

Abstract
Similarities exist between the progressive cerebellar ataxia in ataxia telangiectasia (AT) patients and a number of neurodegenerative diseases in both mouse and man involving specific mutations in ion channels and/or ion channel activity. These relationships led us to investigate the possibility of defective ion channel activity in AT cells. We examined changes in the membrane potential of AT fibroblasts in response to extracellular cation addition and found that the ability of AT fibroblasts to depolarize in response to increasing concentrations of extracellular K+ is significantly reduced when compared with control fibroblasts. Electrophysiological measurements performed with a number of AT cell lines, as well as two matched sets of primary AT fibroblast cultures, reveal that outward rectifier K+ currents are largely absent in AT fibroblasts in comparison with control cells. These K+ current defects can be corrected in AT fibroblasts transfected with the full-length ATM cDNA. These data implicate, for the first time, a role for ATM in the regulation of K+ channel activity and membrane potential.
Keywords: Ataxia telangiectasia, potassium currents, membrane potential, fibroblasts
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by cerebellar degeneration, oculocutaneous telangiectases, thymic deficiencies, increased sensitivity to ionizing radiation, and predisposition to malignancies. The gene responsible for AT (designated ATM, for AT mutated) was identified by positional cloning and encodes a large protein (∼350 kD) with a carboxyl terminal domain sharing some homology with the catalytic domain of phosphatidylinositol 3-kinase (PI3-kinase; Savitsky et al. 1995a,b). Cells derived from AT patients are hypersensitive to ionizing radiation and are defective in the G1, S, and G2 cell cycle checkpoints (Painter and Young 1980; Beamish and Lavin 1994). The ATM protein is involved in a signal transduction pathway responsible for p53 regulation and c-Abl activation in response to DNA damage (Kastan et al. 1992; Khanna and Lavin 1993; Baskaran et al. 1997; Shafman et al. 1997). ATM is localized to the nucleus and is also present in microsomal fractions of cellular extracts (Chen and Lee 1996; Lakin et al. 1996; Watters et al. 1997; Brown et al. 1997).
Most research to date has focused on the role of ATM in cell cycle checkpoints and cancer. However, the phenotype of progressive cerebellar degeneration in AT is, by far, the most debilitating aspect of this disease. A potential mechanism for this degeneration comes from the observation that many other cerebellar ataxias result from defective ion channel activity (Sanguinetti and Spector 1997; Griggs and Nutt 1995). For example, human episodic ataxia (EA) is a rare autosomal dominant disorder characterized by intermittent attacks of ataxia and constant muscle rippling movements (myokymia) brought on by emotional stress or fatigue. This disorder is caused by mutations in a delayed rectifier K+ channel, KCNA1 (Browne et al. 1994). Mutations within this channel result in a decrease in the magnitude of K+ current in neurons, ultimately leading to an inability to efficiently repolarize following an action potential (Adelman et al. 1995). It is known that alterations in ion channel function and/or membrane potential can have dramatic consequences for neuronal function and survival. A previous report has shown that AT lymphocytes have a lower membrane potential than control lymphocytes (Ozer et al. 1989). These relationships between defective ion channel activity in cerebellar ataxia, altered membrane potential in AT cells, and localization of AT in the cytosol led us to examine the possibility of a role for ATM in the regulation of ion channel activity and a potential mechanism for cerebellar degeneration in AT.
Results
Altered membrane potential properties in AT fibroblasts
To investigate potential ion channel defects in AT cells, primary AT fibroblasts and parental controls were stained with the membrane potential-sensitive fluorescent dye, bis-(1,3-dibutylbarbituric acid) trimethine oxonol [DiBAC4(3)], and analyzed for their ability to depolarize in response to increasing concentrations of extracellular K+ (Fig. 1). DiBAC4(3) is a slow-responding fluorescent anion that enters depolarized cells and binds to lipid-rich intracellular components giving rise to an increased fluorescent signal (Brauner et al. 1984; Wilson and Chused 1985). We monitored time-dependent changes in fluorescence using a fluorescence imaging 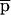 late reader (FLIPR, Molecular Devices Corporation, Sunnyvale, CA). The advantage of the FLIPR system is that it enables simultaneous fluorescence measurements and fluid addition in 96-well plates. Confluent monolayer primary cultures of parental AT heterozygous (GM03397+/−) and AT homozygous (GM03395−/−) fibroblasts were assayed for their ability to depolarize in a dose-dependent manner after exposure to increasing concentrations of extracellular K+ (Fig. 1A).
late reader (FLIPR, Molecular Devices Corporation, Sunnyvale, CA). The advantage of the FLIPR system is that it enables simultaneous fluorescence measurements and fluid addition in 96-well plates. Confluent monolayer primary cultures of parental AT heterozygous (GM03397+/−) and AT homozygous (GM03395−/−) fibroblasts were assayed for their ability to depolarize in a dose-dependent manner after exposure to increasing concentrations of extracellular K+ (Fig. 1A).
Figure 1.




AT fibroblasts are unable to fully depolarize in response to extracellular K+ addition. (A) FLIPR analysis of primary AT heterozygous control cells (GM03397+/−) and homozygous AT cells (GM03395−/−) in response to KCl addition. Time of KCl addition is indicated with an arrow. The final concentration of KCl is shown. (□) Control, 5 mm KCl; (○) control, 15 mm KCl; (▵) control, 25 mm KCl; (×) control, 35 mm KCl; (█) AT, 5 mm KCl; (•) AT, 15 mm KCl; (▴) AT, 25 mm KCl; (|) AT, 35 mm KCl. (B) Quantitation of maximal depolarization response. Maximum fluorescence values were normalized to the parental heterozygous control cells (GM03397; solid bars); (shaded bars) AT cells (GM03395). Data from a minimum of 6 wells (of a 96-well plate) were averaged. (C) FLIPR analysis of SV40-immortalized AT heterozygous (GM637+/−; solid bars) and homozygous (GM9607−/−; shaded bars) cells in response to KCl addition. (D) FLIPR analysis of SV40-immortalized AT heterozygous (GM637+/−) and homozygous cells (GM5849−/−) in response to NaCl addition. (█) Control 130 mm NaCl; (•) control, 140 mm NaCl; (▴) control, 150 mm NaCl; (+) control 170 mm NaCl; (□) AT, 130 mm NaCl; (○) AT, 140 mm NaCl; (▵) 150 mm NaCl; (×) AT, 170 mm.
Primary AT fibroblasts (GM03395−/−) gave rise to much smaller depolarizing responses than did heterozygous control cells (GM03397+/−) when challenged with the same range of extracellular K+ concentrations (Fig. 1A). In AT cells, fluorescence responses to 35 mm KCl were only 32 ± 7% when compared with primary cultures from AT heterozygous parental cells (Fig. 1B). Experiments performed with an SV40-immortalized AT homozygous fibroblast cell line (GM9607−/−) and SV40-immortalized AT heterozygous control cells (GM637+/−) gave similar results (Fig. 1C). Another SV40-immortalized AT cell line (GM5849−/−) also exhibited decreased fluorescence signals in response to 35 mm KCl (58 ± 12%) compared with the heterozygous control (GM637+/−) cells (data not shown). Replacing KCl with other cations that normally do not depolarize cells, such as NaCl (Fig. 1D) or CaCl2 (data not shown), does not result in significant fluorescence responses.
These data demonstrate a fundamental difference in the voltage properties of homozygous AT fibroblasts compared with control cells and lead us to conclude that AT cells have significantly altered membrane potential properties.
K+ channel defects in AT fibroblasts
The defect in the response of AT cells to increases in extracellular K+ suggests that the electrical properties of cells are altered by the loss of ATM function. To examine whether changes in K+ channel functions contribute to the defects in the depolarizing responses seen in the FLIPR, K+ currents were recorded directly by use of whole-cell patch-clamp analysis. K+ channel properties in two matched primary AT fibroblast cultures and three SV40-immortalized AT fibroblast cell lines were compared with AT heterozygous control fibroblasts and wild-type human foreskin fibroblasts (HFF; Fig. 2 and data not shown).
Figure 2.
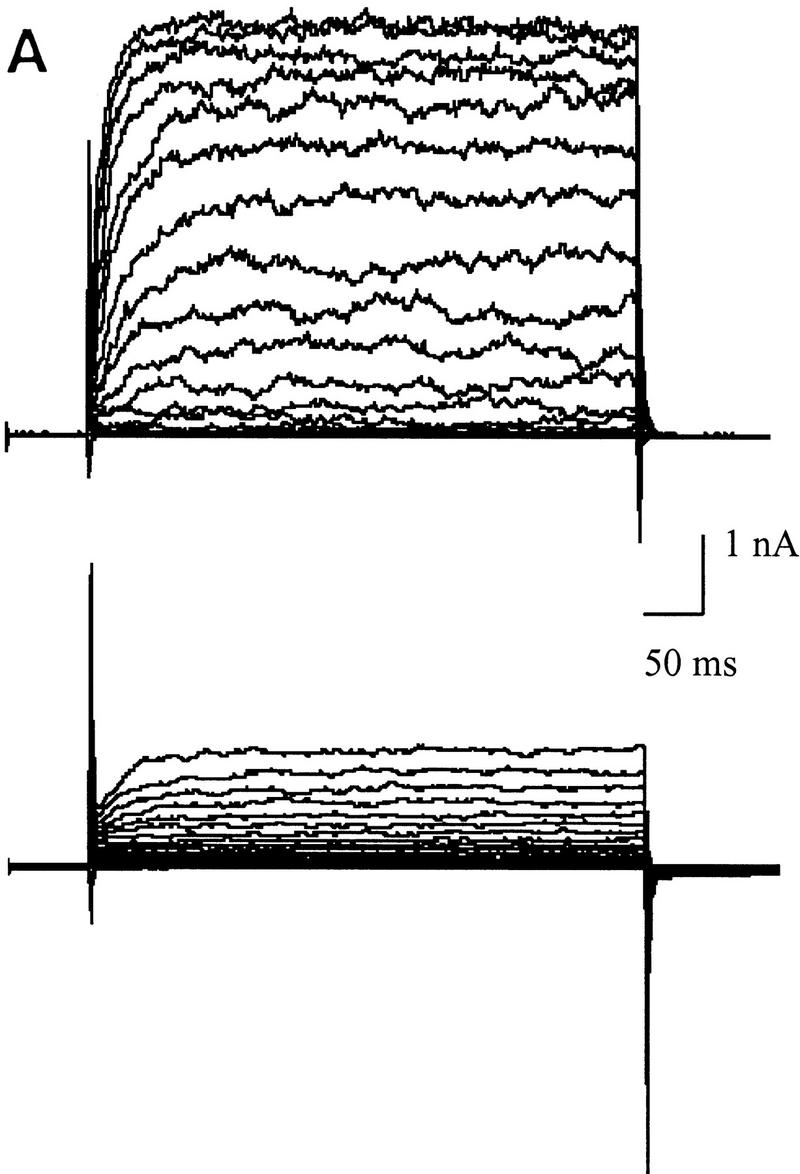





Differences in voltage-activated K+ currents between AT heterozygous and AT homozygous cells. Representative current families and averaged I/V responses evoked by 500-msec voltage pulses from −120 to +110 mV in 10-mV increments. The electrode solution was Ca2+-free, and the holding potential was −90 mV. (A) Currents were recorded from matched AT heterozygous control cells (top, GM03488+/−, n = 6) and AT homozygous primary cultures (bottom, GM03487−/−, n = 5). (B) Currents were recorded from a second set of matched AT heterozygous control cells (top, GM03397 +/− n = 5–7), and AT homozygous primary cultures (bottom, GM03395−/−, n = 6). (C) SV40-immortalized AT heterozygous control cells (top, GM637+/−, n = 7), and AT homozygous (GM5849−/−, n = 7) cells. (D) Averaged current voltage relationships for the three sets of cells [Top I/V curve; (•) GM03488+/−, (○) GM03487−/−; middle I/V curve; (•) GM03397+/− (○) GM03395−/−, (█) HFFs; bottom I/V curve; (•) GM637+/−, (○) GM5849−/−]. The data represent means ± s.e.m.s for five to seven independent experiments. (E) Averaged current voltage relationship recorded from SV40-immortalized AT heterozygous control cells (GM637+/−, n = 5) before (•), and 3 min after (█) the addition of 1 mm TEA.
The data in Figure 2A show the total K+ currents in primary fibroblast cultures from an AT heterozygous parent (GM03488+/−) and their AT homozygous offspring (GM03487−/−). Currents were elicited by 500 msec voltage steps ranging from −120 mV to +110 mV from a holding voltage of −90 mV. Under these conditions, a large outwardly rectifying K+ current was recorded from heterozygous control fibroblasts (2.3 nA at +50 mV) and from wild-type control cells (data not shown; see Fig. 2D, middle). This current was significantly reduced in homozygous AT cells (0.6 nA at +50 mV; Fig. 2D, top). Qualitatively similar data were obtained in a second matched set of primary fibroblast cultures (Fig. 2B,D, middle) comparing currents in cells from wild-type HFFs, an AT heterozygous parent (GM03397+/−), and AT homozygous offspring (GM03395−/−).
Even larger decreases in the magnitude of outward rectifying K+ currents were observed in SV40-immortalized cell lines. Figure 2C shows that the immortalized AT fibroblast cell line (GM5849−/−) passes almost no K+ currents over the complete voltage range from −120mV to +110 mV (see also Fig. 2D, bottom). The AT heterozygous immortalized cell line (GM637+/−) exhibits K+ currents similar to those measured in the two heterozygous primary cell lines. The averaged current voltage relationships for all three pairs of cell cultures are shown in Figure 2D. There is some variability between individual pairs of primary cell cultures, as well as a difference between primary and immortalized cells; however, in all cases, AT cells exhibit significant K+ current defects.
Treatment of control cells (GM637+/−) with the K+ channel blocker, tetraethylammonium chloride (TEA, 1 mm) reduced this current from 2.00 nA at +60 mV to 1.07 nA at +60 mV (Fig. 2E). These results indicate that AT cell lines are defective in a TEA-sensitive delayed-rectifier K+ current. These decreased current levels in homozygous cells are not secondary to a change in cell size, or in plasma membrane surface area. Measurements of the whole-cell membrane capacitance in these cells showed that cell surface areas may be slightly larger, rather than smaller, in homozygous cells (22.3 ± 3.2 pF, n = 22) compared to heterozygous cells (19.7 ± 2.5 pF, n = 21; P < 0.25). Preliminary experiments performed to investigate other types of K+ channels expressed in normal and AT cells indicate the presence of Ca2+-activated K+ (KCa) channels and inward-rectifier K+ channels (data not shown). However, experiments show that there are differences in KCa currents between normal and AT cells (T. D’Souza and P. Reinhart, unpubl.).
Ectopic expression of ATM restores potassium currents in AT fibroblasts
To confirm the relationship between the ATM protein and delayed rectifier K+ channels, we compared channel properties in AT cells and in AT cells complemented with full-length recombinant ATM (Ziv et al. 1997). Potassium currents were measured by use of whole-cell patch clamp analysis in AT fibroblasts transfected with either empty vector DNA (AT22IJE-T pEBS7), or ATM cDNA (AT22IJE-T pEBS7-YZ5). The data in Figure 3 show that the ATM-complemented cell line exhibits much larger K+ currents than do AT cells. The magnitude of K+ currents in these complemented cells is similar to magnitudes measured in wild-type cells. These data are consistent with our previous findings showing that these ATM-complemented cells also exhibit normal sensitivity to ionizing radiation and a normal pattern of post-irradiation DNA synthesis (Ziv et al. 1997).
Figure 3.

Effect on K+ currents in AT cells by complementation with full-length recombinant ATM. Averaged steady-state K+ currents in response to 500-msec voltage steps were measured for either AT cells (ATJ22IJE-T−/−) transfected with empty pEBS7 vector (▵) or with full-length recombinant (pEBS7-YZ5) ATM (•). The data represent means ±s.e.m.s for at least six independent experiments performed for each cell line.
Discussion
Hence, these data for the first time show a functional relationship between the presence of the ATM protein, and the magnitude of K+ currents in the plasma membrane of AT cells. Our findings, that there are large decreases in the magnitude of delayed rectifier K+ currents and subsequent defects in the membrane potential properties of AT cells, are consistent with previous work showing that the membrane potential of AT lymphoblasts is more depolarized than that in control cells (Ozer et al. 1989). These data are also consistent with a previous report showing that fibroblasts express delayed rectifier K+ channels, as well as calcium-activated K+ channels and inward rectifier K+ channels (Estacion 1991).
Fibroblasts are a useful and accessible cell model to study cellular phenotypes associated with human inherited diseases, and defects observed in these cells are often recapitulated in other cell types such as neurons (e.g., see Cox et al. 1997). It is likely that the phenotype associated with AT fibroblasts will also be evident with other cell types, and that likely candidates include excitable cells, particularly neurons.
An alteration in the membrane potential of AT cells may alter not only the electrical properties of these cells, but also lead to secondary changes in the concentration of intracellular Ca2+ and downstream signaling cascades. The link between the membrane potential and intracellular Ca2+ is mediated by voltage-activated Ca2+ channels allowing the entry of extracellular Ca2+ into cells depending on the magnitude of the membrane potential. In this way a defect in one or more K+ channels may lead to a depolarization-induced increase in intracellular Ca2+ (Robitaille et al. 1993; Wisgirda and Dryer 1994; Prakriya et al. 1996). In particularly susceptible neurons, such as cerebellar Purkinje neurons, Ca2+ elevations can lead to neuronal dysfunction and even cell death (Bindokas and Miller 1995; Brorson et al. 1995). Because Purkinje neurons play an important role in many motor functions, defects in the electrical properties of these cells can lead to motor dysfunction including numerous forms of ataxias (Airaksinen et al. 1997; Gomez et al. 1997; Zuo et al. 1997). A progressive cerebellar ataxia in mice, termed weaver, is due to a mutation in the pore of a G-protein gated, inward-rectifying K+ channel, GIRK2 (Patil et al. 1995). These mice have a developmental defect where post-mitotic granule cells fail to migrate out of the external granule cell layer and eventually undergo apoptosis (Rakic and Sidman 1973; Smeyne and Goldwitz 1989; Wood et al. 1993). Fewer Purkinje cells are present in the weaver cerebellum and their dendrites appear to be shrunken and positioned abnormally, similar to defects described in AT patients (Strich 1966; De Leon et al. 1976; Paula-Barbosa et al. 1983). Hence, there is a striking similarity between these previously described ataxias and neuronal defects such as the cerebellar Purkinje cell degeneration and ataxias observed in AT patients. In all cases, the underlying cause of these ataxias is a defect in one or more types of K+ channels (Griggs and Nutt 1995; Sanguinetti and Spector 1997).
These data provide the first mechanistic link between defects in the ATM protein and alterations in the electrical properties of AT cells. Furthermore, these results identify one or more classes of K+ channels as downstream targets of the ATM protein and show that inactivation of ATM down-regulates K+ currents in the membranes of AT cells. Whether or not the ATM protein modulates K+ currents directly or indirectly requires further investigation. Potential mechanisms range from the direct phosphorylation of the channel by the serine/threonine kinase activity of the ATM protein to indirect post-translational modifications, such as tyrosine phosphorylation, or by altering the transcriptional regulation of K+ channels. Certainly many voltage-dependent K+ channels are known substrates for serine/threonine and tyrosine kinases (Levitan 1994; Fadool et al. 1997) and ATM has serine/threonine kinase activity (Baskaran et al. 1997; Banin et al. 1998; Canman et al. 1998). Other potential mechanisms for ATM–K+ channel interactions may involve adaptins. These proteins are known to play a role in vesicle transport in cells (Pearse and Robinson 1990; Robinson 1994). Adaptins, such as the neuron-specific β-NAP (Newman et al. 1995), bind directly to ATM through the conserved amino-terminal portion of the adaptins (Lim et al. 1998). Because the expression of functional K+ channels in the plasma membrane of cells requires their tetramerization and assembly in the endoplasmic reticulum (Nagaya and Papazian 1997), it is possible that the absence of the ATM–adaptin complex contributes to the K+ channel defects observed here. A more mechanistic understanding of how the defective ATM gene can modulate the electrical activity of cells awaits further investigation. Our data have demonstrated the first functional link between the ATM protein and K+ channel activity in AT. These experiments reveal that K+ channels represent novel targets accessible to extracellular therapeutic agents for the treatment of cerebellar degeneration and ataxias in AT, and provide new mechanistic insights into the pathogenesis of this debilitating disease.
Materials and methods
Cells and culture conditions
Primary parental (GM03397+/−, GM03488+/−), primary AT (GM03395−/−, GM03487−/−), SV40-immortalized control (GM637+/−), and SV40-immortalized AT (GM5849−/−, GM9607−/−) fibroblasts were obtained from the Coriell Institute for Medical Research (Camden, NJ). Wild-type HFFs originated from fresh, neonatal foreskins treated with trypsin. Immortalized fibroblasts (GM637+/−, GM9607−/−, and GM5849−/−) and HFFs were grown in DMEM containing 15% fetal bovine serum (FBS) (Hyclone Laboratories Inc.). GM03397+/− and GM03487−/− cells were grown in MEM with Earl’s salts containing 20% FBS, 4 mm l-glutamine, and 2× essential and nonessential amino acids for MEM. GM03488+/− and GM03395−/− cells were grown in Ham’s/F12 medium containing 20% FBS, and 4 mm l-glutamine. Immortalized AT fibroblasts (AT22IJE-T pEBS7 and AT22IJE-T pEBS7-YZ5) were grown in DMEM containing 15% FBS and 100 μg/ml hygromycin B.
FLIPR analysis
For FLIPR analysis, 1–1.5 × 104 cells per well were plated in poly-l-lysine (0.001%) treated, black-walled 96-well plates (Polyfiltronics Inc.) and incubated at 37°C in a humidified chamber with 5% CO2. Later 24–48 hr, the cells were rinsed once with 1× external solution [20 mm HEPES/NaOH (pH 7.4, 130 mm NaCl, 10 mm glucose, 2 mm CaCl2, 1 mm MgCl2, 5 mm KCl) containing 5 μm DiBAC4(3) (Molecular Probes, Inc.; made from a 50 mm stock solution in DMSO) at 37°C. After rinsing, the cells were incubated at 37°C (without CO2) for 0.5–1.5 hr in 180 μl of 1× external solution containing 5 μm DiBAC4(3). The addition solution was prepared by mixing concentrated solutions of KCl, NaCl, or CaCl2 to give 10× the final desired concentration of ions in 1X external solution containing 5 μm DiBAC4(3). All plates, pipettes, and pipette tips were presoaked briefly with 1× external solution containing 5 μm DiBAC4(3) to compensate for dye removal. After placing the assay plate in the FLIPR, a baseline check was performed to ensure that the cells had equilibrated or achieved stable baseline fluorescence. At a specified time, 20 μl of addition solution was dispensed, mixed, and the same volume was re-aspirated from the assay plate to ensure that the volumes remained constant. The fluorescent signal was monitored until the total number of specified measurements had been completed and the raw data were then exported and analyzed.
Electrophysiology
Cells growing on poly-l-lysine treated coverslip fragments were used for all electrophysiological measurements. Whole-cell recordings were performed at room temperature (22°C–24°C) with an Axopatch 200 A patch-clamp amplifier (Axon Instruments, Foster City, CA). Patch electrodes were fire polished to resistances of 1.8–2.4 MΩ. Currents were low-pass filtered at 2–5 kHz with an eight-pole Bessel filter. Voltage commands, data acquisition, data storage, and data analysis were performed by use of pCLAMP version 6.03 (Axon Instruments). The extracellular recording solution consisted of 125 mm NaCl, 2.5 mm KCl, 1.3 mm MgSO4, 1 mm NaH2PO4 · H2O, 26.2 mm NaHCO3, and 2.5 mm CaCl2 · H2O (adjusted to pH 7.3 with KOH). The intracellular recording solution consisted of 130 mm K gluconate, 2 mm NaCl, 20 mm HEPES (adjusted to pH 7.4 with KOH), 4 mm MgCl2 · 6H2O, 4 mm Na2ATP, and 0.4 mm NaGTP. For calcium-free internal saline, EGTA (2 mm) was added prior to the experiments. Desired calcium concentrations were obtained by adding appropriate volumes of a 100 mm EGTA stock solution and 100 mm CaCl2 aided by a calcium electrode (Orion, model 93-20, Boston, MA).
Acknowledgments
We thank Jeff Pfohl and Jennings Worley for technical advice. This work was supported in part by National Institutes of Health grant 2R01NS31253 (P.H.R.). M.B.K. is the Steven Birnbaum Scholar of the Leukemia Society of America. T.D. is a postdoctoral fellow of the American Heart Association.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tmg2457@glaxowellcome.com; FAX (919) 315-3749.
References
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci. 1997;94:1488–1493. doi: 10.1073/pnas.94.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, Barlow C, Baltimore D, Wynshaw-Boris A, Kastan MB, Wang JY. Ataxia-telanigectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387:516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- Beamish H, Lavin MF. Radiosensitivity in ataxia-telangiectasia: Anomalies in radiation-induced cell cycle delay. Int J Radiat Biol. 1994;65:175–184. doi: 10.1080/09553009414550211. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Miller RJ. Excitotoxic degeneration is initiated at non-random sites in cultured rat cerebellar neurons. J Neurosci. 1995;15:6999–7011. doi: 10.1523/JNEUROSCI.15-11-06999.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauner T, Husler DF, Strasser RJ. Comparative measurements of membrane potentials with microelectrodes and voltage-sensitive dyes. Biochim Biophys Acta. 1984;771:208–216. doi: 10.1016/0005-2736(84)90535-2. [DOI] [PubMed] [Google Scholar]
- Brorson JR, Manzolillo PA, Gibbons SJ, Miller RJ. AMPA receptor desensitization predicts the selective vulnerability of cerebellar Purkinje cells to excitotoxicity. J Neurosci. 1995;15:4515–4524. doi: 10.1523/JNEUROSCI.15-06-04515.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genetics. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Brown KD, Ziv Y, Sadanandan SN, Chessa L, Collins FS, Shiloh Y, Tagle DA. The ataxia-telangiectasia gene product, a constitutively expressed nuclear protein that is not upregulated following genome damage. Proc Natl Acad Sci. 1997;94:1840–1845. doi: 10.1073/pnas.94.5.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim D-S, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Chen G, Lee EYP. The product of the ATM gene is a 370-kDa nuclear phosphoprotein. J Biol Chem. 1996;271:33693–33697. doi: 10.1074/jbc.271.52.33693. [DOI] [PubMed] [Google Scholar]
- Cox GA, Lutz CM, Yang CL, Biemesderfer D, Bronson RT, Fu A, Aronson PS, Noebels JL, Frankel WN. Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell. 1997;91:139–148. doi: 10.1016/s0092-8674(01)80016-7. [DOI] [PubMed] [Google Scholar]
- De Leon GA, Grover WD, Huff DS. Neuropathologic changes in ataxia-telangiectasia. Neurology. 1976;26:947–951. doi: 10.1212/wnl.26.10.947. [DOI] [PubMed] [Google Scholar]
- Estacion M. Characterization of ion channels seen in subconfluent human dermal fibroblasts. J Physiol. 1991;436:579–601. doi: 10.1113/jphysiol.1991.sp018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadool DA, Holmes TC, Berman K, Dagan D, Levitan IB. Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- Gomez CM, Thompson RM, Gammack JT, Perlman SL, Dobyns WB, Truwit CL, Zee DS, Clark HB, Anderson JH. Spinocerebellar ataxia type 6: Gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset. Ann Neurol. 1997;42:933–950. doi: 10.1002/ana.410420616. [DOI] [PubMed] [Google Scholar]
- Griggs RC, Nutt JG. Episodic ataxias as channelopathies. Ann Neurol. 1995;37:285–287. doi: 10.1002/ana.410370302. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Lavin MF. Ionizing radiation and UV induction of p53 protein by different pathways in ataxia-telangiectasia cells. Oncogene. 1993;8:3307–3312. [PubMed] [Google Scholar]
- Lakin ND, Weber P, Stankovic T, Rottinghaus ST, Taylor AM, Jackson SP. Analysis of the ATM protein in wild-type and ataxia-telangiectasia cells. Oncogene. 1996;13:2707–2716. [PubMed] [Google Scholar]
- Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- Lim DS, Kirsch DG, Canman CE, Ahn JH, Ziv Y, Newman LS, Darnell RB, Shiloh Y, Kastan MB. ATM binds to β-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaya N, Papazian DM. Potassium channel α and β subunits assemble in the endoplasmic reticulum. J Biol Chem. 1997;272:3022–3027. doi: 10.1074/jbc.272.5.3022. [DOI] [PubMed] [Google Scholar]
- Newman LS, McKeever MO, Okano HJ, Darnell RB. β-NAP, a cerebellar degeneration antigen, is a neuron-specific vesicle coat protein. Cell. 1995;82:773–783. doi: 10.1016/0092-8674(95)90474-3. [DOI] [PubMed] [Google Scholar]
- Ozer NK, Bashford CL, Carter ND, Pasternak CA. Plasma membrane potential of lymphocytes from ataxia telangiectasia patients. Clin Biochem. 1989;22:469–473. doi: 10.1016/s0009-9120(89)80100-6. [DOI] [PubMed] [Google Scholar]
- Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc Natl Acad Sci. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet. 1995;11:126–129. doi: 10.1038/ng1095-126. [DOI] [PubMed] [Google Scholar]
- Paula-Barbosa MM, Ruela C, Tavares MA, Pontes C, Saraiva A, Cruz C. Cerebellar cortex ultrastructure in ataxia-telangiectasia. Ann Neurol. 1983;13:297–302. doi: 10.1002/ana.410130312. [DOI] [PubMed] [Google Scholar]
- Pearse BM, Robinson MS. Clathrin, adaptors, and sorting. Annu Rev Cell Biol. 1990;6:151–171. doi: 10.1146/annurev.cb.06.110190.001055. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Solaro CR, Lingle CJ. [Ca2+]i elevations detected by BK channels during Ca2+ influx and muscarine-mediated release of Ca2+ from intracellular stores in rat chromaffin cells. J Neurosci. 1996;16:4344–4359. doi: 10.1523/JNEUROSCI.16-14-04344.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Sidman RL. Weaver mutant mouse cerebellum: Defective neuronal migration secondary to abnormality of Bergmann glia. Proc Natl Acad Sci. 1973;70:240–244. doi: 10.1073/pnas.70.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS. The role of clathrin, adaptors and dynamin in endocytosis. Curr Opin Cell Biol. 1994;6:538–544. doi: 10.1016/0955-0674(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Spector PS. Potassium channelopathies. Neuropharmacology. 1997;36:755–762. doi: 10.1016/s0028-3908(97)00029-4. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS, Shiloh Y, Rotman G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet. 1995b;4:2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- Shafman T, Khanna KK, Kedar P, Spring K, Kozlov S, Yen T, Hobson K, Gatei M, Zhang N, Watters D, Egerton M, Shiloh Y, Kharbanda S, Kufe D, Lavin MF. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Goldwitz D. Development and death of external granular layer cells in the weaver mouse cerebellum: A quantitative study. J Neurosci. 1989;9:1608–1620. doi: 10.1523/JNEUROSCI.09-05-01608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strich SJ. Pathological findings in three cases of ataxia-telangiectasia. J Neurol Neurosurg Psychiatry. 1966;29:489–499. [Google Scholar]
- Watters D, Khanna KK, Beamish H, Birrell G, Spring K, Kedar P, Gatei M, Stenzel D, Hobson K, Kozlov S, Zhang N, Farrell A, Ramsay J, Gatti R, Lavin MF. Cellular localisation of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogene. 1997;14:1911–1921. doi: 10.1038/sj.onc.1201037. [DOI] [PubMed] [Google Scholar]
- Wilson HA, Chused TM. Lymphocyte membrane potential and Ca2+-sensitive potassium channels described by oxonol dye fluorescence measurements. J Cell Physiol. 1985;125:72–81. doi: 10.1002/jcp.1041250110. [DOI] [PubMed] [Google Scholar]
- Wisgirda ME, Dryer SE. Functional dependence of Ca2+-activated K+ current on L- and N-type Ca2+ channels: Differences between chicken sympathetic and parasympathetic neurons suggest different regulatory mechanisms. Proc Natl Acad Sci. 1994;91:2858–2862. doi: 10.1073/pnas.91.7.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KA, Dipasquale B, Youle RJ. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–167. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in δ2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]