

Abstract
Influenza nucleoprotein (NP) plays multiple roles in the virus life cycle, including an essential function in viral replication as an integral component of the ribonucleoprotein complex, associating with viral RNA and polymerase within the viral core. The multifunctional nature of NP makes it an attractive target for antiviral intervention, and inhibitors targeting this protein have recently been reported. In a parallel effort, we discovered a structurally similar series of influenza replication inhibitors and show that they interfere with NP-dependent processes via formation of higher-order NP oligomers. Support for this unique mechanism is provided by site-directed mutagenesis studies, biophysical characterization of the oligomeric ligand:NP complex, and an X-ray cocrystal structure of an NP dimer of trimers (or hexamer) comprising three NP_A:NP_B dimeric subunits. Each NP_A:NP_B dimeric subunit contains two ligands that bridge two composite, protein-spanning binding sites in an antiparallel orientation to form a stable quaternary complex. Optimization of the initial screening hit produced an analog that protects mice from influenza-induced weight loss and mortality by reducing viral titers to undetectable levels throughout the course of treatment.
Keywords: antiinfluenza, oligomerization, polymerase inhibitor, protein–protein interaction, cooperative inhibition
Global seasonal influenza epidemics are responsible for 250–500,000 deaths annually (1), depending on the virulence and transmissibility of the circulating viral strain. To combat the virus, a number of treatments are currently available. Vaccines, containing killed or attenuated virus or pure viral proteins, can provide prophylactic protection from infection by stimulating the production of antibodies to viral strains predicted to circulate in the ensuing flu season (2). These vaccines must be reformulated and administered annually and generally have lower efficacy in the most susceptible populations, such as the elderly and immunocompromised (3). Direct-acting antiviral agents can be used in either a therapeutic or a prophylactic mode; these include M2 ion channel blockers and neuraminidase inhibitors. However, resistance to M2 blockers is widespread (4–6), and neuraminidase inhibitor resistance is mounting with continued use (7–9). New antiviral agents that act on other targets are clearly needed, to be administered either alone or, preferably, in combination with current modalities (10, 11).
Influenza virus is a single-strand, negative-sense, segmented RNA virus of the Orthomyxovirus family. As such, it uses a heterotrimeric RNA-dependent RNA polymerase in complex with its RNA genome and associated nucleoprotein (NP) to synthesize the requisite viral RNAs (vRNAs), mRNAs, and cRNAs needed for replication (12, 13). NP plays several roles during the viral life cycle, including ribonucleoprotein (RNP) formation, transport of vRNA-polymerase complex to the nucleus, viral replication, and virion assembly (14–17). Consequently, inhibitors of NP function may affect multiple stages of the life cycle (18).
Recently, Kao et al. (19) reported the discovery of nucleozin (compound 1) as a “potent antagonist of NP accumulation in the nucleus.” Similar results were reported by Su et al. (20). Herein, we report the independent discovery and optimization of a related series of compounds with potent antiviral activity both in vitro and in vivo. Moreover, the X-ray structure of a representative compound bound to NP has been solved, revealing an unusual NP_A:NP_B dimeric subunit bearing two protein-bridging ligands in an antiparallel arrangement. This unexpected quaternary complex may portend a unique mode of viral inhibition via ligand-induced formation of stable NP oligomers.
Results
Identification of Hits in a High-Throughput Screen.
High-throughput screening of the Bristol-Myers Squibb compound collection in a whole-cell antiviral assay consisting of Madin–Darby bovine kidney (MDBK) cells infected with H1N1 influenza A/WSN/33 virus (A/WSN) identified compound 1 (Table 1) as a potent inhibitor of viral replication, with EC50 = 0.170 μM. Compound 1 was also recently reported by Kao et al. (19). A substructure search for commercial analogs of compound 1 identified compound 2, with EC50 = 0.060 μM, nearly threefold more potent than compound 1 yet varying only by introduction of an o-Cl substituent on the 3-phenyl ring of the isoxazole. The dose–response curves for compounds 1 and 2 were very steep (Fig. S1), providing EC90/EC50 ratios <1.5 and Hill slopes >5 (21). Steep dose–response curves are characteristic of a cooperative inhibition mechanism that is often observed with oligomeric proteins in which the binding of one ligand enhances the binding of subsequent ligands (21, 22). Additionally, both compounds afforded micromolar IC50 values in a cell-free in vitro polymerase assay (Table 1), evidence that the observed antiviral activity was, in part, due to inhibition of polymerase function. Our interest in identifying influenza antiviral agents with polymerase inhibitory activity prompted us to initiate resistance studies to identify the biological target of compound 1.
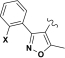
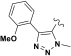
Table 1.
Assay data for compounds 1–5
 |
 |
 |
||||
| Assay | R = | Compound 1 (X=H) | Compound 2 (X=Cl) | Compound 3 (X=OMe) | Compound 4 | Compound 5 |
| EC50, μM | ||||||
| H1N1 A/WSN/33 | 0.17 | 0.06 | 0.04 | 0.07 | 0.07 | |
| H1N1 A/Solomon Islands/3/2006 | 4.9 | 1.0 | 0.66 | 3.5 | 12 | |
| H1N1 A/CA/07/2009 | >20 | >20 | >20 | >20 | >20 | |
| H3N2 A/Brisbane/10/2007 | >20 | >20 | 7.3 | >20 | >20 | |
| H5N1 A/Gull_PA/4175/83 | 0.15 | 0.10 | 0.07 | 0.11 | 0.04 | |
| IC50, μM | ||||||
| H1N1 A/WSN polymerase | 2.5 | 0.61 | 0.17 | 0.26 | 0.90 | |
| Aqueous solubility, μMa | <5 | <5 | 20 | 26 | 29 | |
| MLM stability, % remaining at 10 min | 20 | 0 | 14 | 18 | 45 | |
All EC50 and IC50 values are the mean of at least three experiments.
a20 mM phosphate buffer (pH 7.5), 1.0 M NaCl, and 8% DMSO.
Resistance to Compound 1 Maps to NP.
Influenza virus resistant to compound 1 was generated after multiple passages in the presence of increasing concentrations of compound 1 in cell culture. Sequence analyses of the entire genome of multiple resistant viruses identified NP substitutions Y52C/H, Y289H, and N309K as possible markers for resistance. Reverse genetic analyses using a cell-based plasmid polymerase assay confirmed that these substitutions encode resistance to compound 1 (Table S1). The Y289H and Y52H resistant mutations were identified by Kao et al. (19) and Su et al. (20), respectively, but the N309K mutation has not been previously reported.
Protein alignments indicated that polymorphisms at these positions are found in three recent circulating subtype influenza viruses: H1N1 A/Solomon Islands/3/2006 (N309T), H3N2 A/Brisbane/10/2007 (Y52H), and H1N1 A/CA/07/2009 “pandemic flu” (Y289H). Indeed, as shown in Table 1, compounds 1 and 2 exhibited reduced activity against these three viral strains. In contrast, H5N1 A/Gull_PA/4175/83 avian virus does not contain any of the identified resistant substitutions, and it was found to be sensitive to inhibition by both compounds 1 and 2.
Mapping the locations of these resistant mutations in the previously reported apo X-ray structure of A/WSN NP (23) resulted in the observation that, although the N309 and Y289 residues were in close proximity to one another, they were more than 17 Å apart from Y52. All three residues reside in an inflexible region of NP (23), and thus it is physically impossible for compound 2 (13 Å along the longest axis) to simultaneously interact with both the N309/Y289 and Y52 regions of NP in a single binding pose, which may suggest that NP contains two independent ligand-binding sites.
To better characterize the binding of compound 2 to NP, residues within 8 Å of N309, Y289, and Y52 were systematically mutated, and the effect of these point mutations on the polymerase IC50 values of compound 2 was measured in a cell-based plasmid polymerase assay (24). Table S1 provides a summary of these results, wherein each mutated residue was categorized as “sensitive” (polymerase IC50 fold shift ≥ 3) or “tolerant” (polymerase IC50 fold shift <3) to substitution. In the absence of compound 2, these mutations had a minimal effect on polymerase function, yielding a less than twofold variation from A/WSN WT activity. As illustrated in Fig. 1, mapping the results of these point mutation studies onto the apo X-ray structure of A/WSN NP reported by Ye et al. (23) reveals two regions in which substitution imparts resistance to compound 2. One region comprises residues in close proximity to Y289 and N309 (the “Y289/N309 pocket”), and the other region comprises residues surrounding Y52 (the “Y52 pocket”). The Y289/N309 and Y52 pockets are effectively separated by a well-defined ridge comprising residues that are tolerant to substitution (S310, S283, and D51). These mutation studies provided further evidence of two independent ligand-binding sites on NP.
Fig. 1.

Mapping NP residues that influence inhibitor efficacy. Site-directed mutagenesis of NP and subsequent evaluation in the cell-based plasmid polymerase assay revealed two discontinuous regions of NP that are sensitive to amino acid substitution. Red, sensitive residues (more than threefold increase in IC50 compared with WT); green, tolerant residues (less than threefold change in IC50 compared with WT).
Compounds 3 and 4 Induce Formation of Higher-Order NP Oligomers.
As shown in Table 1, compounds 1 and 2 exhibited limited aqueous solubility (<5 μM), restricting our ability to conduct biophysical studies of the putative ligand:NP complex. Changes to the isoxazole phenyl ring of compound 2 yielded compounds 3 and 4, which possessed improved aqueous solubilities and comparable A/WSN potencies relative to compound 2 (Table 1) and allowed the biophysical interactions of compounds 3 and 4 with purified A/WSN NP to be studied in detail. As shown in Fig. 2, dynamic light scattering (DLS) experiments were used to assess NP oligomerization by measuring the hydrodynamic radius (Rh) and polydispersity (25) of an aqueous solution of NP, alone and in the presence of either compound 3 or compound 4. In the absence of ligand, NP afforded an average Rh = 5.4 nm and a polydispersity of 12%, consistent with the formation of a monodisperse trimeric species. Addition of compound 3 to NP provided a unique, stable oligomeric species with an average Rh = 8.3 nm and a polydispersity of 10%. Similar results were obtained when compound 4 was added to NP: a stable oligomeric species with an average Rh = 8.1 nm and a polydispersity of 14% was observed. These DLS experiments provide strong evidence that compounds 3 and 4 induce the formation of higher-order NP oligomers of well-defined composition. Additional studies of the NP:ligand compound 4 complex have been conducted by using a variety of biophysical techniques, including thermal-shift assays, NMR, analytical ultracentrifugation, and surface plasmon resonance. The results from these studies are consistent with the DLS data.
Fig. 2.

Hydrodynamic radius (Rh) of a solution of NP alone or NP plus compound 3 or compound 4 as measured by DLS. Addition of compound 3 or compound 4 to NP afforded a significant increase in Rh compared with NP alone. In the absence of NP, compounds 3 and 4 provided Rh values <1 nm.
Compound 3 and oseltamivir carboxylate (OC) were compared in a viral yield-reduction assay, as shown in Fig. 3. As expected, OC afforded a 2 log10 gradual decrease in viral yield at micromolar concentrations (26). In contrast, a steep dose–response relationship was observed with compound 3, with viral yields obtained at 0.041 μM but complete inhibition (>4 log10 decrease) observed at the next-highest concentration of 0.123 μM. The viral yield data for compound 3 is consistent with both the EC50 values and steep dose–response curves observed in the whole-cell antiviral assay (Fig. S1) and, coupled with the ligand-induced formation of higher-order oligomers observed via DLS, lends further support to the cooperative inhibition of NP function by compound (21, 22).
Fig. 3.

Efficacy of compound 3 in reducing viral yields in tissue culture. A viral yield assay (A/WSN) was conducted comparing OC (white) and compound 3 (black). At compound 3 concentrations of more than 0.041 μM, the viral titer was below the limit of detection (20 pfu per well).
X-Ray Crystal Structure of Compound 3 Bound to NP Reveals a Protein Dimer.
After an extensive survey of ligands, protein constructs, crystallization conditions, and cryo-preservation methods, the X-ray crystal structure of compound 3 bound to NP was solved with a resolution of 2.7 Å and found to contain two molecules of NP (denoted NP_A and NP_B) and two molecules of compound 3 in the asymmetric unit (PDB ID code 3RO5). As shown in Fig. 4A, expansion of the asymmetric unit revealed symmetry-related molecules consistent with formation of a hexameric NP complex in which an NP_A trimer (comprising three monomers: NP_A, NP_A′, and NP_A′′) and an NP_B trimer (comprising three monomers: NP_B, NP_B′, and NP_B′′) are bridged by six molecules of compound 3 (two molecules per NP_A:NP_B dimeric subunit), forming a dimer of trimers (or hexamer). The NP_A and NP_B trimers closely resemble the trimers observed in the apo A/WSN NP X-ray structure reported by Ye et al. (23) and the apo H5N1 A/Hong Kong/483/97 reported by Ng et al. (27) in that they are formed via interdigitation of the tail loops of each NP_A (or NP_B) monomeric unit (28).
Fig. 4.

X-ray structure of compound 3:NP complex solved with a 2.7-Å resolution. (A) Six molecules of compound 3 (yellow) bridge two NP trimers (NP_A, NP_A′, and NP_A′′ in blue; NP_B, NP_B′, and NP_B′′ in gray) to form a dimer of trimers (or hexamer). (B) Two molecules of compound 3 (yellow) bridge an NP_A:NP_B dimeric subunit (NP_A in blue; NP_B in gray) in an antiparallel orientation. Yellow circles depict the regions of NP that were sensitive to amino acid substitution as described in Fig. 1. (C) Detailed view of compound 3 bound to an NP_A:NP_B dimeric subunit (NP_A in blue; NP_B in gray). Key ligand:protein contacts include a hydrogen bond from the amide carbonyl of compound 3 to the S376 side chain of NP_B and a face-to-face π–π interaction between the 2-Cl-4-NO2-Ph ring of compound 3 and Y289 of NP_A.
The X-ray structure of a single NP_A:NP_B dimeric subunit is shown in Fig. 4B. Two molecules of compound 3 bridge two molecules of NP (i.e., NP_A and NP_B) to form a stable quaternary complex. Two composite, antiparallel binding pockets are formed via combination of the Y289/N309 pocket of NP_A with the Y52 pocket of NP_B and vice versa. Thus, each NP molecule binds two separate halves of compound 3, with the Y289/N309 region occupied by the nitrophenyl moiety and the Y52 region occupied by the isoxazole heterocycle.
A detailed view of one of the two ligand-binding sites present in a single NP_A:NP_B dimeric subunit is provided in Fig. 4C. A network of aromatic residues (Y313 and Y52 from NP_B and Y289, F291, and Y296 from NP_A) extends across the NP_A:NP_B interface and forms a hydrophobic wall around the backside of the ligand-binding pocket. Face-to-face π-stacking interactions are observed between the nitro-aryl moiety of compound 3 and Y289 of NP_A. The piperazine moiety of compound 3 exhibits a hydrophobic interaction with Y52 of NP_B and an intramolecular hydrophobic interaction with the isoxazole-aryl moiety of compound 3. The amide carbonyl of compound 3 makes a strong hydrogen bond to the side chain OH of S376 of NP_B and is conformationally stabilized by intramolecular electrostatic interactions (3.3 Å) with the oxygen of the methoxy group. The isoxazole moiety and its pendent aryl ring fit into a well-defined pocket and engage in both electrostatic and hydrophobic interactions with residues Y52, E53, R99, W104, and Y313 of NP_B.
Identification of Compound 5 for In Vivo Studies.
Compounds 1–4 are potent inhibitors of A/WSN replication in vitro, but they are rapidly metabolized in the presence of mouse liver microsomes (MLMs; Table 1). Consistent with poor MLM stability, isoxazole-containing analogs generally afforded low plasma concentrations upon oral administration to mice and rats, so our synthetic efforts focused on bioisosteric isoxazole replacements. A broad survey of five-membered heterocycles led to the identification of triazole 5, which possesses improved MLM stability and comparable antiviral activity relative to compounds 1–4 (Table 1). Triazole 5 was advanced into mouse pharmacokinetic studies, providing adequate systemic exposure after i.p. doses of 10 and 50 mg/kg (Fig. 5A). Maximum concentration (Cmax) values of 4.7 and 16 μM and area under the plasma concentration–time curve from 0 to 24 h (AUC0–24 h) values of 16.8 and 88.4 μM⋅h, were obtained for the 10 and 50 mg/kg groups, respectively. Similar exposure levels were achieved when compound 5 was administered orally. The Ctrough values at 12 h for both the 10 and 50 mg/kg i.p. doses were below the 70 nM A/WSN EC50 value for compound 5. However, the Ctrough value at 8 h for the 10 mg/kg dose was estimated to be fivefold above the EC50, suggesting the potential for in vivo efficacy. These pharmacokinetic results prompted a proof-of-concept study of compound 5 in a mouse model of influenza.
Fig. 5.

Characterization of inhibitor efficacy in a mouse model of influenza infection. (A) Serum concentration–time curves for compound 5 in mice after a single 10 or 50 mg/kg i.p. dose. (B–D) Mouse cohorts were pretreated with vehicle, compound 5, or OC at 1 h before intranasal infection with 500 pfu of A/WSN influenza virus. Drug or vehicle treatment continued for 5 d. (B) Survival time-course graph. All but two cohorts (vehicle and 2 mg/kg compound 5 t.i.d.) showed 100% survival. (C) Daily body weight loss. (D) Viral titers at days 5 and 8 postinfection. Administration of compound 5 on a 10 mg/kg t.i.d. dosing regimen for 5 d reduced the viral titer below the limit of detection (500 pfu) on days 5 and 8.
Antiviral Activity of Compound 5 in a Balb/C Mouse Model of Influenza Infection.
Mice infected intranasally with 500 pfu of A/WSN were divided into six groups: one group treated with vehicle, one group treated with OC [0.5 mg/kg twice a day (b.i.d.)], and four groups that were administered compound 5 at doses from 2 to 50 mg/kg b.i.d. or three times a day (t.i.d.). A group of uninfected and untreated mice served as controls. The first dose was administered at 1 h preinfection, and then infected mice were treated with drug or vehicle i.p. for 5 d postinfection and monitored for an additional 9 d. Mortality (Fig. 5B) and weight loss (Fig. 5C) were monitored daily throughout the experiment, and viral titers were measured at days 5 and 8 postinfection (Fig. 5D). The vehicle- and OC-treated groups behaved as expected (10): the vehicle-treated mice rapidly lost weight and all mice died by day 9, whereas all of the OC-treated mice survived with minimal weight loss over the 14 d of the study. Viral titers in the OC-treated group were decreased by <1 log10 compared with the vehicle-treated group at day 5 and >2 log10 at day 8. The mice treated with compound 5 responded in a dose-dependent fashion. At the lowest dose (2 mg/kg t.i.d., 6 mg/kg per d), compound 5 delayed but did not prevent mortality: all of the mice survived through day 9, but 60% of the mice died by day 11. The remaining mice in the group gained weight and survived through day 14, although the 5- and 8-d viral titers were similar to vehicle controls. The next-highest dose of compound 5 (10 mg/kg b.i.d., 20 mg/kg per d) protected the mice from weight loss, and all of the mice survived through day 14, but the viral titers dropped only 1 log10 compared with the vehicle-treated animals. The highest dose (50 mg/kg b.i.d., 100 mg/kg per d) protected all mice, with no deaths and a 3 log10 reduction in viral titer on day 5 that rebounded by 1 log10 on day 8. The rebound is likely because of the inability of compound 5 to maintain systemic exposures above the A/WSN EC50 for 12 h (Fig. 5A), even after administration of a 50 mg/kg dose. Interestingly, t.i.d. dosing of 10 mg/kg (30 mg/kg per d) of compound 5 completely protected the mice from infection. Not only did all of the mice survive without any observable weight loss relative to the healthy controls, but the viral titers were below the lower limit of detection (500 pfu) at both the 5- and 8-d time points. Postmortem inspection of the lung tissue indicated that, although the lungs of the 10 mg/kg t.i.d. group showed evidence of infection, the virus did not appear to have spread to neighboring lung tissue.
Discussion
In addition to providing direct confirmation that compounds 1 and 2 are potent influenza antiviral agents that exert their biological effect via binding to NP, this study identified molecules with improved physical properties that enabled biophysical studies of the ligand:NP complex and in vivo studies in a mouse model of influenza. The discovery of compounds 3–5 facilitated three key findings: (i) the addition of either compound 3 or compound 4 to purified NP induced a well-defined, higher-order oligomeric complex; (ii) cocrystallization of compound 3 and NP provided an X-ray structure that revealed two antiparallel ligand-binding pockets spanning the interface between NP_A:NP_B dimeric subunits; and (iii) i.p. administration of compound 5 protected mice from viral infection.
Using fluorescence spectroscopy, Kao et al. (19) observed the accumulation of NP in the cytoplasm when compound 1 was added to cells infected with A/WSN, an effect they ascribed to the formation of “very large NP complexes” that are unable to migrate into the nucleus. As determined via DLS, addition of compound 3 or compound 4 to purified NP provided higher-order NP oligomers (Fig. 2) that are very likely to be related to the oligomeric species observed in cells by Kao et al. In light of the discontinuous ligand-binding domains described in Fig. 1, it remained unclear how a small molecule could induce the formation of higher-order oligomers. We were unable to construct a computational model for the binding of compound 1 or compound 2 to a single NP molecule that could account for the observed resistant residues (Y52C/H, Y289H, and N309K). Similarly, Kao et al. (19) and Su et al. (20) proposed ligand:NP binding models that were inconsistent with our site-directed mutagenesis data, although their efforts may have been hindered by an incomplete set of resistant mutations: Kao et al. did not report the Y52H and N309K resistant mutations (19), and Su et al. did not report the Y289H and N309K resistant mutations (20).
All of the aforementioned ligand:NP binding models were based on the assumption that each NP molecule contained a single, contiguous ligand-binding pocket. However, the X-ray cocrystal structure of compound 3 bound to A/WSN NP (Fig. 4 B and C) revealed that the 17 Å distance between the Y52 and the Y289/N309 pockets in a single NP molecule is accommodated by the antiparallel arrangement of two molecules of compound 3 bridging two NP monomers. In fact, absent the formation of a perfectly aligned NP_A:NP_B dimeric subunit, contiguous ligand-binding sites do not exist. The X-ray crystal structure also revealed a hexameric species (the dimer of trimers shown in Fig. 4A) that offers an explanation for the higher-order oligomers observed via DLS. It should be noted that the DLS and X-ray studies were performed with purified NP in vitro, and further studies will be required to confirm the role (or even the presence) of an NP hexamer in infected cells. Even so, the ligand-induced formation of a previously unreported NP_A:NP_B dimeric subunit offers important clues about the size and structure of the oligomers formed when compound 3 binds to NP. This inhibitor series exhibits steep dose–response relationships in whole-cell (Fig. S1) and viral yield (Fig. 3) assays, suggesting a highly cooperative effect of the compounds with regard to inhibiting target (21, 22). It is very likely that the ligand-induced formation of higher-order NP oligomers is the driving force behind these steep dose–response relationships. In addition, NP plays important roles in polymerase function and RNP assembly and the steep dose–response relationships we observed may also reflect the additive inhibition of several NP-mediated processes. For example, the formation of higher-order NP oligomers is consistent with the observation of “aggregated” forms of NP that are unable to undergo nuclear import in infected cells (19, 20) as well as the inhibition of polymerase activity observed in our cell-free in vitro assay (Table 1).
Before performing in vivo studies, an effort to improve the in vitro potency, solubility, and metabolic stability of the compound series culminated in the identification of compound 5, which was administered in four dosing cohorts to mice infected intranasally with 500 pfu of A/WSN. This in vivo study established that 10 mg/kg of compound 5 administered i.p. on a t.i.d. schedule for 5 d was the most efficacious dose, protecting 100% of the infected mice through 14 d and reducing the viral titer to undetectable levels (a >3 log10 decrease) on days 5 and 8. In contrast, Kao et al. (19) studied the effect of compound 1 in mice infected intranasally with 100 pfu of H5N1 A/Vietnam/1194/04 and observed 50% mortality by day 12 and a 1 log10 decrease in viral titer on day 5. The dose–response relationship observed with compound 5 not only validates NP as an antiviral target but also provides insight into the pharmacokinetic–pharmacodynamic relationship. Specifically, although the 50 mg/kg b.i.d dose (100 mg/kg per d) afforded higher Cmax and AUC values than did the 10 mg/kg t.i.d. dose (30 mg/kg per d), only the t.i.d. dosing regimen reduced viral titers to undetectable levels at days 5 and 8 postinfection (Fig. 5D). These results indicate that optimal in vivo activity of compound 5 is achieved by maintaining a Ctrough level above the in vitro EC50 level (Fig. 5A) and suggest that it is crucial to maintain NP in a higher-order oligomeric state throughout the course of treatment.
As shown in Table 1, compounds 1–5 lack broad influenza strain coverage. Although these compounds exhibit potent activity against historical H1N1 and present-day H5N1 strains, resistance is observed in the circulating strains represented by H1N1 A/Solomon Islands/3/2006, H3N2 A/Brisbane/10/2007, and pandemic H1N1 A/California/07/2009. The observed resistance can be readily explained by mutations found in the ligand-binding pocket. The R305K/N309K (H1N1 A/Solomon Islands/3/2006), Y52H (H3N2 A/Brisbane/10/2007), and E53D/Y289H/Y313V (H1N1 A/CA/07/2009) mutations all impart resistance in the plasmid-based cell-culture polymerase assay (Fig. 1 and Table S1). Although compound 3 exhibits significantly reduced activity against both the H1N1 A/Solomon Islands/3/2006 (EC50 = 0.66 μM) and H3N2 A/Brisbane/10/2007 (EC50 = 7.3 μM) strains, the corresponding dose–response curves (Fig. S2) exhibit the characteristic steep Hill slopes observed with the A/WSN strain (Fig. S1). Modeling and X-ray analysis also suggest that the two ligand-binding pockets remain intact in these drug-resistant NP molecules, and thus overcoming this preexisting resistance may be possible via further optimization of this chemical series.
This article presents compelling evidence that compounds 1–5 inhibit influenza virus replication by binding to a unique NP_A:NP_B dimeric subunit and inducing the formation of higher-order NP oligomers, including the NP hexamer observed in the X-ray structure of compound 3 and NP. As a result, we hypothesize that any two NP molecules—be they monomeric or oligomeric, RNA associated or not, nascent or completely assembled into a polymerase complex—have the potential to form ligand-induced NP_A:NP_B dimeric subunits that yield higher-order NP oligomers. These oligomers have been shown to interfere with RNP transport and viral transcription and may sequester nascent NP from assembling into polymerase complexes and ultimately virions. Furthermore, NP or an analogous protein is found in a number of other viruses (29), and the ligand-induced formation of higher-order NP oligomers may represent a general strategy for the discovery of novel antiviral agents.
Materials and Methods
Analog Synthesis.
Compounds 1 (341001-38-5) and 2 (312537-30-7) were purchased from ChemBridge Corporation. Compounds 3–5 were synthesized and characterized as described in SI Materials and Methods.
Cells and Virus.
Madin–Darby canine kidney (MDCK) cells, MDBK cells, and A/WSN virus were obtained from the American Type Culture Collection. Influenza viruses H1N1 A/Solomon Islands/3/2006 and H3N2 A/Brisbane/10/2007 were obtained from the Centers for Disease Control and Prevention. Cells were maintained with Eagle's MEM supplemented with 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1.5 g/L sodium bicarbonate, and 10% FBS (heat-inactivated).
Whole-Cell Antiviral Assay.
High-throughput screening and antiviral assays were conducted by using neuraminidase activity as the read-out for influenza growth via a method similar to that described in ref. 30, with modifications detailed in SI Materials and Methods.
Cell-Free In Vitro Polymerase Assay.
In vitro polymerase assays were performed by using a scintillation-proximity assay format as described in SI Materials and Methods.
Cell-Based Plasmid Polymerase Assay.
An influenza virus-specific five-plasmid replication system was used to assess the effect of amino acid changes on the sensitivity to the inhibitors in cell culture (24). Mutants of the NP gene of A/WSN were created with the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). Plasmids expressing the PB2, PB1, PA, and NP proteins from A/WSN virus were cloned into pcDNA, and a reporter plasmid expressing a influenza-like humanized Renilla luciferase gene was constructed as described (31). Details on plasmids, protein expression, and NP mutants are provided in SI Materials and Methods. Compounds were tested in a 96-well format at a final DMSO concentration of 1%. The five plasmids (10 ng of each) were transfected into HeLa cells with Lipofectamine 2000 (Invitrogen), and luciferase activity was measured after overnight incubation with EnduRen (Promega) according to the manufacturer's instructions.
Yield-Reduction Assay.
Compounds were evaluated for their ability to reduce tissue culture virus titers as detailed in SI Materials and Methods.
DLS Experiments.
The effect of compounds on the oligomerization of NP was measured with a Wyatt DLS plate reader instrument. The reactions were carried out with a 384-well plate with a 20-μL reaction volume containing 1 μM NP plus 2 μM compound in buffer containing 25 mM Tris (pH 7.5), 1 M NaCl, 10% glycerol, 5 mM DTT, and 8% (vol/vol) DMSO. All reactions were incubated at 25 °C for 1 h before DLS analysis. The samples were then passed through a 0.2-μm filter (Pall), pipetted into the reaction plate, and overlaid with 10 μL of polydimethylsiloxane DC200 oil (Sigma) to prevent evaporation. Each DLS experiment consisted of at least three individual measurements of 20 acquisitions with a 5-s data acquisition time and was carried out at 25 °C. The diffusion coefficients were corrected for the buffer composition by use of the standard conversion equation within Dynamics software version 6 (Wyatt Technologies). Intensity autocorrelation functions were fitted with the Regularization algorithm in the Dynamics software.
Crystallization, Structure Solution, and Refinement.
NP was expressed and purified as described in SI Materials and Methods, then concentrated to 9 mg/mL and complexed with compound (final compound concentration was 2 mM) before crystallization. Crystals were observed in 0.16 M NaCl, 0.1 M sodium citrate (pH 5.25), and 15% PEG 4000. With the Proteros Free-Mounting System, protein crystals were dehydrated during the addition of cryo-solution (25% PEG 550) to the freely mounted crystal by using the Picodropper, resulting in better-diffracting crystals ready for shock freezing in oil. Data were collected at the Swiss Light Source (beamline PXI/X06SA) and processed with XDS (32) and XSCALA (Table S2). The structure was solved by molecular replacement with PDB ID code 2IQH structure used as a search model (MOLREP) (33). As shown in Fig. S3, compound 3 is clearly visible in the electron density map. Refinement was performed with REFMAC (34) with the program MIFit (http://code.google.com/p/mifit/). Results are shown in Table S2. Coordinates have been deposited in the Protein Data Bank, www.pdb.org, under PDB ID code 3RO5.
Metabolic Stability and Mouse Pharmacokinetic Study.
Compounds (final concentration 10 μM) were incubated with MLMs (1 mg/mL), and the enzymatic reaction was initiated with the addition of NADPH (1 mM). After a 10-min incubation at 37 °C, the reaction mixture was quenched with 2 vol of acetonitrile. Animals were given triazole compound 5 either by i.p. injection or oral gavage, and serum samples were collected at various time points. All animal experiments were conducted in accordance to a standard protocol approved by the Animal Care and Use Committee. Both microsomal and serum samples were analyzed via liquid chromatography/tandem MS.
Antiviral Activity Test in Balb/C Mice.
Mice were given the first dose (i.p.) of the vehicle (Labrafil: N-methyl-2-pyrrolidone, 9:1 vol/vol), OC, or triazole 5 and, 1 h later, were infected intranasally with 500 pfu of A/WSN virus. Additional doses were administered i.p. for 5 d as follows: (i) vehicle (b.i.d.), (ii) OC (0.5 mg/kg b.i.d.), (iii) triazole 5 (10 mg/kg b.i.d.), (iv) triazole 5 (50 mg/kg b.i.d.), (v) triazole 5 (2 mg/kg t.i.d.), and (vi) triazole 5 (10 mg/kg t.i.d.). On days 5 and 8 postinfection, five mice from each group were killed to measure viral titers in the lung. The remaining animals were monitored throughout the 14-d experiment for mortality and weight loss, and lung viral titers were measured as described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Mark Cockett, Carl Decicco, John Houston, George Trainor, and Peter Palese for their support and helpful discussions regarding these studies.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3RO5).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1107906108/-/DCSupplemental.
References
- 1.Hedlund M, Larson JL, Fang F. Antiviral strategies for pandemic and seasonal influenza. Viruses. 2010;2:1766–1781. doi: 10.3390/v2081766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.US Food and Drug Administration Influenza Virus Vaccine for the 2010–2011 Season. 2010. Available at http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/post-marketactivities/lotreleases/ucm202750.htm. Accessed August 17, 2011.
- 3.Fiore AE, et al. Centers for Disease Control and Prevention Prevention and control of seasonal influenza with vaccines: Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2009. MMWR Recomm Rep. 2009;58(RR-8):1–52. [PubMed] [Google Scholar]
- 4.Weinstock DM, Zuccotti G. Adamantane resistance in influenza A. JAMA. 2006;295:934–936. doi: 10.1001/jama.295.8.934. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki H, et al. Emergence of amantadine-resistant influenza A viruses: Epidemiological study. J Infect Chemother. 2003;9:195–200. doi: 10.1007/s10156-003-0262-6. [DOI] [PubMed] [Google Scholar]
- 6.Balannik V, et al. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry. 2009;48:11872–11882. doi: 10.1021/bi9014488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moscona A. Oseltamivir resistance—disabling our influenza defenses. N Engl J Med. 2005;353:2633–2636. doi: 10.1056/NEJMp058291. [DOI] [PubMed] [Google Scholar]
- 8.Dharan NJ, et al. Oseltamivir-Resistance Working Group Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. JAMA. 2009;301:1034–1041. doi: 10.1001/jama.2009.294. [DOI] [PubMed] [Google Scholar]
- 9.Yen HL, et al. Importance of neuraminidase active-site residues to the neuraminidase inhibitor resistance of influenza viruses. J Virol. 2006;80:8787–8795. doi: 10.1128/JVI.00477-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krystal MR, Meanwell NA. Influenza—the case for combination therapy. Curr Opin Investig Drugs. 2009;10:746–749. [PubMed] [Google Scholar]
- 11.Govorkova EA, Webster RG. Combination chemotherapy for influenza. Viruses. 2010;2:1510–1529. doi: 10.3390/v2081510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishihama A, Nagata K. Viral RNA polymerases. CRC Crit Rev Biochem. 1988;23:27–76. doi: 10.3109/10409238809103119. [DOI] [PubMed] [Google Scholar]
- 13.Resa-Infante P, Jorba N, Coloma R, Ortin J. The influenza virus RNA synthesis machine: Advances in its structure and function. RNA Biol. 2011;8:207–215. doi: 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem. 1995;270:22701–22704. doi: 10.1074/jbc.270.39.22701. [DOI] [PubMed] [Google Scholar]
- 15.Noda T, et al. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439:490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- 16.Cros JF, García-Sastre A, Palese P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic. 2005;6:205–213. doi: 10.1111/j.1600-0854.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 17.Noton SL, et al. Studies of an influenza A virus temperature-sensitive mutant identify a late role for NP in the formation of infectious virions. J Virol. 2009;83:562–571. doi: 10.1128/JVI.01424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao YJ, Ye Q. Influenza A virus nucleoprotein. In: Wang Q, Tao YJ, editors. Influenza: Molecular Virology. Norfolk, UK: Caister Academic; 2010. pp. 53–68. [Google Scholar]
- 19.Kao RY, et al. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol. 2010;28:600–605. doi: 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su CY, et al. High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity. Proc Natl Acad Sci USA. 2010;107:19151–19156. doi: 10.1073/pnas.1013592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill AV. Proceedings of the Physiological Society: January 22, 1910. J Physiol. 1910;40:iv–vii. [Google Scholar]
- 22.Denisov IG, Frank DJ, Sligar SG. Cooperative properties of cytochromes P450. Pharmacol Ther. 2009;124:151–167. doi: 10.1016/j.pharmthera.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 24.Pleschka S, et al. A plasmid-based reverse genetics system for influenza A virus. J Virol. 1996;70:4188–4192. doi: 10.1128/jvi.70.6.4188-4192.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanlon AD, Larkin MI, Reddick RM. Free-solution, label-free protein-protein interactions characterized by dynamic light scattering. Biophys J. 2010;98:297–304. doi: 10.1016/j.bpj.2009.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K, et al. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir Chem Chemother. 2003;14:235–241. doi: 10.1177/095632020301400502. [DOI] [PubMed] [Google Scholar]
- 27.Ng AK-L, et al. Structure of the influenza virus A H5N1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008;22:3638–3647. doi: 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan W-H, et al. Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. J Virol. 2010;84:7337–7345. doi: 10.1128/JVI.02474-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruigrok RWH, Crepin T. Nucleoproteins of negative strand RNA viruses; RNA binding, oligomerisation and binding to polymerase co-factor. Viruses. 2010;2:27–32. doi: 10.3390/v2010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eichelberger MC, Hassantoufighi A, Wu M, Li M. Neuraminidase activity provides a practical read-out for a high throughput influenza antiviral screening assay. Virol J. 2008;5:109. doi: 10.1186/1743-422X-5-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neumann G, et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci USA. 1999;96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.