 6d) or sequences from a weakly competing peptide from VP16 (EFEQILHRLLGIDE) (data not shown), competed with similar efficiency as the NR-box 2 peptides TSLKEKHKILHRLLQDSS or KHKILHRLLQDSS, respectively. Thus, the preference of GR for NR-box 3 is not specified by its motif-adjacent sequences, and instead appears to reflect differences in the motif itself (NR-box 2, ILHRLL; NR-box 3, LLRYLL). It seems unlikely that the specificity in this case will reside solely with the hydrophobic residues of the motif, so it will be interesting in future studies to pursue this matter with further mutational analyses.
The differences in affinity and preference for NR-box 2 or 3 by GR and TRβ LBD likely reflect receptor-specific structural differences in the proximity of the coactivator interaction surface. For example, GR lacks the carboxy-terminal negatively charged residues (Fig. 6a) that in TRβ LBD stabilized the interaction with NR-box 2, and substitution of these TRβ residues by the corresponding GR residues resulted in decreased affinity for NR-box 2 (Fig. 5f). However, whereas this TRβ mutant interacted with only slight preference (twofold) for NR-box 3 over NR-box 2, the affinity of GR for NR-box 2 is about sixfold lower than for NR-box 3, suggesting that in GR additional components disfavor interaction with NR-box 2. Besides the absence of negatively charged residues carboxy-terminal of helix 12, another obvious difference between GR and TRβ is the so-called F domain, which extends the carboxyl terminus of the GR well beyond the end of H12 and is absent in TRβ. Although the structure of the GR LBD is not known, the structure of the closely related PR LBD (Williams and Sigler 1998) indicates that the F domain may restrict the coactivator interaction surface and displace the amino-terminal residues of the NR box. In any case, the comparison of GR and TRβ demonstrates that at least two components, the LxxLL motif and the sequences flanking the motif, are used differentially to generate distinct specific interactions with the p160 coactivator GRIP1.
Discussion
NR–NR-box interactions
The interaction of NRs with coactivators of the p160 family depends on the presence of NR boxes that contain a hydrophobic LxxLL motif. Various functional and qualitative biochemical studies established that the conserved leucine residues are important for the interaction, demonstrated that different receptors can differ in their interactions, and inferred a putative interaction site (Le Douarin et al. 1996; Heery et al. 1997; Torchia et al. 1997; Ding et al. 1998; Feng et al. 1998; Voegel et al. 1998). In this study we have coupled the first molecular structural analysis and quantitative biochemical analyses of the interactions of NRs with coactivators, and we have identified the structural elements that determine the specificity of those interactions.
Even at the current low-resolution level, the structure of the TRβ LBD:GRIP1 NR-box 2 complex establishes clearly that the NR–coactivator interface is comprised of a hydrophobic groove formed by conserved residues in helices H3, H4, H5, and H12 from the receptor LBD, adjoined to the conserved leucine residues of the LxxLL motif in an amphipathic α-helix from GRIP1; the GRIP1 helix is capped by highly conserved, charged LBD residues. Independently, our quantitative biochemical analyses demonstrate that specific interactions between NRs and GRIP1 require that the integrity of the entire LxxLL motif be strongly affected by immediately adjacent sequences. In addition, we demonstrate that affinity and specificity of the TRβ–GRIP1 interaction can be modulated by sequences outside of the hydrophobic groove of the NR LBD. Different receptors appear to utilize these NID structural elements differentially, and certain receptors are also affected by regions outside of the NID (H. Hong, B.D. Darimont, H. Ma, L. Yang, K.R. Yamamoto, and M.R. Stallcup, in prep.). The combination of these features likely accounts for the differential preference of NR boxes observed even for closely related receptors, such as GR and the mineralocorticoid receptor (Ding et al. 1998). It seems likely that the multiple NR boxes found in p160 coactivators contribute both diversity and specificity to the spectrum of p160–NR interactions.
Notably, our biochemical analyses of the TR and GR interactions with NR-boxes 2 and 3 of GRIP1 provide quantitative information that is independent of our structural studies, and the conclusions derived from the two approaches converge in a consistent and mutually supportive manner.
What is AF-2?
The AF-2 transcriptional activation function requires helix H12 at the carboxyl terminus of the receptor LBD, as mutations within this helix eliminate AF-2 activity (Danielian et al. 1992; Barettino et al. 1994; Durand et al. 1994; Tone et al. 1994; Collingwood et al. 1997; Henttu et al. 1997; Jurutka et al. 1997; Masuyama et al. 1997; Saatcioglu et al. 1997; Feng et al. 1998). Fusions of H12 to the Gal4 DNA-binding domain were reported to activate transcription, leading to the suggestion that H12 alone corresponds to AF-2 (Barettino et al. 1994; Durand et al. 1994; Masuyama et al. 1997; Saatcioglu et al. 1997). However, point mutations in H12 had very different effects on activation in the Gal4 fusion context than in the intact LBD (Saatcioglu et al. 1997). Subsequently, it was inferred from comparisons of the structure of the unliganded hRXRα LBD with that of the liganded TR and RAR LBDs that ligands induced dramatic conformational changes, affecting especially the orientation of H12 (Bourguet et al. 1995; Renaud et al. 1995; Wagner et al. 1995). On that basis, helix H12 was proposed as a ligand-dependent switch that forms, in its functional configuration, a surface for coactivator recognition or binding (Moras and Gronemeyer 1998).
We have demonstrated that residues of H12 are directly involved in coactivator binding. However, our structural analysis reveals that GRIP1 also interacts with helices H3, H4, and H5 of the TRβ LBD, providing a structural basis for mutational studies implicating other parts of the receptor LBDs in AF-2 activity and interaction with coactivators (Renaud et al. 1995; Henttu et al. 1997; Feng et al. 1998; Norris et al. 1998). Thus, AF-2 corresponds to the full coactivator interaction surface, and not to H12 alone.
Ligand effects on LBD structure and function
With respect to the LBD, agonists are ligands that support its interaction with coactivators, presumably by promoting formation of the coactivator interaction surface described here. Antagonists, such as the GR ligand RU486 or the ER ligand raloxifene, do not support LBD interactions with p160 coactivators (Hong et al. 1997; Norris et al. 1998), and conceivably might recruit corepressors instead (Wagner et al. 1998; Zhang et al. 1998). Comparison of the estradiol- and raloxifene-bound structures of the ERα LBD reveals strikingly different positions of helix 12 (Brzozowski et al. 1997). In the ER:raloxifene complex, H12 packs against the hydrophobic residues of helices H3 and H5 in a configuration remarkably similar to that of the NR-box 2 peptide of GRIP1 with the agonist-bound TR LBD. Thus, raloxifene precludes the interaction of ER with coactivators in two ways: (1) eliminating the part of the coactivator interaction surface that involves H12; and (2) occluding with H12 the remaining part of the coactivator interaction surface.
As many NR antagonists like raloxifene appear structurally related to agonists that have been modified by a long ‘central extension,’ it seems likely that antagonists will commonly function by interfering with positioning of H12 in the coactivator interface. It is intriguing to consider whether the position of H12 seen in the ER:raloxifene complex will also prove to be general. In that structure and in the similar ER/tamoxifen structure (A. Shiau, pers. comm.), H12 contacts H3 and H5 through a hydrophobic surface (LxxML) similar to the p160 LxxLL motif. The corresponding face of H12 among other NRs, although less similar to LxxLL, also contains bulky hydrophobic residues, raising the possibility that the H12:H3/H5 interface may be commonly adopted when the ‘agonist conformation’ is precluded.
Common protein–protein interfaces
The structure of the TRβ LBD:GRIP1 interaction shows striking similarities with the other three described protein–protein interfaces that have been defined in transcriptional regulatory complexes: p53:MDM2 (Kussie et al. 1996); VP16:TAFII31 (Uesugi et al. 1997); and CREB:CBP (Radhakrishnan 1997). We note that these complexes share a common theme: the interaction of an amphipathic α-helix containing a conserved hydrophobic motif with a complementary, hydrophobic surface. As with GRIP1, a functional interaction appears to require specific hydrophobic residues; for example, Fxxhh has been proposed as a motif that interacts selectively with TAFII31 (Uesugi et al. 1997).
Consistent with a close fit between the hydrophobic surfaces at the interfaces, even conservative replacements of the hydrophobic residues at the interface typically compromise the affinities of the interactions. However, some factors, such as TAFII31 and MDM2, may interact functionally with more than one hydrophobic motif (Lu and Levine 1995; Martin et al. 1995; Thut et al. 1995; Kussie et al. 1996; Uesugi et al. 1997), suggesting that precise complementarity of the interacting surfaces may be less important for these interactions.
Based on the shared similarities in the structures of these four complexes and the potential for cross-interaction involving different hydrophobic motifs, we tested whether the GRIP1 NID could interact with TAFII31 or MDM2. GRIP1 NID chimeras in which the NR-box 2 LxxLL motif was replaced by the corresponding VP16 (FxxAL) or p53 (FxxLW) sequences were also tested. In no case did we observe GRIP1–NID interactions with TAFII31 or MDM2 (data not shown). Clearly, the presence of the correct motif alone is not sufficient for high-affinity binding, consistent with the notion that specificity in the interactions of these two common structural features is determined by a combination of parameters that include adjacent sequences, and perhaps more remote influences on protein conformation. The use of multiple determinants in defining common interactions provides mechanisms both for flexible assembly and for specificity.
Combinatorial regulation
Transcriptional regulatory complexes are dynamic structures that can assemble differentially from the mixture of components available in different cell and response element contexts but are nevertheless sufficiently precise in structure that they produce specific regulatory consequences. Our results suggest that the combinatorial assembly of these complexes is achieved in part by complementary interactions between two simple structural features, an amphipathic α-helix and a hydrophobic groove, at least one of which is present on each interacting component. Interestingly, the critical contacts at the interaction interfaces are restricted to a surprisingly small area. Thus, it may eventually prove possible to mimic or disrupt those interactions with small molecules.
In the NR LBDs, formation of the coactivator-interacting hydrophobic groove is ligand-dependent, and coactivator specificity is achieved by contributions from several distinct determinants: the α-helical LxxLL motif, the residues immediately adjacent to the motif, and as shown by others, sequences outside the NR interaction domain. This strategy for building specificity into protein–protein interactions, through differential contributions of various features within a common structural interface, may prove to be general for many components of transcriptional regulatory complexes.
Materials and methods
Vectors
hTRβ LBD (His6 E202–D405) was expressed from a pET28a (Novagen)-based construct (A. Shiau, unpubl.). pET GRIP1 563–767His6 (encoding the NID) and pGEX-4T1 GRIP1 563–767His6 (encoding GST–NID) were constructed by cloning a GRIP1 BamHI–XhoI fragment derived from pGEX-2TK GRIP1 563–1121 (H. Hong, B.D. Darimont, H. Ma, L. Yang, K.R. Yamamoto, and M.R. Stallcup, in prep.) into the BamHI–XhoI sites of pET23a or pGEX-4T1His6; the latter was generated by exchanging the XhoI–BsaAI fragment of pGEX-4T1 against a His6–tag-containing XhoI–NaeI fragment of pET23a (Novagen). NID mutants were generated by PCR or single-stranded mutagenesis and confirmed by sequencing.
Protein expression
NID and GST–NID derivatives were expressed in HB101 (37°C, 1 mm IPTG added at OD600 = 0.7, induced 4 hr). hTRβ LBD was expressed in BL21DE3 (14°C, 1 mm IPTG added at OD600 = 0.7, induced 24 hr).
Protein purification
Cell lysis
For GRIP1 derivatives, sonication buffer was 20 mm Tris-HCl at pH 8.0, 0.1 m NaCl, 10% glycerol, 0.1 mm PMSF, protease inhibitors (Complete, EDTA free, Boehringer Mannheim). For TRβ LBD, 50 mm sodium phosphate (pH 8.0), 0.3 m NaCl, 10% glycerol, 25 mm β-mercaptoethanol, and 0.1 mm PMSF were used.
Purification
Freeze–thaw, incubate with 0.1 mg/ml lysozyme (20 min, 0°C); sonicate, clear lysate (Ti45, 36,000 rpm, 1 hr, 4°C). For GST–NID, load lysate on Talon resin (Clontech) equilibrated in sonication buffer; elution with imidazole gradient (12–100 mm) yields 95% pure protein. For NID, the same Talon purification procedure was used as above; protein elutes at 12 mm and at 40–70 mm imidazole; 12 mm fraction was chromatographed on Mono Q (50 mm Tris-HCl at pH 7.5, 10% glycerol, 1 mm EDTA, 1 mm DTT, 0.1 mm PMSF, and protease inhibitors); NID flowed through (>95% pure) and was concentrated by ultrafiltration. For TRβ LBD, the same Talon purification procedure was used as above, except in sodium phosphate buffer, eluted with 12–300 mm imidazole gradient. Isolation of liganded [(3,3′,5-triiodo-l-thyronine (T3; Sigma)] receptor using TSK–phenyl HPLC (TosoHaas, Philadelphia, PA) was as described (Apriletti et al. 1995); yield was 9.5 mg/l bacterial culture. For crystallization, hTRβ LBD was concentrated in 20 mm HEPES (pH 7.4), 3 mm DTT, and 0.1 μm T3 to 9 mg/ml by ultrafiltration (Millipore UFV2BGC10).
Glutathione agarose binding
Imidazole was removed by NAP gel filtration (Pharmacia); protein concentrations determined by Bio-Rad assay. Wild-type or mutant GST–NID was incubated with glutathione agarose (1 hr, 4°C) in binding buffer (sonication buffer + 1 mm DTT, 1 mm EDTA, 0.01% NP-40). Beads were washed with >20 volumes of binding buffer, diluted in binding buffer with 20% glycerol to 40% final bead concentration; aliquots were frozen in liquid nitrogen and stored at −70°C. Loaded beads were analyzed by SDS-PAGE using BSA standard (Pierce) to ensure equal concentrations of bound proteins.
In vitro transcription–translation
rGR was cloned as a BamHI–EagI fragment into the BamHI–BglII site of pSG5 (Stratagene) after blunting the EagI and BglII ends with Klenow. pSG5 hTRβ was constructed as described (Feng et al. 1998). pSG5 rGR, pSG5 hTRβ, and pET28a hTRβ LBD were used to express 35S-labeled rGR, TRβ, and TRβ LBD in a coupled reticulocyte lysate (TNT; Promega). Translations were performed in the presence and absence of 10 μm dexamethasone (GR), or 10 μm T3 (hTRβ and hTRβ LBD). Expression of rGR, hTRβ, and hTRβ LBD yielded 5–50, 20–100, and 100–250 ng/μl reaction, respectively, as computed from the amount of incorporated [35S]methionine.
Interaction assay
Fifty microliters of 20% bead suspension containing 1.6 or 4.0 μm GST–NID or derivatives was incubated with 0.2–2 μl of lysate containing labeled rGR, hTRβ, or hTRβ LBD; final concentration of receptors was 10 nm; binding was independent of lysate concentration. Binding (4°C under rotation, 2 hr) was in binding buffer + 20 μg/ml BSA, with or without 10 μm hormone. For competition experiments, peptides were added before receptors, but altering the order of addition did not affect the results (data not shown), confirming that the reactions reach equilibrium. Beads were washed (4°C, 5×, 200 μl binding buffer), proteins eluted (10 μl 2× SDS loading buffer), subjected to SDS-PAGE, and the fraction of bound receptor determined (Molecular Dynamics PhosphorImager, Storm 860) as percentage relative to input (in direct binding studies) or as percentage relative to bound receptor in the absence of ligand (in competition studies).
Peptides
Peptides (from UCSF Biomolecular Resource Center, Research Genetics, or generously provided by Tularik, Inc.) were HPLC purified and analyzed by mass spectroscopy. Concentrations were determined spectroscopically using the tyrosine signal (ε276 = 1450/m per cm) or by amino acid analysis.
Crystallization
The complex was prepared by mixing 9 mg/ml TRβ LBD (20 mm HEPES at pH 7.4) with 6 mm peptide KHKILHRLLQDSS (0.4 mm ammonium acetate at pH 4.72), at a 1:2 ratio, 0°C, 1 hr. Crystals were obtained (2 days, 4°C) by hanging drop vapor diffusion (drop: 1.5 μl of TRβ LBD:NR-box 2 peptide complex and 0.5 μl 15% PEG-4K, 200 mm sodium citrate at pH 4.9; reservoir: 10% PEG-4K, 100 mm ammonium acetate, and 50 mm sodium citrate at pH 5.6). After 1 hr, 0.2 μl of 10−3 to 10−5 dilutions of microcrystals in reservoir buffer were introduced for nucleation. Crystals were space group P3121 (a = 95.2 Å, c = 137.6 Å) and contained two molecules each of TRβ LBD and NR-box 2 peptide.
Structural analysis
Crystals were soaked overnight in a drop composed as described above plus 10% sucrose; then transferred serially, 5 min each, to drops containing 10% PEG-4K, 100 mm ammonium acetate, and 50 mm sodium citrate (pH 5.6), 10% sucrose, excess NR-box 2 peptide, and increasing concentrations of xylotol (2%–13% by 2% steps). Data were collected from frozen crystals (liquid nitrogen) using Cu Kα radiation (R-axis generator, 50 kV, 300 mA, 0.3-mm collimator, Ni filter); reflections were recorded using an R-Axis II detector, integrated with Denzo, and equivalent reflections (I > −3σ) scaled using Scalepack (Otwinowski and Minor 1997). The resolution limit was chosen as the highest resolution shell with 50% of the reflections with an I/σ > 3, and Rsym < 30%. Possible rotation function solutions were calculated using normalized amplitudes in AMORE from a model of TRβ LBD with ligand omitted (limiting resolution 2.4 Å; Rcryst = 20%) (R.L. Wagner, in prep.); translation function solutions were determined using the program TFFC for the two rotation solutions with the highest correlation coefficients (Collaborative Computational Project 1994). After rigid body refinement of the TRβ LBD molecules, electron density maps were calculated, and positive difference density used to fit the iodine atoms of T3. The iodine atoms were modeled as a rigid body, and refined with the TRβ LBD against a maximum likelihood target with strict noncrystallographic symmetry using CNS (A.T. Brunger, pers. comm.; Adams et al. 1997). 2FoFc and FoFc electron density maps showed interpretable density, related by the noncrystallographic symmetry operator, which was fitted using O (Jones et al. 1991). The electron density was best modeled as a short α-helix with the registration of the peptide fixed by the side chain density for residues H687, I689, and H691; all residues were modeled as low-energy rotamers. Refinement included positional refinement with CNS interspersed with manual rebuilding, monitored using the free-R factor. A flat bulk solvent correction and an overall anisotropic B-factor correction were applied. As partial validation of the refined peptide model, the orientation of the helix was reversed; the sequence was shifted out of register; and the peptide translated 0.3 Å. In each case, after refinement of the new model, the Rfree had increased.
Acknowledgments
We thank S. Mahrus for help in the preparation and assay of PET28aTRβ ED:TN, A. Shiau for the PET28aTRβ LBD plasmid, M. Brasseur and D. Goeddel for synthesis and purification of peptides, P. Kussie and A. Levine for MDM2 expression plasmids, R. Tjian for TAFII31, J. Iñiguez-Lluhi for help with data analysis and for useful discussions, and P. Cherbas, H. Ingraham, J. Iñiguez-Lluhi, C. Jamieson, D. Julius, and W. Lim for critiques of the manuscript. B.D.D. was supported by fellowships from the European Molecular Biology Organization (EMBO) and the Helen Hay Whitney Foundation. This work was supported by grants from the National Science Foundation (K.R.Y.) and the National Institutes of Health (K.R.Y., R.J.F., M.R.S., P.J.K., and J.D.B.). Coordinates have been deposited with the Brookhaven Protein Data Bank (accession no. 1bsx).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
Two other structures of NR LBD:coactivator peptide complexes have recently been solved: PPARγ:SRC-1 (Nolte et al. 1998) and ERα:GRIP-1 (A.K. Shiau, D. Barstad, P.M. Loria, L. Cheng, P.J. Kushner, D.A. Agard, and G.L. Greene, in prep.). Both complexes display features very similar to those described here.
Footnotes
E-MAIL yamamoto@socrates.ucsf.edu; FAX (415) 476-6129.
References
- Adams PD, Pannu NS, Read RJ, Brunger AT. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc Natl Acad Sci. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–958. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- Apriletti JW, Baxter JD, Lau KH, West B. Expression of the rat α1 thyroid hormone receptor ligand binding domain in Escherichia coli and the use of a ligand-induced conformational change as a method for its purification to homogeneity. Protein Expr Purif. 1995;6:363–370. doi: 10.1006/prep.1995.1048. [DOI] [PubMed] [Google Scholar]
- Barettino D, Vivanco Ruiz MdM, Stunnenberg G. Characterization of the ligand-dependent transactivation domain of thyroid receptor. EMBO J. 1994;13:3039–3049. doi: 10.1002/j.1460-2075.1994.tb06603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXRa. Nature. 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, Öhman L, Greene GL, Gustafsson J-A, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with p/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prussoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, No. 4. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Collingwood TN, Rajanayagam O, Adams M, Wagner R, Cavailles V, Kalkhoven E, Matthews C, Nystrom E, Stenlof K, Lindstedt G, Tisell L, Fletterick RJ, Parker MG, Chatterjee VKK. A natural transactivation mutation in the thyroid hormone beta receptor: Impaired interaction with putative transcriptional mediators. Proc Natl Acad Sci. 1997;94:248–253. doi: 10.1073/pnas.94.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, White R, Lees JA, Parker MG. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992;11:1025–1033. doi: 10.1002/j.1460-2075.1992.tb05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding XF, Anderson CM, Ma H, Hong H, Uht RM, Kushner PJ, Stallcup MR. Nuclear receptor binding sites of coactivators GRIP1 and SRC-1: multiple motifs with different binding specificities. Mol Endocrinol. 1998;12:302–313. doi: 10.1210/mend.12.2.0065. [DOI] [PubMed] [Google Scholar]
- Durand B, Saunders M, Gaudon C, Roy B, Losson R, Chambon P. Activation function 2 (AF-2) of retinoic acid receptor and 9-cis retinoic acid receptor: Presence of a conserved autonomous constitutive activation domain and influence of the nature of the response element on AF-2 activity. EMBO J. 1994;13:5370–5382. doi: 10.1002/j.1460-2075.1994.tb06872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Ribeiro RCJ, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747–1750. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr Opin Cell Biol. 1997;9:222–232. doi: 10.1016/s0955-0674(97)80066-x. [DOI] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in the transcriptional co-activators mediate binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- Henttu PMA, Kalkhoven E, Parker MG. AF-2 activity and recruitment of steroid receptor coactivator 1 to the estrogen receptor depend on a lysine residue conserved in nuclear receptors. Mol Cell Biol. 1997;17:1832–1839. doi: 10.1128/mcb.17.4.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Kohli K, Triverdi A, Johnson DL, Stallcup MR. GRIP1, a novel mouse protein that serves as a transcriptional co-activator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci. 1996;93:4948–4952. doi: 10.1073/pnas.93.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Kohli K, Garabedian MJ, Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol. 1997;17:2735–2744. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjelgaard M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallographica. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Jurutka PW, Hsieh JC, Remus LS, Whitfield GK, Thompson PD, Haussler CA, Blanco JC, Ozato K, Haussler MR. Mutations in the 1,25-dihydroxyvitamin D3 receptor identifying carboxy-terminal amino acids required for the transcriptional activation that are functionally dissociated from hormone binding, heterodimeric DNA binding, and interaction with basal transcription factor IIb, in vitro. J Biol Chem. 1997;272:14592–14599. doi: 10.1074/jbc.272.23.14592. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E, Valentine JE, Heery DM, Parker MG. Isoforms of steroid receptor coactivator 1 differ in their ability to potentiate transcription by the oestrogen receptor. EMBO J. 1998;17:232–243. doi: 10.1093/emboj/17.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman R, Rose D, Glass C, Rosenfeld M. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:1–12. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, Chambon P. A possible involvement of TIF1α and TIF1β in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996;15:6701–6715. [PMC free article] [PubMed] [Google Scholar]
- Li H, Gomes PJ, Don Chen J. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF-2. Proc Natl Acad Sci. 1997;94:8479–8484. doi: 10.1073/pnas.94.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Levine AJ. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci. 1995;92:5154–5158. doi: 10.1073/pnas.92.11.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Trouche D, Hagemeier C, Sorensen TS, La Thangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–694. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Brownfield CM, St-Arnaud R, MacDonald PN. Evidence for ligand-dependent intramolecular folding of the AF-2 domain in vitamin D receptor-activated transcription and coactivator interaction. Mol Endocrinol. 1997;11:1507–1517. doi: 10.1210/mend.11.10.9990. [DOI] [PubMed] [Google Scholar]
- Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: Structure and function. Curr Opin Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- Norris JD, Fan D, Stallcup MR, McDonnell DP. Enhancement of the estrogen receptor transcriptional activity by the coactivator GRIP1 highlights the role of activation function 2 in determining estrogen receptor pharmacology. J Biol Chem. 1998;273:6679–6688. doi: 10.1074/jbc.273.12.6679. [DOI] [PubMed] [Google Scholar]
- Oñate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Macromolecular crystallography, Part A. New York, NY: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- Renaud JP, Rachel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RARγ ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins. 1994;19:55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- Saatcioglu F, Lopez G, West BL, Zandi E, Feng W, Lu H, Esmaili A, Apriletti JW, Kushner PJ, Baxter JD, Karin M. Mutations in the conserved carboxy-terminal sequence in thyroid hormone receptor dissociate hormone-dependent activation from interference with AP-1 activity. Mol Cell Biol. 1997;17:4687–4695. doi: 10.1128/mcb.17.8.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW. TRAM-1, a novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid coactivator-1. J Biol Chem. 1997;272:27629–27634. doi: 10.1074/jbc.272.44.27629. [DOI] [PubMed] [Google Scholar]
- Thut CJ, Chen JL, Klemm R, Tjian R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science. 1995;267:100–104. doi: 10.1126/science.7809597. [DOI] [PubMed] [Google Scholar]
- Tone Y, Collingwood TN, Adams M, Chatterjee VK. Functional analysis of a transactivation domain in the thyroid beta receptor. J Biol Chem. 1994;269:31157–31161. [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL. Induced α helix in the VP16 activation domain upon binding to a human TAF. Science. 1997;277:1310–1313. doi: 10.1126/science.277.5330.1310. [DOI] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJS, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667–3675. [PMC free article] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJS, Tini M, Vivat V, Chambon P, Gronemeyer H. The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J. 1998;17:507–519. doi: 10.1093/emboj/17.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BL, Norris JD, Knotts TA, Weigel N, McDonnell DP. The nuclear corepressors NCoR and SMRT are key regulators of both ligand- and 8-bromo-cyclic AMP-dependent transcriptional activity of the human progesterone receptor. Mol Cell Biol. 1998;18:1369–1378. doi: 10.1128/mcb.18.3.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- Williams SP, Sigler PB. Atomic structure of the progesterone/progesterone receptor complex. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- Wurtz JM, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- Yamamoto KR, Godowski PJ, Picard D. Ligand regulated nonspecific inactivation of receptor function: a versatile mechanism for signal transduction. Cold Spring Harb Symp Quant Biol. 1988;53:803–811. doi: 10.1101/sqb.1988.053.01.091. [DOI] [PubMed] [Google Scholar]
- Yamamoto KR, Pearce D, Thomas J, Miner JN. Combinatorial regulation at a mammalian composite response element. In: McKnight SL, Yamamoto KR, editors. Transcriptional regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 1169–1192. [Google Scholar]
- Zhang X, Jeyakumar M, Petukhov S, Bagchi MK. A nuclear receptor corepressor modulates transcriptional activity of antagonist-occupied steroid hormone receptor. Mol Endocrinol. 1998;12:513–524. doi: 10.1210/mend.12.4.0089. [DOI] [PubMed] [Google Scholar]