

Abstract
The T-cell-dependent B-cell response relies on cognate interaction between B cells and CD4+ Th cells. However, the consequences of this interaction for CD4+ T cells are not entirely known. B cells generally promote CD4+ T-cell responses to pathogens, albeit to a variable degree. In contrast, CD4+ T-cell responses to self or tumor antigens are often suppressed by B cells. Here we demonstrated that interaction with B cells dramatically inhibited the function of virus-specific CD4+ T cells in retroviral infection. We have used Friend virus (FV) infection of mice as a model for retroviral infection, in which the behavior of virus-specific CD4+ T cells was monitored according to their TCR avidity. We report that avidity for antigen and interaction with B cells determine distinct aspects of the primary CD4+ T-cell response to FV infection. Virus-specific CD4+ T cells followed exclusive Th1 and T follicular helper (Tfh) differentiation. High avidity for antigen facilitated expansion during priming and enhanced the capacity for IFN-γ and IL-21 production. In contrast, Tfh differentiation was not affected by avidity for antigen. By reducing or preventing B-cell interaction we found that B cells promoted Tfh differentiation, induced programmed death 1 (PD-1) expression and inhibited IFN-γ production by virus-specific CD4+ T cells. Ultimately, B cells protected hosts from CD4+ T-cell-mediated immune pathology, at the detriment of CD4+ T-cell-mediated protective immunity. Our results suggest that B-cell presentation of vaccine antigens could be manipulated to direct the appropriate CD4+ T-cell response.
Introduction
Protective immunity against infection is provided by the multipartite immune system, of which CD4+ Th cells play a coordinating role. CD4+ T-cell priming is initiated by dendritic cells in secondary lymphoid organs. However, the CD4+ T-cell response can be modulated by additional APC types, and B cells in particular (1). Although studies in several experimental systems failed to detect a role for B cells in CD4+ T-cell priming (2-5), the overwhelming majority suggest that B-cell deficiency generally leads to reduced CD4+ T-cell priming following viral infection or immunization (6-15). Thus, antigen presentation by B cells can enhance CD4+ T-cell priming, particularly when antigen is limiting (1). B cells have also been shown to reduce CD4+ T-cell responses, mostly through induction of regulatory T cells or IL-10 production (1), but also directly (16, 17). However, interpretation of many of these studies is confounded by established requirements for B cells in additional physiological processes, including the organization of lymphoid organ architecture (1). Thus, despite the potential of B-cell antigen presentation to either suppress or enhance CD4+ T-cell responses, the factors that determine the ultimate effect of B cells on CD4+ T cells are not completely understood.
B-cell antigen presentation to CD4+ T cells is a necessary step for the T-cell-dependent B-cell response, helped by T follicular helper (Tfh)2 cells, a specialized class of CD4+ T cells (18, 19). In turn, B cells have been implicated as APCs in Tfh differentiation (18, 19). Interaction with B cells has been found necessary for differentiation of antigen-specific CD4+ T cells into Tfh cells (20, 21), whereas early acquisition of Tfh markers or follicular localization have been shown in other studies to be independent of B-cell interaction (22, 23). Continuous antigen presentation has recently been suggested to overcome the requirement for B-cell interaction for Tfh differentiation (5) and antigen presentation by plasma cells negatively regulated the Tfh program (24).
In addition to the type of priming APC, CD4+ T-cell expansion is profoundly affected also by the strength of TCR signaling, which is enhanced by high TCR affinity for antigen (25, 26). Thus, selection following priming of CD4+ T cells favors clones with high TCR affinity (27-29). As well as expansion, CD4+ T-cell differentiation is also affected by TCR affinity and high-affinity CD4+ T-cell clones have been suggested to preferentially develop into Tfh cells (30). Thus, both the overall T-cell avidity for antigen and the nature of the cell presenting it can influence expansion and differentiation of CD4+ T cells, albeit to a variable degree and often with opposite outcomes, largely determined by the experimental system.
To gain insights into the CD4+ T-cell response to chronic retroviral infection we have been using, as a model system, persistent infection of mice with Friend virus (FV), a retroviral complex of Friend murine leukaemia virus (F-MuLV) and spleen-focus forming virus (SFFV) (31). We have also been using polyclonal TCRβ-transgenic CD4+ T cells, in which usage specifically of endogenous TCRVα2 chains creates high avidity for the Ab-restricted env122-141 product of the F-MuLV env gene (32, 33), thus allowing analysis of the impact of T-cell avidity on expansion and differentiation. In addition to helping B cells in this system, env-specific CD4+ T cells provide significant protection against FV-induced disease, independently of a cytotoxic T-cell or antibody response (33), but can also cause bone marrow pathology, particularly when T-cell regulation is incomplete (32). We have previously observed severe immune pathology mediated by virus-specific CD4+ T cells during FV infection of lymphocyte-deficient Rag1−/− or B-cell-deficient Ighm−/− hosts (32). Furthermore, experimental B-cell activation during FV infection, either by coinfection or by direct stimulation, dramatically enhances FV replication (34), which is associated with loss of immune control of FV and premature contraction of env-specific CD4+ T cells (33, 34). These findings raised the possibility that in addition to mounting an FV-neutralizing antibody response, B cells may directly inhibit CD4+ T-cell function.
Materials and Methods
Mice
Inbred C57BL/6 (B6) and CD45.1+ B6 (B6.SJL-Ptprca Pep3b/BoyJ) mice were originally obtained from the Jackson Laboratory (Bar Harbor, Maine, USA) and were subsequently maintained at NIMR animal facilities. The B6 TCR-transgenic strain EF4.1, expressing a transgenic TCRβ chain from a T-cell clone specific to F-MuLV env122-141 presented by MHC class II Ab, has been described (32). B6-backcrossed Rag1-deficient (Rag1−/−) mice (35), B-cell-deficient (Ighm−/−) mice (36), SAP-deficient (Sh2d1a−/−) mice (37), Fv2s-congenic Rag1−/− (Fv2s Rag1−/−) mice (34), hen-egg lysozyme (HEL)-specific B-cell receptor-transgenic (MD4) mice{Goodnow, 1988 267 /id}, T-cell receptor α-deficient (Tcra−/−) mice (39) and IL-10-deficient (Il10−/−) mice (40) were also maintained at NIMR animal facilities. All animal experiments were conducted according to UK Home Office regulations and local guidelines.
Viruses and infections
The FV used in this study was a retroviral complex of a replication-competent B-tropic F-MuLV and a replication-defective SFFV. Stocks were propagated in vivo and prepared as 10% w/v homogenate from the spleen of 12-day infected BALB/c mice. Mice received an inoculum of ~1,000 spleen focus-forming units of FV. The B-tropic helper F-MuLV stock was prepared as culture supernatant harvested from chronically infected Mus dunni cells. Mice received an inoculum of ~104 infectious units of F-MuLV. All viral stocks were free of Sendai virus, Murine hepatitis virus, Parvoviruses 1 and 2, Reovirus 3, Theiler’s murine encephalomyelitis virus, Murine rotavirus, Ectromelia virus, Murine cytomegalovirus, K virus, Polyomavirus, Hantaan virus, Murine norovirus, Lymphocytic choriomeningitis virus, Murine adenoviruses FL and K87, and Lactate dehydrogenase-elevating virus. Viruses were injected via the tail vein in 0.1 ml of phosphate-buffered saline. For the assessment of anemia, mice were bled by a small incision of the tail vein and blood was collected into heparinized capillary tubes. Complete blood counts were measured on a VetScan HMII hematology analyzer (Abaxis, CA, USA), following the manufacturer’s instructions. RBC counts of uninfected mice ranged between 9.56×106 and 10.49×106 per mm3 of blood. FV-induced splenomegaly in infected mice was expressed as spleen index, which is the ratio of the weight of the spleen (in mg) to the weight of the rest of the body (in g).
T-cell purification and adoptive transfer
Single-cell suspensions were prepared from the spleens and lymph nodes of donor CD45.1+ TCRβ-transgenic mice by mechanical disruption. Spleen suspensions were treated with ammonium chloride for erythrocyte lysis. CD4+ T cells were enriched using immunomagnetic positive selection (StemCell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. Purity of the isolated CD4+ T-cell population was routinely higher than 92%. In some experiments, purified cells were labeled with CFSE (Molecular Probes, USA) prior to cell transfer. A total of approximately 3×106 TCRβ-transgenic CD4+ T cells were injected in recipient mice via the tail vein in 0.1 ml of air-buffered Iscove’s Modified Dulbecco’s Media. This resulted in engraftment of 5,000-10,000 env-specific CD4+T cells (4% of all donor CD4+ T cells) per spleen (~0.025% of total host CD4+ T cells), which was routinely confirmed by staining for donor-type (CD45.1+) T cells 1 day post transfer. When adoptive transfer of CD4+ T cells was combined with FV infection, purified CD4+ T cells and virus stocks were injected separately into recipient mice within a 24 hour-period. In indicated experiments, enriched TCRβ-transgenic CD4+ T cells were stained with antibodies to surface markers and then further purified by cell sorting, performed on MoFlo cell sorters (Dako, Fort Collins, CO, USA), according to TCRVα2 expression. A total of approximately 0.35×106 TCRVα2+ or 1.30×106 TCRVα2− purified TCRβ-transgenic CD4+ T cells were injected in recipient mice, resulting in engraftment of a similar number (2,000-4,000) of env-specific CD4+T cells. T cells used for transcriptional profiling were first enriched for total CD4+ or donor-type CD45.1+ T cells, stained with antibodies to surface markers and then further purified by cell sorting. Typical cell purity following cell sorting was higher than 98%.
Flow cytometry
Spleen-cell suspensions were stained with directly-conjugated antibodies to surface markers, obtained from eBiosciences (San Diego, CA, USA), CALTAG/Invitrogen (Carlsbad, CA, USA), BD Biosciences (San Jose, CA, USA) or BioLegend (San Diego, CA, USA). Donor-type env-specific CD4+ T cells were gated either as CFSE− CD45.1+ or CD44hiCD45.1+ CD4+ T cells with comparable results (33). CXCR5 expression was detected with a PE-conjugated anti-mouse CXCR5 (clone 2G8, BD Biosciences) and was further amplified with a biotinylated anti-PE antibody (BioLegend) followed by incubation with a streptavidin-PE conjugate (BioLegend). For detection of cytokine synthesis, cells were stained for surface markers and stimulated for 4 hrs with phorbol 12,13-dibutyrate and ionomycin (both at 500 ng/ml), in the presence of monensin (1 μg/ml). Cells were then fixed and permeabilization using buffers from eBiosciences, before intracellular staining with antibodies specific to IL-17A, IFN-γ (eBiosciences) or IL-21 (R&D Systems, Minneapolis, MN, USA). FV-infected cells were detected by flow cytometry using surface staining for the glycosylated product of the viral gag gene (glyco-Gag), using the matrix (MA)-specific monoclonal antibody 34 (mouse IgG2b), followed by an anti-mouse IgG2b-FITC secondary reagent (BD, San Jose, CA, USA). Four- and 8-color cytometry were performed on FACSCalibur (BD Biosciences) and CyAn (Dako, Fort Collins, CO) flow cytometers, respectively, and analyzed with FlowJo v8.7 (Tree Star Inc., Ashland, OR, USA) or Summit v4.3 (Dako) analysis software, respectively.
FV-neutralizing antibody assay
Serum titers of FV-neutralizing antibodies were measured as previously described (34). The dilution of serum which resulted in 50% neutralization was taken as the neutralizing titer.
TCR re-expression assay
CD45.1+ TCRβ-transgenic env-specific CD4+ T cells were transferred into FV-infected wt recipients and total CD4+ T cells were isolated from the spleens 7 days later using immunomagnetic positive selection (StemCell Technologies). Purified cells were subsequently cultured in the presence of 10 ng/ml recombinant human IL-2 and 50 ng/ml recombinant mouse IL-7. At the indicated time-points, host and donor cells CD4+ T cells were assessed for expression of TCRβ and TCRVα2 by flow cytometry.
RNA extraction, gene expression profiling
Donor-type env-specific effector CD4+ T cells were identified as CD45.1+CD44hiCD4+ (TCRVα2+ or TCRVα2−) and purified by cell sorting on day 7 post transfer into FV-infected recipients. Naïve TCRβ-transgenic CD4+ T-cell precursors were purified as CD44loCD25−CD4+ T cells from uninfected control mice. Between 1.5×106 and 2×106 purified CD4+ T cells were used for RNA extraction using the RNAeasy mini kit, (QIAGEN, Crawley, UK) according to manufacturer’s instructions. For quantitative reverse-transcription (qRT)-PCR analysis, RNA samples were reverse-transcribed with a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA), and used as template for the amplification of target gene transcripts with a SYBR Green PCR Master Mix (Applied Biosystems) and the ABI Prism 7000 Detection System (TaqMan, Applied Biosystems). Primers used for Il2, Il4, Il10 and Tnfa amplification (30), Tbx21, Bcl6, Gata3, Rorc, Foxp3 and Il21 amplification (21) and Ifng amplification (41) have been previously described. The housekeeping gene, Hprt was amplified with forward 5′-TTGTATACCTAATCATTATGCCGAG-3′ and reverse 5′-CATCTCGAGCAAGTCTTTCA-3′ primers and was used to normalize the Critical Threshold (CT) values for the genes of interest. For microarray analysis RNA samples were checked for quality using an Agilent bioanalyser (Agilent, Santa Clara, CA, USA). Synthesis of cDNA, probe labeling and hybridization were performed using MouseGene 1.0 ST oligonucleotide arrays (Affymetrix, Santa Clara, CA, USA). Primary microarray data were analyzed with GeneSpring GX (Agilent Technologies, Inc., Santa Clara, CA, USA) and deposited at http://www.ebi.ac.uk/arrayexpress (E-MEXP-2950).
Statistical analysis
Analysis of variance and statistical comparisons were made using SigmaPlot 11.0 (Systat Software Inc., Germany). Parametric comparisons of normally-distributed values that satisfied the variance criteria were made by unpaired Student’s t-tests. Linear percentages of FV-infected cells, spleen indices and nAb titers, which did not pass the variance test, were compared with non-parametric two-tailed Mann-Whitney Rank Sum or Wilcoxon Signed Rank tests.
Results
Virus-specific CD4+ T-cell differentiation during FV infection
To study the expansion and differentiation of virus-specific CD4+ T cells as a function of functional avidity we used TCRβ-transgenic CD4+ T cells, containing a polyclonal population of env-specific precursors. Approximately 25% of env-specific precursors in these TCRβ-transgenic CD4+ T cells use an endogenous TCRVα2 chain which creates >30-fold higher functional avidity than the use of other endogenous TCRVα chains in the remaining 75% of env-specific precursors (ED50 0.04 μM and >1.24 μM env122-141, respectively) (32). TCRβ-transgenic CD4+ T cells were adoptively transferred into allotypically-marked syngeneic wild-type (wt) hosts that were infected with FV within a 24 hour-period. We monitored a cohort of 5,000-10,000 env-specific CD4+ T cells, which also excluded changes arising from recruitment of new naïve precursors throughout the response. Env-specific CD4+ T cells were identified among donor CD4+ T cells by dilution of prior CFSE labeling or by upregulation of CD44 expression, with comparable results (33). CFSE-labeled CD45.1+ TCRβ-transgenic T cells divided and accumulated substantially (>200-fold) following adoptive transfer into acutely FV-infected, but not into uninfected CD45.2+ B6 wt recipients (Fig. 1A). At this time-point of peak expansion (33), gene expression analysis of env-specific donor CD4+ T cells revealed a transcriptional program compatible with exclusive Th1 and Tfh differentiation (Fig. 1B).
FIGURE 1.

Virus-specific CD4+ T-cell differentiation during FV infection. (A-F) Cohorts of CD45.1+ TCRβ-transgenic CD4+ T cells were adoptively transferred into wt recipients infected with FV the same day and recovered for analysis from the spleen of the recipients 7 days later. (A) Dilution of prior CFSE label in gated CD4+ T cells from acutely FV infected (+ FV) or uninfected (− FV) wt recipients. (B) Fold change in the transcription of indicated genes, grouped according to Th class, in env-specific CD4+ effector T cells, compared with naïve CD4+ TCRβ-transgenic T cells (n = 3-6). (C) Frequency of TCRVα2+ cells in host (CD45.1−) or env-specific (CFSE− CD45.1+) or non-specific (CFSE+CD45.1+) donor CD4+ T cells (Left) and fold expansion of TCRVα2+ or TCRVα2− env-specific donor (CD44hiCD45.1+) CD4+ T cells between days 1 and 7 after transfer (Right). Each symbol is an individual mouse. (D) Fold change in the transcription of the indicated Th class-specific transcription factor (Left) or cytokine (Right) genes in TCRVα2+ or TCRVα2− env-specific donor (CD44hiCD45.1+) CD4+ T cells. (E) Flow cytometric example (Left) and mean frequency (± SEM, n = 9-12) (Right) of IFN-γ and IL-21 production by TCRVα2+ or TCRVα2− env-specific donor CD4+ T cells. (F) CXCR5 and PD-1 expression in TCRVα2+ or TCRVα2− env-specific donor CD4+ T cells (Left). Values within each gate denote the frequency of CXCR5hiPD-1hi cells in that gate, with the dashed gate restricted to cells coexpressing CXCR5 and PD-1 at the highest level. Mean frequency (± SEM, n = 17) of CXCR5hiPD-1hi Tfh cells in either TCRVα2+ or TCRVα2− env-specific donor CD4+ T cells (Right). (G) Serum titers (medians ± SEM, n = 5-6) of FV-neutralizing antibodies in Tcra−/− hosts that received no T cells and those that received either TCRVα2+ or TCRVα2− TCRβ-transgenic CD4+ T cells.
The frequency of high-avidity TCRVα2+ cells in env-specific donor CD4+ T cells rose during the first 7 days from 25% in the naïve repertoire to over 65% (Fig. 1C Left), due to significantly higher expansion of high-avidity cells (500-fold) in comparison with low-avidity ones (50-fold) (Fig. 1C Right). Examination of the earliest time-points of CD4+ T-cell expansion revealed that the numerical advantage of high-avidity TCRVα2+ cells manifested between days 1 and 3 of the response, during which cohorts of low- and high-avidity env-specific cells had already significantly diluted the CFSE label, and after which expansion of both low- and high-avidity env-specific cells followed similar kinetics to reach near peak numbers by day 5 (Fig. S1). Although the potential effect of differential rate of death could not be excluded, the early numerical advantage of high-avidity TCRVα2+ cells indicated faster recruitment and/or proliferation during the expansion phase. Both low- and high-avidity cells that responded were fully activated, suggested by high CD43 expression (Fig. S2A), and expressed comparably higher levels, than those in naïve T cells, of transcription factors Tbx21 and Bcl6, specific to the Th1 and Tfh lineages, respectively (19), and of Th-specific cytokine genes encoding IFN-γ or IL-21 (Fig. 1D). Differences between low- and high-avidity subsets were also noted with high-avidity TCRVα2+ cells expressing lower levels of PD-1 and higher levels of inducible costimulator (ICOS), than low-avidity cells (Fig. S2B, C), and a higher frequency of high-avidity TCRVα2+ cells could be further stimulated in vitro to produced IFN-γ, alone or in combination with IL-21 (Fig. 1E). Importantly, low- and high-avidity populations contained similar frequencies of Tfh cells, defined by high coexpression of PD-1 and the B-cell follicle-homing chemokine receptor CXCR5 (Fig. 1F). Furthermore, purified TCRVα2+ or TCRVα2− naïve TCRβ-transgenic CD4+ T cells, transferred separately into FV-infected T-cell-deficient Tcra−/− hosts, induced comparably high levels of FV-neutralizing antibodies (Fig. 1G), demonstrating that both high- and low-avidity CD4+ T cells provided sufficient help for the T-cell-dependent B-cell response. To control for any potential effects of the TCRVα2 antibody staining used to purify CD4+ T cells, in additional experiments CD4+ T cells were either stained with the TCRVα2 antibody or not, and were then transferred separately into hosts that were either acutely infected with FV or left uninfected. This comparison revealed that TCRVα2 antibody staining did not cause any measurable activation of CD4+ T cells in the absence of infection, nor did it affect their response to FV infection (Fig. S3). Collectively, these results indicated that high-avidity for antigen of TCRVα2+ env-specific CD4+ T cells conferred a substantial numerical advantage and significantly enhanced cytokine production potential, but did not affect Tfh differentiation or function.
Impact of B-cell deficiency on virus-specific CD4+ T-cell expansion and differentiation
We next explored whether virus-specific CD4+ T-cell expansion and differentiation was shaped by interaction with B cells, by comparing wt and B-cell-deficient Ighm−/− hosts. Due to the absence of an FV-neutralizing antibody response, Ighm−/− hosts exhibited dramatically elevated levels of FV replication (Fig. 2A). Nevertheless, expansion of env-specific CD4+ T cells was comparable in wt and Ighm−/− hosts (Fig. 2B). Notably, the proportion of Tfh cells, identified by high expression of CXCR5 and PD-1 in env-specific CD4+ T cells (Fig. 2C) was significantly reduced in Ighm−/− hosts (Fig. 2D). In line with transcriptional analysis, IFN-γ and IL-21 were the major cytokines that could be recalled in env-specific CD4+ T cells, whereas no IL-17A production could be detected (Fig. 2E). Analysis of cytokine production by env-specific CD4+ T cells from either wt or Ighm−/− hosts revealed that B-cell deficiency did not affect IL-21 production (Fig. 2F). In contrast, both the frequency of IFN-γ-producing cells and the amount of IFN-γ produced per cell were significantly increased in env-specific CD4+ T cells from Ighm−/− hosts (Fig. 2G). Thus, in the absence of B cells, env-specific CD4+ T cells displayed a preference for Th1, rather than Tfh differentiation. However, in addition to lack of FV-neutralizing antibodies, B-cell deficiency affects additional physiological processes, which could indirectly affect CD4+ T-cell differentiation. We therefore employed a system where CD4+ T-cell interaction specifically with B cells could be impaired in an otherwise normal host.
FIGURE 2.

Role of B cells in virus-specific CD4+ T-cell expansion and differentiation. (A-G). Cohorts of CD45.1+ TCRβ-transgenic CD4+ T cells were adoptively transferred into wt or B-cell-deficient Ighm−/− hosts and analyzed 7 days later. (A) Mean frequency (± SEM, n = 5-9) of FV-infected (glyco-Gag+) Ter119+ cells in the spleens of FV-infected wt or Ighm−/− hosts. (B) Absolute number of splenic env-specific donor (CD44hi CD45.1+) CD4+ T cells 7 days after transfer. (C) Example of CXCR5 and PD-1 expression in host or donor CD4+ T cells from wt hosts. (D) Frequency of CXCR5hiPD-1hi cells in env-specific donor CD4+ T cells from wt or Ighm−/− hosts. (E) Example of IFN-γ, IL-21 and IL-17A production by env-specific donor CD4+ T cells from wt hosts. (F, G) Frequency of IL-21+ cells (F) or IFN-γ+ cells (F, Left) and MFI of IFN-γ staining (F, Right) in env-specific donor CD4+ T cells from wt or Ighm−/− hosts. (H) Mean frequency (± SEM, n = 8-9) of FV-infected (glyco-Gag+) Ter119+ cells in the spleens of FV-infected wt hosts that received no T cells (−), in comparison with those that received wt (+ wt T cells) or Sh2d1a−/− (+ Sap−/− T cells) TCRβ-transgenic env-specific CD4+ T cells. (I) Absolute number of either wt (+ wt T cells) or Sh2d1a−/− (+ Sap−/− T cells) splenic env-specific donor (CD44hi CD45.1+) CD4+ T cells 7 days after transfer into FV-infected wt hosts. In B, D, F, G and I, each symbol is an individual mouse.
In addition to initial integrin-dependent adhesion, CD4+ T-cell interaction with B cells is uniquely characterized by a longer second phase mediated by multiple signaling lymphocyte activation molecules (SLAMs) (20, 42). Signaling through SLAMs is transduced by SLAM-associated protein (SAP), encoded by Sh2d1a, and SAP deficiency in T cells shortens the duration of CD4+ T-cell contacts with B cells and impairs humoral immunity (5, 20, 22, 42). We generated SAP-deficient env-specific TCRβ-transgenic CD4+ T cells, which we compared with wt env-specific TCRβ-transgenic CD4+ T cells, both transferred into wt hosts infected with FV at the time of T-cell transfer. Importantly, FV replication followed a similar course in wt hosts of both types of CD4+ T cells (Fig. 2H) and peak expansion was also comparable between SAP-deficient or wt CD4+ T cells (Fig. 2I).
Differentiation of high- and low-avidity virus-specific CD4+ T cells when B-cell interaction is absent or shortened
To examine the impact of high- or low-avidity interaction of virus-specific CD4+ T cells with B cells during FV infection we compared the differentiation of wt env-specific TCRβ-transgenic CD4+ T cells transferred into either wt or Ighm−/− hosts, with that of Sh2d1a−/− env-specific TCRβ-transgenic CD4+ T cells transferred into wt hosts. Indicated by the frequency of CXCR5hiPD-1hi cells, Tfh differentiation of Sh2d1a−/− env-specific CD4+ T cells in wt hosts was intermediate between that of wt env-specific CD4+ T cells in wt hosts and in Ighm−/− hosts (Fig. 3A, B). Importantly, the frequency of CXCR5hiPD-1hi cells remained comparable between TCRVα2+ and TCRVα2− CD4+ T cells (Fig. 3A, B), indicating that higher avidity did not favor Tfh differentiation even under suboptimal conditions. IL-21 production by env-specific CD4+ T cells was not significantly affected by the reduction in B-cell interaction, and high-avidity TCRVα2+ CD4+ T cells produced higher IL-21 levels than their low-avidity TCRVα2− counterparts, in all three conditions (Fig. 3C). In comparison with wt CD4+ T cells, Sh2d1a−/− CD4+ T cells from wt hosts showed a significant increase in IFN-γ production, comparable with that of wt CD4+ T cells from Ighm−/− hosts (Fig. 3D). Notably, this increase in IFN-γ production was accompanied by a reciprocal decrease in PD-1 expression levels (Fig. 3D). When analyzed separately, high-avidity env-specific CD4+ T cells maintained increased potential for IFN-γ production than low-avidity CD4+ T cells, independently of B-cell interaction (Fig. 3E). Importantly, however, both the frequency of IFN-γ-producing cells and the amount of IFN-γ produced per cell were comparably increased in both high- and low-avidity Sh2d1a−/− CD4+ T cells from wt hosts and in wt CD4+ T cells from Ighm−/− hosts, in comparison with wt CD4+ T cells from wt hosts (Fig. 3E). These results indicated that low-avidity for antigen and B-cell interaction, independently and comparably reduced IFN-γ production by env-specific CD4+ T cells.
FIGURE 3.

Effect of absent or reduced B-cell interaction on the phenotype of virus-specific CD4+ T cells. (A-G) Cohorts of CD45.1+ wt TCRβ-transgenic CD4+ T cells were transferred into wt (wt → wt) or Ighm−/− hosts (wt → Ighm−/−) and CD45.1+ Sh2d1a−/− TCRβ-transgenic CD4+ T cells were transferred into wt hosts (Sap−/− → wt). All hosts were infected with FV the day of the T-cell transfer and donor CD4+ T cells were recovered for analysis from the spleens of these hosts 7 days later. (A) CXCR5 and PD-1 expression in either TCRVα2+ or TCRVα2− env-specific donor CD4+ T cells. Values within each gate denote the frequency of CXCR5hiPD-1hi cells. (B) Frequency of CXCR5hiPD-1hi cells. (C) Frequency of IL-21+ cells. (D) Flow cytometric measurement of the frequency of IFN-γ+ cells and MFI of intracellular IFN-γ staining (Left), and MFI of PD-1 surface staining (Right) in env-specific donor CD4+ T cells. (E) Frequency of IFN-γ+ cells (Left) and MFI of intracellular IFN-γ staining (Right) in either TCRVα2+ or TCRVα2− env-specific donor CD4+ T cells. (F) MFI of PD-1 surface staining. (G) Correlation between frequency of IFN-γ+ cells and either the MFI of PD-1 expression (Left) or the frequency of CXCR5hiPD-1hi cells (Right) in TCRVα2+ or TCRVα2− env-specific donor CD4+ T cells from the hosts described above. Each symbol is the mean of each condition. In each panel, data show the means ± SEM (n = 6-9).
Although PD-1 expression in Sh2d1a−/− CD4+ T cells from wt hosts was reduced in comparison with wt CD4+ T cells from wt hosts, low-avidity CD4+ T cells expressed significantly higher levels of PD-1 than high-avidity CD4+ T cells, independently of SAP expression (Fig. 3F). Contrastingly, PD-1 expression was similarly reduced in both high- and low-avidity wt CD4+ T cells from Ighm−/− hosts (Fig. 3F), suggesting that PD-1 expression in env-specific CD4+ T cells was regulated by low-avidity interaction preferentially with B cells. Importantly, the frequency of IFN-γ-producing cells in either high- or low-avidity env-specific CD4+ T cells followed a close inverse correlation with the level of PD-1 expression, but not with the proportion of CXCR5hiPD-1hi Tfh cells (Fig. 3G), indicating a mechanistic link. In contrast to regulation of PD-1 expression by B-cell interaction, ICOS expression in several different conditions varying in FV replication levels was regulated strictly by antigen levels (Fig. S4), and did not correlate with the overall Tfh phenotype. Lastly, the reduction of IFN-γ production by CD4+ T cells that interacted with B cells did not depend on IL-10 production by B cells, as expansion, Tfh differentiation, and IFN-γ and IL-21 production by env-specific CD4+ T cells were similar when these cells were transferred into wt or IL-10-deficient hosts (data not shown).
B cells induce TCR downregulation in virus-specific CD4+ T cells
Regulation of the CD4+ T-cell response by B cells has been suggested to involve antigen-induced downregulation of the TCR (16, 43). We have previously observed significant TCR downregulation in env-specific CD4+ T cells at the peak of their expansion during FV infection (33), and it was therefore of interest to determine whether it was induced by B cells. TCR levels were substantially reduced at the peak of the response to FV in both high- and low-avidity env-specific CD4+ T cells to a comparable degree (Fig. 4A). Notably, TCR levels remained significantly reduced throughout infection and were restored when T cells were separated from persisting antigen and cultured in vitro (Fig. 4B, C). In stark contrast, TCR levels were only minimally reduced in env-specific CD4+ T cells recovered from infected Ighm−/− hosts (Fig. 4D). Interestingly, the shortened duration of interaction between B cells and Sh2d1a−/− env-specific CD4+ T cells (22) was sufficient to cause TCR downregulation, comparable to that in wt env-specific CD4+ T cells, during acute FV infection, but not during chronic FV infection (Fig. 4D). Thus, increased antigen availability for presentation by B cells during the acute phase of infection could overcome the requirement for SAP expression in CD4+ T cells for TCR downregulation induced by B-cell interaction. Nevertheless, the finding that Sh2d1a−/− CD4+ T cells from wt hosts produced elevated IFN-γ levels, similarly to wt CD4+ T cells from Ighm−/− hosts, despite profound TCR downregulation, suggested that TCR downregulation was not a requirement inhibition of IFN-γ production.
FIGURE 4.

B cells induce profound but reversible TCR downregulation in virus-specific CD4+ T cells. (A) CD45.1+ TCRβ-transgenic CD4+ T cells were adoptively transferred into FV-infected wt recipients and TCR expression was monitored throughout infection. Levels of TCRβ and TCRVα2 expression in TCRVα2+ env-specific cells donor CD4+ T cells and levels of TCRβ in TCRVα2− env-specific cells donor CD4+ T cells are shown (n = 5-7). (B, C) Purified CD4+ T cells (host and donor) recovered on day 7 from FV-infected wt recipients were subsequently cultured in vitro in the presence of IL-2 and IL-7. TCRβ and TCRVα2 expression in host or env-specific donor (CD45.1+ CD44+) gated CD4+ T cells at the time of ex vivo recovery (t = 0) or after an 18 hr in vitro culture (t = 18h) (B). Dashed lines indicate the MFI of TCRβ or TCRVα2 expression in host CD4+ T cells. Levels of TCR remaining on TCRVα2+ env-specific cells from the same env-specific donor CD4+ T cells after the indicated length of in vitro culture (C) (n = 4). (D) Levels of TCRVα2 remaining on wt TCRβ-transgenic CD4+ T cells transferred into wt (wt → wt) or Ighm−/− hosts (wt → Ighm−/−) and Sh2d1a−/− TCRβ-transgenic CD4+ T cells were transferred into wt hosts (Sap−/− → wt), following FV infection (n = 6-9). In A, C and D, values are the means (± SEM) of TCRβ or TCRVα2 MFI in env-specific donor cells, expressed as a percentage of the same intensity in non-specific donor or host cells.
Virus-nonspecific B cells affect virus-specific CD4+ T-cell function
We next addressed whether the impact of B-cell interaction on differentiation of env-specific CD4+ T cells translated into an effect on either the protective or pathogenic function mediated directly by CD4+ T cells. FV infection in B6 mice is relatively mild due to a strong FV-neutralizing antibody response and to genetic resistance at the Fv2 locus, which prevents splenomegaly (34). We therefore examined if B cells could directly affect CD4+ T-cell-mediated virus control or immune pathology in a severe infection, where FV-neutralizing antibodies could also be precluded. We compared lymphocyte-deficient mice expressing the Fv2 susceptibility (Fv2s) allele (Rag1−/− Fv2s), with those additionally harboring a monoclonal B-cell population specific to the FV-unrelated antigen HEL (Rag1−/− Fv2s MD4). B cells are targets of FV infection and can present viral antigens to T cells in wt mice (44, 45). We further established that HEL-specific B cells were also infected by FV and when isolated from FV-infected MD4 BCR-transgenic mice presented antigen to and stimulated env-specific CD4+ T cells (Fig. 5). However, MD4 BCR-transgenic B cells would not directly contribute to antiviral immunity. Transfer of env-specific TCRβ-transgenic CD4+ T cells in Rag1−/− Fv2s hosts infected with F-MLV alone (which does not cause splenomegaly without SFFV), led to significantly more severe anemia and weight loss than in Rag1−/− Fv2s MD4 hosts (Fig. 6A). When infected with FV, both types of host rapidly developed severe splenomegaly (Fig. 6B). Remarkably, however, transfer of TCRβ-transgenic T cells protected Rag1−/− Fv2s hosts, but failed to protect Rag1−/− Fv2s MD4 hosts against splenomegaly (Fig. 6B). Thus, the presence of FV-neutral B cells negated the antiviral effect and diminished the pathogenic effect of env-specific CD4+ T cells during FV infection.
FIGURE 5.
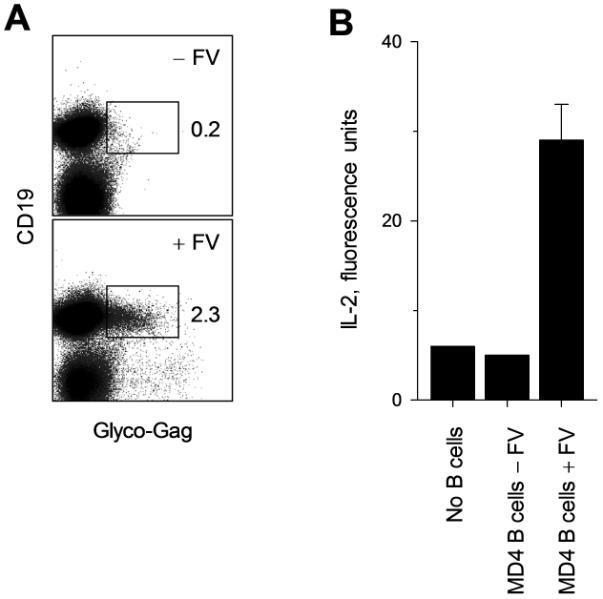
Infection of MD4 BCR-transgenic B cells and presentation of viral antigens to env-specific CD4+ T cells. Splenocytes from MD4 BCR-transgenic (Ighm−/− MD4) mice that were infected with FV 7 days earlier (+FV) or left uninfected (−FV) were isolated. (A) The frequency of FV infected B cells (glyco-Gag+CD19+) in total splenocytes is shown. (B) CD19+ cells were further purified and used for the stimulation of an env-specific T-cell hybridoma cell line. IL-2 production by the T-cell hybridoma cells in culture supernatant is shown. Data are the means ± SEM, n = 2-4.
FIGURE 6.

B cells determine the pathogenic and protective effect of virus-specific CD4+ T cells. (A) Change in red blood-cell (RBC) count (Left) and body weight (Right) of Rag1−/− Fv2s or Rag1−/− Fv2s MD4 hosts 21 days post adoptive transfer of TCRβ-transgenic CD4+ T cells and infection with only the B-tropic helper virus F-MuLV, relative to the average RBC count and body weight of uninfected control groups (dashed lines). (B) Spleen indices (spleen weight, mg/body weight, g) of Rag1−/− Fv2s or Rag1−/− Fv2s MD4 hosts following FV infection, in conjunction with (+ T cells) or without (−) adoptive transfer of TCRβ-transgenic CD4+ T cells. In A and B each symbol is an individual mouse from one representative of two experiments.
Discussion
Upon antigen recognition naïve CD4+ T cells expand and acquire one of many distinct sets of effector functions and migration properties. The final outcome of CD4+ T-cell expansion and differentiation is heavily influenced by T-cell avidity for antigen as well as the type of APC. The results of the current study demonstrated that key aspects of the CD4+ T-cell response to a retroviral antigen were extensively shaped by interaction with B cells. We found that B-cell interaction was dispensable for CD4+ T-cell priming following FV infection. It did, however, contribute significantly to the differentiation of virus-specific CD4+ T cells towards the Tfh phenotype. More importantly, B-cell interaction dramatically reduced the potential of virus-specific CD4+ T cells to produce IFN-γ, in proportion with induction of PD-1 expression, but not with Tfh differentiation, with profound consequences for CD4+ T-cell-mediated antiviral immunity and immune pathology.
B cells have been shown to impact on CD4+ T-cell function by influencing CD4+ T-cell expansion or differentiation in numerous experimental systems, although the results were highly variable and often conflicting (1). B cells have not been consistently found necessary for CD4+ T-cell priming, unless when antigen may be limiting (1). However, several studies of a diverse range of parasites, bacteria and viruses have shown that in most cases B-cell interaction is required for a full Th1 and Th2 effector response (6-15). Similarly, SAP-dependent B-cell interaction has also been shown to contribute to Tfh differentiation, with the amount of this contribution reflecting antigen availability (5, 18-21, 46). Thus, B cells can contribute to a variable degree to CD4+ T-cell differentiation into many functionally distinct subsets.
In contrast, we find that B-cell interaction during FV infection promoted Tfh, at the expense of Th1 differentiation of virus-specific CD4+ T cells. The requirement for B-cell interaction for Tfh differentiation of CD4+ T cells responding to FV infection is seemingly at odds with the suggestion that continuous antigen presentation overcomes this requirement for B-cell interaction (5). More recently, three separate reports demonstrated although B cells may be dispensable for the initial differentiation of Tfh cells, they are critically involved in their maintenance and function (47-49). Although eventually controlled to low levels of replication, FV causes persistent infection in B6 mice and viral antigens are presented chronically. Furthermore, B-cell-deficient mice are unable to control FV replication and thus constantly present very high levels of viral antigens. Nevertheless, both the presence of B cells and SAP expression in CD4+ T cells were necessary for full Tfh differentiation in this model. Based on these seemingly opposing observations we would propose that B cells promote Tfh differentiation when polarizing conditions are suboptimal, in a weakly proinflammatory environment and when antigen is the limiting factor for CD4+ T cells to complete their differentiation program. This contribution of B cells may be overcome with increased antigen availability. At the other end of the spectrum, in a strongly proinflammatory environment and high antigen availability, the limiting factor for Tfh differentiation may be competition with alternative differentiation programs. This is particularly relevant to retroviral infection were we have observed exclusive Th1 and Tfh differentiation of virus-specific CD4+ T cells. In this light, the contribution of B cells to Tfh differentiation of virus-specific CD4+ T cells during FV infection may rely on their ability to suppress otherwise favored Th1 differentiation.
In addition to B-cell interaction, the strength of TCR antigen binding has been suggested to regulate Tfh differentiation (30). This was studied during the CD4+ T effector response to peptide immunization, where CD4+ T cells with stronger peptide-MHC class II complex binding showed increased expression of markers associated with the Tfh subset, although Tfh function was not investigated, and was only inferred by phenotype (30). Our results following retroviral infection did not support this notion and we failed to detect, by any of the read-outs used, any preference for Tfh differentiation in either high- or low-avidity virus-specific CD4+ T cells. Direct comparison of these results is complicated by experimental differences. For example, the relative TCR affinities for the respective antigens used in these studies may differ substantially. Also, kinetics of antigen availability are drastically different, peaking on day 0 following peptide immunization, and on day 7 following viral infection, in relative synchrony with the CD4+ T-cell response. Lastly, preferential Tfh differentiation of high-avidity CD4+ T cells may be particularly pronounced in conditions of limiting antigen availability. Indeed, preferential representation of high-avidity CD4+ T cells in the Tfh subset was accentuated following immunization with less immunogenic peptides (30). These findings suggest that a preference for Tfh differentiation of high-avidity CD4+ T cells may be a result of suboptimal conditions, rather than a general feature of the CD4+ T-cell response. More importantly, we found that high- and low-avidity CD4+ T cells provided comparable help for the T-cell-dependent FV-neutralizing antibody response, which clearly demonstrated that in response to FV infection, low avidity for antigen hinders neither the acquisition nor expression of Tfh function.
Notably, our results revealed that the potential for IFN-γ production by virus-specific CD4+ T cells was significantly reduced if interaction with B cells was allowed, independently of an indirect effect of B cells on antigen availability. It could be argued that B-cell-mediated suppression of the potential for IFN-γ production is a consequence of their effect in promoting Tfh differentiation. Indeed, Tfh cells have been shown to produce reduced amounts of IFN-γ in comparison with Th1 cells (50-52). However, our comparison of high- and low-avidity virus-specific CD4+ T cells revealed significant differences in IFN-γ production, but not Tfh differentiation. Furthermore, the effect of B cells on IFN-γ production by virus-specific CD4+ T cells strongly correlated with PD-1 regulation, with T cells displaying the lowest PD-1 levels expressing the highest IFN-γ amount, but correlated only weakly with Tfh differentiation. These results are in keeping with the established role of PD-1 in inhibiting T-cell activation and effector cytokine production (53, 54). Lastly, PD-1 expression during FV infection was not restricted to Tfh cells, defined by high coexpression of CXCR5, but was also present in non-Tfh virus-specific CD4+ T cells. Thus, B-cell interaction reduced IFN-γ production by CD4+ T cells in line with PD-1 upregulation. These consequences of CD4+ T-cell interaction with B cells may be necessary for the optimization of the B-cell response. Indeed, high PD-1 expression in Tfh cells has been shown to contribute to germinal center B-cell survival and selection and long-lived plasma-cell generation (55).
Interaction with B cells also induced profound TCR downregulation in virus-specific CD4+ T cells. This loss of surface TCR was reversible, indicating it was due to persistent antigenic stimulation during FV infection. B-cell-mediated TCR downregulation in antigen-specific CD4+ T cells has been previously seen in two studies using mice transgenically expressing the cognate antigen (16, 43). It is currently unclear whether antigen presentation by B cells is uniquely capable of inducing TCR downregulation or if this is a consequence of an overabundance (by a factor of 10) of B cells compared to other MHC class II-expressing cells in lymphoid organs. Although not always required, B-cell-induced TCR downregulation has also been linked to CD4+ T-cell unresponsiveness to further stimulation (16, 43). This loss of TCR caused by B cells could indeed further compromise the ability of virus-specific CD4+ T cells to mediate antiviral immunity or immune pathology.
It is interesting to note that, although B cells have been found to promote CD4+ T-cell effector responses against many pathogens, B-cell interaction has also been shown to inhibit the CD4+ T-cell effector response against self-antigens or tumors (1, 16, 17). Indeed, B-cell presentation of tumor antigens may induce a non-protective antibody response, instead of T-cell-mediated antitumor immunity (17). Furthermore, B-cell depletion has been shown to be beneficial in certain T-cell-mediated autoimmune diseases (1). In this respect, our findings suggest that the effect of B cells on the CD4+ T-cell response to retroviral infection is more similar to that on the response to self-antigens, than to pathogens. Thus, in the case of retroviral infection, efficient CD4+ T-cell-B-cell collaboration provides help for B cells at the expense of B-cell-independent CD4+ T-cell functions, and can therefore be seen as a switch between cellular and humoral immunity. Such a switch could be exploited to reduce an excessive CD4+ T-cell response and associated immune pathology. It could also be manipulated to promote B-cell-independent CD4+ T-cell-mediated immunity instead of an ineffective antibody response.
Supplementary Material
Acknowledgments
We thank J. Langhorne for providing the Ighm−/− and Sh2d1a−/− mice; A. O’Garra for providing the Il10−/− mice; and M. Hubank, N. Jina and P. Panchal (UCL Genomics) for assistance with microarrays. We are grateful for assistance from the Division of Biological Services and the Flow Cytometry Facility at NIMR.
Footnotes
This work was supported by the UK Medical Research Council (U117581330).
Abbreviations used in this article: FV, Friend virus; F-MuLV, Friend murine leukaemia virus; PD-1, programmed death 1; Tfh, T follicular helper.
References
- 1.Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4+ T cell immunity. Nat. Rev. Immunol. 2010;10:236–247. doi: 10.1038/nri2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epstein MM, Di Rosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell-deficient mice. J. Exp. Med. 1995;182:915–922. doi: 10.1084/jem.182.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips JA, Romball CG, Hobbs MV, Ernst DN, Shultz L, Weigle WO. CD4+ T cell activation and tolerance induction in B cell knockout mice. J. Exp. Med. 1996;183:1339–1344. doi: 10.1084/jem.183.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topham DJ, Tripp RA, Hamilton-Easton AM, Sarawar SR, Doherty PC. Quantitative analysis of the influenza virus-specific CD4+ T cell memory in the absence of B cells and Ig. J. Immunol. 1996;157:2947–2952. [PubMed] [Google Scholar]
- 5.Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, Tangye SG. Follicular Helper T Cell Differentiation Requires Continuous Antigen Presentation that Is Independent of Unique B Cell Signaling. Immunity. 2010;33:241–253. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergmann CC, Ramakrishna C, Kornacki M, Stohlman SA. Impaired T cell immunity in B cell-deficient mice following viral central nervous system infection. J. Immunol. 2001;167:1575–1583. doi: 10.4049/jimmunol.167.3.1575. [DOI] [PubMed] [Google Scholar]
- 7.Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J. Immunol. 1999;162:5695–5703. [PubMed] [Google Scholar]
- 8.Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J. Immunol. 2006;176:3498–3506. doi: 10.4049/jimmunol.176.6.3498. [DOI] [PubMed] [Google Scholar]
- 9.Iijima N, Linehan MM, Zamora M, Butkus D, Dunn R, Kehry MR, Laufer TM, Iwasaki A. Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J. Exp. Med. 2008;205:3041–3052. doi: 10.1084/jem.20082039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langhorne J, Cross C, Seixas E, Li C, von der WT. A role for B cells in the development of T cell helper function in a malaria infection in mice. Proc. Natl. Acad. Sci U. S. A. 1998;95:1730–1734. doi: 10.1073/pnas.95.4.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linton PJ, Bautista B, Biederman E, Bradley ES, Harbertson J, Kondrack RM, Padrick RC, Bradley LM. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J. Exp. Med. 2003;197:875–883. doi: 10.1084/jem.20021290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Q, Liu Z, Rozo CT, Hamed HA, Alem F, Urban JF, Jr., Gause WC. The role of B cells in the development of CD4 effector T cells during a polarized Th2 immune response. J. Immunol. 2007;179:3821–3830. doi: 10.4049/jimmunol.179.6.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lund FE, Hollifield M, Schuer K, Lines JL, Randall TD, Garvy BA. B cells are required for generation of protective effector and memory CD4 cells in response to Pneumocystis lung infection. J. Immunol. 2006;176:6147–6154. doi: 10.4049/jimmunol.176.10.6147. [DOI] [PubMed] [Google Scholar]
- 14.McClellan KB, Gangappa S, Speck SH, Virgin HW. Antibody-independent control of gamma-herpesvirus latency via B cell induction of anti-viral T cell responses. PLoS Pathog. 2006;2:e58. doi: 10.1371/journal.ppat.0020058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojciechowski W, Harris DP, Sprague F, Mousseau B, Makris M, Kusser K, Honjo T, Mohrs K, Mohrs M, Randall T, Lund FE. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity. 2009;30:421–433. doi: 10.1016/j.immuni.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoechel B, Lohr J, Kahn E, Abbas AK. The link between lymphocyte deficiency and autoimmunity: roles of endogenous T and B lymphocytes in tolerance. J. Immunol. 2005;175:21–26. doi: 10.4049/jimmunol.175.1.21. [DOI] [PubMed] [Google Scholar]
- 17.Qin Z, Richter G, Schuler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat. Med. 1998;4:627–630. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- 18.King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 19.Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity. 2009;30:324–335. doi: 10.1016/j.immuni.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, Wakeland EK, Germain RN, Schwartzberg PL. Optimal germinal center responses require a multistage T cell:B cell adhesion process involving integrins, SLAM-associated protein, and CD84. Immunity. 2010;32:253–265. doi: 10.1016/j.immuni.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature. 2008;455:764–769. doi: 10.1038/nature07345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fillatreau S, Gray D. T cell accumulation in B cell follicles is regulated by dendritic cells and is independent of B cell activation. J. Exp. Med. 2003;197:195–206. doi: 10.1084/jem.20021750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelletier N, McHeyzer-Williams LJ, Wong KA, Urich E, Fazilleau N, McHeyzer-Williams MG. Plasma cells negatively regulate the follicular helper T cell program. Nat. Immunol. 2010;11:1110–1118. doi: 10.1038/ni.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 26.Germain RN, Stefanova I. The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Annu. Rev. Immunol. 1999;17:467–522. doi: 10.1146/annurev.immunol.17.1.467. [DOI] [PubMed] [Google Scholar]
- 27.Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485–492. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 28.Fasso M, Anandasabapathy N, Crawford F, Kappler J, Fathman CG, Ridgway WM. T cell receptor (TCR)-mediated repertoire selection and loss of TCR vbeta diversity during the initiation of a CD4+ T cell response in vivo. J. Exp. Med. 2000;192:1719–1730. doi: 10.1084/jem.192.12.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malherbe L, Hausl C, Teyton L, McHeyzer-Williams MG. Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity. 2004;21:669–679. doi: 10.1016/j.immuni.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasenkrug KJ, Chesebro B. Immunity to retroviral infection: The Friend virus model. Proc. Natl. Acad. Sci. USA. 1997;94:7811–7816. doi: 10.1073/pnas.94.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antunes I, Tolaini M, Kissenpfennig A, Iwashiro M, Kuribayashi K, Malissen B, Hasenkrug K, Kassiotis G. Retrovirus-specificity of regulatory T cells is neither present nor required in preventing retrovirus-induced bone marrow immune pathology. Immunity. 2008;29:782–794. doi: 10.1016/j.immuni.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pike R, Filby A, Ploquin MJ, Eksmond U, Marques R, Antunes I, Hasenkrug K, Kassiotis G. Race between retroviral spread and CD4+ T-cell response determines the outcome of acute Friend virus infection. J. Virol. 2009;83:11211–11222. doi: 10.1128/JVI.01225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marques R, Antunes I, Eksmond U, Stoye J, Hasenkrug K, Kassiotis G. B lymphocyte activation by coinfection prevents immune control of friend virus infection. J. Immunol. 2008;181:3432–3440. doi: 10.4049/jimmunol.181.5.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 36.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 37.Wu C, Nguyen KB, Pien GC, Wang N, Gullo C, Howie D, Sosa MR, Edwards MJ, Borrow P, Satoskar AR, Sharpe AH, Biron CA, Terhorst C. SAP controls T cell responses to virus and terminal differentiation of TH2 cells. Nat. Immunol. 2001;2:410–414. doi: 10.1038/87713. [DOI] [PubMed] [Google Scholar]
- 38.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, Trent RJ, Basten A. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 39.Philpott KL, Viney JL, Kay G, Rastan S, Gardiner EM, Chae S, Hayday AC, Owen MJ. Lymphoid development in mice congenitally lacking T cell receptor alpha beta-expressing cells. Science. 1992;256:1448–1452. doi: 10.1126/science.1604321. [DOI] [PubMed] [Google Scholar]
- 40.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 41.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 42.Schwartzberg PL, Mueller KL, Qi H, Cannons JL. SLAM receptors and SAP influence lymphocyte interactions, development and function. Nat. Rev. Immunol. 2009;9:39–46. doi: 10.1038/nri2456. [DOI] [PubMed] [Google Scholar]
- 43.Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc. Natl. Acad. Sci. USA. 2010;107:20453–20458. doi: 10.1073/pnas.1008437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasenkrug KJ, Brooks DM, Dittmer U. Critical Role for CD4+ T Cells in Controlling Retrovirus Replication and Spread in Persistently Infected Mice. J. Virol. 1998;72:6559–6564. doi: 10.1128/jvi.72.8.6559-6564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bila C, Oberhauser V, Ammann CG, Ejaz A, Huber G, Schimmer S, Messer R, Pekna M, von Laer D, Dittmer U, Hasenkrug KJ, Stoiber H, Banki Z. Complement Opsonization Enhances Friend Virus Infection of B Cells and Thereby Amplifies the Virus-Specific CD8+ T Cell Response. J. Virol. 2011;85:1151–1155. doi: 10.1128/JVI.01821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang Y. h., Watowich SS, Jetten AM, Tian Q, Dong C. Generation of T Follicular Helper Cells Is Mediated by Interleukin-21 but Independent of T Helper 1, 2, or 17 Cell Lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi Y, Kageyama R, Eto D, Escobar T, Johnston R, Monticelli L, Lao C, Crotty S. ICOS Receptor Instructs T Follicular Helper Cell versus Effector Cell Differentiation via Induction of the Transcriptional Repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kerfoot S, Yaari G, Patel J, Johnson K, Gonzalez D, Kleinstein S, Haberman A. Germinal Center B Cell and T Follicular Helper Cell Development Initiates in the Interfollicular Zone. Immunity. 2011;34:947–960. doi: 10.1016/j.immuni.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, Okada T. Bcl6 Protein Expression Shapes Pre-Germinal Center B Cell Dynamics and Follicular Helper T Cell Heterogeneity. Immunity. 2011;34:961–972. doi: 10.1016/j.immuni.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 50.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, Vinuesa CG. The Transcriptional Repressor Bcl-6 Directs T Follicular Helper Cell Lineage Commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Yusuf I, Kageyama R, Monticelli L, Johnston RJ, DiToro D, Hansen K, Barnett B, Crotty S. Germinal Center T Follicular Helper Cell IL-4 Production Is Dependent on Signaling Lymphocytic Activation Molecule Receptor (CD150) J. Immunol. 2010;185:190–202. doi: 10.4049/jimmunol.0903505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 2009;10:385–393. doi: 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 55.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 2010;11:535–542. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.