

Abstract
Past anthrax attacks in the United States have highlighted the need for improved measures against bioweapons. The virulence of anthrax stems from the shielding properties of the Bacillus anthracis poly-γ-d-glutamic acid capsule. In the presence of excess CapD, a B. anthracis γ-glutamyl transpeptidase, the protective capsule is degraded, and the immune system can successfully combat infection. Although CapD shows promise as a next generation protein therapeutic against anthrax, improvements in production, stability, and therapeutic formulation are needed. In this study, we addressed several of these problems through computational protein engineering techniques. We show that circular permutation of CapD improved production properties and dramatically increased kinetic thermostability. At 45 °C, CapD was completely inactive after 5 min, but circularly permuted CapD remained almost entirely active after 30 min. In addition, we identify an amino acid substitution that dramatically decreased transpeptidation activity but not hydrolysis. Subsequently, we show that this mutant had a diminished capsule degradation activity, suggesting that CapD catalyzes capsule degradation through a transpeptidation reaction with endogenous amino acids and peptides in serum rather than hydrolysis.
Keywords: Antibiotics, Enzyme Mutation, Enzymes, Hydrolases, Protein Stability, Anthrax, CapD, Circular Permutation, Rosetta
Introduction
Bacillus anthracis, the causative agent of anthrax, poses a significant bioterrorist threat. This was demonstrated in 2001 by a series of attacks in which B. anthracis spores were sent via the United States Postal Service to various media outlets and government officials. Although a safe and effective vaccine is available (1), there remain concerns about the potential for use of naturally occurring or genetically engineered antibiotic- and vaccine-resistant strains of B. anthracis (2, 3) as bioterrorist weapons. Anthrax inhalation of aerosolized spores seems the most likely scenario for a bioterrorist attack. The last case of inhalational anthrax reported in the United States prior to the 2001 attacks occurred in 1976 (4). The rarity of this particular type of infection makes it difficult to study the efficacy of various response strategies. These concerns and the historically effective use of B. anthracis to cause disease intentionally have led the biomedical community to focus on developing novel therapeutics as prophylactic countermeasures to the potential threat of a bioterrorist attack.
The potent virulence of B. anthracis is thought to be a result primarily of exotoxins (lethal and edema toxins) as well as an antiphagocytic γ-linked poly-d-glutamic acid (PDGA)2 capsule (5, 6). Although the precise mechanism by which the capsule inhibits phagocytosis is unknown, it has been well established that strains lacking the PDGA capsule have significantly diminished virulence (7).
The capsule is created by a γ-glutamyl transpeptidase, CapD, attached to the outer cell wall, which covalently anchors PDGA to the B. anthracis peptidoglycan layer (8). Although the function of native CapD is to build the antiphagocytic capsule, it was shown recently that administration of high doses of recombinant CapD significantly decreased the mortality rate of mice infected with B. anthracis (9). The ability of recombinant CapD to counteract virulence (the opposite of its native function) is likely due to the removal of the outer antiphagocytic capsule of B. anthracis in vivo, as it is no longer attached to the outer cell wall and localized by the peptidoglycan layer. The ability to degrade the capsule has been demonstrated in vitro previously (7, 9, 10). Because of the ability to confer antibiotic resistance to anthrax in a laboratory setting, significant focus has been placed on the further development of CapD into a robust and effective next generation protein therapeutic.
The structure of CapD was solved recently and has opened the possibility of applying computational protein design techniques to further enhance the potential therapeutic properties of this protein (11). In this study, we used the Rosetta software suite (12, 13) to create a circularly permuted variant of CapD (termed CP) with significantly improved production properties and kinetic thermostability. In addition, we computationally designed a variant of CP that altered the reaction specificity of the enzyme such that the transpeptidation reaction rate was reduced, whereas the rate of its hydrolytic side reaction was essentially maintained. We then utilized the mutant to examine the mechanism of capsule degradation. The improved physical properties of CP, in addition to a more complete understanding of the native mechanism of capsule degradation, should lead to the development of improved protein-based therapeutics that can better withstand environmental stress and that have improved pharmacokinetic properties.
EXPERIMENTAL PROCEDURES
DNA Manipulations
The genes encoding proteins were synthesized to optimize codon usage for Escherichia coli by GenScript. The genes were inserted into the pET29b+ vector between the NdeI and XhoI restriction endonuclease sites, and a genetically encoded His6 tag was added to the C terminus for ease of purification. Mutants of these enzymes were generated using site-directed mutagenesis as described by Kunkel (14).
Protein Production and Purification
Proteins were produced by transforming the constructs into electrocompetent E. coli BL21(DE3) cells. Single colonies were picked and grown in 1-ml cultures of lysogeny broth with 50 μg/ml kanamycin overnight at 37 °C. These cultures were used to inoculate a 50-ml culture of Terrific broth with 50 μg/ml kanamycin, which was grown to A600 = 0.3–0.5, at which point protein expression was induced by the addition of isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mm. The temperature was then reduced to 18 °C, and overexpression of protein was allowed to continue for another 24 h. The cells were harvested via centrifugation and stored at −20 °C until lysis. To lyse the cells, frozen pellets were thawed and resuspended in 50 mm HEPES (pH 7.4), 500 mm NaCl, 25 mm imidazole, 2× BugBuster (Novagen), 2 mg/ml lysozyme, and 0.2 mg/ml DNase I. The resuspended pellets were then incubated while rocking for 1 h at 4 °C, after which the lysate was clarified by centrifugation at 20,000 × g for 30 min. The supernatant was then flowed over TALON resin (Clontech, Mountain View, CA); washed with 20 column volumes of 50 mm HEPES (pH 7.4), 500 mm NaCl, and 25 mm imidazole; and eluted with 5 column volumes of 50 mm HEPES (pH 7.4), 500 mm NaCl, and 500 mm imidazole. Protein purity was determined by SDS-PAGE analysis. The relative level of processing for heterogeneous samples of CapD was quantified using NIH ImageJ gel densitometry software.
Kinetics Assay and Mutant Screen
The enzymatic activities of CapD and CP were assayed using the Förster resonance energy transfer substrate 5-FAM-(d-γ-Glu)5-K(QXLTM 520)-NH2 (AnaSpec, Fremont, CA). 5 nm enzyme was incubated with 2-fold serially diluted substrate from 1000 to 1 nm in a 100-μl reaction containing 25 mm HEPES and 0.1% Tween 20 and with 1 mg/ml BSA at pH 7.4 and 21 °C. No significant differences in reaction rates were observed regardless of whether or not BSA was present, suggesting that BSA does not act as an acceptor substrate for CapD. The addition of BSA appeared to result in increased reproducibility of the results when enzyme concentrations were <1 nm. This is likely due to the prevention of adsorption of CapD to the plastic tubes used in the experiments. l-Glutamic acid was added to a final concentration of 5 mm to measure transpeptidation. The difference in activity observed in the presence versus absence of an acceptor amino acid (such as l-glutamic acid) was used to determine final transpeptidation activity. To measure hydrolytic activity, the reaction rate was measured in the absence of added acceptors. The reaction rates were determined by continuously measuring an increase in relative fluorescence units at an excitation wavelength of 490 nm and an emission wavelength of 520 nm in a SpectraMax M5e microplate reader (Molecular Devices, Sunnyvale, CA) for 1 h. The change in fluorescence intensity was converted to product concentration by creating a standard curve with the unquenched synthetic product 5-FAM-(d-γ-Glu)5 (AnaSpec). To account for the inner filter effect, 12 separate standard curves were created in which the product standard was varied in the presence of each substrate concentration used for the kinetics assays (supplemental Table S1).
Amino Acid Specificity Assay
Enzymatic activity was assayed using a modified protocol of the kinetics assay. Specifically, 0.5 nm enzyme was incubated with 50 nm substrate and 5 mm amino acid. l-Tyrosine was used at 1.8 mm due to its limited solubility.
Capsule Degradation Assay
Capsule degradation gel electrophoresis assays were performed using a modification of a previously described method (7). Briefly, 500 μg of purified capsule was incubated with CapD or derivative enzyme for 30 min at 37 °C in 0.5× PBS with 50% mouse serum. Digested samples were run on a 1% agarose gel and stained with methylene blue.
Circular Dichroism
Far-UV CD spectra were taken in PBS at 25 °C with a Jasco-810 spectropolarimeter and a stoppered 1-mm path length rectangular quartz cell (Starna, Atascadero, CA). Protein solutions were 0.1 mg/ml. Four scans were accumulated and averaged, without smoothing. Melting curves were done by heating at 1 °C/min.
Stability Assay
Purified enzyme was incubated in PBS at a concentration of 100 nm and at temperatures ranging from 30 to 50 °C. Aliquots were removed at 0, 1, 2, 5, 10, 20, and 30 min and diluted 10-fold into a 4 °C solution of PBS with 1 mg/ml BSA. Activity was assayed at 0.5 nm enzyme, 50 nm substrate, and 5 mm l-glutamate in buffer as described under “Kinetics Assay and Mutant Screen.”
Protein Modeling
A model of the tetrahedral intermediate of the hydrolysis reaction transition state was built in the program Spartan (15) and overlaid onto the active site of the crystal structure of CapD (Protein Data Bank code 3G9K). The protein-ligand interface was then minimized using the Rosetta modeling suite (12) such that the geometric constraints (supplemental Fig. S1) were satisfied. This model was used as a starting point to identify mutations aimed to change the protein's reaction specificity. Mutations were evaluated using the RosettaDesign algorithm (16) and were accepted if they resulted in a decrease in energy or if they increased the predicted energy by <5 units.
Circular Permutation Design
To design CP, the first amino acid of the small subunit of CapD (normally residue 325) was set to correspond to the first amino acid at the N terminus of the new protein, CP. A short peptide linker (SGGSG) predicted to be capable of bridging the C terminus of the small subunit (residue 521 in CapD) to the N terminus of the large subunit (residue 1 in CapD) was modeled into the protein using the FoldIt interface to the Rosetta software suite. This change is graphically depicted in Fig. 1.
FIGURE 1.

Protein structures of CapD and CP. For both CapD and CP, the N termini are highlighted in blue, and C termini are highlighted in red. A, the crystal structure of the CapD heterodimer (Protein Data Bank code 3G9K) in the small subunit (cyan) and the large subunit (green) are highlighted. B, circular permutation of CapD results in a single continuous monomer (purple) that links the C terminus of the small subunit to the N terminus of the large subunit using an SGGSG linker (lime). The linker was modeled using the FoldIt interface to RosettaDesign. All images were generated using PyMOL (22).
RESULTS
Circular Permutation of CapD
Recombinant expression and purification of CapD resulted in an inconsistent and heterogeneous mixture of soluble protein consisting of unprocessed (and therefore inactive) enzyme, as well as the processed large and small subunits (Fig. 2). In contrast, expression and purification of CP resulted in a homogeneous soluble protein of the expected size upon SDS-PAGE analysis. Because translation is initiated with the AUG start codon, one concern with respect to the circular permutation is that the actual N-terminal residue of CP is methionine, whereas the N terminus of the autocatalytically processed small subunit of CapD is the catalytic threonine (11). It is well established that, in E. coli, the N-terminal methionine is often cleaved immediately after translation, especially if the N+1 amino acid is small (e.g. threonine) (17). Therefore, we examined whether the N-terminal methionine had been cleaved in CP. After expression and purification, the molecular mass of CP was determined using electrospray ionization mass spectrometry. The observed mass of CP was 55,275 Da, and the expected mass of the enzymes without the methionine is 55,285 Da, whereas the predicted mass of CP with an N-terminal methionine is 55,417 Da. The observed mass is within the reported 0.02% machine error of the expected size of the protein from which the N-terminal methionine has been removed. This suggests that the catalytic threonine represents the actual N terminus of CP (supplemental Fig. S2).
FIGURE 2.

SDS-PAGE of CapD and CP. Both CapD (lanes 2–4) and CP (lanes 5–7) were purified using immobilized metal ion affinity chromatography as described under “Experimental Procedures” and then analyzed on a 4–20% gradient SDS gel. Lane 1 contains Precision Plus Protein Kaleidoscope standards (Bio-Rad), and molecular masses are indicated kilodaltons. Each lane represents an independent purification of the protein. The upper band on CapD at 57.5 kDa represents the fraction of the protein that has not undergone autocatalytic processing, whereas the two lower bands at 35.7 and 21.8 kDa are the products of CapD after it has been processed. CP forms a single homogeneous band at 55.3 kDa as expected.
Kinetic Characterization of CP
To determine whether the circular permutation of CapD had affected catalytic activity, the reaction velocity was measured as a function of substrate concentration to determine the kinetic constants of the transpeptidation and hydrolysis reactions for CapD and CP (Table 1 and supplemental Fig. S3). Both enzymes exhibited standard Michaelis-Menten behavior for the transpeptidation reactions, and the data were fit to the standard Michaelis-Menten model (Equation 1).
 |
For the hydrolysis reactions, substrate inhibition was observed for both enzymes, and the data were therefore fit to Equation 2.
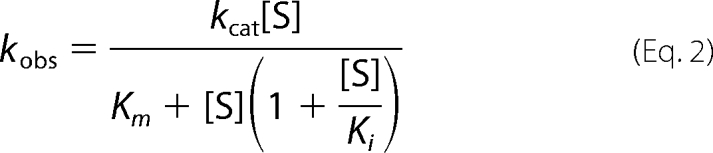 |
Because of the heterogeneity of purified soluble CapD, it is difficult to determine exact kinetic constants. Therefore, a sample that was >85% processed, as determined by gel electrophoresis (supplemental Fig. S4), was used for the assays. No corrections for purity were made to the calculations, as the level of additional processing that takes place over the course of the measured reaction is unclear. The additional corrections would not significantly change the calculated kinetic constants. This was not a concern for CP, as it appears homogeneous by both gel electrophoresis and mass spectrometry (Fig. 2 and supplemental Fig. S2).
TABLE 1.
Kinetic constants for CapD, CP, and F24H
Each reaction was carried out with three independent measurements as described under “Experimental Procedures.” The errors represent the 95% confidence interval determined from nonlinear regression fit of the data. No product inhibition was observed for the transpeptidation reaction and was fit to Equation 1, whereas product inhibition was observed for the hydrolysis reaction and was fit to Equation 2. Error in kcat/Km was calculated using δ(kcat/Km) = |kcat/Km| . NA, not applicable.
| Transpeptidation | Hydrolysis | |
|---|---|---|
| CapD | ||
| kcat (h−1) | 68.07 ± 1.34 | 3.16 ± 0.31 |
| Km (nm) | 65.45 ± 4.65 | 3.04 ± 1.00 |
| Ki (nm) | NA | 377.59 ± 126.48 |
| kcat/Km (m−1 s−1 × 103) | 288.90 ± 21.30 | 288.74 ± 99.11 |
| CP | ||
| kcat (h−1) | 63.37 ± 2.67 | 3.33 ± 0.35 |
| Km (nm) | 43.72 ± 7.20 | 2.83 ± 1.00 |
| Ki (nm) | NA | 319.46 ± 112.35 |
| kcat/Km (m−1 s−1 × 103) | 402.63 ± 6.84 | 326.86 ± 120.50 |
| F24H | ||
| kcat (h−1) | 4.42 ± 0.20 | 1.72 ± 0.04 |
| Km (nm) | 57.49 ± 9.54 | 6.92 ± 0.58 |
| Ki (nm) | NA | 3583.2 ± 726.98 |
| kcat/Km (m−1 s−1 × 103) | 21.36 ± 3.67 | 69.04 ± 6.00 |
CapD and CP both exhibited a 20-fold increase in the catalytic rate (kcat) for the transpeptidation reaction relative to hydrolysis (Table 1). Analysis of the hydrolytic activity for both enzymes suggested that each had at least a 15-fold higher Km in the transpeptidation reaction. Interestingly, in the case of transpeptidation, a 1.5-fold decrease in Km was observed relative to CapD despite the fact that no mutations were made within the active site of CP. For the hydrolysis reaction, the calculated substrate inhibition constants (Ki) were ∼100-fold higher than the Michaelis constant (Km) for both enzymes. The cause of substrate inhibition for this enzyme is unclear and has not been previously reported.
The amino acid acceptor specificity in the transpeptidation reaction for CapD and CP was characterized by measuring the specific activity for these enzymes against all 20 amino acids (Fig. 3). Both enzymes were able to utilize most amino acids as substrates for the transpeptidation reaction. There was a systematic increase in activity for CP over CapD, consistent with the shift in Km (Table 1). Proline showed the lowest transpeptidation activity with an 11-fold drop relative to glutamate, the most active of the acceptor amino acids tested. The trends for specific activity across the 20 amino acids for both CapD and CP were consistent, suggesting that no significant structural changes had occurred within the active site after the circular permutation.
FIGURE 3.

Amino acid specificity of CapD, CP, and F24H. The specific activity for each enzyme, CapD (gray), CP (light gray), and F24H (dark gray), was determined using 5 mm amino acid acceptor as described under “Experimental Procedures.” The specific activity for l-Tyrosine (Tyr*) was assayed at 1.8 mm amino acid due to the limited solubility of tyrosine. Each activity was measured in triplicate, and S.D. values are given in supplemental Table S2.
CD Analysis of CapD and CP
The far-UV CD spectra of CapD and CP showed no significant differences (supplemental Fig. S5A), suggesting that their secondary structures are similar, as expected. The melting temperature of CapD was 48.4 °C, whereas CP had a melting temperature of 51.1 °C, 2.7 ° higher (supplemental Fig. S5B). Both proteins denatured irreversibly and aggregated with heating above the melting temperatures.
Kinetic Thermostability
To determine the effect of temperature on enzymatic activity (i.e. kinetic thermostability), both CapD and CP were incubated at temperatures ranging from 30 to 50 °C, and the rate of decay of enzymatic activity was measured. The loss of activity for both proteins over time exhibited first-order kinetics, as shown in Fig. 4A. At higher temperatures, a clear increase in thermostability of CP relative to CapD was observed, as incubation at 45 °C for 30 min resulted in complete inactivation of CapD, whereas CP was still 90% active (Fig. 4A).
FIGURE 4.

Kinetic thermostability of CapD, CP, and F24H. A, shown is a plot of the fraction of remaining activity versus time for CapD (■) and CP (◢) at 45 °C. The data were fit to a first-order exponential decay to determine the rate of decay. Errors bars represent S.D. from three independent measurements. B, the rates of decay were determined by fitting five individual time points using nonlinear regression to an exponential decay function for temperatures ranging from 30 to 50 °C for CapD (♦), CP (■), and F24H (◢). These data were then fit using nonlinear regression to the Arrhenius equation. The calculated free energy of inactivation and 95% confidence interval for CapD, CP, and F24H are 56.2 ± 11, 144 ± 15, and 163 ± 13 kilocalories/mol, respectively.
Computational Redesign of CP Reaction Specificity
To determine whether the in vivo mechanism of capsule degradation was due to hydrolysis or transpeptidation, we aimed to engineer an enzyme that exhibited a decreased level of transpeptidation activity without loss of the ability to catalyze the hydrolytic side reaction. Using the FoldIt interface to the Rosetta software suite, a library of 84 CP enzyme variants was designed such that the hydrolysis transition state was either stabilized or not significantly affected. Although the mutations were not explicitly modeled in the presence of the substrate or transpeptidation product transition states (as the software is not currently set up for multistate modeling), we hypothesized that some of the enzymes in the designed library would prevent an amino acid from binding in the acceptor site while still allowing the substrate to bind, resulting in a switch in reaction specificity. Each design was then produced and experimentally characterized with respect to its hydrolytic and transpeptidation activities (supplemental Fig. S6).
One mutant, F24H (which corresponds to residue 374 in CapD), exhibited a decrease in transpeptidation activity without a substantial effect on hydrolytic activity (Fig. 5). Kinetic characterization of F24H showed a 15-fold reduction in the catalytic rate of the transpeptidation reaction relative to CP, whereas the rate of the hydrolysis reaction decreased only 2-fold (Table 1).
FIGURE 5.

Active site mutation alters reaction specificity. Shown is the hydrolysis transition state model, in which PDGA (teal) is attached to the catalytic threonine (red). Two glycine backbone nitrogen atoms form an oxyanion hole to stabilize the tetrahedral intermediate (dashed black lines) formed during the hydrolysis reaction. A, the wild-type residue Phe24 (orange) is highlighted in CP. B, the point mutation of Phe24 to His (orange) is modeled in F24H. All images were generated using PyMOL (22).
To ensure that the observed effect was general, the specific activity of F24H-catalyzed transpeptidation for all 20 amino acids was then measured. As depicted in Fig. 3, transpeptidation activity was significantly decreased for all amino acid acceptors. In addition, the F24H mutation did not affect kinetic thermostability (Fig. 4B).
Mechanism of Capsule Degradation
To model the mechanism of capsular degradation in vivo, the degradation efficiency of F24H and CP in mouse serum was analyzed. If the mechanism of action in serum was transpeptidation, the activity of F24H should be significantly crippled relative to CP. Alternatively, if the mechanism of action in serum was hydrolysis, only a minimal difference in degradation activity between CP and F24H would be expected. The level of capsular degradation by both enzymes was determined using a gel shift assay as depicted in Fig. 6. F24H was significantly less efficient at degrading the capsule in serum compared with CP. Although the gel analysis is only qualitative, the levels of degradation for 12.5 μg/ml F24H are roughly equivalent to 0.78 μg/ml CP, representing a 16-fold change in activity (Fig. 6). These data agree well with the 15-fold drop in transpeptidation activity measured using the synthetic PDGA substrate.
FIGURE 6.

Capsule degradation by CapD, CP, and F24H. Each protein was tested for capsule degradation activity when incubated in normal mouse serum. The proteins were serially diluted from 12.5 down to 0.78 μg/ml in a solution of 50% 1× PBS and 50% normal mouse serum. After titration, the capsule at a total concentration of 500 μg (in water) was added to each sample. Lane 1, negative control, capsule only; lanes 2–6, capsule with 12.5, 6.25, 3.125, 1.56, and 0.78 μg/ml F24H; lanes 7–11, capsule with 12.5, 6.25, 3.125, 1.56, and 0.78 μg/ml CapD; lanes 12–16, capsule with 12.5, 6.25, 3.125, 1.56, and 0.78 μg/ml CP; lane 17, negative control, capsule only. As the capsule is degraded, it will migrate farther down the gel.
DISCUSSION
The B. anthracis γ-linked d-glutamyl transpeptidase CapD is expressed as an inactive linear peptide chain, which must undergo autocatalytic processing to form the active enzyme. This post-translational processing results in a heterodimer with a large subunit (residues 1–324) and a small subunit (residues 325–521), as depicted in Fig. 1A. The N-terminal threonine of the small subunit serves as the catalytic residue for the degradation of the PDGA substrate. Recombinant expression of CapD results in a heterogeneous mixture of processed and unprocessed soluble proteins (Fig. 2). This production property is not ideal for a protein therapeutic because consistent structural integrity is required for Food and Drug Administration (FDA) approval (18). Specifically, the FDA requires that a combination of SDS-PAGE, isoelectric focusing, high-performance liquid chromatography, and mass spectrometry be used to show that proteins are not fragmented, aggregated, or otherwise modified (e.g. partial autocatalytic processing). To address the issue of partial and variable autocatalytic processing of CapD, the protein was circularly permuted such that it would be produced as a homogeneous monomeric protein.
As depicted in Fig. 1A, the N terminus of the large subunit and the C terminus of the small subunit of CapD are in proximity to one another in the folded processed heterodimer. We hypothesized that the termini could be connected to create a single-chain monomeric protein, circumventing the need for autocatalytic processing. Using the FoldIt interface of the Rosetta software suite, we designed a short peptide linker that was predicted to connect the C terminus of the small subunit to the N terminus of the large subunit (Fig. 1B). In the circularly permuted variant, the catalytic N-terminal threonine is natively translated as the protein's N-terminal residue, abrogating the need for CapD to undergo autocatalytic cleavage to form an active enzyme.
Experimental characterization confirmed that autocatalytic processing was no longer required for the CP variant (Fig. 2). The activities of CP for both hydrolysis and transpeptidation were analyzed and were observed to remain the same as for CapD, if not slightly improved (Table 1).
It has been previously demonstrated that circular permutation of proteins can result in changes in thermostability (19). To characterize kinetic thermostability, CapD and CP were incubated at a range of temperatures, after which the remaining activity was measured at a series of time points. As depicted in Fig. 4 (A and B), the circular permutation resulted in a significant increase in overall kinetic thermostability. At 45 °C, CapD was rendered inactive in a manner of minutes, whereas CP remained 90% active after 0.5 h. However, the structural thermostability was determined by CD for CapD and CP. Both enzymes exhibited similar melting temperatures of 48.4 and 51.1 °C, respectively (supplemental Fig. S5B). In this case, there was a much smaller change in stability. We hypothesize that this difference in stability is due to a loss of quaternary structure (i.e. dissociation of the two subunits), resulting in a decrease in catalytic activity for CapD. Such a disruption of quaternary structure would result in an inactive enzyme without significantly affecting the signal observed by CD. At significantly higher temperatures, the secondary structures of both proteins were disrupted, resulting in the observed changes in the CD signal (supplemental Fig. S5B). The designed peptide linker that connects the two subunits in CP altered the quaternary structure of the enzyme such that it was converted from a heterodimeric protein into a single-chain monomeric protein. This change would be predicted to have a more significant effect on the dissociation of the two subunits than the protein's secondary structure. This is consistent with the circular permutation of CapD having an increased kinetic thermostability. The increase in stability is likely to improve the ease of handling and may enhance its in vivo pharmacokinetic properties.
Another feature of CapD that has remained a mystery is the mechanism through which it catalyzes the capsular degradation of B. anthracis in mouse serum. The primary function of CapD is transpeptidation, in which the polyglutamyl chain is cleaved, resulting in an acyl-enzyme intermediate. An acceptor with a free amine (such as an amino acid or a peptide from the peptidoglycan layer) must then bind within the active site and subsequently accept the polyglutamyl chain. If no acceptor is present, hydrolysis of the acyl-enzyme intermediate occurs instead. In this case, a water molecule acts as the primary acceptor, resulting in the formation of a free acid and the regenerated enzyme (supplemental Fig. S7). The balance of transpeptidation versus hydrolysis is well established for this class of enzymes (20, 21).
Although the native function of CapD is to build the antiphagocytic outer coat of B. anthracis through a transpeptidation reaction, the mechanism of capsular degradation could proceed through either a hydrolysis or transpeptidation reaction. In the case of transpeptidation, endogenous amino acids and peptides in the host serum could act as acceptors instead of the peptidoglycan layer. If CapD levels are elevated beyond a particular threshold, the low-level but irreversible hydrolysis side reaction could eventually overwhelm the dominant but reversible transpeptidation reaction to the peptidoglycan layer. In an effort to develop protein-based anthrax therapeutics, it will be useful to know through which of the two mechanisms the capsular degradation is occurring. One potential method to determine this is to re-engineer the reaction specificity of CapD such that the transpeptidation reaction is diminished but the hydrolytic activity of the enzyme remains intact.
Using the Rosetta software suite, we created a computationally designed active site library of 84 mutants predicted to be compatible with the hydrolysis transition state. Each of the 84 mutants was generated and screened for both hydrolytic and transpeptidation activities. From the initial screen, a general preference of disrupting transpeptidation with concomitant maintenance of hydrolytic activity was observed (supplemental Fig. S6). One mutant, F24H, showed a particularly interesting property in that it essentially maintained hydrolytic activity while significantly decreasing the transpeptidation reaction rate. Kinetic characterization of the F24H variant suggested that this mutation accomplished the desired alteration of reaction specificity, as shown in Table 1.
To use this protein as a tool to elucidate the in vivo mechanism of capsular degradation, it was important to validate that the rate of the transpeptidation reaction was diminished not only with respect to the amino acid acceptor glutamate but also for all of the 20 amino acids that may be present in serum. As depicted in Fig. 3, this mutation significantly decreased the transpeptidation activity for every amino acid. In addition, the mutation did not compromise kinetic thermostability (Fig. 4B), suggesting that the change of the reaction was due to an alteration of the chemistry occurring in the active site and not simply due to a global structural destabilization. The F24H model does not clearly indicate why this particular mutation would result in such a drastic change in reaction specificity, as the closest nitrogen atom of His24 is 7.1 Å away from the oxygen atom of the catalytic Thr1 (Thr1 of CP corresponds to Thr352 of CapD) (Fig. 5B). The change in specificity could be the result of a modulation of the electrostatic environment of the active site such that the attack of the acyl-enzyme intermediate by a neutral amine within the active site is disfavored. Further experimentation is required to develop a more complete understanding of how this mutation alters the reaction specificity.
Assays were conducted to explore the mechanism of capsular degradation in serum using CapD, CP, and F24H. Degradation efficiency in the presence of serum was measured as a function of protein concentration, as shown in Fig. 6. F24H exhibited a >10-fold decrease in its ability to degrade the capsule relative to CP and CapD. This change in reaction specificity is consistent with the mechanism of CapD-mediated capsule degradation being transpeptidation. If a hydrolytic mechanism were employed, this assay would not have shown such a significant difference between the ability of F24H to degrade the capsule relative to CP. The abilities of CP and F24H to catalyze the degradation of a synthetic PDGA substrate in the presence or absence of serum was then explored. Again, F24H showed an ∼2-fold increase in activity in the presence of 25% serum, whereas CP and CapD showed a >8-fold increase in activity in the presence of 25% serum (supplemental Fig. S8). Assuming that capsular degradation in mouse serum is representative of an in vivo environment, these experimental results comparing F24H with CP are consistent with the in vivo capsule degradation mechanism being transpeptidation (likely using endogenous amino acids and peptides as PDGA acceptors), as opposed to hydrolysis. Although F24H exhibits a significant change in reaction specificity and has provided an essential tool for the investigation of the in vivo mechanism of capsular degradation, additional mutants with larger changes in reaction specificity will be required to conclude that the in vivo reaction catalyzed by CapD is done primarily through transpeptidation and not hydrolysis.
It is important to generate improved anthrax therapeutics in the near future. The native B. anthracis protein CapD is a promising candidate for such a protein-based therapeutic but currently suffers from limitations that may prevent its general use. In this study, we have applied computational protein modeling techniques both to improve the production properties of CapD and to significantly increase its kinetic thermostability. In addition, we generated a novel enzyme that was then used to help elucidate how this protein functions in vivo, and our results will inform future studies aimed at converting this protein into a potent protein-based therapeutic. The improved properties of CP, as well as the enhanced knowledge of this protein's function, should lead to the more efficient development of anthrax therapeutics.
Supplementary Material
Acknowledgments
We thank the International Genetically Engineered Machine (iGEM) Competition for promoting undergraduate research projects and providing a rich academic environment. Alan Weiner, Dominic Chung, and Ling Lin Liu (Department of Biochemistry, University of Washington) generously provided laboratory space for the undergraduates involved in this work. We thank Drs. Patricia Legler and Brock Siegel for insightful comments that helped refine the manuscript.
This work was supported by a grant from the Defense Advanced Research Projects Agency and the Howard Hughes Medical Institute.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8 and Tables S1–S3.
- PDGA
- γ-linked poly-d-glutamic acid.
REFERENCES
- 1. Institute of Medicine (United States). Committee to Assess the Safety and Efficacy of the Anthrax Vaccine (2002) The Anthrax Vaccine: Is It Safe? Does It Work?, National Academy Press, Washington, D.C [Google Scholar]
- 2. Pomerantsev A. P., Staritsin N. A., Mockov YuV., Marinin L. I. (1997) Vaccine 15, 1846–1850 [DOI] [PubMed] [Google Scholar]
- 3. Stepanov A. V., Marinin L. I., Pomerantsev A. P., Staritsin N. A. (1996) J. Biotechnol. 44, 155–160 [DOI] [PubMed] [Google Scholar]
- 4. Inglesby T. V., O'Toole T., Henderson D. A., Bartlett J. G., Ascher M. S., Eitzen E., Friedlander A. M., Gerberding J., Hauer J., Hughes J., McDade J., Osterholm M. T., Parker G., Perl T. M., Russell P. K., Tonat K. (2002) JAMA 287, 2236–2252 [DOI] [PubMed] [Google Scholar]
- 5. Green B. D., Battisti L., Koehler T. M., Thorne C. B., Ivins B. E. (1985) Infect. Immun. 49, 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mikesell P., Ivins B. E., Ristroph J. D., Dreier T. M. (1983) Infect. Immun. 39, 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scorpio A., Chabot D. J., Day W. A., O'Brien D. K., Vietri N. J., Itoh Y., Mohamadzadeh M., Friedlander A. M. (2007) Antimicrob. Agents Chemother. 51, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Candela T., Fouet A. (2005) Mol. Microbiol. 57, 717–726 [DOI] [PubMed] [Google Scholar]
- 9. Scorpio A., Tobery S. A., Ribot W. J., Friedlander A. M. (2008) Antimicrob. Agents Chemother. 52, 1014–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scorpio A., Chabot D. J., Day W. A., Hoover T. A., Friedlander A. M. (2010) Microbiology 156, 1459–1467 [DOI] [PubMed] [Google Scholar]
- 11. Wu R., Richter S., Zhang R. G., Anderson V. J., Missiakas D., Joachimiak A. (2009) J. Biol. Chem. 284, 24406–24414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simons K. T., Kooperberg C., Huang E., Baker D. (1997) J. Mol. Biol. 268, 209–225 [DOI] [PubMed] [Google Scholar]
- 13. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y. E., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P. (2011) Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kunkel T. A. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 488–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zakharian T. Y., Coon S. R. (2001) Comput. Chem. 25, 135–144 [DOI] [PubMed] [Google Scholar]
- 16. Kuhlman B., Baker D. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lowther W. T., Matthews B. W. (2000) Biochim. Biophys. Acta 1477, 157–167 [DOI] [PubMed] [Google Scholar]
- 18. U. S. Department of Health and Human Services Food and Drug Administration (1997) Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use (Docket No. 94D-0259), www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/OtherRecommendationsforManufacturers/UCM153182.pdf
- 19. Yu Y., Lutz S. (2011) Trends Biotechnol. 29, 18–25 [DOI] [PubMed] [Google Scholar]
- 20. Tate S. S., Meister A. (1981) Mol. Cell. Biochem. 39, 357–368 [DOI] [PubMed] [Google Scholar]
- 21. Silverman R. B. (2002) The Organic Chemistry of Enzyme-catalyzed Reactions, Academic Press, London [Google Scholar]
- 22. Delano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC, San Carlos, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.