

Abstract
Currently, it is unknown whether defects in stem cell growth and differentiation contribute to myocardial aging and chronic heart failure (CHF), and whether a compartment of functional human cardiac stem cells (hCSCs) persists in the decompensated heart. To determine whether aging and CHF are critical determinants of the loss in growth reserve of the heart, the properties of hCSCs were evaluated in 18 control and 23 explanted hearts. Age and CHF showed a progressive decrease in functionally competent hCSCs. Chronological age was a major predictor of five biomarkers of hCSC senescence: telomeric shortening, attenuated telomerase activity, telomere dysfunction-induced foci, and p21Cip1 and p16INK4a expression. CHF had similar consequences for hCSCs, suggesting that defects in the balance between cardiomyocyte mass and the pool of nonsenescent hCSCs may condition the evolution of the decompensated myopathy. A correlation was found previously between telomere length in circulating bone marrow cells and cardiovascular diseases, but that analysis was restricted to average telomere length in a cell population, neglecting the fact that telomere attrition does not occur uniformly in all cells. The present study provides the first demonstration that dysfunctional telomeres in hCSCs are biomarkers of aging and heart failure. The biomarkers of cellular senescence identified here can be used to define the birth date of hCSCs and to sort young cells with potential therapeutic efficacy.
The recognition that the human heart possesses a compartment of c-Kit-positive cardiac stem cells (CSCs) that can regenerate myocytes and coronary vessels offers the unique opportunity to reconstitute the damaged myocardium, restoring in part the physiological and anatomical characteristics of the normal heart. Human CSCs (hCSCs) can be isolated from small tissue samples and, after their expansion in vitro, can be delivered back to the same patient in an attempt to repair the failing heart.1–5 This discovery has laid the groundwork for the use of hCSCs in the treatment of the human disease. Preclinical studies have been completed, and two phase I clinical trials are in progress [NCT00474461 (last accessed December 22, 2010) and NCT00893360 (last accessed January 18, 2011) at http://www.clinical trials.gov]. Nonetheless, aging, cardiac hypertrophy, ischemic myocardial injury, and metabolic disorders, together with genetic and environmental factors, may dramatically affect the growth and differentiation behavior of resident hCSCs.6–9 This possibility raises two critical questions. First, is heart failure a stem cell disease characterized by severe depletion of the hCSC pool? Second, does a compartment of functionally competent hCSCs persist in the decompensated heart and do these cells have potential therapeutic implications?
As is the case for other stem cells,10 the life cycle of hCSCs is regulated by telomerase activity and telomere length.2 Telomerase is a reverse transcriptase that extends the 3′ chromosomal ends by using its own RNA as a template.11 Telomerase activity delays but cannot prevent telomere erosion, which is dictated by down-regulation of telomerase, oxidative stress, and loss of telomere-related proteins.12 Shortening of telomeres beyond a critical length triggers cellular senescence, which corresponds to irreversible growth arrest in the G1 phase with loss of specialized functions, including stem cell proliferation, migration, and differentiation. Despite suggestive evidence in humans and in genetically manipulated mice13–15 that telomere shortening is a major variable of cellular senescence and organ aging, it remains to be demonstrated whether hCSCs with critically shortened or dysfunctional telomeres undergo replicative senescence and apoptosis, and whether loss of telomere integrity is one of the variables involved in the decline of stem cell function in the failing human heart.
These issues were addressed in the present study, and the consequences of aging and ventricular decompensation on the clonogenicity, multipotency, and migratory properties of hCSCs were characterized, based on analysis of normal donor hearts and explanted hearts in end-stage heart failure. Moreover, the telomere-telomerase axis and the presence of telomere dysfunction-induced foci (TIFs), in combination with markers of DNA damage response and replicative senescence, were determined, to obtain a common denominator for the processes that regulate the growth and death of hCSCs.
Materials and Methods
Human Samples
Discarded atrial specimens, weighing 3 to 6 g, were collected over a period of 5 years from donor hearts at the time of transplantation and from explanted hearts of patients undergoing cardiac transplantation at the Cardiac Surgery Unit of the University Hospital of Udine in Italy. The clinical data are presented in Supplemental Tables S1 and S2 (available at http://ajp.amjpathol.org). Informed consent was obtained in accordance with the Declaration of Helsinki and with approval by the Independent Ethics Committee of the University of Udine.
Histological analysis of samples from donor hearts did not show pathological changes. Samples were used for the isolation of c-Kit-positive hCSCs.2–4 Fragments were also fixed in formalin and embedded in paraffin2–4 for the identification of stem cell niches and for the quantitative analysis of lineage-negative hCSCs.8,16,17 Lineage-negative hCSCs were defined as cells positive for c-Kit and negative for CD45, to exclude the bone marrow origin and the mast cell phenotype, and negative for GATA4 and α-sarcomeric actin (α-SA), to exclude commitment to cardiac lineages. The localization of OCT3/4 and NANOG was determined in CD45-negative, c-Kit-positive hCSCs. Magnitude of sampling is presented in Supplemental Table S3 (available at http://ajp.amjpathol.org); antibodies are listed in Table 1.
Table 1.
Antibodies for FACS, Immunolabeling, and Western Blotting
| Antigen | Antibody | Source | Fluorochrome | Pseudocolor |
|---|---|---|---|---|
| FACS | ||||
| c-kit | APC-conjugated mouse mAb | BD | APC | |
| c-kit | PE-conjugated mouse mAb | BD | PE | |
| CD34 | FITC-conjugated mouse mAb | BD | FITC | |
| CD44 | FITC-conjugated mouse mAb | BD | FITC | |
| CD45 | FITC-conjugated mouse mAb | BD | FITC | |
| CD45 | Pacific Blue-conjugated mouse mAb | Dako | Pacific Blue | |
| CD49a | PE- conjugated mouse mAb | BD | PE | |
| CD73 | PE-conjugated mouse mAb | BD | PE | |
| CD90 | PE-conjugated mouse mAb | BD | PE | |
| CD133 | PE-conjugated mouse mAb | Miltenyi Biotec | PE | |
| CD146 | PE-conjugated mouse mAb | BD | PE | |
| CD105 | PE-conjugated mouse mAb | Serotec | PE | |
| ABCG2 | PE-conjugated mouse mAb | BD | PE | |
| KDR | PE-conjugated mouse mAb | Serotec | PE | |
| Immunolabeling | ||||
| c-kit | Goat pAb | R&D | A488, A555, | Green, red |
| c-kit | Rabbit pAb | Dako | A488, A555 | Green, red |
| Oct3/4 | Rabbit pAb | Abcam | A488 | Yellow, red |
| Oct3/4 | Goat pAb | Santa Cruz | A555 | Yellow, red |
| Oct3/4 | Mouse mAb | Santa Cruz | Cy5 | Yellow, red |
| Nanog | Mouse mAb | Abcam | A555, Cy5 | Red, white |
| Nanog | Rabbit pAb | Abcam | A488 | Magenta |
| Nkx2.5 | Goat pAb | Santa Cruz | Cy5 | White |
| GATA-6 | Rabbit pAb | Santa Cruz | Cy5 | Yellow |
| Ets1 | Mouse mAb | Santa Cruz | Cy5 | Magenta |
| vWF | Rabbit pAb | Sigma-Aldrich | A488 | Yellow |
| α-SMA | Mouse mAb | Dako | A488 | Magenta |
| α-CA | Mouse mAb | Sigma | TRITC | Red |
| α-SA | Mouse mAb | Sigma-Aldrich | TRITC, Cy5 | Red, white |
| connexin 43 | Rabbit pAb | Santa Cruz | A488 | Green |
| p16INK4A | Mouse mAb | Novocastra | A488 | Green |
| p21 | Mouse mAb | Calbiochem | A555 | Yellow |
| phospho-p53ser15 | Mouse mAb | Cell Signaling | A555 | Red |
| γH2AX | Mouse mAb | Upstate | A488 | Green |
| 53BP1 | Rabbit pAb | Cell Signaling | A555 | Red |
| Western blotting | ||||
| phospho-p53ser15 | Mouse mAb | Cell Signaling | ||
| Ran | Mouse mAb | Sigma-Aldrich |
A488 and A555, Alexa Fluor 488 and 555; APC, allophycocyanin; Cy5, cyanine 5; FITC, fluorescein isothiocyanate; mAb, monoclonal antibody; pAb, polyclonal antibody; PE, phycoerythrin; TRITC, tetramethylrhodamine isothiocyanate; vWf, von Willebrand factor; α-CA, α-cardiac actinin; α-SA, α-sarcomeric actin; α-SMA, α-smooth muscle actin.
Spectral Analysis
Spectral analysis was performed with a TCS-SP2 confocal microscope (Leica, Wetzlar, Germany) using the lambda acquisition mode; myocardial sections were labeled by c-Kit, OCT3/4, and NANOG conjugated with Alexa Fluor 555, Cy5, and Alexa Fluor 555, respectively. Myocytes were stained with α-SA (Cy5) and nuclei with DAPI. The emission signal for Alexa Fluor 555 was excited at 543 nm with an argon laser, and the fluorescence intensity was recorded, generating a lambda stack ranging from 558 to 798 nm at 4.9-nm intervals. The emission signal for Cy5 was excited at 633 nm with a helium/neon laser, and the fluorescence intensity was recorded, generating a lambda stack ranging from 653 to 798 nm at 6-nm intervals. Lens and corresponding numerical aperture were 63× and 1.4, respectively. Sampling consisted of 30 c-Kit-positive, OCT3/4-positive, and NANOG-positive cells. Additionally, 10 cells negative for these markers and present in the same samples were used as control, to discriminate background autofluorescence from specific labeling. Each determination was restricted to the region of interest within each hCSC. For each region of interest, a graph plotting mean pixel intensity and the emission wavelength of the lambda stack was generated. This protocol has repeatedly been used in our laboratory.17–20
Human Cardiac Stem Cells
In all cases, atrial samples were used for the collection of hCSCs; this approach was followed to obtain a direct comparison between control and failing hearts. Two protocols were used for the isolation of hCSCs: enzymatic dissociation of the samples with collagenase and primary explant technique. These protocols were developed in the original documentation of hCSCs2 and were retained here for continuity and comparison. As previously,2 these two methodologies yielded comparable results up to 20 to 25 population doublings; efficiency and viability of hCSCs were superimposable. Collagenase treatment was not found to affect these variables. The phenotype of the small cell population was defined by flow-cytometry and immunolabeling.2,4 Single-cell suspensions were labeled with directly conjugated antibodies (Table 1) specific for hematopoietic and endothelial cell (EC) lineages. Conjugated isotype antibodies were used as negative control. To exclude the presence of erythrocytes, the samples of freshly isolated cells were exposed to BD fluorescence-activated cell sorting (FACS) lysing solution; the flow cytometric analysis was performed with a BD FACScan system (BD Biosciences, San Jose, CA) or a CyAn system (Beckman Coulter, Fullerton, CA). For immunolabeling, hCSCs were fixed in 4% paraformaldehyde for 15 minutes at room temperature and the expression of c-Kit, OCT3/4, NANOG, and SOX2 was evaluated.2,4,17 For clonal analysis, a FACSAria cell sorter (BD-Biosciences) was used to seed single c-Kit-positive hCSCs into 96-well Terasaki plates.2,4
RT-PCR
Total RNA was extracted from nonconfluent cultures of hCSCs at passage 3 using TRIzol reagent (Invitrogen, Carlsbad, CA). After treatment with DNase I (Ambion; Applied Biosystems, Austin, TX), first-strand cDNA synthesis was performed with 1 μg total RNA using random hexanucleotides and M-MLV reverse transcriptase (Invitrogen). PCR amplification was performed in a final volume of 50 μL, using 80 to 150 ng cDNA, 10 mmol/L Tris-HCl pH 9.0, 1.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, 25 pmol of each primer, and 2 U TaqI polymerase (Amersham; GE Healthcare, Little Chalfont, UK). The PCR conditions were as follows: 94°C for 2 minutes; 30 to 45 cycles at 94°C for 60 seconds, 60°C for 60 seconds, and 72°C for 90 seconds. The optimal conditions and the number of cycles were determined to allow amplification of samples within the linear phase of the PCR. The reaction products were analyzed on 1.8% agarose gels.19 Primers are listed in Table 2.
Table 2.
PCR Primers and Product Length
| Gene | Forward primer | Reverse primer | Amplicon size (bp) |
|---|---|---|---|
| KIT⁎ | 5′-CGTGGAAAAGAGAAAACAGTCA-3′ | 5′-CACCGTGATGCCAGCTATTA-3′ | 70 |
| POU5F1† | 5′-GAGGATCACCCTGGGATATAC-3′ | 5′-CGATACTGGTTCGCTTTCTCTTT-3′ | 295 |
| NANOG | 5′-AATACCTCAGCCTCCAGCAGATG-3′ | 5′-CTGCGTCACACCATTGCTATTCT-3′ | 149 |
| SOX2 | 5′-GTTACCTCTTCCTCCCACTCCA-3′ | 5′-CACCCCTCCCATTTCCCTCGTT-3′ | 260 |
| KLF4 | 5′-GGGAGAAGACACTGCGTCA-3′ | 5′-GGAAGCACTGGGGGAAGT-3′ | 88 |
| GAPDH | 5′-CGACCACTTTGTCAAGCTCA-3′ | 5′-AGGGGTCTACATGGCAACTG-3′ | 228 |
| HPRT | 5′-CGTGATTAGTGATGATGAACCAG-3′ | 5′-CGAGCAAGACGTTCAGTCCT-3′ | 129 |
The protein product for the KIT gene is known also as c-kit.
The protein product for POU5F1 is known also as OCT3, OCT4, or OCT3/4.
Expression of c-Kit (KIT) was measured by quantitative real-time PCR, performed using a LightCycler 480 real-time PCR system (Roche Applied Sciences, Indianapolis, IN). Primers were designed from available human sequences using primer analysis software Primer3 version 0.4.0 (available at http://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp?id=UP030000) (Table 2). A dual-color assay with Hydrolysis Probe 2 and HPRT as internal control was used for normalization (Universal Probe Library, Roche). LightCycler 480 Basic software (Roche) used the second-derivative maximum method to identify the crossing point (Cp). Glioma cell line A172 was used as positive control.
Immunofluorescence and Confocal Microscopy
The expression of pluripotency genes and cardiac cytoplasmic proteins was determined by immunolabeling (Table 1). Image acquisition was performed with a Leica TCS-SP2 confocal laser microscope, using a 63× oil immersion objective (numerical aperture, 1.40) or a 40× oil immersion objective (numerical aperture, 1.25).
Migration Assay
Migration analysis was performed using a Cultrex 24-well cell migration assay (Trevigen, Gaithersburg, MD). Approximately 200,000 cells were seeded per well. Growth medium containing 10% fetal bovine serum was used as chemoattractant.
Tumorigenicity of hCSCs
The tumorigenic potential of hCSCs was established by in vitro and in vivo protocols. In vitro, the soft agar assay was used; 1 × 105 hCSCs were seeded in a 0.5% soft agar solution placed on top of a 1% hard agar. The number of colonies grown within the semisolid medium was measured 28 days later. For the in vivo analysis, 1 × 106 hCSCs were injected subcutaneously in NOD/Scid mice (HARLAN Italy S.R.L., San Pietro al Natisone, Italy). SKOV-3 ovarian cancer cells were used as positive control (Sigma-Aldrich, St. Louis, MO). Animals were kept in pathogen-free conditions and tumor growth was evaluated monthly for a period of 6 months, or until SKOV-3 ovarian cancer cells generated a tumor 1 cm in diameter.
Measurements of Ca2+ Oscillations in hCSCs
hCSCs were loaded with 10 μmol/L Fluo-3 AM dye (Invitrogen) and were placed on the stage of a two-photon microscope: a BX51WI Olympus microscope (Olympus, Tokyo, Japan) coupled with a Bio-Rad Radiance 2100MP system (Bio-Rad Laboratories, Hercules, CA). Cells were bathed with Tyrode's solution containing (in mmol/L) NaCl 140, KCl 5.4, MgCl2 1, HEPES 5, glucose 5.5, and CaCl2 2.0 (pH 7.4, adjusted with NaOH). The Fluo-3 was excited at 900 to 960 nm with a Tsunami mode-locked Ti:sapphire femtosecond laser (Spectra-Physics; Newport Corporation, Irvine, CA), and the emission signal was collected at 535 nm. Series of images were acquired at 10-second intervals for a period of 33 minutes. Changes of intracellular Ca2+ in individual hCSCs were determined by measuring the fluorescent signal of Fluo-3. In each cell, the oscillations in fluorescence with time were graphically visualized using ImageJ (NIH, Bethesda, MD) and Microsoft Office Excel 2003 software. These traces were used to assess the number, amplitude, and duration of Ca2+ oscillations in hCSCs. Fluo-3 signals were expressed as normalized fluorescence (F/F0). The duration of Ca2+ oscillations was measured from the onset of the rising phase to the return to baseline.21 Caffeine (10 mmol/L; Sigma-Aldrich) was used to assess ryanodine receptor function.22,23
Telomere Repeat Amplification Protocol Assay
The detection of telomerase activity was performed using a TRAPeze kit (Chemicon–Millipore, Temecula, CA) for the telomere repeat amplification protocol (TRAP) assay, with a radiolabeling detection method. Samples were labeled with 32P. Radiographic films were digitized with a film scanner (SnapScan 1236; Agfa-Gevaert, Mortsel, Belgium) and a G3 Macintosh computer (Apple, Cupertino, CA). Telomerase activity was measured on digital images by ImageJ software version 1.42q (NIH) as units of total product generated (TPG), calculated as TPG = [(x − x0)/c × 100]/[(r − r0)/cR], where x is the signal of the region of the gel lane corresponding to the TRAP product ladder bands from non-heat-treated samples, x0 is the signal of the region of the gel lane corresponding to the TRAP product ladder bands from heat-treated samples, r is the signal of the region of the gel lane corresponding to the TRAP product ladder bands from TSR8 quantitation control, r0 is the signal of the region of the gel lane corresponding to the TRAP product ladder bands from 1× CHAPS lysis buffer-only control, c is the signal from the internal standard (S-IC) in non-heat-treated samples, and cR is the signal from the internal standard (S-IC) in TSR8 quantitation control. Each unit of TPG corresponds to the number of TS primers in a quantity of 1 × 10−3 amol or 600 molecules extended with at least four telomeric repeats by telomerase enzyme present in the extract.
Telomere Length in hCSCs
Telomere length was measured by flow FISH (a combination of flow cytometry and fluorescence in situ hybridization).24 Suspensions consisting of a 1:1 mixture of hCSCs and control cells were hybridized in the presence or absence of fluorescein-conjugated peptide nucleic acid (PNA) telomere probe (DakoCytomation, Glostrup, Denmark). The average telomeric fluorescence per genome in the two cell classes was calculated as follows: relative telomere length RTL = [(mean FL1 sample cells with probe − mean FL1 sample cells without probe) × DNA index of control cells × 100]/[(mean FL1 control cells with probe − mean FL1 control cells without probe) × DNA index of sample cells]
Telomere Dysfunction-Induced Foci
Telomere dysfunction-induced foci, which were defined by the colocalization of 53BP1 with telomeres, were analyzed using a Leica DMI 6000B microscope connected to a Leica DFC350FX camera. hCSCs were considered to be TIF-positive when at least 50% of 53BP1 spots colocalized with telomere hybridization signals. The number of TIFs per hCSC was also measured. Sampling consisted of more than 100 hCSCs in each case (see Supplemental Table S3 at http://ajp.amjpathol.org).
Microarray Analysis
RNA Extraction, cDNA Labeling, and Microarray Hybridization
hCSCs obtained from age- and sex-matched donor and explanted hearts (n = 3 each) were used for the analysis of the transcriptional profile. Total RNA was extracted with TRIzol reagent (Invitrogen) and subjected to DNase I treatment. After RNA quality control, microarray cDNA targets were prepared according to the two-step indirect fluorescent labeling procedure,25 starting from 10 μg total RNA. First-strand cDNA synthesis reaction was performed with unmodified random primers in the presence of aminoallyl-dUTP. The resulting primary amino groups were then coupled to the N-hydroxysuccinimide ester of the fluorescent dye. For each age- and sex- matched pair, one sample was labeled with Cy5 and the other with Cy3.
LNCIB 28K cDNA microarray slides (supporting 28,000 cDNA clones, hereafter called features) were used in the large-scale transcript expression profiling studies; these were prepared by spotting onto Amersham Type 7 Star reflective microarray slides the purified PCR products of 19,741 LNCIB human cDNA clones26 and 8160 cDNA clones, selected from the human sequence-verified I.M.A.G.E. clone collection (Research Genetics/Invitrogen).27 This clone set consisted of approximately 13,620 unique human genes. Purified cDNA inserts were resuspended in 50% dimethyl sulfoxide (Sigma-Aldrich) and printed on the slides using a SDDC-2 ChipWriter Pro microarrayer (Virtek Vision International, Waterloo, ON, Canada). GAPDH cDNA and β-actin cDNA were used as positive array controls, and the corresponding purified PCR products were printed at different concentrations on the slides. Plant cDNAs and printing buffer, 50% dimethyl sulfoxide, were used as negative controls.
Competitive hybridizations were performed under coverslips at 63°C, overnight, in the presence of 3× standard saline citrate, 0.2% SDS, 100 ng/μL human Cot-1 DNA, and 330 ng/μL yeast tRNA, using a semiautomated hybridization station (Advalytix ArrayBooster; Beckman Coulter Biomedical, Munich, Germany). After a washing, slides were scanned at a resolution of 10 μm per pixel using an Axon GenePix 4000B microarray scanner (Axon Instruments–Molecular Devices, Sunnyvale, CA). Images for the Cy3 and Cy5 channel were acquired in a 16-bit tagged-image file format (TIFF) and were analyzed using GenePix 4.0 analysis software (Axon Instruments). Images were manually flagged to identify artifacts and bad spots.
Low-level analysis of microarray data were performed in the R (version 2.8)/Bioconductor (version 2.3) environment using the limma microarray package (available at http://www.R-project.org, last accessed January 2009).28 Expression data were loaded from the GenePix image analysis output files. Features displaying, in at least one channel, median fluorescence intensity lower than background plus two standard deviations were flagged as bad spots. Fluorescent intensity of the red and green channels was background-subtracted,29 and the corresponding expression ratio (Cy5/Cy3) was log-transformed (base 2). Within each array, normalization was accomplished by the print-tip LOWESS method, independently from each cDNA collection. Normalized ratios from replicate expression measurements within the same array were averaged to produce a final expression matrix (FEM) to be used in the subsequent analyses. The FEM was composed of 21,069 features and three age- and sex-matched paired samples.
Unsupervised hierarchical cluster analysis (average-linkage clustering)30 was applied to the filtered expression matrix. Flagged features, features not present in all samples, and features with fold changes equal to one in at least one sample were filtered out. By this approach, 7868 features were identified. For both genes and samples, uncorrelated distance was used as dissimilarity measure. Cluster results were visualized using Java TreeView.31
Differential Gene Expression
The differential gene expression between hCSCs obtained from donor and explanted hearts was established by functions and methods implemented in the R/Bioconductor environment. A fixed-effects linear model was fitted for each individual feature to estimate expression differences. An empirical Bayes approach was applied to moderate standard errors of M values.32 For each analyzed feature, moderated t-statistics, log-odds ratios of differential expression (B-statistics), and raw and adjusted P values (false discovery rate control by the Benjamini and Hochberg method33) were obtained.
Functional Annotation Analysis
Differentially expressed gene lists were subjected to functional analysis using the DAVID/ease Web-based tool version 2008 (NIH-NIAID, Bethesda, MD); LNCIB 28K unique associated genes were used as a background list. To highlight biological processes relevant to the investigated sample groups, an Ingenuity Pathways analysis was performed, using IPA software version 7 (Ingenuity Systems, Redwood City, CA). For core analysis, only genes with absolute expression value >1.7-fold change (donor versus explanted heart-derived hCSCs) were considered. The resulting annotations (networks, biofunctions, canonical pathways) were ranked, depending on the number of significantly expressed genes, and were ordered by the score representing the probability of finding the observed molecules enriched in a given annotation by chance: score = −log(P value). The graphical representation of the biofunctions was obtained by use of the IPA Knowledge Base according to full-text articles and information databases available online (http://www.ingenuity.com/products/pathways_analysis.html, last accessed June 2009). The spotted genes were determined to be up-regulated or down-regulated from the microarray low-level analysis.
Statistical Analysis
Data are reported as means ± SD. Differences were determined by two-tailed unpaired Student's t-test. Linear regressions were calculated by the least squares method. Comparisons between slopes were made by the analysis of covariance.34 Microarray analysis was performed as described above.
Results
Human Myocardium
Over a period of 5 years, atrial tissue was obtained from 63 donor hearts at the time of transplantation and from 121 explanted failing hearts. Atrial samples were collected in anatomical areas lacking apparent macroscopic alterations. By enzymatic digestion and primary explant technique,2,4 a pool of small cardiac cells was obtained in 166 of 184 samples tested, which corresponds to a 90% rate of success. Cell isolation was not feasible in samples processed 48 hours after collection. In successful isolations, the cells were expanded and stored at passage 2 to passage 3. For the present study, the properties of hCSCs were analyzed in a subset of 41 consecutive samples acquired from 18 donor and 23 explanted hearts (see Supplemental Table S1 at http://ajp.amjpathol.org). These hearts were collected during the period from 2007 to 2009 at the University Hospital of Udine. In these cases, cell isolation was performed within 48 hours. Cell isolation and expansion was similar, and no difficulty was encountered at any stage of the culture protocol.
Men were more numerous than women in the study samples, and ischemic cardiomyopathy predominated. These variables may have introduced a partially biased sampling; however, this reflects the sex differences and etiology of the disease in patients undergoing transplantation. Donor subjects were younger than patients undergoing transplantation, who were diagnosed with ischemic, hypertrophic, and idiopathic dilated cardiomyopathy or valvular defects and acromegaly. All patients undergoing transplantation were class IV in the New York Heart Association functional classification (NYHA IV). The duration of the underlying disease and the time from the onset of heart failure to transplantation varied from 1 to 30 years and from 1 to 12 years, respectively (see Supplemental Table S1 at http://ajp.amjpathol.org). With the exception of one patient affected by hypertrophic cardiomyopathy, the echocardiographic and hemodynamic parameters were similar across subjects, documenting severe ventricular dysfunction (see Supplemental Table S2 at http://ajp.amjpathol.org).
Human Cardiac Stem Cells
Structures with the characteristics of stem cell niches were identified in tissue sections of human atrial myocardium (Figure 1A), in which stem cells are preferentially located.16 Importantly, c-Kit-positive hCSCs expressed OCT3/4, NANOG, KLF4, and SOX2, which are implicated in the self-renewing and pluripotent state of stem cells.35 These properties were identified in c-Kit-positive hCSCs distributed in atria from both donor and explanted hearts (Figures 1, B–D, and 2E), strengthening support for the notion that hCSCs retain a highly immature embryonic-like cell phenotype.
Figure 1.
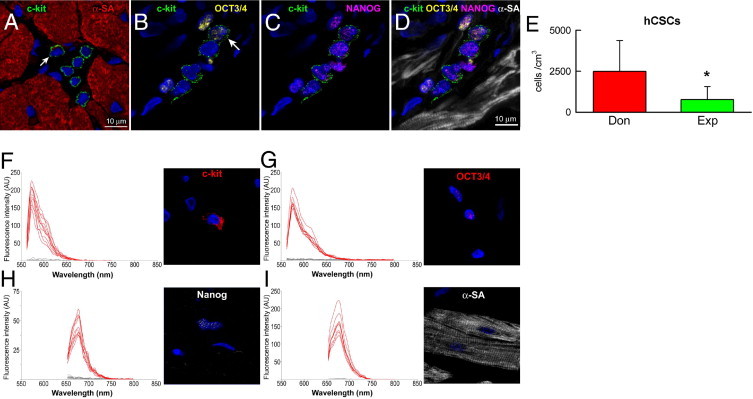
Human cardiac stem cell (hCSC) niches. A: Atrial myocardium containing a cluster of six c-Kit-positive cells (green). Five are negative for α-sarcomeric actin (α-SA; red), and one has a minimal amount of this contractile protein (red spot indicated by arrow), a myocyte precursor. B–D: Four c-Kit-positive cells are present in this section of atrial myocardium. One cell is positive for OCT3/4 (yellow, indicated by arrow) (B); all four c-Kit-positive cells express NANOG (magenta). Myocytes are labeled by α-SA (white). E: Number of lineage-negative (Linneg), c-Kit-positive (c-Kitpos) hCSCs in the atria of donor (Don) and explanted (Exp) hearts. Data are reported as means ± SD. *P < 0.05 versus Don. F–I: Validation of immunolabeling by spectral analysis of c-Kit-positive cells (F), OCT3/4-positive nuclei (G), NANOG-positive nuclei (H), and α-SA-positive myocytes (I); corresponding images show examples of cells used in this assay.
Figure 2.
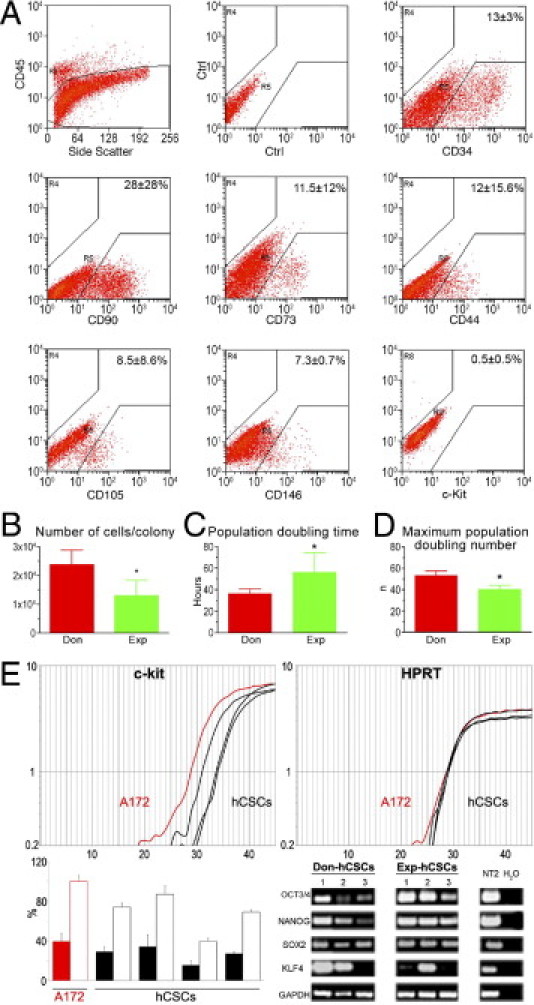
Properties of hCSCs. A: Surface phenotype of small cardiac cells isolated from donor and explanted hearts. B–D: Growth properties of hCSCs. Data are reported as means ± SD. *P < 0.05 versus Don. E: Representative amplification curves of c-Kit and HPRT (housekeeping gene) expression in cell line A172 (red), which was used as positive control and in hCSC samples (black). In the histogram, solid bars indicate the relative expression of c-Kit in A172 cells (red) and in hCSCs (black), and open bars indicate the expression of c-Kit in hCSCs normalized versus A172 cells. Transcripts for OCT3/4, NANOG, SOX2, and KLF4 are present in hCSCs obtained from donor (Don-hCSCs) and explanted (Exp-hCSCs) hearts. The neuronally committed human teratocarcinoma cell line NT2 was used as positive control. PCR products were run on 1.8% agarose gel.
hCSCs were negative for CD45, as well as for the transcription factor GATA4 and the myocyte cytoplasmic protein α-SA. Quantitatively, the number of c-Kit-positive, lineage-negative hCSCs was 3.2-fold higher in atria of donor hearts than of explanted hearts (Figure 1E). This difference was at variance with previous findings in the ventricular myocardium of control and failing human hearts, in which cardiac decompensation was coupled with an increase in hCSC number.8 Thus, regional differences in hCSC number appear to be present in the failing human heart.
In the atria of donor hearts, there was 1 hCSC per 2000 myocytes, a frequency that is approximately fivefold higher than in the normal human left ventricle.8 In severe failure, the number of hCSCs in the atria was approximately threefold lower than in the left ventricle.8 The former difference may reflect the site of storage of hCSCs in the atria,16 with the latter difference reflecting growth activation of ventricular hCSCs in proximity of areas of injury. Scarring and acute damage are distributed exclusively to the failing ventricle.8
To validate the specificity of the recorded signals, spectral analysis was performed.17,20 The emission spectra for c-Kit, OCT3/4, NANOG, and α-SA were clearly distinct from the emission spectra for tissue autofluorescence, confirming the accuracy of the immunolabeling protocol (Figure 1, F–I). Collectively, these findings support the notion that human atria contain a pool of primitive hCSCs expressing surface and nuclear markers of stemness.
Properties of Human Cardiac Stem Cells
hCSCs were analyzed in 41 consecutive specimens acquired from 18 donor and 23 explanted hearts. The analysis by flow cytometry documented that the freshly isolated unselected cardiac cell population was heterogeneous and included cells that expressed the bone marrow and endothelial epitopes CD45, CD34, CD90, CD73, CD44, CD105, and CD146. The CD45-negative fraction contained 0.5% c-Kit-positive cells (Figure 2A), which were negative for hematopoietic and EC markers.
Over a period of 1 to 2 weeks, clusters of adherent spindle-shaped cells were found. Cell aggregates derived from control hearts (2.4 × 104 cells) were larger than those generated from explanted hearts (1.3 × 104 cells) (Figure 2B). However, the frequency of cell clusters was similar, whether cells came from control or explanted hearts (see Supplemental Figure S1 at http://ajp.amjpathol.org). Cell clusters consisted of a uniform population of hCSCs that expressed c-Kit, OCT3/4, and NANOG (see Supplemental Figure S2 at http://ajp.amjpathol.org). The fraction of cells positive for c-Kit, OCT3/4, and NANOG did not vary with time in culture.
Subsequently, clusters were disaggregated and the growth properties of c-Kit-positive hCSCs were determined at passage 3. Population doubling time was 1.6-fold shorter in c-Kit-positive cells from control than from explanted hearts (Figure 2C). Similarly, the maximum number of population doublings reached by hCSCs, before the onset of senescence and irreversible growth arrest, was 1.3-fold higher in hCSCs from donor hearts (Figure 2D). In comparison with hCSCs from explanted hearts, these variables led in 30 days to a 117-fold greater expansion of hCSCs collected from donor myocardium. Despite the lower growth kinetics of hCSCs from explanted hearts, it was still possible to expand this pool and to obtain a large quantity of primitive cells. Thus, heart failure affected the growth reserve of resident hCSCs only in part.
Under FACS analysis, both classes of hCSCs displayed a comparable surface phenotype. Markers of the hematopoietic/EC lineage, including CD133, CD34, CD45, ABCG2, and KDR, were not detected (see Supplemental Figure S3 at http://ajp.amjpathol.org). CD49a/α1 integrin was 4.4-fold more represented in hCSCs from failing hearts. The α1 integrin isoform increases with cardiac aging and disease.36 The expression of stem cell-related genes was detected at the protein level (see Supplemental Figure S3 at http://ajp.amjpathol.org) and at the mRNA level. Transcripts for c-Kit, OCT3/4, NANOG, SOX2, and KLF-4 were detected in both categories of hCSCs (Figure 2E). The expression of pluripotency-associated genes in hCSCs was consistent with previous results obtained in small and large animals and in humans.4
Clonogenicity and Multipotency
The properties of mother cells can be retrospectively defined by the number of daughter cells generated with clonogenic growth. Founder cells that form large multicellular clones correspond to true hCSCs, whereas cells that give rise to colonies of small size represent early committed cells, transit amplifying cells (TACs), or senescent cells at the end of their proliferative life span. FACS-sorted individual hCSCs were deposited in single wells of Terasaki plates. Wells with more than one cell were excluded. Of 3682 seeded wells, hCSCs derived from explanted hearts gave rise to 237 colonies of approximately 100 to 200 cells. The majority of these small cell clusters underwent growth arrest, however, and only 39 actively proliferating clones (Figure 3A) were passaged to 24-well-plates, where they formed large clusters of 40,000 to 60,000 cells. Nearly 30% of these colonies stopped growing; the remaining 70% were transferred to 6-well-plates and continued to replicate, undergoing an additional 18 population doublings without reaching replicative senescence. Thus, the actual cloning efficiency of hCSCs from failing hearts was 0.8% (Figure 3B). A comparable approach and magnitude of sampling was used with hCSCs from normal hearts. A cloning efficiency nearly threefold higher was found (Figure 3B). These clones expanded exponentially for more than 20 population doublings. Clonal cells maintained a stable phenotype and continued to express c-Kit and NANOG.
Figure 3.
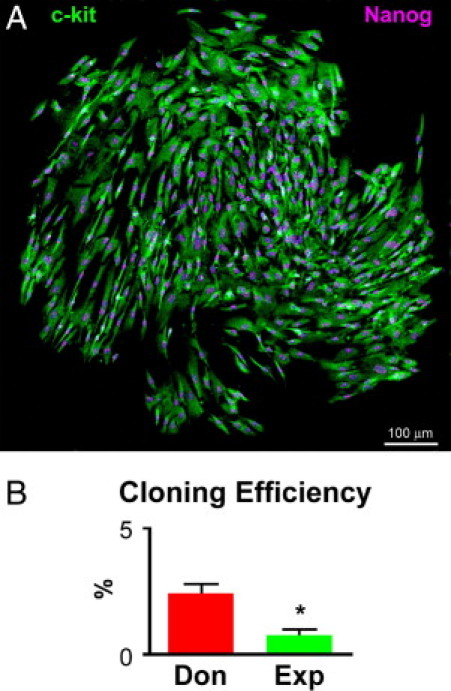
Clonal growth of hCSCs. A: Single cell-derived clone composed of c-Kit-positive hCSCs (green) expressing NANOG in their nuclei (magenta). B: Cloning efficiency of hCSCs from donor (Don) and explanted (Exp) hearts, reported as means ± SD. *P < 0.05 versus Don.
To determine whether hCSCs from control and explanted hearts had a comparable ability to differentiate into cardiac cell lineages, cells were exposed to differentiating medium. With both classes of hCSCs, cells expressing transcription factors and cytoplasmic proteins specific of smooth muscle cells (SMCs), ECs, and cardiomyocytes were identified by immunolabeling. Myofibrillar structures with sarcomere striations were observed together with the expression of connexin 43 at the site of myocyte coupling (Figure 4, A–H). Differentiating hCSCs derived from donor and explanted hearts formed similar proportions of myocytes, SMCs, and ECs (Figure 4I). These maturing cells may reflect TACs in vitro.
Figure 4.
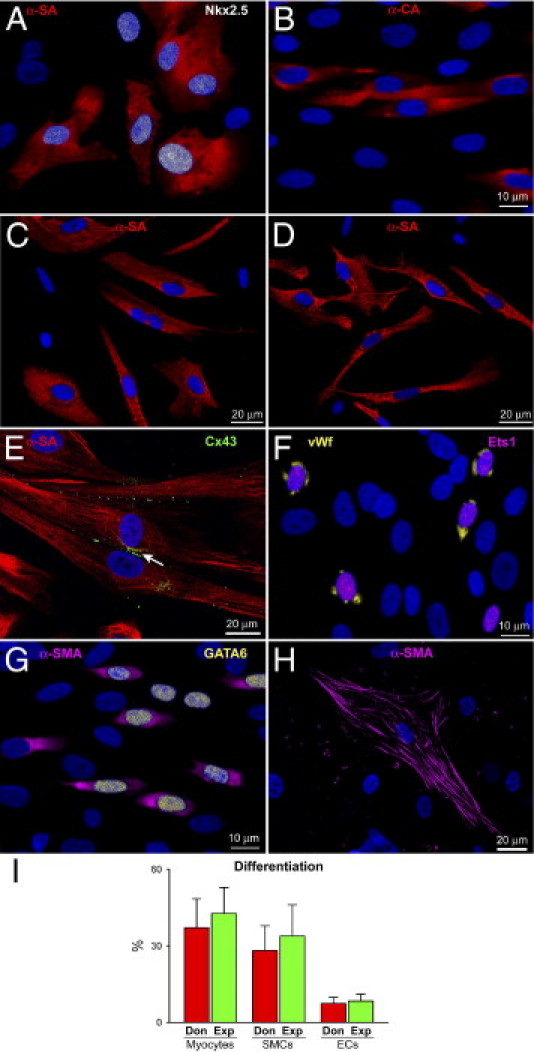
hCSC differentiation. A–H: Differentiating hCSCs acquire the myocyte lineage and express Nkx2.5 (white) (A), α-SA (red) (A, C, and D), α-cardiac actinin (red) (B), and connexin 43 (green, marked by arrow) (E). They also express the EC markers Ets1 (magenta), and von Willebrand factor (vWf, yellow) (F) and the SMC proteins GATA6 (yellow) (G) and α-smooth muscle actin (α-SMA) (magenta) (G and H). I: Differentiation of hCSCs from donor (Don) and explanted (Exp) hearts, reported as means ± SD. Scale bars: 10 μm (A, B, F, G); 20 μm (C, D, E, H).
hCSCs and myocytes are equipped with distinct Ca2+ handling machinery.21,23 These properties were used to establish whether hCSCs acquired the physiological myocyte phenotype. Intracellular Ca2+ was monitored in primitive and differentiating hCSCs and the effect of caffeine, an agonist of ryanodine receptors, was tested. In primitive hCSCs, caffeine had no effect on Ca2+ oscillations, a response consistent with the lack of ryanodine receptor function in these cells.22 In contrast, the presence of caffeine in differentiating hCSCs led to an increase in the number of active cells, and in the amplitude and duration of Ca2+ oscillatory events, indicating their commitment to the myocyte lineage (Figure 5, A and B). Importantly, there were no differences in these parameters between cells collected from control and failing hearts after differentiation with dexamethasone (Figure 5C). However, hCSCs from explanted hearts showed a lower migratory capacity, compared with hCSCs from donor hearts (Figure 5D).
Figure 5.
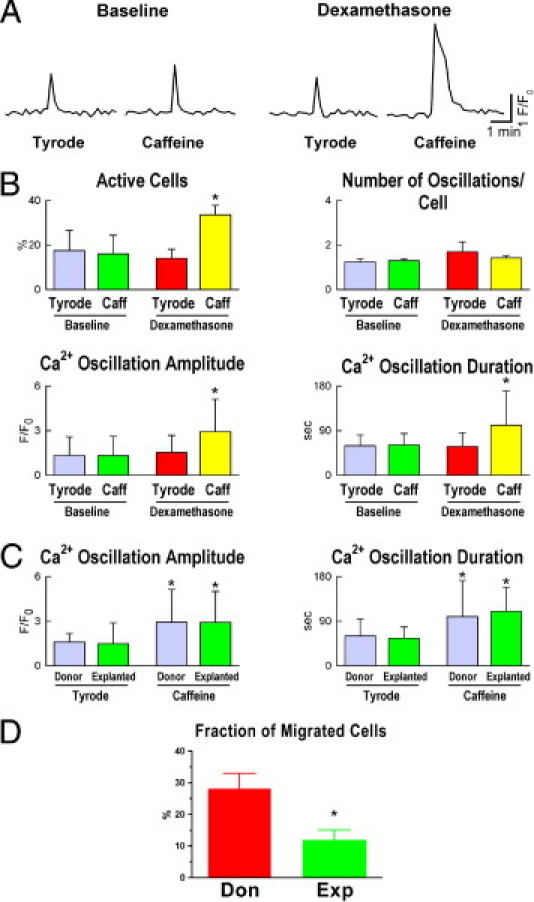
Properties of differentiating myocytes . A: Ca2+ oscillations in hCSCs at baseline (undifferentiated) and after exposure to dexamethasone (differentiating) in the absence (Tyrode) and presence of caffeine (Caffeine). B: Fraction of cells displaying Ca2+ oscillation (Active Cells) and frequency of oscillatory events over a period of 33 minutes, with amplitude and duration of the oscillatory episodes. Data are reported as means ± SD. *P < 0.05 versus Tyrode. C: Ca2+ events in differentiating myocytes from donor and explanted hearts. *P < 0.05 versus Tyrode. D: Fraction of migrated hCSCs from donor (Don) and explanted (Exp) hearts. Data are reported as means ± SD. *P < 0.05 versus Don.
The presence of nuclear proteins typical of highly primitive cells raised the question of whether hCSCs could give rise to cancer cells and/or malignant neoplasms.35 To evaluate anchorage-independent growth, hCSCs were seeded in soft agar. Under this condition, hCSCs did not divide and did not form spheroids. Similarly, the subcutaneous injection of hCSCs in NOD/Scid mice did not result in tumor formation over a 6-month period (data not shown). Collectively, these observations indicate that clonogenic, multipotent hCSCs can be isolated from the atrial myocardium of human hearts in end-stage failure. Although these cells have lost to a certain degree their replicative potential, a large progeny can nonetheless be formed. Importantly, these hCSCs do not have tumorigenic potential.
Telomerase Activity, Telomere Length, and hCSC Senescence
Stem cells possess elevated levels of telomerase protein and activity, and down-regulation of telomerase expression and function is consistent with stem cell commitment and terminal differentiation.37 By TRAP assay, hCSCs from donor hearts displayed a fivefold higher level of telomerase activity than hCSCs from explanted hearts (Figure 6A). The reduced activity of this ribonucleoprotein in the latter class of cells was consistent with a 25% shorter telomere length (Figure 6B).
Figure 6.
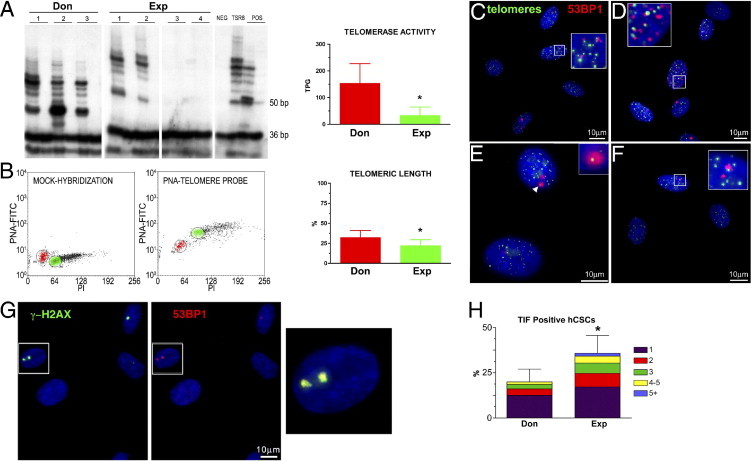
Telomere-telomerase axis in hCSCs from donor (Don) and explanted (Exp) hearts. A: In hCSC classes, products of telomerase activity display a 6-bp periodicity. Heat-inactivated lysates were used as negative control (lane 2 for each sample). TSR8 indicates telomerase control template; the 1301 cell line was used as a positive control (POS), and the primer-dimer lane as negative control (NEG). B: Dot plots of hCSCs hybridized without (mock-hybridization) and with the peptide nucleic acid (PNA) telomere probe. Gates were set around cells in the G0/G1 phase for both hCSCs (red) and control cells, tetraploid 1301 cell line (green). PI, propidium iodide. Relative telomeric length was computed as the ratio of the telomere signal of hCSCs and control cells. C–F: Four examples of telomere dysfunction-induced foci (TIFs) in which 53PB1 (red) colocalizes with telomere hybridization spots (green). Detail of interest (arrowhead, inset) is shown at higher magnification in the corresponding inset. G: γ-H2AX (green) and 53BP1 (red) colocalize at sites of DNA damage. The area included in the rectangles is shown at higher magnification on the right (yellow merged signals). In histograms, data are reported as means ± SD. H: Fraction of hCSCs with 1 to 5 TIFs. *P < 0.05 versus Don. Scale bars: 10 μm.
Although telomerase activity and average telomere length are valid indicators of the age of a cell compartment, the fraction of cells with critically shortened telomeres is the major determinant of the growth reserve of a cell population. Telomere erosion beyond a critical value38 and/or loss of telomere integrity elicits a DNA damage response that enables cells to block cell cycle progression and initiate DNA repair. Alternatively, cells undergo senescence, irreversible growth arrest, and apoptosis.39,40 The DNA damage response involves interaction of the adaptor protein p53 binding protein 1 (53BP1) and the chromatin modifier phosphorylated histone H2AX (γ-H2AX).41 The localization of these proteins within telomeric sequences reflects TIFs. The TIFs activate the ataxia telangiectasia mutated (ATM) kinase, which phosphorylates p53 at serine 15.42
By quantitative fluorescence in situ hybridization and immunolabeling, the colocalization of telomere hybridization spots and 53BP1 was established. Relative to hCSCs from donor hearts, a 75% larger fraction of hCSCs from explanted hearts showed TIFs in their telomeres. Also, the number of TIFs per cell was higher in these hCSCs (Figure 6, C–F and H). 53BP1 was detected in proximity of γ-H2AX, further documenting the presence of TIFs in hCSCs from explanted hearts (Figure 6G). Consistent with these observations, the quantity of phospho-p53Ser15 and the expression of the p53 target gene p21Cip1 were nearly twofold higher in this hCSC category (Figure 7, A–C). Importantly, a twofold larger fraction of hCSCs from explanted hearts was positive for the senescence-associated protein p16INK4a (Figure 7D); p16INK4a rarely colocalizes with DNA double-strand breaks and represents a delayed response,43 which follows the induction of p53 and p21Cip1. Thus, prolonged pathology promotes dysfunctional telomeres and senescence of hCSCs, a phenomenon that is less apparent in adult donor hearts.
Figure 7.
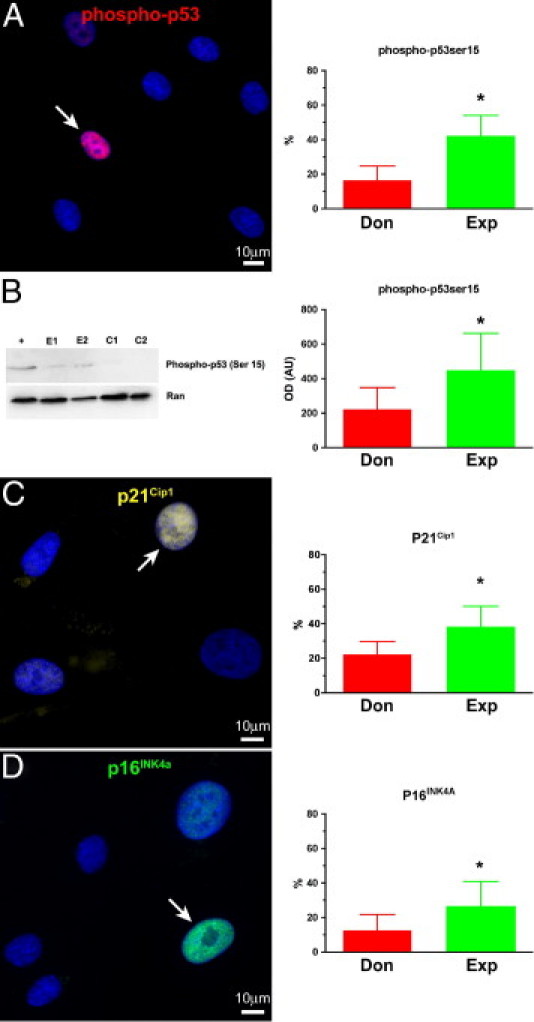
Senescence-associated proteins in hCSCs from donor (Don) and explanted (Exp) hearts. A and B: Phosphorylation of p53 at serine 15 by immunolabeling (red) (A) and Western blotting (B); Ran indicates loading conditions. C and D: Expression of p21Cip1 (yellow) (C) and p16INK4a (green) (D) in nuclei of hCSCs. Arrows represent positive nuclei. In histograms, data are reported as means ± SD. *P < 0.05 versus Don. Scale bars: 10 μm.
Age, Disease, and Cellular Senescence
A relevant question concerned whether chronological age, and the presence and duration of cardiac disease, were coupled with the expression of biomarkers of hCSC senescence and, thereby, with growth arrest and impaired therapeutic import. Based on the assumption that telomere length, telomerase activity, TIFs, and p16INK4a and p21Cip1 are biomarkers of stem cell function, their interrelation in hCSCs was established. Telomere length was directly related to telomerase activity and inversely correlated with TIFs, p16INK4a, and p21Cip1 in hCSCs from both control and diseased hearts, analyzed separately and in combination (Figure 8). Additionally, the level of catalytic activity of telomerase decreased with increased number of hCSCs showing TIFs, p16INK4a, and p21Cip1 (Figure 9). Positive relationships were also found among TIFs, p16INK4a, and p21Cip1 (Figure 10), indicating that these parameters of stem cell behavior were not independent; each of them could be used to predict the growth reserve of hCSCs.
Figure 8.
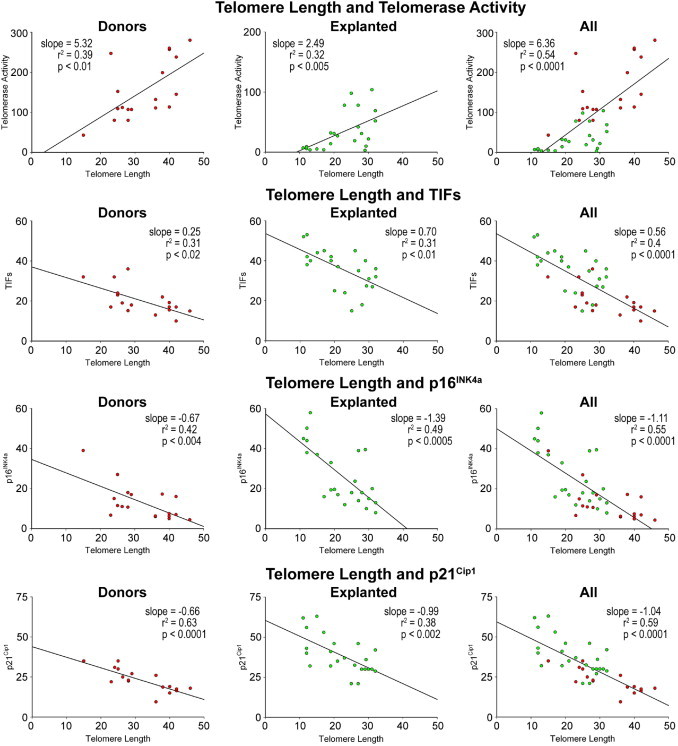
Biomarkers of senescence: telomere length in hCSCs from explanted and donor hearts. Linear regression of various parameters of hCSC function impairment.
Figure 9.
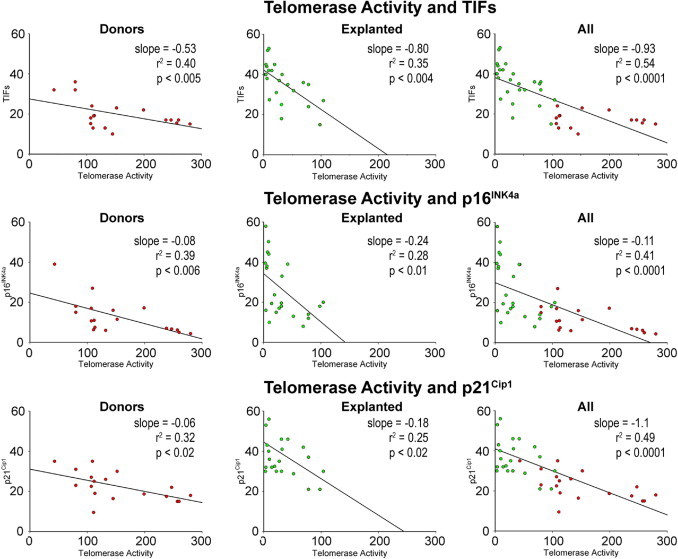
Biomarkers of senescence: telomerase activity in hCSCs from explanted and donor hearts. Linear regression of various parameters of hCSC function impairment.
Figure 10.
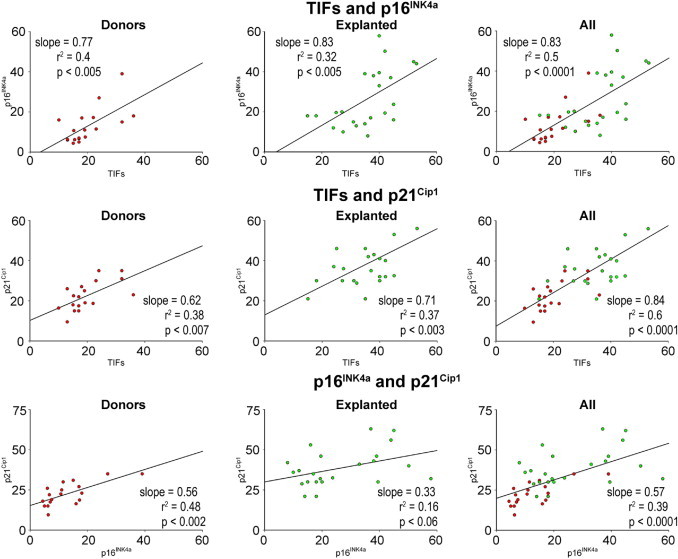
Biomarkers of hCSC senescence: telomere dysfunction-induced foci (TIFs) in hCSCs from explanted and donor hearts. Linear regression of various parameters of hCSC function impairment.
Age was identified as a major predictor of telomere shortening, attenuation in telomerase activity, and increased incidence of TIFs, p16INK4a, and p21Cip1 in hCSCs. Cardiac disease influenced these cellular parameters even further (Figure 11). However, there was no correlation between these indices of hCSC senescence and the duration of the cardiac diseases from clinical diagnosis to transplantation, or from the onset of heart failure to transplantation (see Supplemental Figure S4 at http://ajp.amjpathol.org).
Figure 11.
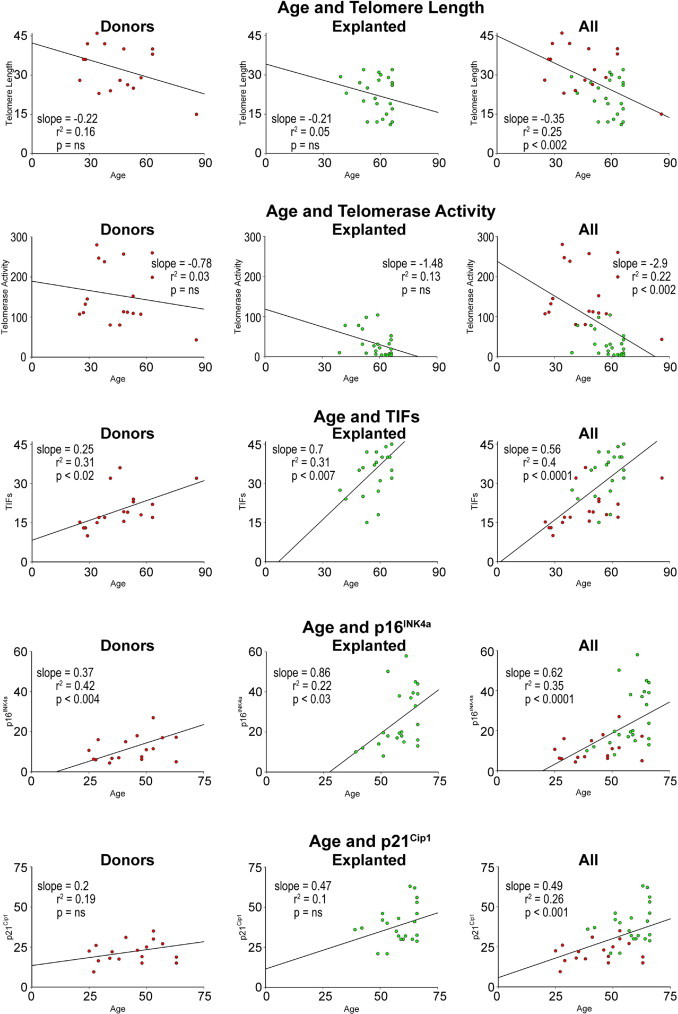
Biomarkers of hCSC senescence as a function of age in hCSCs from explanted and donor hearts. Linear regression of various parameters of hCSC function impairment.
To establish whether cardiac disease negatively affected hCSC function, hCSCs from subjects of comparable age were studied. In comparison with hCSCs from donor hearts, hCSCs from age-matched explanted hearts had shorter telomere length, lower telomerase activity, higher frequency of TIFs, and enhanced expression of p16INK4a and p21Cip1 (Figure 12A). These results suggest that both aging and pathological insults trigger DNA lesions at the level of the telomeric repeats, with initiation of the DNA repair response. Activation of telomerase was apparent in hCSCs from control hearts, and this molecular mechanism may in part preserve telomere integrity.
Figure 12.
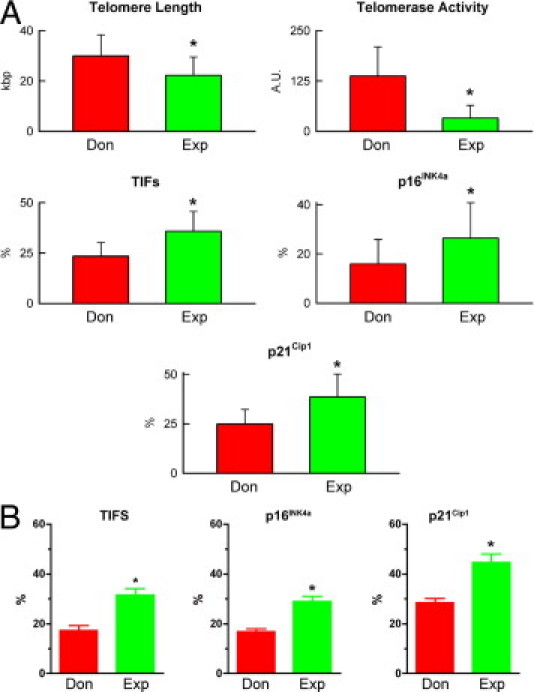
Senescence of hCSCs from donor (Don) and explanted (Exp) hearts. A: Biomarkers of cellular senescence in hCSCs obtained from age-matched donor and explanted hearts are shown quantitatively. Data are reported as means ± SD. *P < 0.05 versus Don. B: Fraction of clonogenic hCSCs with TIFs, or expressing p16INK4a and p21Cip1.
To characterize further the role of TIFs, p16INK4a, and p21Cip1 in hCSC growth, the expression of these markers was determined in clonal cells (Figure 3). The fraction of cells positive for TIF, p16INK4a, and p21Cip1 was lower in clonogenic hCSCs obtained from donor than from explanted hearts (Figure 12B). However, these differences were relatively small, indicating that clonogenic hCSCs from failing hearts contained a pool of young cells with significant growth reserve, as inferred from their telomere integrity.
Gene Expression Profile of hCSCs
The transcriptional profile of hCSCs from donor and explanted hearts was determined by microarray analysis. Age- and sex-matched hCSCs were used. Unsupervised hierarchical cluster analysis was applied to the expression matrix; 7868 genes were initially considered, on the basis of their presence in age- and sex-matched hCSC pairs and differential expression in at least one pair. By applying more stringent criteria and including only genes that showed a significant (P < 0.05) expression difference of at least 1.7-fold between hCSCs from donors and explanted hearts, 398 genes were found to be up-regulated and 452 genes were found to be down-regulated in hCSCs from failing hearts (see Supplemental Figure S5 at http://ajp.amjpathol.org).
These results were then subjected to functional annotation using gene ontology (DAVID/ease) for the identification of molecular pathways showing different state of activation or repression in the two hCSC classes (see Supplemental Table S4 at http://ajp.amjpathol.org). Transcripts of genes involved in wound healing and response to stress were up-regulated in hCSCs from explanted hearts. Similarly, a group of chemokines and inflammatory factors were more represented in these stem cells, possibly reflecting their proficiency to engraftment in the microenvironment of the failing heart. The expression of a subset of genes implicated in lipid metabolism was decreased in hCSCs from failing hearts, suggesting a metabolic shift from fatty acid to carbohydrate metabolism, together with a reduction in mitochondrial-encoded gene expression and protein synthesis. An attenuated response of genes involved in the adaptation to oxidative stress was also observed.
Highly differentially expressed genes in hCSCs from donor and explanted hearts were then analyzed using Ingenuity Pathway Analysis (IPA) software. This independent approach highlighted the presence of two functional gene cores differentially represented in the two classes of hCSCs (see Supplemental Figure S6 at http://ajp.amjpathol.org). The first core included the components of the molecular systems that regulate cell growth, proliferation, and motility, lipid and carbohydrate metabolism, and cell-to-cell signaling. The second core, involving genes implicated in cardiac development, homeostasis, and repair, was less abundant in hCSCs from failing hearts, suggesting that the efficiency of cell turnover and regeneration varied between normal and diseased hearts. Molecular targets of therapeutic agents used in the treatment of cardiac diseases were up-regulated in hCSCs from explanted hearts. As noted under Materials and Methods, the transcriptional profile was studied in hCSCs obtained from three donor and three explanted hearts, matched for age and sex, using a linear model-based analysis for differentially expressed genes. The output provided statistically significant results, and the global profile of the differential genes was nearly identical in the three biological replicates. Importantly, the three patients received the same pharmacological treatment consisting of β-blockers, diuretics, and angiotensin-converting enzyme inhibitors. Thus, a distinct global expression profile characterized the two cell classes.
Discussion
The results of the present study indicate that chronological age is a major determinant of the loss in growth reserve of the adult heart, dictated by a progressive decrease in the number of functionally competent hCSCs. Five biomarkers of hCSC senescence have been identified: telomeric shortening, attenuated telomerase activity, increased number of TIFs, and enhanced expression of p16INK4a and p21Cip1. Severe heart failure has similar effects on the hCSC compartment, suggesting that alterations in the balance between the cardiomyocyte mass and the pool of nonsenescent hCSCs may condition the evolution of the decompensated myopathy. Importantly, these biomarkers can be used to define the birth date of the hCSC population and to sort the fraction of young cells that retain potential therapeutic efficacy.
Work from our laboratory2,4 and from others1,3,5,44 has documented that the human heart is a constantly renewing organ in which the ability to replace old dying cells depends on the persistence of a stem cell compartment. In addition to stem cells, the adult myocardium contains a second population of replicating cells, the transit amplifying cells (TACs). These TACs correspond to the stem cell progeny that is destined to reach terminal differentiation. Before cell cycle withdrawal, TACs undergo a limited number of divisions45 that expand the pool of differentiated daughter cells resulting from activation and lineage commitment of each stem cell. This mechanism allows hCSCs to divide infrequently, maintaining their compartment intact while ensuring the formation of an adequate progeny.
In the young healthy heart, this pattern of stem cell growth efficiently preserves organ homeostasis. In the old and diseased heart, however, the structural and functional decline may be coupled with defects in the hierarchical growth of the organ, suggesting that quantitative and qualitative alterations occur in resident hCSCs and/or in the pool of TACs. In stem cell-regulated organs, both possibilities have been found to be true. The number of epithelial stem cells in the small intestine is gradually reduced with age,46 and the function of hematopoietic stem cells changes with time.47 Whether aging of the skin is conditioned by a decreased frequency of epidermal stem cells, or by alterations in the kinetics of the TAC pool, remains controversial.45
The relationship between aging hCSCs and the myocyte progeny was not investigated in the present study, although results suggest that hCSCs with shortened telomeres give rise to progenitors, precursors, and amplifying cells that inherit the characteristics of the mother cell and generate myocytes that rapidly acquire the senescent cell phenotype and express p16INK4a.48 The link between the past history of CSCs, their telomere length, and the age of the formed progeny has been documented in a rigorous manner not only in humans,48 but also in small-animal models of physiological aging, supporting the notion that myocardial biology and function may be determined largely by the state of the controlling cell (ie, the hCSC).49,50 However, current information on the lifespan of hCSCs is lacking, although this cell compartment is affected by replicative senescence and irreversible growth arrest together with apoptosis.48 Both the female and the male heart contain undifferentiated hCSCs that undergo symmetric and asymmetric division in vitro and in vivo2,4 throughout the lifespan of the organ.48 This characteristic indicates that hCSCs can be continuously formed, regulating niche homeostasis.16 Aging is coupled with an increase in senescent and apoptotic hCSCs, progressively reducing the pool of functionally competent primitive cells within the myocardium. Importantly, men are more affected than women.48
In the pathological heart, the homeostatic control of stem cell growth and differentiation is defective, resulting in decline of the hCSC pool, inefficient formation of myocytes, cellular senescence, and activation of apoptosis.6–8 For several decades, the inability of the heart to sustain hemodynamic performance with age and disease has been interpreted as the consequence of the loss of regenerative capacity of the myocardium after birth.51 However, the effects of age and disease on the number of tissue-specific parenchymal cells are similar in organs with high and low turnover rates,45–48 in which the degree of cell loss consistently exceeds the magnitude of cell replication. Thus, whether heart failure is characterized by depletion of the hCSC compartment, which is uniformly affected by the pathological condition, or whether a stem cell subset maintains its youth throughout the organism natural life, independently from the severely depressed cardiac behavior, was unknown.
Data from the present study strongly suggest that the latter mechanism is operative and that hCSCs with preserved growth reserve are present in explanted hearts at the end of their physiological lifespan. This possibility has important clinical implications, because these functionally competent hCSCs can be isolated and expanded in vitro 2,4 for subsequent delivery to the diseased heart. The hCSCs collected from the explanted hearts contained a fraction of cells carrying dysfunctional telomeres with accumulation of sites of DNA lesion and activation of the cell cycle inhibitors p53, p21Cip1, and p16INk4a. However, the clones formed by these cells were only partly senescent, most likely because of selective growth of cells with predominantly intact telomeres.
In the present study, the senescent state of hCSCs was defined by telomere shortening, incidence of TIFs, and up-regulation of p21Cip1, p53, and p16INK4a. Senescent hCSCs were characterized by the expression of phosphorylated histone H2AX and p53 binding protein. Both γ-H2AX and 53BP1 are markers of DNA double-strand breakage,52,53 and their presence in old hCSCs from donor and explanted hearts identified DNA lesions at the sites of telomere hybridization spots. TIF-positive hCSCs reflected old cells in which the DNA damage response was activated. This process may precede the onset of irreversible growth arrest dictated by the expression of p16INK4a.39,40 Telomere capping is monitored at the G2/M transition by the p53/p21Cip1 effector pathway. A p53-dependent signaling mechanism monitors telomere capping after DNA replication and delays G2/M progression in the presence of unprotected telomeres.53 This process prevents cell cycle progression and avoids the formation of end-to-end fusion by uncapped telomeres. However, telomere-initiated senescence reflects a permanent cell-cycle arrest, enforced when the DNA damage checkpoint pathway is triggered by critically shortened telomeres.41 TIFs have been detected in vitro nearly 2 months after cells have entered senescence,54 suggesting that the DNA damage response is maintained in the senescent state.
As we have discussed, cellular senescence is triggered by multiple mechanisms that include telomeric shortening, unrepaired DNA damage, and epigenetic derepression of the INK4a/ARF locus.55 These three pathways converge on the transcription factor p53, which is a critical determinant of cellular aging. Up-regulation of p53 by epigenetic activation of the ARF pathway is involved specifically in tumor-suppression activity. In the presence of stress, p53 induces cell-cycle arrest and DNA repair or promotes cellular senescence and apoptosis.40 Overexpression of constitutively active p53 leads to premature organism aging and decreased longevity, which points to p53 as a positive modulator of senescence.56 However, an extra copy of nonactivated p53 or reduced expression of Mdm2 prevents cancer and does not affect life expectancy.57,58 These conflicting findings suggest that premature aging is mediated by improper regulation of p53 and preferential activation of a restricted group of target genes. The role of p53 may be cell-dependent and may vary in postmitotic myocytes and stem cells. Whether p53-responsive genes condition hCSC senescence and organ aging is currently unknown. The transcriptional profiles of hCSCs from control and failing hearts suggest that p53 may be implicated in the changes in the expression of genes governing the adaptation to oxidative stress, cell growth, cell motility, and tissue repair.
Although a correlation has been found between telomere length in subsets of bone marrow-derived circulating cells and cardiovascular diseases,9,59 the present study provides the first demonstration that dysfunctional telomeres in hCSCs are biomarkers of aging and heart failure. Previous studies have restricted their analysis to average telomere length in a cell population,59 neglecting the fact that telomere attrition does not occur uniformly in all cells and at all chromosomal ends.60 The shortest telomere (rather than mean telomere length) determines the function and fate of the cell.38 Importantly, TIFs have been used to identify lesion foci in individual chromosomal ends in single hCSCs with defective growth and enhanced proficiency to undergo apoptosis. Additionally, the TIF assay does not require the identification of end-to-end chromosome fusions and anaphase bridges, which necessitate the progression of cells into mitosis, despite telomere dysfunction. The recognition that TIFs define a population of hCSCs that increases with chronological age and with the concomitant presence of heart failure emphasizes the critical role that intrinsic cellular factors at the level of resident stem cells have in the characterization of the human disease. Theoretically, TIF incidence may discriminate eugeric from pathogeric processes (ie, physiological aging as such versus physiological aging in the presence of cardiovascular diseases commonly found in the elderly population). However, the lack of overlap between the age of donor and explanted hearts in the present study made this comparison problematic, and actual distinction of physiological and pathological aging was not possible. Donor hearts are generally obtained from young individuals, and heart failure affects mostly patients 65 years of age and older.
Footnotes
Supported by Regione Friuli Venezia Giulia (art. 23 LR 26/2005, Progetto di ricerca SeND–Search for New Drugs to C.A.B.), MIUR (PRIN 2006 pr. 2006060854 to C.A.B.), and by NIH grants P01-AG-023071 (P.A.), R01-AG-026107 (A.L.), R01-HL-65577 (A.L.), P01-HL-092868 (P.A.), and R01-AG-37490 (A.L.).
D.C. and A.P.B. contributed equally to the present work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.03.036.
A guest editor acted as editor-in-chief for this manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition for this article.
Supplementary data
Growth of hCSCs. Colonies formed by hCSCs isolated by donor (A) and explanted (B) hearts. Staining, crystal violet. C: Colony forming efficiency (means ± SD) of hCSCs from donor (Don) and explanted (Exp) hearts. Scale bars: 20 μm.
Colony characteristics. Expression of c-Kit (green) (A), OCT3/4 (yellow) (B), and NANOG (red) C) in hCSCs obtaioned from a donor heart.
Characteristics of expanded hCSCs. A: Surface phenotype of expanded hCSCs. B-D: Localization of c-Kit (green) (B), OCT3/4 (yellow) (C), and NANOG (red) (D) in hCSCs obtaioned from a donor heart. Scale bars: 20 μm.
Cardiac disease and biomarkers of hCSC senescence.
Transcriptional profile of hCSCs. Heatmap showing the 7868 differentially expressed genes in hCSCs from donor and explanted hearts. Each column (C1, C2 and C3) represents the direct comparison of hCSCs collected from age- and sex-matched donor and failing hearts. C1, C2 and C3 reflect three different sex- and age-matched pair. Genes down-regulated and up-regulated in hCSCs from donor hearts are shown in green and red, respectively. Fold changes are shown for a selected list of genes. The complete list of genes is available on request.
Ingenuity pathway analysis (IPA). A: Biofunctions with score, −log(P value), >1.5 are listed. B and C: Graphical representation of the interactions among distinct genes within two functional cores: cardiac development network and cardiovascular disease biofunction. Red and green define genes up-regulated and down-regulated in hCSCs from donor versus explanted hearts, respectively. The color gradient corresponds to the degree of difference. A solid line represents a direct interaction between two genes and a dotted line indicates an indirect interaction.
References
- 1.Smith R.R., Barile L., Cho H.C., Leppo M.K., Hare J.M., Messina E., Giacomello A., Abraham M.R., Marbán E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 2.Bearzi C., Rota M., Hosoda T., Tillmanns J., Nascimbene A., De Angelis A., Yasuzawa-Amano S., Trofimova I., Siggins R.W., Lecapitaine N., Cascapera S., Beltrami A.P., D'Alessandro D.A., Zias E., Quaini F., Urbanek K., Michler R.E., Bolli R., Kajstura J., Leri A., Anversa P. Human cardiac stem cells. Proc Natl Acad Sci USA. 2007;104:14068–14073. doi: 10.1073/pnas.0706760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltrami A.P., Cesselli D., Bergamin N., Marcon P., Rigo S., Puppato E., D'Aurizio F., Verardo R., Piazza S., Pignatelli A., Poz A., Baccarani U., Damiani D., Fanin R., Mariuzzi L., Finato N., Masolini P., Burelli S., Belluzzi O., Schneider C., Beltrami C.A. Multipotent cells can be generated in vitro from several adult human organs (heart, liver, and bone marrow) Blood. 2007;110:3438–3446. doi: 10.1182/blood-2006-11-055566. [DOI] [PubMed] [Google Scholar]
- 4.Bearzi C., Leri A., Lo Monaco F., Rota M., Gonzalez A., Hosoda T., Pepe M., Qanud K., Ojaimi C., Bardelli S., D'Amario D., D'Alessandro D.A., Michler R.E., Dimmeler S., Zeiher A.M., Urbanek K., Hintze T.H., Kajstura J., Anversa P. Identification of a coronary vascular progenitor cell in the human heart. Proc Natl Acad Sci USA. 2009;106:15885–15890. doi: 10.1073/pnas.0907622106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Itzhaki-Alfia A., Leor J., Raanani E., Sternik L., Spiegelstein D., Netser S., Holbova R., Pevsner-Fischer M., Lavee J., Barbash I.M. Patient characteristics and cell source determine the number of isolated human cardiac progenitor cells. Circulation. 2009;120:2559–2566. doi: 10.1161/CIRCULATIONAHA.109.849588. [DOI] [PubMed] [Google Scholar]
- 6.Chimenti C., Kajstura J., Torella D., Urbanek K., Heleniak H., Colussi C., Di Meglio F., Nadal-Ginard B., Frustaci A., Leri A., Maseri A., Anversa P. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93:604–613. doi: 10.1161/01.RES.0000093985.76901.AF. [DOI] [PubMed] [Google Scholar]
- 7.Urbanek K., Quaini F., Tasca G., Torella D., Castaldo C., Nadal-Ginard B., Leri A., Kajstura J., Quaini E., Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urbanek K., Torella D., Sheikh F., De Angelis A., Nurzynska D., Silvestri F., Beltrami C.A., Bussani R., Beltrami A.P., Quaini F., Bolli R., Leri A., Kajstura J., Anversa P. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005;102:8692–8697. doi: 10.1073/pnas.0500169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dimmeler S., Leri A. Aging and disease as modifiers of efficacy of cell therapy. Circ Res. 2008;102:1319–1330. doi: 10.1161/CIRCRESAHA.108.175943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahin E., Depinho R.A. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackburn E.H. The end of the (DNA) line. Nat Struct Biol. 2000;7:847–850. doi: 10.1038/79594. [DOI] [PubMed] [Google Scholar]
- 12.de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lansdorp P.M. Lessons from mice without telomerase. J Cell Biol. 1997;139:309–312. doi: 10.1083/jcb.139.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xin Z.T., Beauchamp A.D., Calado R.T., Bradford J.W., Regal J.A., Shenoy A., Liang Y., Lansdorp P.M., Young N.S., Ly H. Functional characterization of natural telomerase mutations found in patients with hematologic disorders. Blood. 2007;109:524–532. doi: 10.1182/blood-2006-07-035089. [DOI] [PubMed] [Google Scholar]
- 15.Alter B.P., Baerlocher G.M., Savage S.A., Chanock S.J., Weksler B.B., Willner J.P., Peters J.A., Giri N., Lansdorp P.M. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110:1439–1447. doi: 10.1182/blood-2007-02-075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urbanek K., Cesselli D., Rota M., Nascimbene A., De Angelis A., Hosoda T., Bearzi C., Boni A., Bolli R., Kajstura J., Anversa P., Leri A. Stem cell niches in the adult mouse heart. Proc Natl Acad Sci USA. 2006;103:9226–9231. doi: 10.1073/pnas.0600635103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosoda T., D'Amario D., Cabral-Da-Silva M.C., Zheng H., Padin-Iruegas M.E., Ogorek B., Ferreira-Martins J., Yasuzawa-Amano S., Amano K., Ide-Iwata N., Cheng W., Rota M., Urbanek K., Kajstura J., Anversa P., Leri A. Clonality of mouse and human cardiomyogenesis in vivo. Proc Natl Acad Sci USA. 2009;106:17169–17174. doi: 10.1073/pnas.0903089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Alessandro D.A., Kajstura J., Hosoda T., Gatti A., Bello R., Mosna F., Bardelli S., Zheng H., D'Amario D., Padin-Iruegas M.E., Carvalho A.B., Rota M., Zembala M.O., Stern D., Rimoldi O., Urbanek K., Michler R.E., Leri A., Anversa P. Progenitor cells from the explanted heart generate immunocompatible myocardium within the transplanted donor heart. Circ Res. 2009;105:1128–1140. doi: 10.1161/CIRCRESAHA.109.207266. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Boni A., Urbanek K., Nascimbene A., Hosoda T., Zheng H., Delucchi F., Amano K., Gonzalez A., Vitale S., Ojaimi C., Rizzi R., Bolli R., Yutzey K.E., Rota M., Kajstura J., Anversa P., Leri A. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008;105:15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Kajstura J., Urbanek K., Perl S., Hosoda T., Zheng H., Ogórek B., Ferreira-Martins J., Goichberg P., Rondon-Clavo C., Sanada F., D'Amario D., Rota M., Del Monte F., Orlic D., Tisdale J., Leri A., Anversa P. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–315. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Ferreira-Martins J., Rondon-Clavo C., Tugal D., Korn J.A., Rizzi R., Padin-Iruegas M.E., Ottolenghi S., De Angelis A., Urbanek K., Ide-Iwata N., D'Amario D., Hosoda T., Leri A., Kajstura J., Anversa P., Rota M. Spontaneous calcium oscillations regulate human cardiac progenitor cell growth. Circ Res. 2009;105:764–774. doi: 10.1161/CIRCRESAHA.109.206698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rota M., Boni A., Urbanek K., Padin-Iruegas M.E., Kajstura T.J., Fiore G., Kubo H., Sonnenblick E.H., Musso E., Houser S.R., Leri A., Sussman M.A., Anversa P. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res. 2005;95:1332–1341. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 23.Rota M., Hosoda T., De Angelis A., Arcarese M.L., Esposito G., Rizzi R., Tillmanns J., Tugal D., Musso E., Rimoldi O., Bearzi C., Urbanek K., Anversa P., Leri A., Kajstura J. The young mouse heart is composed of myocytes heterogeneous in age and function. Circ Res. 2007;101:387–399. doi: 10.1161/CIRCRESAHA.107.151449. [DOI] [PubMed] [Google Scholar]
- 24.Baerlocher G.M., Vulto I., de Jong G., Lansdorp P.M. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1:2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 25.Xiang C.C., Kozhich O.A., Chen M., Inman J.M., Phan Q.N., Chen Y., Brownstein M.J. Amine-modified random primers to label probes for DNA microarrays. Nat Biotechnol. 2002;20:738–742. doi: 10.1038/nb0702-738. [DOI] [PubMed] [Google Scholar]
- 26.Dalla E., Mignone F., Verardo R., Marchionni L., Marzinotto S., Lazarević D., Reid J.F., Marzio R., Klarić E., Licastro D., Marcuzzi G., Gambetta R., Pierotti M.A., Pesole G., Schneider C. Discovery of 342 putative new genes from the analysis of 5′-end-sequenced full-length-enriched cDNA human transcripts. Genomics. 2005;85:739–751. doi: 10.1016/j.ygeno.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Lennon G., Auffray C., Polymeropoulos M., Soares M.B. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- 28.Smyth G.K. Limma: linear models for microarray data: Bioinformatics and Computational Biology Solutions using R and Bioconductor. In: Gentleman R.C., Carey V.J., Huber W., Irizarry R.A., Dudoit S., editors. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 29.Edwards D. Non-linear normalization and background correction in one-channel cDNA microarray studies. Bioinformatics. 2003;19:825–833. doi: 10.1093/bioinformatics/btg083. [DOI] [PubMed] [Google Scholar]
- 30.Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saldanha A.J. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 32.Smyth G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 33.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 34.Berenson M.L., Levine D.M., Rindskopf D. Simple linear regression and correlation: Applied Statistics. In: Berenson M.L., Levine D.M., Rindskopf D., editors. Prentice Hall; Englewood Cliffs: 1998. pp. 362–418. [Google Scholar]
- 35.Beltrami A.P., Cesselli D., Beltrami C.A. Pluripotency rush! Molecular cues for pluripotency, genetic reprogramming of adult stem cells, and widely multipotent adult cells. Pharmacol Ther. 2009;124:23–30. doi: 10.1016/j.pharmthera.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Burgess M.L., McCrea J.C., Hedrick H.L. Age-associated changes in cardiac matrix and integrins. Mech Ageing Dev. 2001;122:1739–1756. doi: 10.1016/s0047-6374(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 37.Hiyama E., Hiyama K. Telomere and telomerase in stem cells. Br J Cancer. 2007;96:1020–1024. doi: 10.1038/sj.bjc.6603671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hemann M.T., Strong M.A., Hao L.Y., Greider C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/s0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 39.Sharpless N.E., DePinho R.A. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 40.Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 41.d'Adda di Fagagna F., Reaper P.M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N.P., Jackson S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 42.Gire V., Roux P., Wynford-Thomas D., Brondello J.M., Dulic V. DNA damage checkpoint kinase Chk2 triggers replicative senescence. EMBO J. 2004;23:2554–2563. doi: 10.1038/sj.emboj.7600259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs J.J., de Lange T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005;4:1364–1368. doi: 10.4161/cc.4.10.2104. [DOI] [PubMed] [Google Scholar]
- 44.Castaldo C., Di Meglio F., Nurzynska D., Romano G., Maiello C., Bancone C., Müller P., Böhm M., Cotrufo M., Montagnani S. CD117-positive cells in adult human heart are localized in the subepicardium, and their activation is associated with laminin-1 and alpha6 integrin expression. Stem Cells. 2008;26:1723–1731. doi: 10.1634/stemcells.2007-0732. [DOI] [PubMed] [Google Scholar]
- 45.Blanpain C., Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–217. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin K., Kirkwood T.B., Potten C.S. Age changes in stem cells of murine small intestinal crypts. Exp Cell Res. 1998;241:316–323. doi: 10.1006/excr.1998.4001. [DOI] [PubMed] [Google Scholar]
- 47.Rossi D.J., Bryder D., Zahn J.M., Ahlenius H., Sonu R., Wagers A.J., Weissman I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci USA. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kajstura J., Gurusamy N., Ogórek B., Goichberg P., Clavos-Rondon C., Hosoda T., D'Amario D., Bardelli S., Beltrami A.P., Cesselli D., Bussani R., del Monte F., Quaini F., Rota M., Beltrami C.A., Buchholz B., Leri A., Anversa P. Myocyte turnover in the aging human heart. Circ Res. 2010;107:1374–1386. doi: 10.1161/CIRCRESAHA.110.231498. [DOI] [PubMed] [Google Scholar]
- 49.Torella D., Rota M., Nurzynska D., Musso E., Monsen A., Shiraishi I., Zias E., Walsh K., Rosenzweig A., Sussman M.A., Urbanek K., Nadal-Ginard B., Kajstura J., Anversa P., Leri A. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–524. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez A., Rota M., Nurzynska D., Misao Y., Tillmanns J., Ojaimi C., Padin-Iruegas M.E., Müller P., Esposito G., Bearzi C., Vitale S., Dawn B., Sanganalmath S.K., Baker M., Hintze T.H., Bolli R., Urbanek K., Hosoda T., Anversa P., Kajstura J., Leri A. Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circ Res. 2008;102:597–606. doi: 10.1161/CIRCRESAHA.107.165464. [DOI] [PubMed] [Google Scholar]
- 51.Murry C.E., Field L.J., Menasché P. Cell-based cardiac repair: reflections at the 10-year point. Circulation. 2005;112:3174–3183. doi: 10.1161/CIRCULATIONAHA.105.546218. [DOI] [PubMed] [Google Scholar]
- 52.Herbig U., Ferreira M., Condel L., Carey D., Sedivy J.M. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 53.Thanasoula M., Escandell J.M., Martinez P., Badie S., Muñoz P., Blasco M.A., Tarsounas M. p53 prevents entry into mitosis with uncapped telomeres. Curr Biol. 2010;20:521–526. doi: 10.1016/j.cub.2010.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takai H., Smogorzewska A., de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 55.Collado M., Blasco M.A., Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 56.Maier B., Gluba W., Bernier B., Turner T., Mohammad K., Guise T., Sutherland A., Thorner M., Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.García-Cao I., García-Cao M., Martín-Caballero J., Criado L.M., Klatt P., Flores J.M., Weill J.C., Blasco M.A., Serrano M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21:6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mendrysa S.M., O'Leary K.A., McElwee M.K., Michalowski J., Eisenman R.N., Powell D.A., Perry M.E. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20:16–21. doi: 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brouilette S.W., Whittaker A., Stevens S.E., van der Harst P., Goodall A.H., Samani N.J. Telomere length is shorter in healthy offspring of subjects with coronary artery disease: support for the telomere hypothesis. Heart. 2008;94:422–425. doi: 10.1136/hrt.2007.139675. [DOI] [PubMed] [Google Scholar]
- 60.Aubert G., Lansdorp P.M. Telomeres and aging. Physiol Rev. 2008;88:557–579. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth of hCSCs. Colonies formed by hCSCs isolated by donor (A) and explanted (B) hearts. Staining, crystal violet. C: Colony forming efficiency (means ± SD) of hCSCs from donor (Don) and explanted (Exp) hearts. Scale bars: 20 μm.
Colony characteristics. Expression of c-Kit (green) (A), OCT3/4 (yellow) (B), and NANOG (red) C) in hCSCs obtaioned from a donor heart.
Characteristics of expanded hCSCs. A: Surface phenotype of expanded hCSCs. B-D: Localization of c-Kit (green) (B), OCT3/4 (yellow) (C), and NANOG (red) (D) in hCSCs obtaioned from a donor heart. Scale bars: 20 μm.
Cardiac disease and biomarkers of hCSC senescence.
Transcriptional profile of hCSCs. Heatmap showing the 7868 differentially expressed genes in hCSCs from donor and explanted hearts. Each column (C1, C2 and C3) represents the direct comparison of hCSCs collected from age- and sex-matched donor and failing hearts. C1, C2 and C3 reflect three different sex- and age-matched pair. Genes down-regulated and up-regulated in hCSCs from donor hearts are shown in green and red, respectively. Fold changes are shown for a selected list of genes. The complete list of genes is available on request.
Ingenuity pathway analysis (IPA). A: Biofunctions with score, −log(P value), >1.5 are listed. B and C: Graphical representation of the interactions among distinct genes within two functional cores: cardiac development network and cardiovascular disease biofunction. Red and green define genes up-regulated and down-regulated in hCSCs from donor versus explanted hearts, respectively. The color gradient corresponds to the degree of difference. A solid line represents a direct interaction between two genes and a dotted line indicates an indirect interaction.