

Abstract
Accumulating evidence shows that obesity is associated with doxorubicin cardiac toxicity in the heart, but the molecular mechanisms that contribute to this pathological response are not understood. Adiponectin is an adipose-derived, cardioprotective factor that is down-regulated in obesity. Here, we investigated the effect of adiponectin on doxorubicin (DOX)-induced cardiotoxicity and assessed the mechanisms of this effect. A single dose of DOX was intraperitoneally injected into the abdomen of adiponectin knock-out (APN-KO) and wild-type (WT) mice. APN-KO mice had increased mortality and exacerbated contractile dysfunction of left ventricle compared with WT mice. APN-KO mice also showed increased apoptotic activity and diminished Akt signaling in the failing myocardium. Systemic delivery of adenoviral vector expressing adiponectin improved left ventricle dysfunction and myocardial apoptosis following DOX injection in WT and APN-KO mice but not in Akt1 heterozygous KO mice. In cultured rat neonatal cardiomyocytes, adiponectin stimulated Akt phosphorylation and inhibited DOX-stimulated apoptosis. Treatment with sphingosine kinase-1 inhibitor or sphingosine 1-phosphate receptor antagonist diminished adiponectin-induced Akt phosphorylation and reversed the inhibitory effects of adiponectin on myocyte apoptosis. Pretreatment with anti-calreticulin antibody reduced the binding of adiponectin to cardiac myocytes and blocked the adiponectin-stimulated increase in Akt activation and survival in cardiomyocytes. Interference of the LRP1/calreticulin co-receptor system by siRNA or blocking antibodies diminished the stimulatory actions of adiponectin on Akt activation and myocyte survival. These data show that adiponectin protects against DOX-induced cardiotoxicity by its ability to promote Akt signaling.
Keywords: Adipocyte, Akt PKB, Apoptosis, Cardiac Muscle, Heart, Adiponectin, Cardiotoxicity, Doxorubicin
Introduction
Doxorubicin (DOX)3 is the widely used anticancer agent. However, its usefulness for this application can be limited by its ability to promote cardiotoxicity and heart failure (1, 2). Obesity is associated with poor outcome in breast cancer patients treated with DOX-based chemotherapy (3). Experimental studies showed that high fat diet-induced obese rats are highly sensitive to DOX-induced cardiotoxicity (4) and that moderate diet restriction protects against this cardiotoxicity (5). However, the mechanism by which obese organisms display enhanced sensitivity to DOX-mediated toxicity is unknown. Accumulating evidence indicates that dysregulation of adipocyte-derived hormones, also known as adipocytokines, promotes the development of various obesity-linked diseases (6–9). These findings allowed us to hypothesize that obese states affect DOX-induced heart function through modulation of adipocytokine production.
Adiponectin is an adipocytokine that is exclusively produced by adipocytes (6, 7). Plasma adiponectin levels are decreased in obese subjects (10), and adiponectin is down-regulated in association with obesity-linked disorders including type 2 diabetes, hypertension, atherosclerosis, and ischemic heart disease (6, 7). Consistent with these clinical observations, experimental studies demonstrate that adiponectin deficiency contributes to exacerbation of insulin resistance, hypertension, vascular dysfunction, and cardiac remodeling under conditions of stress (11–13), indicating the protective actions of adiponectin on various obesity-inducible complications.
Several experimental studies have shown that adiponectin protects against acute cardiac injury through its ability to attenuate myocyte apoptosis (14, 15). However, the signaling pathways leading to cardioprotection and receptors that mediate these actions of adiponectin are not well defined (16, 17). Because increased myocyte apoptosis contributes to the development of DOX-induced cardiomyopathy (2), we hypothesized that adiponectin could modulate cardiac injury in response to DOX. Here, we investigated the effect of adiponectin on DOX-induced cardiomyopathy with loss- and gain-of-function genetic manipulations and assessed its molecular mechanisms.
EXPERIMENTAL PROCEDURES
Materials
Recombinant human adiponectin protein obtained from a mammalian cell expression system was purchased from BioVendor. The following primary antibodies were purchased from Cell Signaling Technology: phospho-Akt (Ser-473) antibody, Akt antibody, cleaved caspase-3 antibody, caspase-3 antibody, and α-tubulin antibody. Calreticulin (CRT) antibody was purchased from Affinity BioReagents. Sarcomeric actinin antibody was from Sigma. LY294002 was purchased from Calbiochem. Sphingosine kinase (SphK)-1 inhibitor was purchased from Cayman Chemical. VPC23019 was purchased from Avanti Polar Lipids. DOX was provided as a generous gift by Kyowa Hakko Kirin Co., Ltd. (Tokyo, Japan). Adenovirus vectors containing the gene for β-galactosidase (Ad-β-gal) and full-length mouse adiponectin (Ad-APN) were prepared as described previously (14).
Animals and Experimental Model
Male wild-type (WT), adiponectin knock-out (APN-KO) (11), and Akt1 heterozygous knock-out (Akt1-KO) mice (The Jackson Laboratory) in C57/BL6J background at 8–10 weeks of age were used in the present study. Mice were intraperitoneally injected with single dose of DOX (20 mg/kg) as described previously (18). In some experiments, 2 × 108 plaque-forming units (pfu) of Ad-APN or Ad-β-gal were systemically injected into the tail vein of mice 3 days before DOX injection (14). Heart rate and blood pressure were determined using a tail cuff pressure analysis system (Softron, Tokyo, Japan). Survival of mice after DOX administration was evaluated using the Kaplan-Meier method. The study protocol was approved by the Institutional Animal Care and Use Committee of the Nagoya University School of Medicine.
Echocardiographic Analysis
We subjected surviving mice to transthoracic echocardiography to determine cardiac function and structure in the conscious state at day 5 after DOX injection (19). To measure left ventricular (LV) systolic function and chamber dimensions, echocardiogram analysis was performed with an Acuson Sequoia C-256 machine using a 15-MHz probe. We quantified LV end systolic diameter (LVDs), LV end diastolic diameter (LVDd), and percent LV fractional shortening (%FS) from M-mode images.
Cell Culture
Primary cultures of neonatal rat ventricular myocytes (NRVMs) were prepared as described previously (19). Isolated myocytes were cultured in DMEM containing 10% fetal calf serum. Before each experiment, cells were placed in serum-free DMEM for 24 h. For the adiponectin stimulation studies, cells were treated with recombinant human adiponectin protein (10 μg/ml) for the indicated lengths of time. To determine the degree of apoptotic cells, NRVMs were preincubated with adiponectin protein (10 μg/ml) or vehicle for 60 min followed by treatment with DOX (1 μm) or vehicle for 24 h. Cell number and TUNEL-positive cells were examined as described previously (14). In some experiments, NRVMs were pretreated with LY294002 (50 μm) or CRT-neutralizing antibody (5 μg/ml) before adiponectin stimulation.
For gene ablation studies, NRVMs were transfected with small interfering RNAs (siRNAs) targeting SphK-1, CRT, LDL receptor-related protein 1 (LRP1), AdipoR1, and AdipoR2 or unrelated siRNAs (Dharmacon Inc., Lafayette, CO) by Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. Forty-eight hours after transfection, NRVMs were incubated with recombinant adiponectin protein for the indicated length of times.
For detection of CRT, NRVMs were incubated with anti-CRT antibody (5 μg/ml) or control chicken IgY (5 μg/ml) for 60 min, stained with FITC-conjugated anti-chicken IgY, and analyzed by flow cytometric analysis. In some experiments, NRVMs were preincubated with 6 μg/ml FAM-labeled adiponectin (Phoenix Pharmaceuticals) for 60 min followed by incubation with anti-CRT antibody for 60 min. Fluorescence intensity was measured with a fluorescence microplate reader.
Histological Analysis
LV tissues were obtained 5 days after DOX administration, embedded in OCT compound (Miles, Elkhart, IN), and frozen in liquid nitrogen. To determine myocardial apoptosis, we performed TUNEL staining using the In Situ Cell Death Detection kit (Roche Applied Science) as described previously (14). DAPI was used for nuclear staining. Anti-sarcomeric actinin antibody was used for determination of myocytes followed by TUNEL and nuclear staining. Ten randomly chosen microscopic fields from four different sections in each tissue block were examined for the presence of TUNEL-positive cells.
Western Blot Analysis
Heart tissue samples obtained at 5 days after DOX administration were homogenized in lysis buffer containing 20 mm Tris-HCI (pH 8.0), 1% Nonidet P-40, 150 mm NaCl, 0.5% deoxycholic acid, 1 mm sodium orthovanadate, and protease inhibitor mixture (Sigma). NRVMs were lysed in the same component element buffer. Equal amounts of protein were separated with denaturing SDS, 10% polyacrylamide gels. The membranes were immunoblotted with the primary antibodies at a 1:1000 dilution followed by secondary antibody at a 1:5000 dilution. Bands were visualized using the ECL Western Blotting Detection kit (Amersham Biosciences).
Measurement of mRNA
Total RNA from cultured cells was prepared by using an RNA isolation kit (Qiagen, Valencia, CA) according to the manufacturer's protocols. Complementary DNA (cDNA) from 500 ng of total RNA was synthesized by using SuperScript RT-PCR Systems (Invitrogen) according to the manufacturer's instructions. Quantitative real time PCR (RT-PCR) analysis was performed on a CFX-96 system using EvaGreen as a double-stranded DNA-specific dye according to the manufacturer's instructions (Bio-Rad). Primers were designed as follows: 5′-TCCCTGACCCTGATGCTAAGAAG-3′ and 5′-ATTGTGCCAGACTTGACCTGCC-3′ for rat CRT, 5′-GGACTTCAGTTATGCCAACG-3′ and 5′-GACTCAGGGAGATGTTGA-3′ for rat LRP1, 5′-CGTGGCCTTTATGCTGCTCG-3′ and 5′-TCTAGGCCGTAACGGAATTC-3′ for rat AdipoR1, 5′-CCACAACCTTGCTTCATCTA-3′ and 5′-GATACTGAGGGGTGGCAAAC-3′ for rat AdipoR2, and 5′-TCAAGAAGGTGGTGAAGCAG-3′ and 5′-AGGTGGAAGAATGGGAGTTG-3′ for GAPDH. The expression levels of examined transcripts were compared with that of GAPDH and normalized to the mean value of controls.
Statistical Analysis
Data are presented as mean ± S.D. Statistical analysis was performed by analysis of variance followed by Tukey's honestly significant difference test. Calculation and statistical tests including Kaplan-Meier estimates of survival were performed using SPSS version 17 (SPSS Japan Inc.). A value of p < 0.05 was accepted as statistically significant.
RESULTS
APN-KO Mice Had Enhanced LV Dysfunction after DOX Injection
To test whether adiponectin modulates DOX-induced cardiomyopathy, we intraperitoneally injected APN-KO or WT mice with a single dose of DOX (20 mg/kg). Mortality after DOX injection is shown in Fig. 1A. APN-KO mice exhibited a significant increase in mortality after DOX injection compared with WT mice by Kaplan-Meier estimates (p = 0.02; Fig. 1A).
FIGURE 1.

Adiponectin improves DOX-induced cardiac dysfunction. A, Kaplan-Meier survival analysis of WT and KO mice following DOX or vehicle administration (n = 11 in each group). B, representative M-mode echocardiograms for WT or KO mice at 5 days after DOX or vehicle injection. Vertical scale bar, 1 mm; horizontal scale bar, 200 ms. C–E, quantitative analysis of the LVDd (C), LVDs (D), and %FS (E) for WT and KO mice at 5 days after DOX injection (n = 5). F, quantitative analysis of %FS in WT and APN-KO mice treated with Ad-β-gal or Ad-APN at 5 days after DOX injection. Ad-APN or Ad-β-gal (2 × 108 pfu total) was delivered intravenously via the tail vein 3 days before DOX injection (n = 5). Results are presented as mean ± S.D.
Echocardiographic analysis at 5 days showed that DOX injection led to an increase in LVDs and a decrease in %FS in both APN-KO and WT mice without affecting LVDd (Fig. 1, B–E). APN-KO mice showed increased LVDs and decreased %FS compared with WT mice (Fig. 1, B, D, and E). LVDd did not differ after DOX injection between the two strains of mice (Fig. 1C). There were no significant differences in LVDd, LVDs, and %FS after vehicle injection between APN-KO and WT mice (Fig. 1, B–E).
To test whether overexpression of adiponectin modulates LV contractile dysfunction in response to DOX injection, we systemically treated APN-KO and WT mice with Ad-APN or Ad-β-gal as a control via the tail vein 3 days before DOX injection. At the time of DOX injection, adiponectin levels were 21.2 ± 4.8 μg/ml in WT/Ad-APN, 12.9 ± 1.9 μg/ml in WT/control, 11.5 ± 1.5 μg/ml in APN-KO/Ad-APN, and <0.05 μg/ml in APN-KO/control. Ad-APN treatment significantly increased %FS following DOX injection in both WT and APN-KO mice (Fig. 1F).
Adiponectin Inhibits DOX-induced Apoptosis in Heart
A previous study indicated that an increase in myocyte apoptosis contributes to the pathogenesis of DOX-induced cardiomyopathy (2). To investigate the extent of apoptosis in the heart, we performed TUNEL staining on hearts isolated from the different experimental sets of mice. Representative photographs of TUNEL-positive nuclei in the heart at day 5 after DOX injection are shown in Fig. 2A. Quantitative analysis revealed a significantly higher proportion of TUNEL-positive cells in the myocardium of APN-KO mice compared with WT mice after DOX injection, whereas few or no TUNEL-positive cells could be detected in the hearts of WT or APN-KO mice following vehicle injection (Fig. 2B). Administration of DOX led to an increase in the conversion of the proapoptotic proenzyme caspase-3 to the active cleaved form in WT hearts, but the magnitude of this induction was greater in APN-KO than WT mice (Fig. 2C).
FIGURE 2.


Adiponectin suppresses DOX-induced apoptosis in heart. A, representative photographs of heart sections stained with TUNEL from WT and APN-KO mice at 5 days after DOX or vehicle injection. Apoptotic nuclei were determined by TUNEL staining (green). Myocytes were determined by sarcomeric actinin (red), and total nuclei were counterstained with DAPI (blue). High magnification representative photographs of heart sections are shown in the bottom panels. Scales bars, 40 μm (top) and 10 μm (bottom). B, quantitative analysis of apoptotic nuclei from WT (n = 5) and APN-KO mouse (n = 5) hearts following DOX or vehicle injection. TUNEL-positive nuclei were counted in several randomly selected fields and expressed as a percentage of the total number of nuclei. C, detection of cleaved caspase-3 from WT (n = 5) and APN-KO mouse (n = 5) hearts following DOX or vehicle injection by Western blot analysis. D, quantitative analysis of apoptotic nuclei from WT (n = 5) and APN-KO (n = 5) mouse hearts treated with Ad-APN or Ad-β-gal following DOX injection. TUNEL-positive nuclei were counted in several randomly selected fields and expressed as a percentage of the total number of nuclei. E, detection of cleaved caspase-3 from WT (n = 5) and APN-KO (n = 5) mouse hearts treated with Ad-APN or Ad-β-gal following DOX injection by Western blot analysis. F, effect of DOX on cultured myocytes. Representative photographs of TUNEL-positive cardiac myocytes under conditions of serum deprivation with or without DOX stimulation for 24 h in the presence or absence of adiponectin protein are shown. Apoptotic nuclei were identified by TUNEL staining (green), and total nuclei were identified by DAPI counterstaining (blue). G, quantitative analysis of TUNEL-positive cells with DOX or vehicle exposure in the presence or absence of adiponectin. TUNEL-positive nuclei were counted in several randomly selected fields and expressed as a percentage of the total number of nuclei (n = 4). H, detection of cleaved caspase-3 with DOX or vehicle exposure in the presence or absence of adiponectin.
To examine whether an increase in adiponectin levels has antiapoptotic actions in vivo, we assessed myocardial cell viability by TUNEL assay in tissue sections following DOX injection in both WT and APN-KO mice treated with Ad-APN or Ad-β-gal. Ad-APN treatment decreased the frequencies of TUNEL-positive cells in both WT and APN-KO mice (Fig. 2D). The expression level of cleaved caspase-3 was also decreased by Ad-APN treatment (Fig. 2E).
To analyze the antiapoptotic actions of adiponectin at the cellular level, primary cultures of rat neonatal ventricular myocytes were subjected to DOX treatment under conditions of serum deprivation for 24 h in the presence or absence of recombinant adiponectin protein, and then we performed TUNEL staining. Treatment with a physiological concentration of adiponectin diminished the frequency of TUNEL-positive cardiomyocytes in the absence of DOX (Fig. 2, F and G). DOX treatment increased the frequency of TUNEL-positive cardiomyocytes, which was inhibited by treatment with adiponectin (Fig. 2, F and G). Treatment with adiponectin also diminished the expression of cleaved caspase-3 in the presence or absence of DOX (Fig. 2H).
Akt Is Involved in Inhibitory Action of Adiponectin on Myocyte Apoptosis
Because it has been reported that DOX promotes cardiomyocyte apoptosis through modulation of Akt activity (20, 21), the effect of adiponectin on phosphorylation of Akt in the heart was investigated by Western blotting in vivo and in vitro. Consistent with previous observations (22, 23), administration of DOX decreased the phosphorylation levels of Akt in WT hearts. The DOX-induced declines in Akt phosphorylation occurred to a greater degree in APN-KO mice than in WT mice (Fig. 3A). The expression level of total Akt protein did not differ between sham-treated WT and APN-KO mice. In cultured cardiac myocytes, treatment with adiponectin stimulated the phosphorylation of Akt in a time-dependent manner with maximal Akt phosphorylation occurring at 16 h (Fig. 3B). To test whether Akt is involved in the inhibitory effects of adiponectin on myocyte apoptosis, cardiac myocytes were pretreated with the phosphatidylinositol 3-kinase inhibitor LY294002, and DOX-induced apoptosis was assessed. Treatment with LY294002 reversed the inhibitory effects of adiponectin on the frequency of TUNEL-positive cells and caspase-3 activation after DOX stimulation (Fig. 3, C and D).
FIGURE 3.


Adiponectin inhibits DOX-induced myocyte apoptosis via Akt signaling. A, the representative immunoblots in phosphorylated Akt (p-Akt) in heart tissues from WT and APN-KO mice at 5 days after DOX or vehicle injection (left). Akt phosphorylation levels in myocardium were analyzed by Western blotting. Right, quantitative analysis of relative changes in phosphorylated Akt. Phosphorylation levels of Akt were normalized to the α-tubulin signal and expressed as the percentage of the signal intensity of vehicle-injected WT mice (n = 5). B, time-dependent changes in the phosphorylation of Akt in rat cultured cardiac myocytes after adiponectin treatment (10 μg/ml). Quantitative analysis of relative changes in phosphorylated Akt (n = 4) (*, p < 0.05 versus control) is shown. C and D, involvement of Akt in adiponectin inhibition of DOX-stimulated myocyte apoptosis. Cells were pretreated with the phosphatidylinositol 3-kinase inhibitor LY294002 or vehicle for 60 min and treated with or without adiponectin (10 μg/ml) followed by stimulation with DOX for 24 h. Quantitative analysis of TUNEL-positive nuclei (C) and caspase-3 activity (D) is shown. TUNEL-positive nuclei were counted in several randomly selected fields and expressed as a percentage of the total number of nuclei (n = 4). E, F, and G, effects of adiponectin on DOX-induced cardiomyopathy in Akt-1 KO mice. Quantitative analysis of %FS (E), TUNEL-positive nuclei in hearts (F), and caspase-3 activity (G) in Ad-β-gal- or Ad-APN-treated WT and Akt-1 KO mice at 5 days after DOX injection (n = 5) is shown. Ad-APN or Ad-β-gal (2 × 108 pfu total) was delivered intravenously via the tail vein 3 days before DOX injection. n.s., not significant.
To analyze the involvement of Akt signaling in the cardioprotective action of adiponectin in vivo, Akt1-KO mice that received an intravenous infusion of Ad-APN or Ad-β-gal were injected with a single dose of DOX. At the time of DOX injection, circulating adiponectin levels increased to a level 1.9 ± 0.4 times higher in Ad-APN-treated Akt1-KO mice than in Ad-β-gal-treated Akt1-KO mice (20.6 ± 5.0 and 11.9 ± 1.9 μg/ml, respectively; p < 0.05). Thus, adiponectin levels in the bloodstream of Ad-APN-treated Akt1-KO mice were similar to those of Ad-APN-treated WT mice at the time of DOX administration. In contrast to WT mice, treatment with Ad-APN did not improve the DOX-induced reduction of %FS in Akt1-KO mice (Fig. 3E). In addition, treatment with Ad-APN did not attenuate the increase in the extent of TUNEL-positive cardiomyocytes and caspase-3 activation in Akt-1 KO mice (Fig. 3, F and G).
Sphingosine Kinase Is Essential for Adiponectin-induced Antiapoptotic Effect
Sphingosine 1-phosphate (S1P) exerts protective effects on cell survival through Akt signaling in various cell types including cardiac myocytes (24–26). Therefore, we assessed whether adiponectin regulates Akt activation through SphK, which converts sphingosine to S1P. Cardiac myocytes were pretreated with SphK-1 inhibitor followed by incubation with adiponectin or vehicle. Adiponectin-induced Akt phosphorylation was diminished by pretreatment with SphK-1 inhibitor (Fig. 4A). S1P receptors 1, 2, and 3 are expressed in cardiac myocytes (25). Thus, to test whether S1P receptors participate in adiponectin-induced Akt activation, myocytes were treated with the S1P receptor antagonist VPC23019. Pretreatment with VPC23019 partially diminished adiponectin-induced Akt phosphorylation (Fig. 4A). The inhibitory actions of adiponectin on the DOX-induced increase in TUNEL-positive cells and caspase-3 activation were reversed by pretreatment with SphK-1 inhibitor or VPC23019 (Fig. 4, B and C). Furthermore, to corroborate these data, knockdown experiments were performed with siRNA against SphK-1. Transfection with siRNA targeting SphK-1 resulted in a 79% reduction of SphK-1 expression. Knockdown of SphK-1 reduced Akt phosphorylation caused by adiponectin (Fig. 4D). Collectively, these data suggested that adiponectin suppresses DOX-induced apoptosis through an Akt/SphK-1-dependent pathway.
FIGURE 4.

SphK-dependent Akt signaling is essential for inhibitory effects of adiponectin on DOX-induced apoptosis. A, Western blot analysis for adiponectin-induced phosphorylation of Akt in the presence of the SphK-1 inhibitor, the S1P receptor antagonist VPC23019, or vehicle (DMSO). Quantitative analysis of relative changes in phosphorylated Akt (P-Akt) is shown. Phosphorylation of Akt was normalized to the α-tubulin signal (n = 4 in each group). B and C, effect of SphK-1 inhibitor or VPC23019 on the inhibitory effects of adiponectin on DOX-induced myocyte apoptosis. Cells were pretreated with SphK-1 inhibitor, VPC23019, or vehicle (DMSO) for 60 min and treated with or without adiponectin (10 μg/ml) for 24 h. Quantitative analysis of TUNEL-positive nuclei (B) and caspase-3 activity (C) is shown. TUNEL-positive nuclei were counted in several randomly selected fields and expressed as a percentage of the total number of nuclei (n = 4). D, representative immunoblots of phosphorylated Akt following treatment with adiponectin for 16 h in the presence of siRNA targeting SphK-1 or unrelated siRNA in cardiac myocytes (top). Bottom, quantitative analysis of relative changes in phosphorylated Akt. SK-I, SphK-1 inhibitor. (*, p < 0.05 versus control; #, p < 0.05 versus APN+/DOX+/DMSO+/SK−I−/VPC23019−.)
Adiponectin Suppresses DOX-induced Myocyte Apoptosis through LRP1/CRT-mediated Akt Activation
Recently, we have shown that the LRP1/CRT co-receptor system mediates adiponectin stimulation of vascular cells (27). Thus, to test whether this receptor system is involved in the protective actions of adiponectin on myocyte apoptosis, we first assessed whether adiponectin binds to cell surface CRT of myocytes. As shown in Fig. 5A, CRT was detected on the cell surface of cardiac myocytes by flow cytometric analysis. Furthermore, pretreatment with anti-CRT antibody diminished the binding of fluorescence-labeled adiponectin to cardiac myocytes (Fig. 5B).
FIGURE 5.

LRP1/CRT co-receptor system is involved in inhibitory effects of adiponectin on DOX-induced myocyte apoptosis. A, detection of CRT on the cell surface of cardiac myocytes by flow cytometric analysis. Myocytes were incubated with anti-CRT antibody or control IgY for 60 min. B, anti-CRT antibody (ab) diminishes the binding of fluorescence-labeled adiponectin to cardiac myocytes. Myocytes were preincubated with anti-CRT antibody (200 μg/ml) or control IgY (200 μg/ml) for 60 min followed by incubation with FAM-labeled adiponectin (6 μg/ml) for 60 min. C, Western blot analysis for adiponectin-induced phosphorylation of Akt in the presence of anti-CRT antibody or control IgY. Myocytes were pretreated with anti-CRT antibody or control IgY for 60 min and treated with adiponectin (10 μg/ml) or vehicle for 16 h. Quantitative analysis of relative changes in phosphorylated Akt (P-Akt) is shown. Phosphorylation of Akt was normalized to the α-tubulin signal. D and E, effect of the anti-CRT antibody on adiponectin inhibition of myocyte apoptosis after DOX stimulation. Cells were pretreated with anti-CRT antibody or control IgY for 60 min and treated with adiponectin or vehicle for 24 h. Quantitative analysis of TUNEL-positive nuclei (D) and caspase-3 activity (E) is shown. F, the representative immunoblots of phosphorylated Akt (p-Akt) following treatment with adiponectin for 16 h in the presence of siRNA targeting CRT, LRP1, AdipoR1, or AdipoR2 or unrelated siRNA in cardiac myocytes (top). Bottom, quantitative analysis of relative changes in phosphorylated Akt. G and H, effect of knockdown of CRT, LRP1, AdipoR1, or AdipoR2 on adiponectin inhibition of TUNEL-positive myocytes (G) and caspase-3 activity (H) after DOX stimulation (n = 4–6).
To determine whether adiponectin activates Akt through an LRP1/CRT-dependent pathway in vitro, cardiac myocytes were incubated with recombinant adiponectin in the presence or absence of anti-CRT antibody. The adiponectin-induced increase in Akt phosphorylation was reduced by pretreatment with anti-CRT antibody (Fig. 5C). Preincubation with anti-CRT antibody also reversed the inhibitory effects of adiponectin on TUNEL-positive cells and caspase-3 activation induced by DOX (Fig. 5, D and E). To corroborate these data, knockdown experiments were performed with siRNAs targeting adiponectin receptors including AdipoR1 and AdipoR2. After transfection of cardiomyocytes with siRNA against CRT, CRT mRNA expression was reduced by 81%. Transfection with siRNA against LRP1 led to a 76% reduction of LRP1 expression. Following transfection with siRNA against AdipoR1 or AdipoR2, AdipoR1 mRNA was decreased by 78%, and AdipoR2 mRNA was decreased by 71%. The phosphorylation of Akt stimulated by adiponectin was reduced by knockdown of CRT and LRP1 with siRNA but not siRNA against AdipoR1 and AdipoR2 (Fig. 5F). The inhibitory actions of adiponectin on the DOX-induced increase in TUNEL-positive cells and caspase-3 activation were reversed by ablation of CRT, LRP1, or AdipoR1 but not by ablation of AdipoR2 (Fig. 5, G and H).
DISCUSSION
The present work provides the first evidence that adiponectin confers resistance to DOX-induced myocardial damage through activation of Akt signaling involving the LRP1/CRT co-receptor system within cardiomyocytes. APN-KO mice showed exacerbated LV contractile dysfunction following DOX injection, whereas exogenous adiponectin improved DOX-induced LV dysfunction in WT and APN-KO mice. Obese states have been shown to be associated with an increased sensitivity to DOX-induced cardiotoxicity (4, 5). Because obesity is associated with low adiponectin levels, our data suggest that adiponectin can function as a crucial adipocytokine that affects DOX-induced heart function under obese conditions.
A number of animal studies demonstrated that adiponectin has beneficial effects on the heart in various disease models. We have shown that APN-KO mice developed larger infarcts in the heart following ischemia-reperfusion injury (14). Administration of adiponectin to mice, rats, or pigs leads to improvement of myocardial injury and function after ischemia-reperfusion (14, 15, 28, 29). Adiponectin also improves systolic dysfunction following permanent coronary ligation (30) and reduces concentric cardiac hypertrophy after pressure overload (19). However, the mechanism of the cardioprotective actions of adiponectin is incompletely understood. Our present data document for the first time using a mouse genetic model that adiponectin protects against cardiac apoptosis and dysfunction through its ability to promote Akt-dependent myocyte survival. Mechanistic studies in cultured myocytes provide further evidence that Akt signaling protects against DOX-induced cardiac toxicity. Furthermore, these mechanistic studies implicate the LRP1/CRT co-receptor system and the production of S1P in the activation of Akt signaling by adiponectin.
The ability of adiponectin to ameliorate acute cardiac injury is due at least in part to its ability to protect the myocardium from cell death. In the present study, APN-KO mice showed increased apoptotic activity in the failing myocardium after DOX treatment. Systemic delivery of adiponectin reduced cardiomyocyte apoptosis in the DOX-treated hearts in WT and APN-KO mice. Adiponectin inhibited DOX-stimulated apoptosis in cultured cardiomyocytes. Similarly, adiponectin deficiency causes enhanced apoptosis in the heart following ischemia-reperfusion (14, 15). Adiponectin protein prevents hypoxia-reoxygenation-induced apoptotic activity in cardiomyocytes (14). Moreover, adiponectin suppresses apoptosis of endothelial cells under conditions of serum starvation (31).
Akt acts as a crucial regulator of cell survival in the heart (20, 32). It has been shown that elevated myocardial Akt signaling ameliorates DOX-induced cardiomyopathy (21) and that Akt signaling protects against myocyte apoptosis induced by cardiac ischemia-reperfusion injury (32). Furthermore, Akt protects against DOX-induced apoptosis in cardiac myocytes in vitro (20). It is shown here that adiponectin stimulated Akt phosphorylation in cardiac myocytes and that inhibition of Akt signaling abrogated the inhibitory effects of adiponectin on DOX-induced apoptosis. APN-KO mice displayed a reduction of Akt phosphorylation levels in the heart after DOX injection. Of importance, adiponectin improved DOX-induced cardiac apoptosis and dysfunction in WT mice but not in Akt1-KO mice. Therefore, our genetic data indicate that the protective action of adiponectin on DOX-induced cardiomyopathy is mediated by its ability to promote Akt-dependent survival of cardiac myocytes.
SphK-1 converts sphingosine to S1P, which has various bioactivities including suppression of apoptosis (24, 33). S1P inhibits apoptosis through Akt signaling pathway in cardiac cells (25, 26). Previously, we have reported that adiponectin stimulates cyclooxygenase-2 expression in cardiac myocytes through an Shpk-1-dependent mechanism (34). A recent study has also shown that overproduction of adiponectin decreases caspase-8-mediated cell death in vivo through a sphingolipid-mediated pathway (35). Here, we report that inhibition of Shpk-1-dependent pathways abolished adiponectin-stimulated Akt activation in cardiomyocytes and blocked the suppressive effects of adiponectin on DOX-induced myocyte apoptosis. Collectively, SphK-1-dependent Akt activation may be one of the crucial pathways involved in adiponectin-induced myocyte survival.
Recently, Konishi et al. (36) reported that adiponectin knockdown with antisense RNA exacerbates DOX-induced cardiac toxicity, and this was correlated with changes in AMP-activated protein kinase phosphorylation and levels of the antiapoptosis factor Bcl2. We and others reported that adiponectin can suppress myocyte apoptosis in vitro (14, 15, 36), and prosurvival actions of adiponectin are mediated by its ability to promote AMP-activated protein kinase signaling (14, 36). AdipoR1 has been shown to mediate adiponectin-induced AMP-activated protein kinase activation in various cells including hepatocytes, skeletal muscle, cardiac cells, and endothelial cells (37, 38). Taken together, these observations suggest that adiponectin protects against myocardial apoptosis through at least two mechanisms, the LRP1/CRT/SphK-1/Akt and AdipoR1/AMP-activated protein kinase pathways.
The roles of adiponectin receptors in mediating the actions of adiponectin are controversial and incompletely understood. In this regard, adiponectin exists in the bloodstream at high concentrations and can account for 0.01% of the total plasma protein. Furthermore, adiponectin is structurally similar to members of the collectin family, and it predominantly occurs as a high molecular weight multimer (39). Thus, it is difficult to rationalize that adiponectin as a ligand has a classical high affinity interaction with a receptor because of issues of saturated receptor occupancy. Recently, we have shown that some of the functions of adiponectin can be mediated by the LRP1/CRT co-receptor system that binds high abundance macromolecules (27, 39). This receptor system has also been reported to mediate the regulation of intracellular signaling by other collectin family proteins (27, 40–43). Our present data indicate that CRT is expressed on the cell surface of cardiac myocytes, and ablation of CRT or LRP1 by siRNA or pretreatment with anti-CRT blocking antibody reduces the stimulatory actions of adiponectin on Akt activation and survival activity in cultured myocytes. Although AdipoR1 and AdipoR2 expression can be detected on the surface of cardiac myocytes (44), ablation of these receptors by siRNA had no detectable effects on the adiponectin-stimulated Akt activation. Collectively, these data suggest that LRP1/CRT on cardiac myocytes functions as a mediator of Akt-dependent antiapoptotic signals by adiponectin.
In conclusion, we show that adiponectin protects against DOX-induced cardiac dysfunction and damage. Adiponectin activates SphK-1/Akt signaling via a receptor-dependent pathway involving LRP1/CRT, and this signaling pathway protects against DOX-induced myocardial apoptosis. The hypothetical signaling pathways are schematically shown in Fig. 6. These data suggest that decreased adiponectin levels may contribute to the increased susceptibility to DOX-inducible cardiotoxicity.
FIGURE 6.
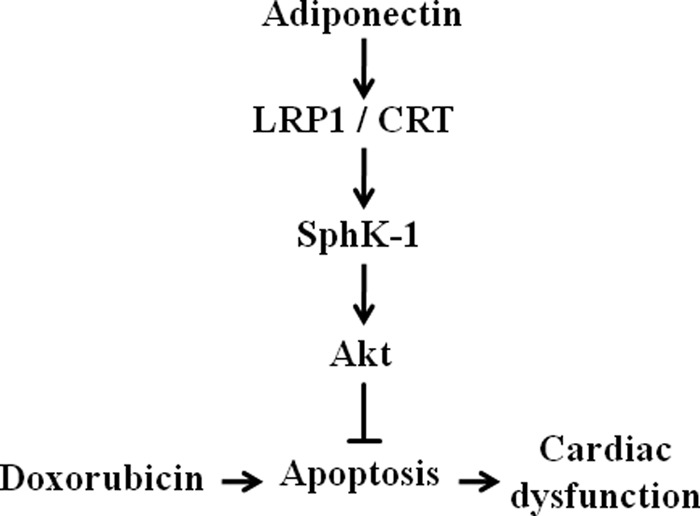
Proposed scheme for protective actions of adiponectin on DOX-induced cardiomyopathy. Adiponectin activates SphK-1/Akt signaling via an LRP1/CRT-dependent pathway, and this pathway protects against DOX-induced cardiac dysfunction by its ability to attenuate myocardial apoptosis.
Acknowledgments
We gratefully acknowledge the technical assistance of Megumi Kondo, Rie Miura, and Takashi Enomoto.
This work was supported in part by a grant-in-aid for young scientists B, the Uehara Memorial Foundation, and the Japan Heart Foundation/Novartis Grant for Research Award on Molecular and Cellular Cardiology 2011 (to R. S.).
- DOX
- doxorubicin
- APN
- adiponectin
- CRT
- calreticulin
- LRP1
- LDL receptor-related protein 1
- SphK
- sphingosine kinase
- S1P
- sphingosine 1-phosphate
- LV
- left ventricle
- Ad
- adenovirus
- LVDs
- LV end systolic diameter
- LVDd
- LV end diastolic diameter
- %FS
- percent LV fractional shortening
- NRVM
- neonatal rat ventricular myocyte
- FAM
- mono-5-(and-6)-carboxyfluorescein.
REFERENCES
- 1. Singal P. K., Iliskovic N. (1998) N. Engl. J. Med. 339, 900–905 [DOI] [PubMed] [Google Scholar]
- 2. Arola O. J., Saraste A., Pulkki K., Kallajoki M., Parvinen M., Voipio-Pulkki L. M. (2000) Cancer Res. 60, 1789–1792 [PubMed] [Google Scholar]
- 3. de Azambuja E., McCaskill-Stevens W., Francis P., Quinaux E., Crown J. P., Vicente M., Giuliani R., Nordenskjöld B., Gutiérez J., Andersson M., Vila M. M., Jakesz R., Demol J., Dewar J., Santoro A., Lluch A., Olsen S., Gelber R. D., Di Leo A., Piccart-Gebhart M. (2010) Breast Cancer Res. Treat. 119, 145–153 [DOI] [PubMed] [Google Scholar]
- 4. Mitra M. S., Donthamsetty S., White B., Mehendale H. M. (2008) Toxicol. Appl. Pharmacol. 231, 413–422 [DOI] [PubMed] [Google Scholar]
- 5. Mitra M. S., Donthamsetty S., White B., Latendresse J. R., Mehendale H. M. (2007) Toxicol. Appl. Pharmacol. 225, 90–101 [DOI] [PubMed] [Google Scholar]
- 6. Ouchi N., Kihara S., Funahashi T., Matsuzawa Y., Walsh K. (2003) Curr. Opin. Lipidol. 14, 561–566 [DOI] [PubMed] [Google Scholar]
- 7. Shibata R., Ouchi N., Murohara T. (2009) Circ. J. 73, 608–614 [DOI] [PubMed] [Google Scholar]
- 8. Berg A. H., Scherer P. E. (2005) Circ. Res. 96, 939–949 [DOI] [PubMed] [Google Scholar]
- 9. Ouchi N., Parker J. L., Lugus J. J., Walsh K. (2011) Nat. Rev. Immunol. 11, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arita Y., Kihara S., Ouchi N., Takahashi M., Maeda K., Miyagawa J., Hotta K., Shimomura I., Nakamura T., Miyaoka K., Kuriyama H., Nishida M., Yamashita S., Okubo K., Matsubara K., Muraguchi M., Ohmoto Y., Funahashi T., Matsuzawa Y. (1999) Biochem. Biophys. Res. Commun. 257, 79–83 [DOI] [PubMed] [Google Scholar]
- 11. Maeda N., Shimomura I., Kishida K., Nishizawa H., Matsuda M., Nagaretani H., Furuyama N., Kondo H., Takahashi M., Arita Y., Komuro R., Ouchi N., Kihara S., Tochino Y., Okutomi K., Horie M., Takeda S., Aoyama T., Funahashi T., Matsuzawa Y. (2002) Nat. Med. 8, 731–737 [DOI] [PubMed] [Google Scholar]
- 12. Ohashi K., Kihara S., Ouchi N., Kumada M., Fujita K., Hiuge A., Hibuse T., Ryo M., Nishizawa H., Maeda N., Maeda K., Shibata R., Walsh K., Funahashi T., Shimomura I. (2006) Hypertension 47, 1108–1116 [DOI] [PubMed] [Google Scholar]
- 13. Shibata R., Ouchi N., Kihara S., Sato K., Funahashi T., Walsh K. (2004) J. Biol. Chem. 279, 28670–28674 [DOI] [PubMed] [Google Scholar]
- 14. Shibata R., Sato K., Pimentel D. R., Takemura Y., Kihara S., Ohashi K., Funahashi T., Ouchi N., Walsh K. (2005) Nat. Med. 11, 1096–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tao L., Gao E., Jiao X., Yuan Y., Li S., Christopher T. A., Lopez B. L., Koch W., Chan L., Goldstein B. J., Ma X. L. (2007) Circulation 115, 1408–1416 [DOI] [PubMed] [Google Scholar]
- 16. Denzel M. S., Scimia M. C., Zumstein P. M., Walsh K., Ruiz-Lozano P., Ranscht B. (2010) J. Clin. Investig. 120, 4342–4352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Ge G., Spooner E., Hug C., Gimeno R., Lodish H. F. (2009) FASEB J. 23, 241–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weinstein D. M., Mihm M. J., Bauer J. A. (2000) J. Pharmacol Exp. Ther. 294, 396–401 [PubMed] [Google Scholar]
- 19. Shibata R., Ouchi N., Ito M., Kihara S., Shiojima I., Pimentel D. R., Kumada M., Sato K., Schiekofer S., Ohashi K., Funahashi T., Colucci W. S., Walsh K. (2004) Nat. Med. 10, 1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Negoro S., Oh H., Tone E., Kunisada K., Fujio Y., Walsh K., Kishimoto T., Yamauchi-Takihara K. (2001) Circulation 103, 555–561 [DOI] [PubMed] [Google Scholar]
- 21. Taniyama Y., Walsh K. (2002) J. Mol. Cell. Cardiol. 34, 1241–1247 [DOI] [PubMed] [Google Scholar]
- 22. Bian Y., Sun M., Silver M., Ho K. K., Marchionni M. A., Caggiano A. O., Stone J. R., Amende I., Hampton T. G., Morgan J. P., Yan X. (2009) Am. J. Physiol. Heart Circ. Physiol. 297, H1974–H1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ikeda Y., Aihara K., Akaike M., Sato T., Ishikawa K., Ise T., Yagi S., Iwase T., Ueda Y., Yoshida S., Azuma H., Walsh K., Tamaki T., Kato S., Matsumoto T. (2010) Mol. Endocrinol. 24, 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Osawa Y., Banno Y., Nagaki M., Brenner D. A., Naiki T., Nozawa Y., Nakashima S., Moriwaki H. (2001) J. Immunol. 167, 173–180 [DOI] [PubMed] [Google Scholar]
- 25. Zhang J., Honbo N., Goetzl E. J., Chatterjee K., Karliner J. S., Gray M. O. (2007) Am. J. Physiol. Heart Circ. Physiol. 293, H3150–H3158 [DOI] [PubMed] [Google Scholar]
- 26. Gonzalez E., Kou R., Michel T. (2006) J. Biol. Chem. 281, 3210–3216 [DOI] [PubMed] [Google Scholar]
- 27. Ohashi K., Ouchi N., Sato K., Higuchi A., Ishikawa T. O., Herschman H. R., Kihara S., Walsh K. (2009) Mol. Cell. Biol. 29, 3487–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kondo K., Shibata R., Unno K., Shimano M., Ishii M., Kito T., Shintani S., Walsh K., Ouchi N., Murohara T. (2010) Circ. Cardiovasc. Interv. 3, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonon A. T., Widegren U., Bulhak A., Salehzadeh F., Persson J., Sjöquist P. O., Pernow J. (2008) Cardiovasc. Res. 78, 116–122 [DOI] [PubMed] [Google Scholar]
- 30. Shibata R., Izumiya Y., Sato K., Papanicolaou K., Kihara S., Colucci W. S., Sam F., Ouchi N., Walsh K. (2007) J. Mol. Cell. Cardiol. 42, 1065–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kobayashi H., Ouchi N., Kihara S., Walsh K., Kumada M., Abe Y., Funahashi T., Matsuzawa Y. (2004) Circ. Res. 94, e27–e31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fujio Y., Nguyen T., Wencker D., Kitsis R. N., Walsh K. (2000) Circulation 101, 660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hait N. C., Oskeritzian C. A., Paugh S. W., Milstien S., Spiegel S. (2006) Biochim. Biophys. Acta 1758, 2016–2026 [DOI] [PubMed] [Google Scholar]
- 34. Ikeda Y., Ohashi K., Shibata R., Pimentel D. R., Kihara S., Ouchi N., Walsh K. (2008) FEBS Lett. 582, 1147–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holland W. L., Miller R. A., Wang Z. V., Sun K., Barth B. M., Bui H. H., Davis K. E., Bikman B. T., Halberg N., Rutkowski J. M., Wade M. R., Tenorio V. M., Kuo M. S., Brozinick J. T., Zhang B. B., Birnbaum M. J., Summers S. A., Scherer P. E. (2011) Nat. Med. 17, 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Konishi M., Haraguchi G., Ohigashi H., Ishihara T., Saito K., Nakano Y., Isobe M. (2011) Cardiovasc. Res. 89, 309–319 [DOI] [PubMed] [Google Scholar]
- 37. Yamauchi T., Kamon J., Ito Y., Tsuchida A., Yokomizo T., Kita S., Sugiyama T., Miyagishi M., Hara K., Tsunoda M., Murakami K., Ohteki T., Uchida S., Takekawa S., Waki H., Tsuno N. H., Shibata Y., Terauchi Y., Froguel P., Tobe K., Koyasu S., Taira K., Kitamura T., Shimizu T., Nagai R., Kadowaki T. (2003) Nature 423, 762–769 [DOI] [PubMed] [Google Scholar]
- 38. Yamauchi T., Nio Y., Maki T., Kobayashi M., Takazawa T., Iwabu M., Okada-Iwabu M., Kawamoto S., Kubota N., Kubota T., Ito Y., Kamon J., Tsuchida A., Kumagai K., Kozono H., Hada Y., Ogata H., Tokuyama K., Tsunoda M., Ide T., Murakami K., Awazawa M., Takamoto I., Froguel P., Hara K., Tobe K., Nagai R., Ueki K., Kadowaki T. (2007) Nat. Med. 13, 332–339 [DOI] [PubMed] [Google Scholar]
- 39. Takemura Y., Ouchi N., Shibata R., Aprahamian T., Kirber M. T., Summer R. S., Kihara S., Walsh K. (2007) J. Clin. Investig. 117, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boucher P., Li W. P., Matz R. L., Takayama Y., Auwerx J., Anderson R. G., Herz J. (2007) PLoS One 2, e448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi Y., Mantuano E., Inoue G., Campana W. M., Gonias S. L. (2009) Sci. Signal. 2, ra18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orr A. W., Pallero M. A., Xiong W. C., Murphy-Ullrich J. E. (2004) J. Biol. Chem. 279, 48983–48992 [DOI] [PubMed] [Google Scholar]
- 43. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., Strickland D. K. (2008) Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujioka D., Kawabata K., Saito Y., Kobayashi T., Nakamura T., Kodama Y., Takano H., Obata J. E., Kitta Y., Umetani K., Kugiyama K. (2006) Am. J. Physiol. Heart Circ. Physiol. 290, H2409–H2416 [DOI] [PubMed] [Google Scholar]