

Abstract
The epidermal growth factor receptor (EGFR) gene is amplified or mutated in 30%–50% of human gliobastoma multiforme (GBM). These mutations are associated usually with deletions of the INK4a–ARF locus, which encodes two gene products (p16INK4a and p19ARF) involved in cell-cycle arrest and apoptosis. We have investigated the role of EGFR mutation in gliomagenesis, using avian retroviral vectors to transfer a mutant EGFR gene to glial precursors and astrocytes in transgenic mice expressing tv-a, a gene encoding the retrovirus receptor. TVA, under control of brain cell type-specific promoters. We demonstrate that expression of a constitutively active, mutant form of EGFR in cells in the glial lineage can induce lesions with many similarities to human gliomas. These lesions occur more frequently with gene transfer to mice expressing tv-a from the progenitor-specific nestin promoter than to mice expressing tv-a from the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter, suggesting that tumors arise more efficiently from immature cells in the glial lineage. Furthermore, EGFR-induced gliomagenesis appears to require additional mutations in genes encoding proteins involved in cell-cycle arrest pathways. We have produced these combinations by simultaneously infecting tv-a transgenic mice with vectors carrying cdk4 and EGFR or by infecting tv-a transgenic mice bearing a disrupted INK4a–ARF locus with the EGFR-carrying vector alone. Moreover, EGFR-induced gliomagenesis does not occur in conjunction with p53 deficiency, unless the mice are also infected with a vector carrying cdk4. The gliomagenic combinations of genetic lesions required in mice are similar to those found in human gliomas.
Keywords: EGFR, gliomagenesis, cell cycle arrest
Gliomas are the most common forms of primary brain tumors and are classified into four clinical grades (Kleihues et al. 1993). The most aggressive tumors, grade 4, also known as glioblastoma multiforme (GBM), appear to arise either de novo as GBMs or present initially as lower grade tumors that progress to higher grades over time (von Deimling et al. 1993). Certain mutations and other properties are highly correlated with one or the other of these two pathogenic pathways. Gliomas that are first detected as grade 4 tumors usually lack an intact INK4a–ARF tumor suppressor locus (Schmidt et al. 1994; He et al. 1995; Ichimura et al. 1996); and they are thus unable to make the two INK4a–ARF gene products, p16INK4a and p19ARF, which arrest the cell cycle by different pathways in G1 and in both G1 and G2, respectively (Fig. 1; Serrano et al. 1993; Quelle et al. 1995). In addition, these tumors occur frequently in older patients, contain mutations or amplification of the EGFR gene, tend to be more aggressive, and are generally diploid (Louis et al. 1993; He et al. 1994; Schlegel et al. 1994; Ono et al. 1996; Hayashi et al. 1997). In contrast, gliomas that progress from lower to higher grade lesions are usually wild type for INK4a–ARF, but have an amplified CDK4 locus or have lost the Rb gene, and frequently also display either loss of p53 function or amplification of mdm2. Therefore, in both types of tumors the same two cell-cycle arrest pathways are disrupted by different mutations (Fig. 1). Furthermore, the tumors retaining a wild-type INK4a–ARF gene arise at an earlier age, are less aggressive, and are usually aneuploid (Lang et al. 1994; Rasheed et al. 1994; van Meyel et al. 1994; Watanabe et al. 1996).
Figure 1.

Proposed roles of INK4a–ARF in cell-cycle arrest and human gliomagenesis. (A) Cell-cycle arrest pathways involving the INK4a–ARF gene products p16INK4a and p19ARF. (B) Subdivision of human gliomas into those with mutations in INK4a–ARF and those with wild-type INK4a–ARF loci. Mutations of other genes associated frequently with these two tumor populations are indicated. The gliomas with mutations INK4a–ARF are more common and associated with abnormalities in the EGFR gene, whereas the INK4a–ARF wild-type gliomas disrupt the G1 arrest pathways downstream of the INK4a–ARF gene products.
We have previously designed a system for glia-specific gene transfer in vivo that allows us to investigate the effects of both individual mutations and combinations of mutations on gliomagenesis in mice (Holland and Varmus 1998). This system utilizes replication-competent ALV splice acceptor (RCAS) viral vectors, derived from the avian retrovirus, ALV subgroup A, and a transgenic mouse line (Gtv-a) producing TVA, the receptor for ALV-A, from the astrocyte-specific promoter for the gene encoding glial fibrillary acidic protein (GFAP). Astrocytes from Gtv-a mice are susceptible to infection and gene transfer by RCAS vectors both in vivo and in vitro. We have used this system previously to show that infection of GFAP+ cells with RCAS carrying the coding sequence for basic fibroblast growth factor (bFGF) causes glial to proliferate, migrate over long distances, and assimilate into the normal brain structure without tumor formation (Holland and Varmus 1998). In this study we describe a second transgenic mouse line (Ntv-a), expressing tv-a from the nestin promoter, which is active in cells early in the glial lineage. We also describe the construction and use of an RCAS vector that transfers a potent oncogene encoding a constitutively active form of EGFR (RCAS–EGFR*) to tv-a+ cells. With these and other tools, we have compared the effects of gene transfer to Ntv-a mice (presumably to glial progenitors) and to Gtv-a mice (presumably to terminally differentiated astrocytes) and asked whether EGFR*, alone or in combination with other genetic lesions observed in human gliomas, can initiate gliomagenesis.
We show here that transfer of the EGFR* gene into either Gtv-a or Ntv-a transgenic animals induces glioma-like lesions in mice deficient for INK4a –ARF, but not in transgenic mice wild type at the INK4a –ARF locus. The frequency of gliomagenesis is higher after infection of Ntv-a mice than Gtv-a mice, consistent with the possibility that cells earlier in the glial lineage may be more susceptible to transformation than terminally differentiated astrocytes. The cooperativity between EGFR* and INK4a –ARF deficiency appears specific, as transfer of EGFR* to p53+/− mice does not result in gliomatous changes. Coinfection with RCAS–cdk4 and RCAS-EGFR* induces lesions in mice with wild-type INK4a –ARF and p53 loci at low frequency. However, this combination induces glioma-like lesions at a higher frequency in p53-heterozygous mice than in wild-type mice, implying that cdk4 overexpression and p53 deficiency also cooperate in this model system as they seem to do in human tumors.
Results
Construction of Ntv-a transgenic mice
The intermediate filament protein nestin is expressed in central nervous system (CNS) progenitors during embryonic development and in glial progenitors after birth (Tohyama et al. 1992). A transgene (NES 1689/lacZ), in which lacZ expression is under the control of the nestin promoter, produces β-galactosidase in glial and neuronal progenitors throughout the developing CNS (Lothian and Lendhal 1997). We have constructed a nestin–tv-a transgene (Ntv-a) by replacing the lacZ gene in NES 1689/lacZ with the 800-bp quail cDNA encoding the glycosylphosphatidylinositol-linked form of TVA (Fig. 2A; Bates et al. 1993; Hunter 1997). Ntv-a is designed to be expressed in glial progenitors and to allow gene transfer with RCAS vectors to this cell population. In contrast, the original Gtv-a transgenic line allows genes to be transferred primarily to terminally and near terminally differentiated astrocytes (Holland and Varmus 1998).
Figure 2.

tv-a transgenes and RCAS vectors. (A) Transgenes expressing the ALV subgroup A receptor, TVA. The astrocyte-specific Gtv-a transgene was described previously (Holland and Varmus 1998). The Ntv-a transgene utilizes a modified nestin promoter including a portion of its second intron and thymidine kinase promoter sequences that have been shown to direct expression to CNS progenitor cells (Lothian and Lendahl 1997). Transcription start sites are indicated with arrows. (B) RCAS vectors carrying an exogenous gene 3′ of env. RCAS–EGFR* expresses a constitutively active, mutant form of human EGFR (top) with deletions of intra- and extracellular sequences as indicated (TM). The region coding for the transmembrane domain; (δ) the position of the novel junction formed by the deletion in the coding region for the extracellular domain of EGFR. RCAS–AP, RCAS–bFGF, and RCAS–cdk4 carry human placental alkaline phosphatase cDNA, mouse bFGF cDNA, and human cdk4 cDNA, respectively. Although these vectors replicate only in avian cells, the exogenous genes are expressed in both avian and mammalian cells from a spliced message as illustrated.
Three independent lines of Ntv-a mice transmitted the Ntv-a transgene in a simple Mendelian inheritance pattern and were infectable with RCAS vectors. To demonstrate appropriate gene transfer, we infected both Gtv-a and Ntv-a mice at birth with RCAS–AP, which carries a gene encoding the histologic marker, alkaline phosphatase (AP) (Fig. 2B). Three weeks after infection of newborn Ntv-a mice, AP+ cells surrounded the needle track. The morphology of the AP+ cells was similar to that seen with RCAS–AP infection of Gtv-a mice (Holland and Varmus 1998; Fig. 4, below; data not shown). This is expected, because the mitotically active nestin+ cell population present at birth gives rise primarily to astrocytes and oligodendrocytes. Although nestin is expressed embryonically in neuronal progenitors, the majority of these are postmitotic after birth (Jacobson 1991), when the infection with RCAS vectors takes place and infection with avian retroviruses requires a round of cell division. Consistent with these features, we have not detected AP+ neurons in Ntv-a mice infected with RCAS–AP at birth.
Figure 4.
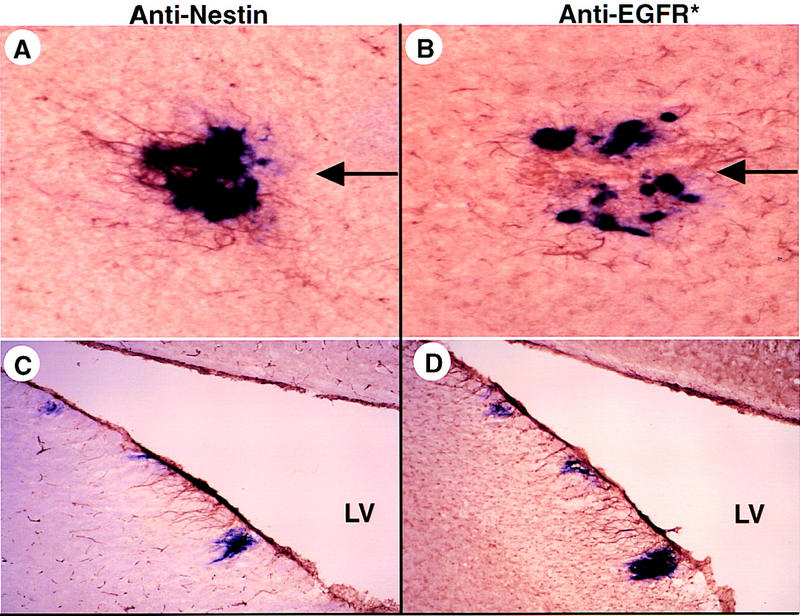
Intracerebral infection of Ntv-a mice with RCAS–AP and RCAS–EGFR*. (A) Simultaneous transfer of AP and EGFR* genes to Ntv-a astrocytes in vivo. Newborn Ntv-a mice were injected in the right frontal lobe with a mixture of DF-1 cells producing RCAS–AP or RCAS–EGFR* and sacrificed at 8 weeks of age. Sequential brain sections (frozen, 40 μm) were stained for AP activity and with either anti-nestin (A,C) or anti-EGFR* antibodies (B,D). A and B include the injection track used to introduce producer cells (arrows); C and D represent the margin of the lateral ventricle (LV). Cells with a strocytic morphology stain for nestin and EGFR* in the vicinity of AP+ cells.
Development and characterization of a vector, RCAS–EGFR*, for expression of a mutant constitutively active EGF receptor
Increased EGFR kinase activity in gliomas is associated with amplification of the EGFR gene and three general classes of mutations that result in constitutively active gene products. The most common mutation (vIII) deletes part of the extracellular domain of EGFR, causing the receptor to signal through its protein–tyrosine kinase activity in the absence of ligand (Wong et al. 1992; Ekstrand et al. 1994). A second deletion removes an intracellular kinase-regulatory domain. A third group of alleles has both kinds of deletions (Ekstrand et al. 1992). Because efficient packaging of viral RNA genomes into RCAS vectors requires inserts of 2.6 kb or less, we constructed an RCAS vector carrying the shortest (doubly deleted, EGFR*) form of the EGFR cDNA (Fig. 2B). Using a monoclonal antibody specific for the junction peptide generated by the extracellular deletion, we demonstrated with the Western blot method that the chicken fibroblast line DF-1 infected with RCAS–EGFR* produced a shortened form of EGFR (Fig. 3A), and after repeated passage, the infected cells appeared transformed morphologically (data not shown). Anti-EGFR* immunostaining of DF-1 cells infected with RCAS–EGFR* demonstrated that the expression of EGFR* could be detected specifically in a distribution consistent with a plasma-membrane location (Fig. 3B). We also confirmed that the expression of EGFR* by Gtv-a transgenic astrocytes infected with RCAS–EGFR* in vitro could be detected specifically by immunohistochemistry (Fig. 3C).
Figure 3.
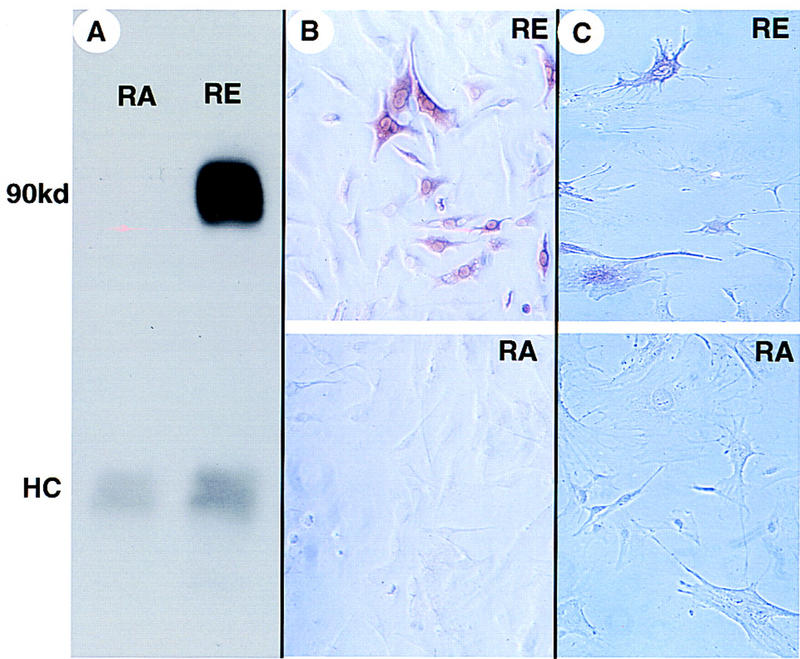
Characterization of an RCAS vector carrying a gene for activated EGFR (RCAS–EGFR*). (A) Immunoprecipitation and Western blot analysis of extracts of the chicken cell line DF-1 infected with and producing either RCAS–EGFR* (RE) or RCAS–AP (RA). A band is detected migrating at the position expected for a glycosylated protein with the sequence predicted from the EGRF* cDNA (molecular mass ∼90 kD). (HC) The position of the immunoglobulin heavy chain. Immunochemical staining with antibodies specific for EGFR* in DF-1 cells (B) or Gtv-a astrocytes (C) infected with either RCAS–AP (RA) or RCAS–EGFR* (RE).
EGFR* gene transfer in vivo
To demonstrate the effects of transferring the EGFR* gene to glia in vivo, we injected a mixture of DF-1 cells producing RCAS–AP and RCAS–EGFR* into the frontal cerebral lobes of newborn Ntv-a mice. Eight weeks later, cells adjacent to the needle track stained positive for AP activity and with antibodies to EGFR* and nestin (Fig. 4A,B). The nestin- and EGFR*-positive cells resembled mature astrocytes with small nuclei and long processes. Such cells were detected occasionally adjacent to the ventricles (Fig. 4C,D), presumably a result of infection via cerebrospinal fluid. These nestin- and EGFR*-expressing cells in Ntv-a; INK4a–ARF+/+ mice appeared normal histologically by hematoxylin and eosin (H&E) staining in five such animals. As documented in Table 1, no structural abnormalities were seen by H&E staining in 21 Gtv-aand 24 Ntv-a mice infected with a mixture of RCAS–AP and RCAS–EGFR*. Cells near the needle track or ventricles of nontransgenic littermates infected with these vectors did not stain for AP, nestin, or EGFR* (data not shown) as anticipated from earlier findings (Holland and Varmus 1998).
Table 1.
Induction of gliomas by RCAS vectors in tv-a transgenic mice
|
tv-a transgenes
|
Genes introduced with RCAS vectorsa
|
Genetic backgroundb
|
|||||
|---|---|---|---|---|---|---|---|
|
EGFR*
|
cdk4
|
bFGF
|
wild type
|
INK4a+/−
|
INK4a−/−
|
p53+/−
|
|
| Gtv-a | + | − | − | 0/21 | 1/17 | 2/6 | |
| Ntv-a | + | − | − | 0/24 | 13/25 | 8/19 | 0/16 |
| Ntv-a | + | + | − | 1/8 | 5/19 | 4/10 | 3/8 |
| Ntv-a | + | + | + | 3/29 | 4/7 | 6/16 | 17/39 |
Glioma formation, as measured by either histologic inspection of frozen brain sections or by clinical appearance of hydrocephalus, is tabulated for Gtv-a and Ntv-a transgenic mice from four different genetic backgrounds with respect to the tumor suppressors INK4a–ARF and p53, after infection of newborns with RCAS vectors carrying the indicated genes.
EGFR* (RCAS–EGFR*); cdk4 (RCAS–cdk4); bFGF (RCAS–bFGF). Tumors induced by infections with RCAS–EGFR* alone were scored at 8–10 weeks by histologic analysis of fixed frozen sections. Mice infected with combinations of RCAS–EGFR* and RCAS–cdk4 were scored for hydrocephalus between 3 and 8 weeks of age, after determining in >15 mice that early hydrocephalus is highly correlated with histologic features of gliomas.
INK4a+/− and INK4a−/− are heterozygous and homozygous for a targeted deletion of the INK4a–ARF tumor suppressor locus; and p53+/− is heterozygous for a targeted deletion of the p53 tumor suppressor gene.
Features of gliomas are induced by EGFR* gene transfer in INK4a –ARF-deficient mice
Otherwise normal tv-a transgenic mice infected with RCAS–EGFR* did not develop histologic changes characteristic of gliomas. In contrast, tv-a transgenic mice carrying targeted deletions in the INK4a –ARF locus frequently displayed glioma-like lesions after infection with RCAS–EGFR*. The properties of these lesions were varied but included several characteristics also observed in human gliomas: (1) structural abnormalities seen histologically after H&E staining (distortion of normal structures by cell mass or hydrocephalus); (2) increased cell density; (3) immunopositivity for expression of the mutant form of EGFR; and (4) immunopositivity for GFAP (identifying astrocytic character) and nestin (indicating a more primitive expression pattern). For the purpose of this study lesions were defined as gliomas if they displayed all four characteristics.
One such lesion in the hippocampus of an Ntv-a mouse infected with RCAS–AP and RCAS–EGFR* is illustrated in Figure 5 (A–C). This lesion demonstrates increased cell density, immunohistochemical staining for both EGFR* and GFAP, increased vascularity and tumor cells surrounding blood vessels at a distance from the tumor mass. A second lesion generated by EGFR* gene transfer into a Gtv-a mouse homozygous for the mutant INK4a–ARF allele (Fig. 5D–I) shows evidence of previous hemorrhages, increased vascularity and cell density, as well as invasion of adjacent normal tissue. This lesion was sufficiently large to obtain numerous sections for immunohistochemical staining for other gene products. Similar to what has been reported for high-grade human gliomas (Takahashi et al. 1990; Plate et al. 1994), cells in the implicated region overexpress endogenous bFGF and vascular endothelial growth factor (VEGF). The cells comprising this lesion also stain with antibodies to GFAP and nestin, indicating an immature astrocytic cell type.
Figure 5.
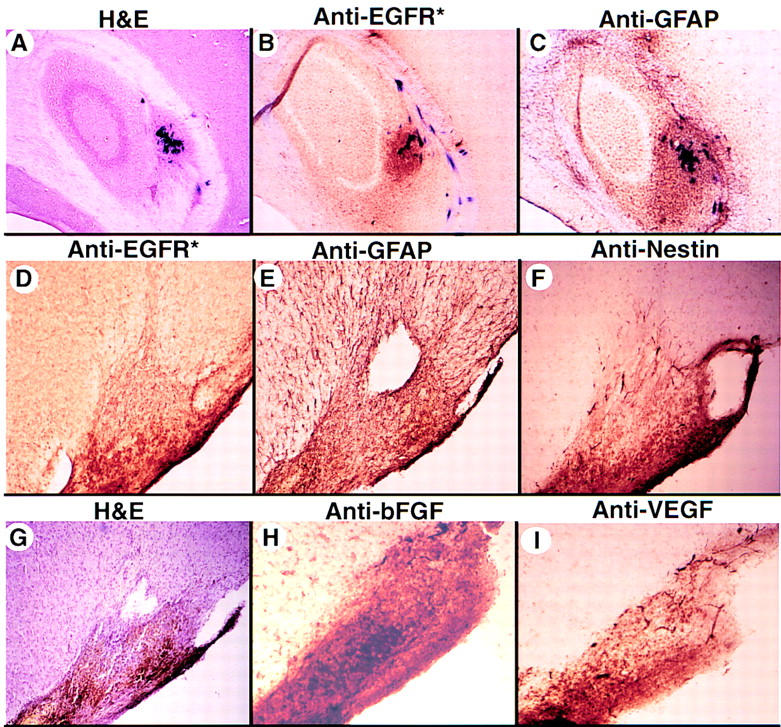
EGFR*-induced lesions in INK4a–ARF+/− and INK4a–ARF−/− mice. (A–C) Eight weeks after injection with RCAS–AP and RCAS–EGFR*, the brain from a Ntv-a; INK4+/− mouse was sectioned (frozen, 40 μm), stained for AP activity, and then stained with either H&E (A), or monoclonal antibodies recognizing EGFR* (B) or GFAP (C), as indicated. (A–C) Magnification, 40×. This tumor, located in the hippocampus, is a mixture of AP+ and AP− cells that express EGFR* and GFAP, indicating an astrocytic cell origin. (D–I) Brain sections from a 10-week-old Gtv-a; INK4a–ARF−/− mouse infected with RCAS–EGFR*. The brain was fixed, and frozen sections (40 μm) were either stained with H&E, illustrating high cell density and previous hemorrhage (G) or stained with polyclonal antibodies to EGFR* (D), GFAP (E), nestin (F), bFGF (H), or VEGF (I). Magnification, 40× (A–C); 100× (D–I).
Another histopathological feature is illustrated in sections from a Gtv-a; INK4a –ARF+/− mouse infected with RCAS–AP and RCAS–EGFR* (Fig. 6). This mouse developed hydrocephalus at 6 weeks of age (recognized clinically by macrocephaly, lethargy, and dehydration) and was sacrificed. The lesion was comprised of EGFR*- and GFAP-positive cells spread diffusely throughout the brain stem and thalamus, leading to an increased cell density in these regions. Areas of intraparenchymal hemorrhage in brains with similar lesions were adjacent to astrocytic cells staining for EGFR* and nestin, but a distinct tumor mass was usually absent (data not shown).
Figure 6.
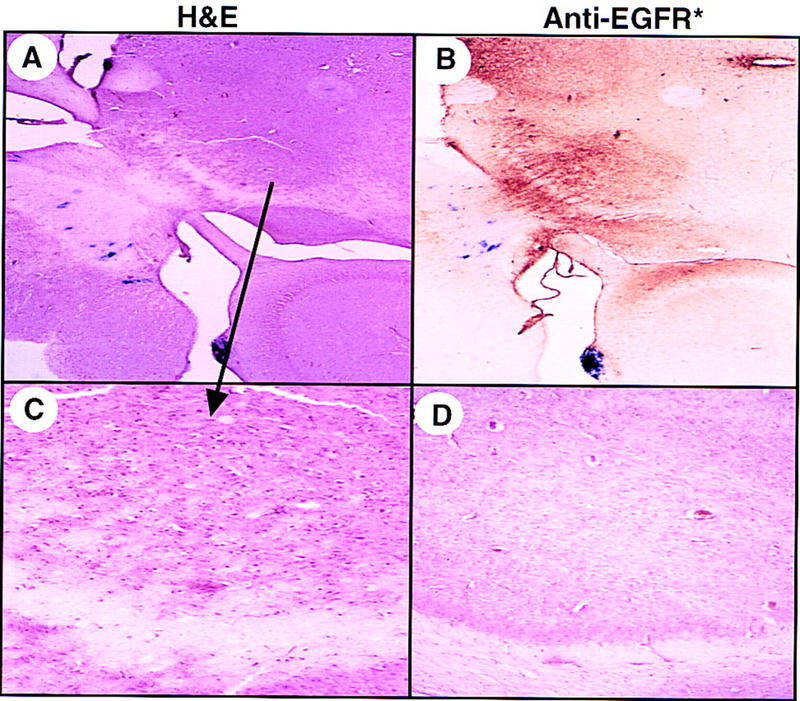
Diffuse thalamic glioma associated with hydrocephalus. Frozen brain sectcions (40 μm) from a Gtv-a;INK4a–ARF+/− mouse infected at birth with both RCAS–AP and RCAS–EGFR* and analyzed after the development of hydrocephalus at 6 weeks of age. Brain sections were stained for alkaline phosphatase activity, and then stained with either H&E (A) or monoclonal antibodies to EGFR* (B). (C) A higher magnification of the H&E section in A and compared to the same anatomic region of an infected littermate that had not developed hydrocephalus (D). Magnification, 40× (A,B); 100× (C,D).
EGFR*-induced gliomagenesis requires disruption of cell-cycle arrest pathways and occurs more frequently after infection of Ntv-a than Gtv-a mice
As described earlier, we did not detect any gliomas after RCAS–EGFR* infection of Gtv-a or Ntv-a transgenic mice wild type for the INK4a –ARF locus. Only one glioma was observed after infection of 17 Gtv-a transgenic; INK4a –ARF+/− mice with RCAS–EGFR*, and two mice developed gliomas after infection of six Gtv-a; INK4a –ARF−/− mice (see Table 1). Gliomas were induced at a much higher frequency in Ntv-a; INK4a –ARF-heterozygous and -nullizygous animals (13 of 25 and 8 of 19, respectively). Furthermore, infection of Ntv-a and Gtv-a mice with RCAS–AP indicate that the efficiency of viral uptake and expression of these two mouse lines are similar (data not shown). These results suggest two signficant conclusions: (1) induction of gliomagenesis by those producing a EGFR* requires additional genetic lesions, such as deficiency of p16INK4a, p19ARF, or both; and (2) gliomas arise with greater efficiency when EGFR* is introduced into Ntv-a mice, presumably into cells relatively early in the glial lineage. The second conclusion may imply that either glial progenitors are generally more prone to malignant transformation or that the specific interaction between EGFR signaling and G1 and cell-cycle arrest is more efficient in cells earlier in the glial lineage.
The data summarized in Table 1 suggest that INK4a –ARF+/− mice are as susceptible to RCAS–EGFR*-induced gliomas as are INK4a –ARF−/− mice. The most obvious explanation for this is that either the remaining INK4a –ARF allele in tumors arising in INK4a –ARF+/ − mice is inactivated at high frequency or that haploinsufficiency of the INK4a –ARF locus is sufficient for glioma formation. We attempted to demonstrate the loss of p16INK4a expression with an anti-p16 monoclonal antibody in a histochemical assay; unfortunately, we found the level of p16INK4a in the normal adult mouse brain below the level of detection. Therefore, we were unable to measure loss of expression. The small size and diffuse character of the gliomas prevented dissection of tumor from normal tissue in most of the mice heterozygous for INK4a –ARF. In one sample large enough to permit dissection, PCR analysis demonstrated maintenance of the heterozygous INK4a –ARF genotype in the tumor tissue (data not shown). This single finding is difficult to interpret, but could indicate either contamination of the tumor tissue with normal cells or inactivation of the remaining wild-type allele in some other way, such as methylation.
bFGF gene transfer does not enhance EGFR*-induced glioma formation
bFGF is overexpressed in most high-grade gliomas (Takahashi et al. 1990), and we have shown previously that RCAS–bFGF induces proliferation and migration of astrocytes without tumor formation in Gtv-a mice (Holland and Varmus 1998). The addition of RCAS–bFGF to the combination of RCAS–EGFR* and RCAS–cdk4 for infection of Ntv-a mice did not change either the type of lesion or the age at onset of symptoms appreciably in any of the genetic backgrounds tested (Table 1; data not shown). One explanation for this finding is that sufficient expression of bFGF may be induced in gliomas secondary to EGFR* gene expression, as in the tumor illustrated in Fig. 4. If bFGF expression is essential for gliomagenesis in mice, its induction appears not to be rate-limiting because the addition of virally transduced bFGF did not appear to affect glioma formation.
CDK4 overexpression can cooperate with EGFR* and is required for EGFR*-induced gliomagenesis in p53+/− mice
In human gliomas, mutations and amplification of the EGFR gene are found more frequently in gliomas that lack INK4a –ARF (and hence cannot make p16INK4a or p19ARF) than in tumors with abnormalities of cdk4 or Rb (Hayashi et al. 1997). Furthermore, EGFR and p53 mutations appear to be mutually exclusive in these tumors (Watanabe et al. 1996). These observations are consistent with recent data demonstrating that loss of p19ARF and p53 mutations appear to be mutually exclusive in mouse fibroblast culture (Kamijo et al. 1997), and that RAS-induced INK4a –ARF-deficient melanomas remain consistently wild type for p53 (Chin et al. 1997). Gliomas with amplification of the CDK4 gene most frequently harbor p53 mutations, implying synergy between the effects of these two genetic alterations in gliomagenesis. One interpretation of these findings might be that the combination of CDK4 amplification and p53 loss (or mdm2 amplification) inactivates both the Rb and p53 cell-cycle arrest pathways, similar to the effect of INK4a –ARF loss (see Fig. 1), and that inactivation of both pathways promotes gliomagenesis strongly (see Discussion).
In human gliomas, EGFR and p53 mutations are found in the same tumor rarely, implying a lack of cooperation between the effects of these two abnormalities. To investigate the interaction of EGFR mutations and p53 loss in our model, we infected Ntv-a; p53+/− mice with RCAS–EGFR*. No glioma-like lesions or other intracranial structural abnormalities appeared in 16 p53+/−; Ntv-a mice after infection with RCAS–EGFR*.
Multiple genes can be transferred to specific cell types in tv-a transgenic mice. Therefore, we used mixtures of RCAS vectors (RCAS–cdk4 and RCAS–EGFR* or RCAS–cdk4, RCAS–EGFR*, and RCAS–bFGF) to infect tv-a transgenic mice, including mice that also carry mutations in tumor suppressor genes. As demonstrated above, RCAS–EGFR* alone was unable to generate gliomas in wild-type mice; however, the addition of RCAS–cdk4 to RCAS–EGFR* produced hydrocephalus in ∼10% of Ntv-a mice with an otherwise wild-type genetic background. We examined two of these hydrocephalic brains from Ntv-a mice infected with RCAS–cdk4 and RCAS–EGFR* and found histologic features of diffuse gliomas. These gliomas depend presumably on the cooperative effects of excessive EGFR signaling and disruption of the G1 arrest pathway caused by overexpression of CDK4. However, we did not determine the status of p53, Mdm2, and p16ARF in these tumors.
Strikingly, although RCAS–EGFR* alone caused no abnormalities in Ntv-a; p53+/− mice, the addition of RCAS–cdk4 to RCAS–EGFR* caused hydrocephalus and death within the first 3–5 weeks of life in 40% of p53 heterozygous mice. In the hydrocephalic mouse brain, the cerebrospinal fluid (CSF) was frequently frankly bloody and associated with subdural hematomas, intraparenchymal hemorrhage, vascular proliferation, and increased cell density and gliotic changes (Fig. 7). A periventricular proliferation of glia was also observed, with astrocytic cells that expressed GFAP, nestin, and EGFR* diffusely infiltrating the thalamus and striatum. Thus, in our mouse model, overexpression of CDK4 appears to be required for either efficient induction or progression of gliomas by EGFR* in p53-deficient mice. Although it is probable that gliomas arising in p53+/− mice have lost the remaining wild-type p53 allele, similar to what has been reported for other tumor models arising in p53+/− mice (Donehower 1995), because of the diffuse nature of the tumors and heavy contamination with normal cells we were unable to determine the status of the p53 locus in these lesions by PCR or Southern blot analysis.
Figure 7.
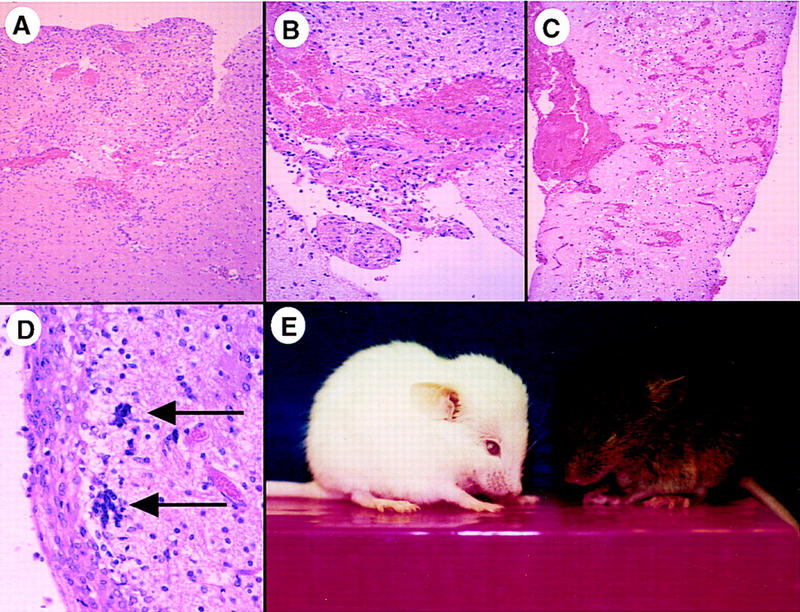
Characteristics of brains harboring lesions induced by multiple mutations. (A–D) Microscopic findings after RCAS–EGFR* and RCAS–cdk4 infection of Ntv-a; INK4a–ARF−/− mice. (A) Increased cell density and vascularity relative to adjacent brain parenchyma (40×). (B) Vascular endothelial hyperproliferation (100×). (C) Intraparenchymal hemorrhage throughout the cortex (40×). (D) Multinucleated cells (arrows) and nuclear pleomorphism (200×). (A–D) Paraffin-embedded sections stained with H&E. (A,D) The same mouse sacrificed at 5 weeks of age; (B,C) Two other mice sacrificed at 4 and 5 weeks, respectively. (E) The appearance of two hydrocephalic Ntv-a mice infected with RCAS vectors at birth. The white mouse was Ntv-a; p53+/−, infected with RCAS–cdk4, RCAS–bFGF, and RCAS–EGFR* and photographed at 4 weeks of age; the brown mouse was Ntv-a; INK4a–ARF−/−, infected with RCAS–bFGF and RCAS–EGFR*, and photographed at 10 weeks of age.
Discussion
We have generated an animal model for gliomagenesis that recapitulates the genetic abnormalities found in human gliomas. The lesions generated by this model have many characteristics similar to those found in human gliomas, including expression of glial markers and induction of angiogenic growth factors such as bFGF and VEGF. Human gliomas have a number of histologic characteristics; we find many of these histologic characteristics in the lesions induced in our mouse model.
All the tumors we have generated thus far require expression of the activated form of EGFR (EGFR*) as well as combinations of lesions affecting cell-cycle regulation. EGFR* induces gliomas in conjunction with overexpression of CDK4 only occasionally, whereas INK4a–ARF loss, which eliminates both the Rb- and p53-mediated pathways for cell-cycle arrest, cooperates efficiently with EGFR* in gliomagenesis. In addition, EGFR* appears incapable of generating gliomas in a p53-deficient background unless CDK4 is also overproduced. These patterns of mutations—especially the cooperation between EGFR* and INK4a–ARF deficiency and the apparent inability of EGFR* alone to stimulate gliomagenesis in p53-deficient mice—resemble those seen in human gliomas.
Infection of tv-a transgenic mice with RCAS vectors as a method for modeling human cancer
There are various methods for modeling the contribution that combinations of mutations make to oncogenesis. Cell-type-specific gene transfer using RCAS vectors has a number of advantages over previously reported strategies. First, with the use of transgenic mouse lines expressing tv-a in specific cell types, combinations of genes can be tested by the use of easily constructed or previously existing viral vectors. Therefore, the cost and time required to test complex combinations of genetic lesions are significantly less than for strategies in which each genetic alteration requires a separate line of mice, and multiple matings are necessary to observe interactions between mutations (e.g., Kwan et al. 1992; Donehower 1995). The tv-a system can also combine gain-of-function (by infection with RCAS vectors carrying oncogenes) and loss-of-tumor-suppressor-gene function, either as shown here by breeding to mice with targeted germ-line deletions or in the future by deleting alleles that contain lox recombination sites by infecting with an RCAS vector carrying cre, encoding a lox-specific recombinase. Second, classic transgenic strategies express oncogenes in all cells utilizing the transgenic promoter. Cells giving rise to tumors from such a population have acquired further (usually unknown) mutations resulting in tumor formation. In contrast, when injected into the brain, RCAS vectors infect only a few hundred cells in the vicinity of the injection track (Holland and Varmus 1998). Therefore, any observed phenotypes are more likely to be direct effects of the transferred genes, since a relatively small number of cells would be at risk initially of secondary mutations. Finally, retroviral insertional mutagenesis has been used to identify genes that cooperate with oncogenic transgenes (e.g., van Lohuizen et al. 1991; Shackleford et al. 1993). This strategy can detect novel genes, but it is likely to recognize the combinations that result in the most rapidly growing cell populatios. In contrast, because of the limited number of cells infected initially by RCAS vectors, the tv-a system will be informative mainly about known genes carried by RCAS vectors, but it can be used to assess weakly oncogenic combinations.
Gliomagenesis requires multiple mutations
Combinations of mutations appear to be essential for the induction of gliomas in mice, because in our study single mutations were incapable of generating gliomas with any measurable frequency. We have shown previously that neither bFGF (Holland and Varmus 1998) nor CDK4 gene transfer alone (Holland et al. 1998) results in glioma formation in mice, and p53-deficient mice have not been reported to develop gliomas (Donehower et al. 1992). In this report we show that EGFR* gene transfer alone does not induce gliomagenesis either. Furthermore, INK4a–ARF-deficient mice develop normally without glioma formation (Serrano et al. 1996); therefore loss of both p16INK4a and p19ARF also appears insufficient to induce gliomagenesis.
Similar to the experience of others with INK4a–ARF-deficient human gliomas (Louis et al. 1993; Lang et al. 1994; Rasheed et al. 1994; Schlegel et al. 1994; He et al. 1995; Hayashi et al. 1997), we find cooperation between loss of INK4a–ARF and expression of mutant EGFR during gliomagenesis. In our study the percent of mice developing EGFR*-induced gliomas was similar in INK4a–ARF+/− and INK4a–ARF−/− genetic backgrounds. We were unable to test conclusively whether tumors arising in INK4a–ARF+/− mice retained a functional INK4a–ARF allele. It is possible either that haploinsufficiency of the INK4a–ARF gene allows EGFR* to induce gliomas in these mice or that the tumorigenic cells are nullizygous physically or functionally. In one published series of 120 human glioblastomas, 38% of the tumors demonstrated deletion of both copies of INK4a–ARF, whereas 33% maintained one wild-type allele of INK4a–ARF. Furthermore, the majority of gliomas in this series with hemizygous deletion of INK4a–ARF did not have mutations in the remaining INK4a–ARF allele (Ichimura et al. 1996). Methylation of the INK4a–ARF locus in human gliomas is one possible mechanism for loss of p16INK4a protein production in gliomas with hemizygous deletions of INK4a–ARF, although in most tumors in this series there was evidence for neither methylation nor mutation of the remaining INK4a–ARF locus (Schmidt et al. 1994).
INK4a–ARF appears to be normal in ∼30%–50% of gliomas in adults. These gliomas usually contain p53 mutations and display EGFR mutations rarely; by the time they reach grade 4 status, they frequently overexpress cdk4 or lose Rb (von Deimling et al. 1993). In Holland et al. (1998), we show that CDK4-immortalized astrocyte cultures sometimes acquire p53 mutations. Although the p53 mutations are not required to escape senescence or destabilize the genome—a phenomenon that we attribute to the varied consequences of expressing high levels of CDK4—they may enhance growth potential. These observations in cell culture are further evidence that overexpression of CDK4 and loss of p53 may cooperate in some way to achieve more rapid proliferation of astrocytes in vivo or in vitro.
Our data indicate that RCAS–EGFR* can efficiently induce glioma-like changes either in INK4a–ARF−/− animals or in animals carrying an inactive p53 gene and coinfected with RCAS–cdk4. These results are consistent with the view that INK4a–ARF governs both the RB and p53 pathways and that the inactivation of both pathways is essential for efficient EGFR*-induced gliomagenesis. In this sense, the mutations that promote gliomagenesis by EGFR* resemble the action of SV40 T antigen, which is known to achieve its oncogenic effect by disrupting both p53 and RB function (Fanning 1992).
In the accompanying paper, we show that either loss of INK4a–ARF or overexpression of CDK4 can lead to immortalization of astrocytes in culture (Holland et al. 1998). Clearly, there must be additional constraints on proliferation in vivo to prevent cells with inactivated G1 arrest pathways from proliferating abnormally, as neither infection withRCAS–cdk4 n or the INK4a–ARF−/− genotype is sufficient to induce gliomas. In this study we show that if cells with aberrant G1 arrest pathways also express a constitutively active EGFR, they are capable of escaping these apparent constraints. Many signaling pathways function downstream of EGFR, and some involve proteins governing progression through the G1 phase of the cell cycle. However, it is not clear which pathways are involved in gliomagenesis or how they cooperate with the inactivation of G1 arrest mechanisms.
Previous work has indicated that although bFGF and VEGF are overexpressed in most high-grade human gliomas, these genes are not mutated or amplified. These findings have been interpreted to suggest that augmented activity of these genes is secondary to tumor initiation and progression. The mouse glioma illustrated in Figure 5 was initiated by infection with RCAS–EGFR*, but also produces high levels of endogenous bFGF and VEGF. These findings imply that overexpression of bFGF and VEGF can be induced secondarily and support the notion that regulation of growth factors in human gliomas can be regulated in response to other genetic alterations. For example, an active form of Ras, a signaling protein also activated by EGFR, has been shown to enhance VEGF expression in glial cell culture (Okata et al. 1998).
Thus far, our gene transfer model for gliomagenesis reflects the patterns of mutations seen in human gliomas and, therefore, represents a useful tool for understanding the molecular basis of gliomagenesis. Using glioma formation as an assay, it is possible to identify the tumorigenic components of signaling pathways downstream of EGFR or implicated in G1 arrest. These components may prove to be targets suitable for interventions directed against human gliomas.
Materials and methods
Constructs
The Gtv-a transgene (with a 2.2-kb fragment of the GFAP promoter driving expression of the quail tv-a cDNA and a fragment from the mouse protamine gene (MP-1) supplying an intron and signal for polyadenylation), RCAS–AP, RCAS–bFGF, and RCAS–cdk4 have been described (Holland and Varmus 1998; Holland et al. 1998). RCAS–puro was obtained from Steve Hughes (National Cancer Institute). The Ntv-a transgene utilizes a modified nestin promoter, including portions of its second intron and thymidine kinase promoter sequences that have been shown to direct expression to CNS progenitor cells; polyadenylation sequences are provided by SV40 DNA. This transgene was constructed by NotI digestion of the NES 1689/lacZ (Lothian and Lendhal 1997) to release the lacZ gene, followed by incubation with Klenow fragment of DNA polymerase and ligation to the tv-a cDNA from pSPKE 0.8 (Bates et al. 1993), which had been digested with EcoRV and SmaI to achieve blunt, compatible ends. RCAS–EGFR* was constructed by ClaI digestion of RCAS–puro to remove the puromycin-resistance gene and replacement with a fragment from pIND5′3′EGFR (Ekstrand et al. 1992; gift of David James, Mayo Clinic Rochester, and Chris Thomas, Mayo Clinic Jacksonville). This fragment contains a human EGFR cDNA lacking sequences corresponding to exons 2–7 (bases 275–1075) and sequences 3′ of base 3133.
Mice
Production of the Gtv-a mouse line has been described (Holland and Varmus 1998). The Gtv-a mouse line was generated originally from an FVB/N crossed with a C57B6 X BALB/C F1. The Gtv-a founder was then bred to an FVB/N to generate F1 progeny which have been interbred subsequently to maintain the transgenic line. Production of the FVB/N Ntv-a mouse line was by pronuclear injection of the Ntv-a transgene, which had been released from the plasmid by digestion with HinDIII. Mice with mixed C57BL/6 and 129 genetic background and with targeted, inactivated mutations of INK4a–ARF and p53 have been described previously (Donehower et al. 1992; Serrano et al. 1993). The genetic backgrounds of the tv-a transgenic mice used for infection were therefore mixes of FVB/N, 129, and C57BL6.
Cell culture
Primary brain cell cultures from newborn transgenic mice were obtained by mechanical dissociation of the whole brain, followed by digestion with 0.25% trypsin for 15 min at 37°C. Large debris was allowed to settle, and single cells were plated and grown in DMEM with 10% fetal calf serum (GIBCO–BRL). DF-1 cells, an immortalized line of chicken cells were a gift from Doug Foster, University of Minnesota, and were grown in DMEM with 5% fetal calf serum, 5% calf serum, 1% chicken serum, and 10% tryptose phosphate broth (GIBCO–BRL).
Infection in cell culture
The supernatant from DF-1 cells infected with and producing RCAS vectors was filtered through a 0.45-μm filter and plated directly onto primary brain cells cultures from Gtv-a mice. These primary brain cultures were infected with filtered medium from RCAS–puro-producing cells and then selected in 4 μg/ml puromycin.
Immunoprecipitation, Western blot analysis, and immunoperoxidase staining of cultured cells
Proteins were isolated from cultured cells using Tris-buffered saline (pH 8.0) with 1.0% Tween (TBST) and precipitated overnight at 4°C with 0.4 μg of mouse monoclonal anti-mutant EGFR (gift of Darrel Bigner, Duke University, Durham, NC; Wikstrand et al. 1995) using anti-mouse immunoglobulin–Sepharose (Sigma). The products were separated by gel electrophoresis and transferred to nictrocellulose. The blot was incubated in TBST with the same anti-mutant EGFR antibody, washed extensively in TBST, incubated with a peroxidase-conjugated goat anti-mouse antibody (Boehringer), and then detected with ECL (Amersham).
For immunoperoxidase analysis, cultured cells were fixed in 100% methanol and blocked in TBST with 1% goat serum. They were then incubated for 1 hr at room temperature after the addition of 0.5 μg of anti-mutant EGFR antibody, washed extensively with TBST, and detected with peroxidase-conjugated anti-mouse antibody (ABC, Vector).
Infection of transgenic mice
DF-1 cells infected with and producing RCAS vectors were harvested by trypsin digestion and pelleted by centrifugation, the cell pellets were resuspended in ∼ 50 μl of medium and placed on ice. Using a 10-μl gas-tight Hamilton syringe, a single intracranial injection of 1 μl containing 104 cells was made in the right frontal region of newborn mice, just anterior to the striatum, with the tip of the needle just touching the skull base.
Brain sectioning and immuno- and histochemical staining
Animals were sacrificed at 4–10 weeks of age, the brains fixed in 4% formaldehyde, 0.4% glutaraldehyde, 1× PBS for 36 hr, and then dehydrated in 20% sucrose, 2% glycerol, 1× PBS. Frozen sections (40 μm) were obtained using a sledge microtome (Zeiss). The sections were then stained in solution for alkaline phosphatase activity using 5-bromo-4-chloro-indolyl-phosphate and 4-nitro-blue-tetrazolium-chloride (Boehringer) after treatment at 65°C (pH 9.5) for 3 min to remove endogenous alkaline phosphatase activity. The sections were then mounted on glass slides and counterstained with hematoxylin and eosin. For immunostaining, fixed frozen sections (40 μm) were stained in solution for AP as described. The sections were then treated with 10% hydrogen peroxidase/70% methanol for 15 min to inactivate endogenous peroxidases. The sections were then blocked with 1% goat serum in Tris-buffered saline (pH 8.0), with 0.1% Tween (TBST) solution for 20 min followed by a 1-hr incubation at room temperature after the addition of mouse monoclonal antibodies to human GFAP (Boehringer), rat nestin (Pharmingen), bovine bFGF (Calbiochem), mouse VEGF (Upstate Biochemicals), or to the vIII mutant form of human EGFR (gift of Darell Bigner; Wiksrand et al. 1995) or rabbit polyclonal antibodies to the human vIII mutant EGFR (gift of Albert Wong, Thomas Jefferson University, Philadelphia, PA). The sections were washed extensively with TBST and antibody staining was then visualized with peroximase-conjugated anti-mouse antibody (ABC, Vector) and sections then mounted on glass slides.
Acknowledgments
We thank Doug Foster for the DF-1 cells, Han-Woong Lee for providing the INK4–ARF null mice, Chris Thomas for suggesting and supplying the mutant EGFR cDNA, Steve Hughes for the RCAS-puro vector, Urban Lendhal for the nestin promoter used to generate the Ntv-a mouse line, Darell Bigner for the mouse monoclonal anti-mutant EGFR antibody, Albert Wong for the rabbit polyclonal anti-mutant EGFR antibody, Ed Harlow for the anti-p16 antibody, Rod Bronson for helpful discussion, and Martin Raff for urging us to infect cells early in the glial lineage. Lisa Garrett and Amy Chen performed the injections of fertilized eggs that generated the Ntv-a mouse line, Gene Elliott and Theresa Hernandez provided outstanding animal husbandry. R.A.D. is supported by grants from the National Institutes of Health (grant EY11267) as well as a recipient of the Irma T. Hirschl Career Scientist Award. E.C.H. was a Howard Hughes Physician Postdoctoral Fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
We have detected aberrant transcripts that could encode the amino-terminal portion of p19ARF from the INK4a–ARF exon 1β in astrocytes derived from the mice used in this study having targeted deletions in the INK4a–ARF locus (W.P. Hively, E.C. Holland, R.A. DePinho, H.E. Varmus, and M. Serrano, unpubl.). However, cells from these mice and from mice with a mutation that specifically eliminates exon 1β (Kamijo et al. 1997) have very similar phenotypic properties with respect to p19ARF functions in apoptosis, transformation, and ploidy (Serrano et al. 1996; Zindy et al. 1998; W.P. Hively, E.C. Holland, R.A. DePinho, H.E. Varmus, and M. Serrano, unpubl.). Therefore, the INK4a–ARF mutants we have used in this paper and in Holland et al. (1998) are likely to be severely hypomorphic or null with respect to p19ARF function.
Footnotes
E-MAIL eholland@notes.mdacc.tmc.edu; FAX (713) 794-4950.
References
- Bates P, Young JA, Varmus HE. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell. 1993;74:1043–1051. doi: 10.1016/0092-8674(93)90726-7. [DOI] [PubMed] [Google Scholar]
- Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, Horner JWS, DePinho RA. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes & Dev. 1997;21:2822–2834. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel J, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, Grey J, Bradley A, Medina D, Varmus HE. Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes & Dev. 1995;9:882–895. doi: 10.1101/gad.9.7.882. [DOI] [PubMed] [Google Scholar]
- Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci. 1992;89:4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, Collins VP, James CD. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9:2313–2330. [PubMed] [Google Scholar]
- Fanning E. Modulation of cellular growth control by SV40 large T antigen. In: Doerfler W, Bohm P, editors. Malignant transformation by DNA viruses. Weinheim, Germany: VCH; 1992. pp. 1–19. [Google Scholar]
- Hawkins RA, Killen E, Whitte IR, Jack WJ, Chetty U, Prescott RJ. Epidermal growth factor receptors in intracranial and breast tumours: Their clinical significance. Br J Cancer. 1991;63:553–560. doi: 10.1038/bjc.1991.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol. 1997;7:871–875. doi: 10.1111/j.1750-3639.1997.tb00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Allen JR, Collins VP, Allalunis-Turner MJ, Godbout R, Day RS, James CD. CDK4 amplification is an alternative mechanism to p16 gene homozygous deletion in glioma lines. Cancer Res. 1994;54:5804–5807. [PubMed] [Google Scholar]
- He J, Olson JJ, James CD. Lack of p16INK4 or retinoblastoma protein (pRb) or amplification-associated overexpression of cdk4 is observed in distinct subsets of malignant glial tumors and cell lines. Cancer Res. 1995;55:4833–4836. [PubMed] [Google Scholar]
- Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci. 1998;95:1218–1223. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, E.C., W.P. Hively, V. Gallo, and H.E. Varmus. 1998. Overexpression of cdk4 but not loss of INK4a–ARF induces hyperploidy in cultured mouse astrocytes, modeling mutations in the G1 arrest pathway in human gliomas. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Hunter E. Viral entry and receptors. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor, NY: Cold Spring Laboritory Press; 1997. pp. 71–120. [PubMed] [Google Scholar]
- Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–1072. [PubMed] [Google Scholar]
- Jacobson M. Developmental neurobiology. 3rd ed. New York, NY: Plenum Press; 1991. pp. 41–89. [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Dowling JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Burger PC, Scheithauer BW. Histological typing of tumours of the central nervous system. Germany: Springer-Verlag Berlin; 1993. [Google Scholar]
- Kwan H, Pecenka V, Tsukamoto A, Parslow T, Guzman R, Lin T, Muller WJ, Lee FS, Leder P, Varmus HE. Transgenes expressing the Wnt-1 and int-2 proto-oncogenes cooperate during mammary carcinogenesis in doubly transgenic mice. Mol Cell Biol. 1992;12:147–154. doi: 10.1128/mcb.12.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang FF, Miller DC, Koslow M, Newcomb EW. Pathways leading to glioblastoma multiforme: A molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg. 1994;81:427–436. doi: 10.3171/jns.1994.81.3.0427. [DOI] [PubMed] [Google Scholar]
- Lothian C, Lendahl U. An evolutionarily conserved region in the second intron of the human nestin gene directs gene expression to CNS progenitor cells and to early neural crest cells. Eur J Neurosci. 1997;9:452–462. doi: 10.1111/j.1460-9568.1997.tb01622.x. [DOI] [PubMed] [Google Scholar]
- Louis DN, von Deimling A, Chung RY, Rubio M, Waley JM, Eibl RH, Ohgaki H, Wiestler OD, Thor AD, Seizinger BR. Comparative study of p53 gene and protein alterations in human astrocytic tumors. J Neuropathol Exp Neurol. 1993;52:31–38. doi: 10.1097/00005072-199301000-00005. [DOI] [PubMed] [Google Scholar]
- Okata F, Rak JW, St. Croix B, Lieubeau B, Kaya M, Roncari L, Shirasawa S, Sasazuki T, Kerbel RS. Impact of oncogenes in tumor angiogenesis: Mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenesity of human colorectal carcinoma cells. Proc Natl Acad Sci. 1998;95:3609–3614. doi: 10.1073/pnas.95.7.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono Y, Yamiya T, Ichikawa T, Kunishio K, Matsumoto K, Furuta T, Ohmoto T, Ueki K, Loius DN. Malignant astrocytomas with homozygous CDKN2/p16 gene deletions have higher Ki-67 proliferation indices. J Neuropathol Exp Neurol. 1996;55:1026–1031. [PubMed] [Google Scholar]
- Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor and glioma angiogenesis: Coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer. 1994;59:520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encoding two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Rasheed BK, McLendon RE, Herndon JE, Friedman HS, Friedman AH, Bigner DD, Bigner SH. Alterations of the TP53 gene in human gliomas. Cancer Res. 1994;54:1324–1330. [PubMed] [Google Scholar]
- Schlegel J, Merdes A, Stumm G, Albert FK, Forsting M, Hynes N, Kiessling M. Amplification of the epidermal-growth-factor-receptor gene correlates with different growth behavior in human glioblastoma. Int J Cancer. 1994;56:72–77. doi: 10.1002/ijc.2910560114. [DOI] [PubMed] [Google Scholar]
- Schaefer-Klein J, Givol I, Barsov EV, Witcomb JM, VanBrocklin M, Foster DN, Federspiel MJ, Hughes SH. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology. 1998;248:305–311. doi: 10.1006/viro.1998.9291. [DOI] [PubMed] [Google Scholar]
- Schmidt EE, Ichimura K, Reifenberger G, Collins VP. CDKN2 (p16/MTS1) gene deletion or cdk4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321–6324. [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell motility. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Shackelford GM, MacArthur CA, Kwan HC, Varmus HE. Mouse mammary tumor virus infection accelerates mammary carcinogenesis in Wnt-1 transgenic mice by insertional activation of int-2/Fgf3 and hst/Fgf-4. Proc Natl Acad Sci. 1993;90:740–744. doi: 10.1073/pnas.90.2.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J A, Hirotaka M, Fukumoto M, Igarashi K, Jaye M, Oda Y, Kikuchi H, Hatanaka M. Gene expression of fibroblast growth factors in human gliomas and meningiomas: Demonstration of cellular source of basic fibroblast growth factor mRNA and peptide in tumor tissues. Proc Natl Acad Sci. 1990;87:5710–5714. doi: 10.1073/pnas.87.15.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama T, Lee V, Rorke LB, Marvikn M, McKay RD, Trojanowski JQ. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumors. Lab Invest. 1992;66:303–313. [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Guilden H, Berns A. Identification of cooperating oncogenes in Eu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- van Meyel DJ, Ransay DA, Casson AG, Keeney M, Chambers AF, Cairncross JG. P53 mutation, expression and DNA ploidy in evolving gliomas: Evidence for two pathways of progression. J Natl Cancer Inst. 1994;86:1011–1017. doi: 10.1093/jnci/86.13.1011. [DOI] [PubMed] [Google Scholar]
- von Deimling A, von Ammon K, Schoenfeld D, Wiestler OD, Seizinger BR, Louis DN. Subsets of glioblastoma multiforme defined by molecular genetic analysis. Brain Pathol. 1993;3:19–23. doi: 10.1111/j.1750-3639.1993.tb00721.x. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Tachibana O, Sato K, Yonekawa Y, Kleihues P, Ohgaki H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996;6:217–224. doi: 10.1111/j.1750-3639.1996.tb00848.x. [DOI] [PubMed] [Google Scholar]
- Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humprhey PA, Kurpad SN, McLendon RE, Moscatello D, Pegram CN, Reist CJ, Traweek ST, Wong AJ, Zalutsky MR, Bigner DD. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinoma and malignant gliomas. Cancer Res. 1995;55:3140–3148. [PubMed] [Google Scholar]
- Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DD, Vogelstein B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]