

Abstract
The photosynthetic light-harvesting systems of purple bacteria and plants both utilize specific carotenoids as quenchers of the harmful (bacterio)chlorophyll triplet states via triplet-triplet energy transfer. Here, we explore how the binding of carotenoids to the different types of light-harvesting proteins found in plants and purple bacteria provides adaptation in this vital photoprotective function. We show that the creation of the carotenoid triplet states in the light-harvesting complexes may occur without detectable conformational changes, in contrast to that found for carotenoids in solution. However, in plant light-harvesting complexes, the triplet wavefunction is shared between the carotenoids and their adjacent chlorophylls. This is not observed for the antenna proteins of purple bacteria, where the triplet is virtually fully located on the carotenoid molecule. These results explain the faster triplet-triplet transfer times in plant light-harvesting complexes. We show that this molecular mechanism, which spreads the location of the triplet wavefunction through the pigments of plant light-harvesting complexes, results in the absence of any detectable chlorophyll triplet in these complexes upon excitation, and we propose that it emerged as a photoprotective adaptation during the evolution of oxygenic photosynthesis.
Introduction
During the first steps of the photosynthetic process, the absorption of photons by antenna proteins and the subsequent transfer of excitation energy to the reaction centers are both intimately linked with the potential production of dangerous oxidative species. To avoid photooxidative damage, photosynthetic organisms have developed a number of strategies to minimize the formation of reactive oxygen; these processes often involve carotenoid molecules (e.g., (1–7).). The best characterized of the photoprotective functions of carotenoid molecules in photosynthetic organisms involves quenching of (bacterio)chlorophyll ((B)Chl) triplets. Excited singlet states of (B)Chl molecules may decay to (B)Chl triplet states, with a low but significant yield. Since the energy of these triplet states is above that of the singlet state of oxygen, they may sensitize the formation of singlet oxygen, one of the most dangerous chemical species for living organisms (8). In photosynthetic proteins, the efficiency of this reaction is impaired by the competing transfer of the triplet state from (B)Chls to carotenoid molecules, which display a triplet-state energy below that of singlet oxygen. This quenching reaction reduces the lifetime of the (B)Chl triplet state by more than three orders of magnitude (9).
In light-harvesting (LH) proteins from purple bacteria, the triplet-triplet transfer from (B)Chl to carotenoid molecules has a characteristic lifetime in the nanosecond range (10,11). In most of these complexes, formation of the carotenoid triplet state has a significant, but limited, influence on the electronic transitions of (B)Chl molecules (10). The energy of the lowest electronic transition of the carotenoid triplet state is proportional to the inverse of the number of carotenoid C=C conjugated bonds, as predicted by theory (12,13). In contrast, carotenoid triplet states in the LH complexes (LHCs) of higher plants exhibit properties largely different from those found in LH proteins ((14) and Table 1). Formation of carotenoid triplet states in these complexes induces an intense bleaching of Chl transitions, and the triplet-triplet transfer lifetime, although not determined with precision, has been proposed to be ultrafast (15). In this article, we show that in the major LH protein from higher plants, LHCII, this transfer is fast enough that chlorophyll triplet is not accumulated in these complexes. Application of infrared absorption and resonance Raman spectroscopies provide a precise picture of the carotenoid triplet states in these complexes, which can be related to their electronic and functional properties.
Table 1.
Electronic transitions of triplet carotenoid states in purple bacteria and higher plant LHC complexes
| Carotenoid (complex) | Number of C=C bonds | S0→S2 transition (nm) | T1→Tn transition (nm) | ΔE (cm−1) |
|---|---|---|---|---|
| Neurosporene (LH2) | 9 | 495 | 516 | 822 |
| Spheroidene (LH2) | 10 | 514 | 537 | 833 |
| Rhodopin (LH2) | 11 | 529 | 556 | 918 |
| Lutein 1 (LHCII trimers) | 10 | 494 | 506 | 480 |
| Lutein 2 (LHCII trimers) | 10 | 510 | 525 | 560 |
| Lutein 1 and 2 (LHCII monomers, CP29) | 10 | 494, 494 | 508, 505 | 558, 441 |
Materials and Methods
Protein and carotenoid purification
The LH2 protein from Rhodoblastus acidophilus 10050 was purified as described previously (16,17). LHC complexes were prepared from Spinacia oleracea photosystem-II-enriched particles by isoelectric focusing followed by sucrose gradient centrifugation (18). The carotenoid lutein was purified as described previously (19).
Femtosecond transient absorption spectroscopy
These measurements were carried out with a spectrometer described in a previous study (20). The output of the Ti:sapphire laser at 800 nm was frequency-doubled and used to excite the sample at 400 nm; the pulse energy was ∼20 nJ. The data were globally analyzed (21) using a kinetic model consisting of sequentially interconverting evolution-associated difference spectra (EADS), e.g., 1→2→3→…, in which the arrows indicate successive monoexponential decays with increasing time constants, which can be regarded as the lifetime of each EADS. Note that EADS generally does not represent pure states. The number of kinetic components corresponds to the minimum required to eliminate any correlated structure in the residuals. The instrument response function was fitted to a Gaussian of 120 fs (full width at half-maximum), similar to the value obtained from the analysis of the induced birefringence in CS2.
Time-resolved step-scan and steady-state Fourier-transform infrared absorption measurements
Time-resolved interferograms were recorded using a step-scan Fourier transform infrared (FTIR) spectrometer (IFS 66s, Bruker, Billerica, MA) placed on an air-bearing table (Kinetic Systems, Boston, MA). The experimental setup included an IR light source and a fast preamplified photovoltaic MCT detector (20 MHz, KV 100, Kolmar Technologies, Newburyport, MA). The IR light impinging on the sample was sent through 4000 cm−1 low-pass and 1850–1200 cm−1 band-pass filters, which blocked the laser light before the interferometer and the detector. The detector signal was recorded with the internal digitizer (200 kHz, 16-bit A/D converter), allowing measurements with an instrument response function of ∼3 μs. A 20 Hz Nd:YAG laser (5 ns, 100 mJ at 355 nm, Continuum, Santa Clara, CA) was used to pump an optical parametric oscillator (Panther, Continuum), producing tunable visible light from 400 to 700 nm, with a pulse duration of 5–7 ns. This light was attenuated to 2 mJ/cm2 at 475 nm and was weakly focused to a spot of 4 mm in diameter and overlapped with the IR probe beam, which was slightly smaller. Direct excitation of chlorophyll using laser radiation at 670 or 625 nm, resonant with the Qy or the Qx Chl a transitions, respectively, led to similar results (data not shown). A digital delay generator (DCR 35, Stanford Research Systems, Sunnyvale, CA) was used to vary the time delay between the pump laser pulse and the trigger of the detection electronics. Each three-dimensional IR interferogram is composed of 660 points in the spectral region 1800–1200 cm−1, giving a spectral resolution of 8 cm−1. The data presented are an average of three data sets; each set is an average of 20 time-resolved interferograms of which each point is a time slice that is the average of nine coadditions. The time-resolved interferograms have been further Fourier-transformed into time-resolved IR difference spectra (OPUS software, Bruker). The step-scan FTIR sample was made with a drop of 2 μL of OD 670 nm of ∼500 spread between two tightly fixed CaF2 windows, without any spacer and greased for tightness. All experiments were performed at room temperature. The obtained time-resolved IR difference spectra were analyzed by global analysis (21) with a sequential kinetic model, which simultaneously fitted the dynamics at every point of a spectral data set. This analysis leads to decay-associated difference spectra (DADS), with associated lifetimes. The FTIR spectroscopy of lutein was performed on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA), equipped with an MCTA photoconductive detector (SensIR) in ATR mode.
Raman spectroscopy
Spectra, in resonance with the carotenoid transitions, were recorded with a Jobin Yvon U1000 spectrometer equipped with a UV-coated CCD camera (Spectraview 2D, Jobin Yvon, Longjumeau, France). For these experiments, spectra were recorded at 90° geometry from samples maintained at 77 K in a flow cryostat (Air Liquide, Sassenage, France) cooled with liquid nitrogen. An argon laser (Innova 100, Coherent, Santa Clara, CA) provided excitation wavelengths of 495.6, 501.7, 514.5, and 528.7 nm. To minimize systematic errors, no subtraction of the background signal was made. Laser power was controlled at the sample using a photon-counting device (Vector H410, Scientech, Boulder, CO).
Results
Fig. 1 shows the result of a global analysis of the time-resolved absorption spectroscopy data obtained upon nonselective excitation of trimeric LHCII at 400 nm. The first EADS (black line) shows the bleaching of the S0→S2 absorption transition arising from carotenoids below 515 nm, and of the transitions of Chl b and Chl a at ∼650 nm and ∼675 nm, respectively. It decays in 100 fs to the next spectrum (red trace), which is characterized by a pronounced carotenoid excited-state absorption at 537 nm, corresponding to the S1→Sn electronic transition of this molecule, a partial restoration of the S0→S2 carotenoid transition, and a strong bleaching of the chlorophyll transitions. The bleaching of the chlorophylls results from energy transfers, either intramolecular or from the carotenoid molecules. This red spectrum decays in 1 ps to the blue one, which is characterized by a more pronounced bleaching of the Chl a Qy transitions, mainly due to excitation energy transfer from Chl b and carotenoids. The next evolution takes place in 14.9 ps and results in the green spectrum, which shows a decrease in bleaching of the Chl a Qy transition, due to singlet-singlet annihilation processes, and the complete decay of the carotenoid S1 state(s). The final evolution, which occurs in 3.9 ns, results in the magenta spectrum. This spectrum clearly shows that the decay of the Chl singlet excited state is complete in 3.9 ns, and that an absorption transition with properties typical of that of a carotenoid triplet state (displaying a maximum at 514 nm, together with a shoulder at ∼525 nm) (13,14) appears concomitantly with the disappearance of the Chl excited state. The inset in Fig. 1 displays kinetic traces at 511 nm, i.e., close to the maximum of the T1→Tn electronic transition of carotenoid molecules, and at 675 nm, which corresponds to the Qy maximum of Chl molecules. A comparison of the two traces shows that the rise of the carotenoid triplet state occurs in parallel with the decay of the Chl singlet excited state. This unambiguously demonstrates that no chlorophyll triplet state is appreciably accumulated in these complexes after excitation, and thus that a fast and extremely efficient triplet-triplet channel exists, which very effectively quenches the chlorophyll triplet states as soon as they are formed. The results in this work are fully consistent with a recent EADS study on the peridinin-chlorophyll protein (PCP) from the dinoflagellate Amphidinium carterae, where the Chl to carotenoid transfer is similarly fast and where no significant transient formation of Chl triplet was observed (22).
Figure 1.
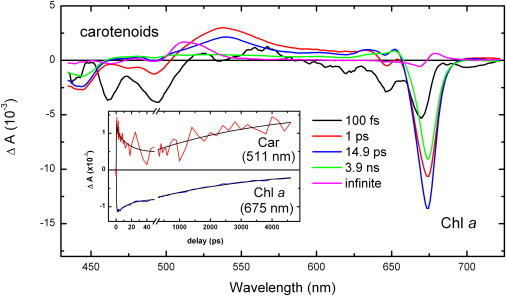
Evolution-associated difference spectra (EADS) of LHCII excited at 400 nm. The EADS spectra correspond to the 100 fs (black), 1 ps (red), 14.9 ps (blue), 3.9 ns (green; Chl excited-state lifetime), and infinite (magenta) lifetimes. The Chl a singlet excited signal that is characterized by ground-state bleach at ∼675 nm within 3.9 ns is replaced by the carotenoid triplet spectrum (magenta). (Inset) Kinetic traces at 511 nm, the maximum of the T1→Tn transition, and at 675 nm, corresponding to the maximum of the Chl a bleach signal; for clarity, the magnitude of the latter trace has been reduced 10-fold.
To understand the mechanisms underlying this very efficient triplet-triplet transfer, we investigated LHCII by step-scan time-resolved FTIR spectroscopy. Upon excitation of LHCII carotenoids at 475 nm, the triplet decay was satisfactorily fitted with two components only, using global analysis. The first component decays with a time constant of 20 μs, whereas the second component, which does not account for more than 10% of the signal, does not decay within the time window of the measurement (∼320 μs). Fig. 2 A displays the first DADS normalized to the contribution of the keto group at 1653 cm−1. Taking into account the time-resolved absorption data displayed above, the first decay-associated spectrum must be assigned to the carotenoid triplet (Fig. 2 A, black line, DADS1). In view of its long time, the second spectrum is attributed to unquenched Chl, i.e., a small proportion of triplet chlorophyll states that have not been transferred to the carotenoid molecules (Fig. 2 A, blue line; DADS2). It has already been observed that a small fraction of chlorophyll triplet may not be quenched by the carotenoid molecules (23). Considering the results of time-resolved absorption, DADS1 should contain positive contributions of the carotenoid triplet state and negative contributions of its ground state. Although such contributions clearly appear in the difference spectrum (region termed luteins), additional bands are obviously present in this spectrum. Indeed, no intense contribution in the higher-frequency region is expected from carotenoid molecules (see Fig. 2 B) (24). On the contrary, in this region, DADS1 is typical of the spectrum of a chlorophyll triplet in solvent, which is plotted in Fig. 2 C for comparison (25,26). The negative contributions around 1700 cm−1 represent bleaching of bands arising from the stretching modes of conjugated keto carbonyl groups. These groups, when conjugated with the Chl macrocycle, experience large downshifts upon triplet formation, which results in positive contributions at lower frequencies. Nevertheless, DADS1 has a lifetime characteristic of carotenoid triplets, and so we conclude that in LHCII these chlorophyll infrared modes decay with the same lifetimes as carotenoid modes. Recently, an identical result was observed for the triplets in PCP (26,27).
Figure 2.
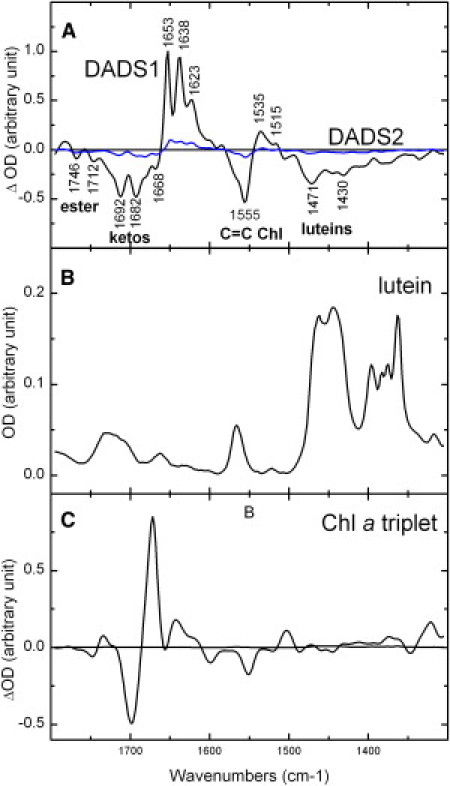
Global analysis of step-scan FTIR data of LHCII excited at 475 nm showing the lutein-Chl shared triplet state. (A) The DADS have a 20 μs component (black line, DADS1), with an amplitude of 90% (lutein-Chl shared triplet) and a nondecaying component (blue line, DADS2) that has an amplitude of 10% (unquenched Chls). For clarity, the spectra have been normalized to the keto modes. (B and C) For comparison, the FTIR of the lutein (B) and Chl a (C) triplet (redrawn from Bonetti et al. (26)) in THF are also plotted.
In resonance Raman spectra of carotenoid molecules, in either their ground or triplet excited states, two groups of bands yield precise, important information on conformation and molecular structure: the so-called ν1 and ν4 regions. The ν1 vibrational mode is usually the most intense band in carotenoid resonance Raman spectra. It arises from stretching modes of the conjugated C=C bonds of the molecule and its position, in the 1480–1550 cm−1 range reflects the bond order of the conjugated C=C bonds. The ν4 band(s) arises from out-of-plane wagging modes of the carotenoid C-H groups, which, for symmetry reasons, are not coupled with the electronic transition in the case of a perfectly planar carotenoid molecule. Their resonance Raman activity, as well as their precise frequency, tightly depend on any out-of-plane distortions these molecules experience.
Fig. 3 displays resonance Raman spectra (800–1650 cm−1) of the LH2 complex from Rbl. acidophilus probed with an excitation wavelength of 514.5 nm, using three different incident intensities (0.5, 5.4, and 21.5 μW). The ν4 region of the Raman spectra indicates that the carotenoid rhodopin glucoside is distorted (28). With increasing laser power, a number of small bands in the spectrum are seen to increase in relative intensity. The observed power dependence of the appearance of these features is consistent with progressive, dynamic accumulation of a transient state (see the Supporting Material), and the bands observed at high laser intensity are characteristic of resonance Raman spectra of carotenoid molecules in their triplet states (29–33). We therefore conclude that Fig. 3 B is the resonance Raman spectrum of LH-bound rhodopin glucoside in its triplet state.
Figure 3.
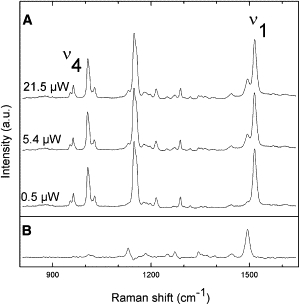
Raman spectra (800–1650cm−1) of the LH2 complex from Rbl. acidophilus in resonance with the carotenoid rhodopin glucoside. (A) The subpopulation attributed to triplet carotenoid increases with laser power. (B) The deduced triplet-state Raman spectrum of rhodopin glucoside. T = 77 K, λex =514.5 nm.
In LHCII trimers from higher plants, both the bound luteins may accept triplet states from chlorophyll. The T1→Tn triplet absorptions of these carotenoids peak (13,14) near the 514.5 and 528.0 nm excitation lines from an argon laser. The positions of these transitions are separated enough so that each of these triplets can be selectively observed in resonance Raman. This is in contrast to LHCII monomers and CP29, where the absorption spectra of the central luteins are degenerate, so that they both contribute equally to the measured spectra. Fig. 4 displays the ν1 and ν4 Raman bands of the lutein molecules in LHCII trimers, obtained when excited at 514.5 nm and 528.7 nm with low (black lines) and high (blue lines) laser intensities. It has been previously shown that the lutein 2 in higher plant LHCs is distorted, as seen by its Raman spectrum in the ν4 region (34). As for the LH2-bound carotenoid (Fig. 3 B), power-induced spectra obtained by difference highlight the characteristic downshifted triplet signal of the v1 mode (Fig. 4 A, red traces).
Figure 4.
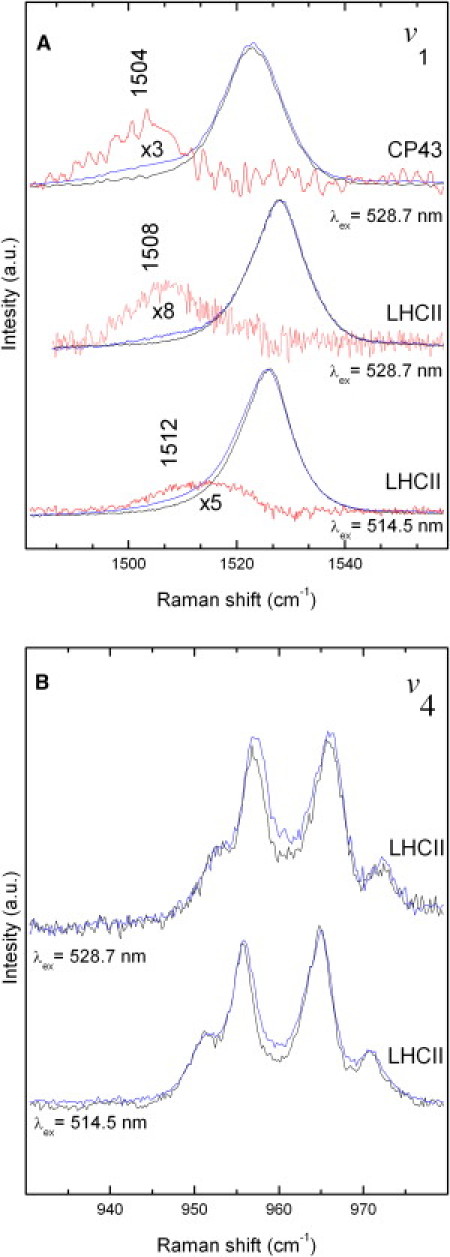
Power-induced triplet excited-state resonance Raman spectra in the v1 (A) and v4 (B) regions of the carotenoid molecules in LHCII trimers excited at 514.5 and 528.7 nm. The blue and black traces were obtained after excitation with 30 μW and 200 μW, respectively. The red traces are the difference spectra and are ascribed to the positions of the triplet states (these are magnified in the v1 region). Also shown is the power-induced triplet excited-state resonance Raman spectra in the v1 region of CP43 trimers when excited at 528.7 nm. T = 77 K.
As discussed in detail in Ruban et al. (35), the lowest electronic transitions of lutein 1 and 2 are located at 494 and 510 nm, respectively, and the triplet-triplet transitions of these molecules should accordingly contribute at 514 and 528 nm, respectively. The spectra of the ground- and triplet-states of lutein 1 are thus optimally obtained with excitation at 496 and 514 nm, respectively, whereas the contributions of the ground- and triplet states of lutein 2 require excitation at 510 and 528 nm. Thus, with an excitation wavelength of 514.5 nm at low power, the Raman spectra mainly arise from lutein 2 in its ground state, whereas the additional triplet bands observed at higher power arise from the lutein 1 molecule (cf. Table 1). When exciting at 528.7 nm, the triplet contribution observed at high power must arise only from lutein 2. The resonance Raman spectra of carotenoids bound to monomeric LHCII, the minor antenna CP29 (not shown; see Table 2) and the inner PSII antenna CP43 (Fig. 4 A) were also extracted using the same method. It is worth noting that for carotenoids in all antenna complexes, the frequencies and the shape of the bands in the ν4 region of resonance Raman spectra remain similar in both the ground and triplet states, in sharp contrast to the case of all-trans spheroidene in solution, where ν4 downshifts from 959 to 944 cm−1 when converted to the triplet state (33). This indicates that in these antenna complexes, carotenoid molecules do not change configuration when promoted to their triplet state, i.e., that their configuration is tightly locked by the protein environment.
Table 2.
Comparison of ν1 vibrational band position in resonance Raman spectra of ground and triplet states from protein-bound and other carotenoids
| Carotenoid | ν1 (cm−1) | Δν1 (cm−1) |
|---|---|---|
| LH2 rhodopin glucoside ground state | 1517 | 24 |
| LH2 rhodopin glucoside triplet | 1493 | |
| LHCII trimer Lutein1 ground state | 1530 | 18 |
| LHCII trimer Lutein 1 triplet | 1512 | |
| LHCII trimer Lutein 2 ground state | 1526 | 18 |
| LHCII trimer Lutein 2 triplet | 1508 | |
| LHCII monomer lutein ground state | 1526 | 18 |
| LHCII monomer lutein triplet | 1508 | |
| CP29 lutein ground state | 1526 | 18 |
| CP29 lutein triplet | 1508 | |
| CP43 lutein ground state | 1522 | 18 |
| CP43 lutein triplet | 1504 | |
| All-trans β-carotene singlet state in THF | 1522 | 25 |
| All-trans β-carotene triplet in THF | 1497 | |
| All-trans-spheroidene singlet state in n-hexane | 1523 | 23 |
| All-trans-spheroidene triplet in n-hexane | 1500 |
In solution, upon triplet-state formation, the frequency of stretching modes of the conjugated C=Cs of the molecule dramatically downshifts from 1522 to 1494 cm−1 for β-carotene in tetrahydrofuran (THF) (30,36), or from 1529 to 1500 cm−1 for all-trans spheroidene in hexane (33). throughout the conjugated system (triplet high spin density hot spot is mainly in the center of the conjugated system) (33,37), which causes a reduction in the C=C bond order. The frequencies of the ν1 resonance Raman bands observed in protein-bound carotenoid triplet spectra are reported in Table 2 and are there compared to those observed for triplet states of other carotenoids, including β-carotene in solution and spheroidene bound to the bacterial reaction center. The downshift depends on the carotenoid configuration, and it is always larger in cis configurations (29–33). For rhodopin glucoside in LH2, this downshift of the band from 1517 to 1493 cm−1 is very similar to that observed for all-trans β-carotene and spheroidene in solution (24 cm−1 vs. 23–25 cm−1). In contrast, the observed downshifts for this band for both luteins in LHCII trimers, LHCII monomers, CP29, and CP43 are much smaller and in all cases close to 18 cm−1, i.e., ∼75% of the 23–25 cm−1shift that is observed for all-trans carotenoid triplets in solution (see Table 2). Such a reduction of the ν1 band's downshift reflects a dramatic alteration of the nature of the triplet state, which correlates very well with a reduction in the energy gap between the S0/S2 and T1/Tn transitions observed for these carotenoids in LHCII (see Table 1; it is harder to determine this gap with precision for CP43 due to the larger number of carotenoid molecules in this complex).
Discussion
Our results are quite paradoxical at first sight. On the one hand, time-resolved absorption spectra show that no Chl a triplet is accumulated in LHCII, whereas, on the other hand, the FTIR difference spectra strongly suggest the presence of Chl a contributions long after the initial excitation. To explain this apparent contradiction, the simplest hypothesis is that the triplet could be shared between carotenoid and chlorophyll molecules. This question has already been addressed in the literature (10). To explain the influence of the carotenoid triplet state on the absorption bands of the BChl molecules in LH complexes from purple bacteria, Angerhofer et al. proposed “a small delocalization of the carotenoid triplet over an adjacent BChl molecule” (10). They also noted that the apparent rate of (B)Chl to carotenoid triplet-triplet transfer seems to correlate with how much the carotenoid triplet state is able to influence the (B)Chl transition. However, it should be pointed out that in LH complexes from purple bacteria, the carotenoid and (B)Chl molecules are located very close to each other (an essential condition for triplet/triplet transfer), and therefore they each constitute a sizeable part of the environment (or solvation) of the other. It would thus be expected that the dielectric changes after the appearance of the carotenoid triplet state would have a measurable influence on the (B)Chl absorption transitions. Hence, these previous results (10) do not formally demonstrate a sharing of the triplet. In contrast, the resonance Raman spectra reported in this work provides a direct and unambiguous measurement of the sharing of the triplet of the carotenoid molecule. Indeed, upon sharing, the carotenoid triplet should progressively loose its pure triplet character, and the Raman signature of this state should become intermediate between the ground and triplet states. In the case of rhodopin glucoside in LH2 from Rbl. acidophilus, the downshift of the ν1 band is quite similar to that of β-carotene or spheroidene in solution. We may thus safely conclude that there is very little, if any, wavefunction sharing between carotenoid and (B)Chl triplet states in the LH2 complex from Rbl. acidophilus.
In LHCs from higher plants, the presence of the triplet state of the lutein molecules is known to induce a net bleaching of the electronic absorption transition of the neighboring chlorophyll molecules (14). However, delocalization of the triplet was for a long time considered unlikely, due to the large energy gap between the triplet states of carotenoid and chlorophyll molecules (14). More recently, this position was challenged by time-resolved FTIR studies on peridinin-chlorophyll complexes, which demonstrated a co-existence of chlorophyll and carotenoid triplets throughout the entire triplet lifetime (27). Although the authors concluded that there could be a sharing of the triplet wavefunction between these two pigments, there is no other evidence for this conclusion apart from the contribution of chlorophyll spectral features during the carotenoid triplet lifetime. Here, we describe a similar situation in the LHCII of higher plants. In this case, however, resonance Raman spectroscopy provides additional information on the nature of the carotenoid triplet in these complexes. The sensitivity of the v1 bands of the ground and triplet states is expected to exhibit similar responses to the environment. However, in LHCII we observe a reduction of the energy gap between the v1 bands of the ground and triplet states by >30%. This indicates unambiguously that the electronic state gained by the carotenoid has lost a fraction of its carotenoid triplet character, and consequently, part of the triplet must be localized on another molecule. From the results of the step-scan time-resolved FTIR measurements (Fig. 2), we can safely conclude that a neighboring chlorophyll molecule is the acceptor of the carotenoid triplet. The fact that the same effect was found for both luteins to the same extent strongly substantiates this conclusion. Each lutein experiences a different protein environment (which induces the red shift of the electronic transitions of lutein 2 (38)) but is surrounded by a number of similarly positioned chlorophylls. The fact that both luteins exhibit the same 75/25 sharing of the triplet is thus most easily explained as a result of the pseudosymmetry of the position of these chlorophyll molecules (38). The same conclusion can be drawn for monomeric LHCII, CP29, and CP43.
A closer analysis of the recently obtained time-resolved FTIR data obtained for the PCP peridinin triplet supports this analysis. In FTIR, the intensity and frequency of the bands arising from the vibrational modes of the triplet state should reflect the triplet sharing. Since most of the bands are distorted by the differential method and by overlapping contributions, an accurate determination of their precise intensity and frequency is difficult except in the case of well-isolated bands. In the case of LHCII (Fig. 2), no carotenoid band can safely be used for that purpose. In the PCP spectra, the band arising from the stretching mode of the lactone carbonyl of peridinin is well isolated and contributes at ∼1745 cm−1. From FTIR steady-state measurements of peridinin mixed with Chl a in THF (data not shown), the peridinin C=O lactone extinction coefficient was estimated to be similar to that of the Chl a C=O keto group. This allows us to estimate the extent of the triplet sharing between peridinin and Chl a. It can be seen in Fig. 1 of Bonetti et al. (26) that the negative band area assigned to 9-keto C=O corresponds to ∼25% and 40% of the negative band area assigned to lactone C=O for A-PCP and H-PCP, respectively. Taking into account the similar C=O extinction coefficient of peridinin lactone C=O and Chl a 9-keto C=O, ∼25% and 40% of the 3peridinin is shared with Chl a in A-PCP and H-PCP, respectively. This conclusion is in good agreement with the relative amplitude of the Qy bleach as compared to the peridinin bleach of ∼20% in the T-S spectra of A-PCP (39). This estimate of a 25% and 40% triplet sharing in A-PCP and H-PCP is also in line with the smaller observed bandshift of the carbonyl lactone for H-PCP (i.e., 20 cm−1 as compared to 25 cm−1 for A-PCP).
Thus, in contrast to photosynthetic bacteria, our results provide compelling evidence of triplet sharing between carotenoid and chlorophyll molecules in plant and algal light-harvesting complexes. It is obviously tempting to try to unravel which molecular mechanisms may be at the origin of this difference. In LH2, contacts between carotenoid and bacteriochlorophyll molecules essentially occur at the very end of the C=C conjugated chain of the carotenoid (Fig. 5 A, and McDermott et al. (40)), and the minimum distance between these molecules is 3.42 Å (41). In strong contrast, the closest contacts between Chl a and luteins in LHCII occur at the middle of the C=C polyenic chain (Fig. 5 B), and the pigments are slightly more closely packed in these complexes. Taking into account the expected molecular structure of the carotenoid triplet state, the relative positioning of carotenoid and chlorophyll molecules in LHCII appears definitely more favorable for triplet sharing between these molecules
Figure 5.
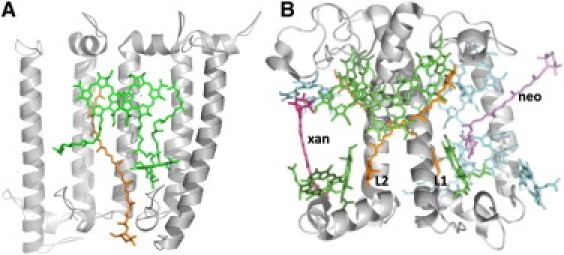
Organization of the (bacterio)chlorophyll and carotenoid molecules in LH2 and LHCII. (A) A slice of the nonameric structure of the LH2 complex from Rbl. acidophilus viewed in parallel with the membrane plane and from the outside of the protein. For clarity the central outer helix from three α/β-apoprotein dimers has been removed, allowing the interaction of the carotenoid (orange) with its nearest neighbor, (B)Chl a (green) molecules to be visualized. The contacts between Car and (B)Chl molecules essentially occur at the very end of the C=C conjugated chain of the carotenoid. Protein Data Bank accession number 1KZU. (B) View of a monomer of LHCII from Spinacia oleracea in parallel with the membrane plane. The colors of the two luteins (L), neoxanthin (neo), and xanthophyll (xan) cycle carotenoids are orange, purple, and magenta, respectively. The Chl a and Chl b molecules are colored green and blue, respectively. The closest contacts between Chl a and luteins in LHCII occur at the middle of the C=C polyenic chain. Although the Chl molecules have a pseudosymmetry within the monomer, lutein 1 (L1) and lutein 2 (L2) experience a different protein environment. Protein Data Bank accession number 1RWT.
To summarize, our results clearly show that the nature of the triplet excited state of carotenoid molecules are fundamentally different in the LH2 from Rbl. acidophilus and the antenna isolated from spinach. In the former case, the triplet state is mainly (if not totally) localized on the rhodopin glucoside molecule. According to the work of Angerhofer et al. (10), this is the case for the vast majority of light-harvesting proteins from purple photosynthetic bacteria. This localization of the triplet state is associated with a relatively slow triplet-triplet transfer between the BChl and carotenoid molecules, on the 20–200 ns timescale. In LHCII complexes from higher plants, there is a sharing of the triplet between luteins and their neighboring chlorophylls. As this is also the case in CP43 and CP29, as well as in PCP, it is likely that this triplet sharing exists in all light-harvesting proteins from plants and algae. This delocalization is associated with an ultrafast transfer/equilibration of the triplet between the chlorophyll and carotenoid molecules, which results in the absence of any measurable accumulation of pure triplet chlorophyll in these complexes. However, the price paid to avoid the accumulation of this species is that sharing of the triplet state between the chlorophyll and the carotenoid lasts for several microseconds. Apparently, such a mechanism drags the energy of the shared triplet below that of singlet oxygen. The resultant decrease in the probability of production of singlet oxygen thereby optimizes photoprotection of these complexes. It is striking that this tuning of photoprotection was found only in those organisms that perform photosynthesis in the presence of large amounts of molecular oxygen. We propose that triplet sharing represents an adaptation of the molecular mechanisms of protection against photooxidative stress, associated with the evolution of oxygenic photosynthesis.
Acknowledgments
This work was supported by the French Agence Nationale de la Recherche (ANR) through the BIOPHYSMEMBPROTS (A.G.) and CAROPROTECT (A.G., L.B., A.A.P., and B.R.) research grants, the French Foreign Ministry (ECONET contract 18905YD (B.R., A.G., and L.V.)), the European Union INTRO2 Marie Curie Research Training Network MRTN-CT-2003-505069 (M.T.A., R.B., P.H., J.T.M.K., A.A.P., A.R., B.R., and R.vG.), the Ile-de-France Region (France) through a Blaise Pascal International Research Chair (R.vG.), the European Research Council (ERC) through an Advanced Grant 2010 (R.vG. and B.R.), and the Earth and Life Sciences Council of the Netherlands Foundation for Scientific Research (NWO-ALW) through a Rubicon grant (R.B.) and a VIDI grant (J.T.M.K.).
Footnotes
T. A. Alexandre's present address is CEA, Institute of Biology and Technology of Saclay, and CNRS, Gif sur Yvette, France.
Katerina V. Stoitchkova's present address is Sofia University, Faculty of Physics, Department of Condensed Matter Physics, Sofia, Bulgaria.
Supporting Material
References
- 1.Truscott T.G., Land E.J., Sykes A. The in vitro photochemistry of biological molecules. 3. Absorption spectra, lifetimes and rates of oxygen quenching of the triplet states of β-carotene, retinal and related polyenes. Photochem. Photobiol. 1973;17:43–51. doi: 10.1111/j.1751-1097.1973.tb06329.x. [DOI] [PubMed] [Google Scholar]
- 2.Palozza P., Krinsky N.I. Antioxidant effects of carotenoids in vivo and in vitro: an overview. Methods Enzymol. 1992;213:403–420. doi: 10.1016/0076-6879(92)13142-k. [DOI] [PubMed] [Google Scholar]
- 3.Niyogi K.K., Björkman O., Grossman A.R. The roles of specific xanthophylls in photoprotection. Proc. Natl. Acad. Sci. USA. 1997;94:14162–14167. doi: 10.1073/pnas.94.25.14162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pascal A.A., Liu Z., Ruban A. Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature. 2005;436:134–137. doi: 10.1038/nature03795. [DOI] [PubMed] [Google Scholar]
- 5.Ruban A.V., Berera R., van Grondelle R. Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature. 2007;450:575–578. doi: 10.1038/nature06262. [DOI] [PubMed] [Google Scholar]
- 6.Ishikita H., Loll B., Knapp E.W. Function of two β-carotenes near the D1 and D2 proteins in photosystem II dimers. Biochim. Biophys. Acta. 2007;1767:79–87. doi: 10.1016/j.bbabio.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Wilson A., Punginelli C., Kirilovsky D. A photoactive carotenoid protein acting as light intensity sensor. Proc. Natl. Acad. Sci. USA. 2008;105:12075–12080. doi: 10.1073/pnas.0804636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foote C.S. Photosensitizatized oxidation and singlet oxygen: consequences in biological systems. In: Pryor W.A., editor. Free Radicals and Biological Systems. Academic Press; New York: 1976. pp. 85–133. [Google Scholar]
- 9.Monger T.G., Cogdell R.J., Parson W.W. Triplet states of bacteriochlorophyll and carotenoids in chromatophores of photosynthetic bacteria. Biochim. Biophys. Acta. 1976;449:136–153. doi: 10.1016/0005-2728(76)90013-x. [DOI] [PubMed] [Google Scholar]
- 10.Angerhofer A., Bornhaeuser F., Cogdell R.J. Optical and optically detected magnetic resonance investigation on purple photosynthetic bacterial antenna complexes. Chem. Phys. 1995;194:259–274. [Google Scholar]
- 11.Bittl R., Schlodder E., Cogdell R.J. Transient EPR and absorption studies of carotenoid triplet formation in purple bacterial antenna complexes. J. Phys. Chem. B. 2001;105:5525–5535. [Google Scholar]
- 12.Tavan P., Schulten K. Electronic excitations in finite and infinite polyenes. Phys. Rev. B. 1987;36:4337–4358. doi: 10.1103/physrevb.36.4337. [DOI] [PubMed] [Google Scholar]
- 13.Christensen R.L. The electronic states of carotenoids. In: Frank H.A., Young A.J., Britton G., Cogdell R.J., editors. Advances in Photosynthesis. Kluwer Academic; Dordrecht, The Netherlands: 1999. pp. 137–157. [Google Scholar]
- 14.Peterman E.J.G., Dukker F.M., van Amerongen H. Chlorophyll a and carotenoid triplet states in light-harvesting complex II of higher plants. Biophys. J. 1995;69:2670–2678. doi: 10.1016/S0006-3495(95)80138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schödel R., Irrgang K.D., Renger G. Rate of carotenoid triplet formation in solubilized light-harvesting complex II (LHCII) from spinach. Biophys. J. 1998;75:3143–3153. doi: 10.1016/S0006-3495(98)77756-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cogdell R.J., Durant I., Schmidt K. The isolation and partial characterization of the light-harvesting pigment-protein complement of Rhodopseudomonas acidophila. Biochim. Biophys. Acta. 1983;722:427–435. [Google Scholar]
- 17.Gall A., Fraser N.J., Cogdell R.J. Bacteriochlorin-protein interactions in native B800-B850, B800 deficient and B800-Bchla(p)-reconstituted complexes from Rhodopseudomonas acidophila, strain 10050. FEBS Lett. 1999;449:269–272. doi: 10.1016/s0014-5793(99)00410-x. [DOI] [PubMed] [Google Scholar]
- 18.Ruban A.V., Lee P.J., Horton P. Determination of the stoichiometry and strength of binding of xanthophylls to the photosystem II light harvesting complexes. J. Biol. Chem. 1999;274:10458–10465. doi: 10.1074/jbc.274.15.10458. [DOI] [PubMed] [Google Scholar]
- 19.Phillip D., Ruban A.V., Young A.J. Quenching of chlorophyll fluorescence in the major light-harvesting complex of photosystem II: a systematic study of the effect of carotenoid structure. Proc. Natl. Acad. Sci. USA. 1996;93:1492–1497. doi: 10.1073/pnas.93.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gradinaru C.C., van Stokkum I.H.M., van Amerongen H. Identifying the pathways of energy transfer between carotenoids and chlorophylls in LHCII and CP29. A multicolor, femtosecond pump-probe study. J. Phys. Chem. B. 2000;104:9330–9342. [Google Scholar]
- 21.van Stokkum I.H., Larsen D.S., van Grondelle R. Global and target analysis of time-resolved spectra. Biochim. Biophys. Acta. 2004;1657:82–104. doi: 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Bonetti C., Alexandre M.T., Kennis J.T. Identification of excited-state energy transfer and relaxation pathways in the peridinin-chlorophyll complex: an ultrafast mid-infrared study. Phys. Chem. Chem. Phys. 2010;12:9256–9266. doi: 10.1039/b923695c. [DOI] [PubMed] [Google Scholar]
- 23.Mozzo M., Dall'Osto L., Croce R. Photoprotection in the antenna complexes of photosystem II: role of individual xanthophylls in chlorophyll triplet quenching. J. Biol. Chem. 2008;283:6184–6192. doi: 10.1074/jbc.M708961200. [DOI] [PubMed] [Google Scholar]
- 24.Jensen S.L., Hertzber S. Selective preparation of lutein monomethyl ethers. Acta Chem. Scand. 1966;20:1703. [Google Scholar]
- 25.Breton J., Nabedryk E., Leibl W. FTIR study of the primary electron donor of photosystem I (P700) revealing delocalization of the charge in P700+ and localization of the triplet character in 3P700. Biochemistry. 1999;38:11585–11592. doi: 10.1021/bi991216k. [DOI] [PubMed] [Google Scholar]
- 26.Bonetti C., Alexandre M.T.A., Grondelle R.v. Chl-a triplet quenching by peridinin in H-PCP and organic solvent revealed by step-scan FTIR time-resolved spectroscopy. Chem. Phys. 2009;357:63–69. [Google Scholar]
- 27.Alexandre M.T.A., Lührs D.C., van Grondelle R. Triplet state dynamics in peridinin-chlorophyll-a-protein: a new pathway of photoprotection in LHCs? Biophys. J. 2007;93:2118–2128. doi: 10.1529/biophysj.107.106674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi H., Noguchi T., Tasumi M. Studies on the interrelationship among the intensity of a Raman marker band of carotenoids, polyene chain structure, and efficiency of the energy transfer from carotenoids to bacteriochlorophyll in photosynthetic bacteria. Photochem. Photobiol. 1989;49:337–343. [Google Scholar]
- 29.Rondonuwu F.S., Taguchi T., Watanabe Y. The energies and kinetics of triplet carotenoids in the LH2 antenna complexes as determined by phosphorescence spectroscopy. Chem. Phys. Lett. 2004;384:364–371. [Google Scholar]
- 30.Hashimoto H., Koyama Y., Mataga N. S1 and T1 species of β-carotene generated by direct photoexcitation from the all-trans, 9-cis, 13-cis, and 15-cis isomers as revealed by picosecond transient absorption and transient Raman spectroscopies. J. Phys. Chem. 1991;95:3072–3076. [Google Scholar]
- 31.Ohashi N., KoChi N., Koyama Y. The structures of S-0 spheroidene in the light-harvesting (LH2) complex and S-0 and T-1 spheroidene in the reaction center of Rhodobacter sphaeroides 2.4.1 as revealed by Raman spectroscopy. Biospectroscopy. 1996;2:59–69. [Google Scholar]
- 32.Hashimoto H., Koyama Y. Time-resolved resonance Raman-spectroscopy of triplet β-carotene produced from all-trans, 7-cis, 9-cis, 13-cis, and 15-cis isomers and high-pressure liquid-chromatography analyses of photoisomerization via the triplet-state. J. Phys. Chem. 1988;92:2101–2108. [Google Scholar]
- 33.Mukai-Kuroda Y., Fujii R., Koyama Y. Changes in molecular structure upon triplet excitation of all-trans-spheroidene in n-hexane solution and 15-cis-spheroidene bound to the photo-reaction center from Rhodobacter sphaeroides as revealed by resonance-Raman spectroscopy and normal-coordinate analysis. J. Phys. Chem. A. 2002;106:3566–3579. [Google Scholar]
- 34.Ruban A.V., Pascal A.A., Robert B. Xanthophylls of the major photosynthetic light-harvesting complex of plants: identification, conformation and dynamics. FEBS Lett. 2000;477:181–185. doi: 10.1016/s0014-5793(00)01799-3. [DOI] [PubMed] [Google Scholar]
- 35.Ruban A.V., Pascal A., Horton P. Molecular configuration of xanthophyll cycle carotenoids in photosystem II antenna complexes. J. Biol. Chem. 2002;277:42937–42942. doi: 10.1074/jbc.M207823200. [DOI] [PubMed] [Google Scholar]
- 36.Fujiwara M., Yamauchi K., Hashimoto H. Energy dissipation in the ground-state vibrational manifolds of β-carotene homologues: a sub-20-fs time-resolved transient grating spectroscopic study. Phys. Rev. B. 2008;77:205118. [Google Scholar]
- 37.Di Valentin M., Ceola S., Carbonera D. Pulse ENDOR and density functional theory on the peridinin triplet state involved in the photo-protective mechanism in the peridinin-chlorophyll a-protein from Amphidinium carterae. Biochim. Biophys. Acta. 2008;1777:295–307. doi: 10.1016/j.bbabio.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z., Yan H., Chang W. Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature. 2004;428:287–292. doi: 10.1038/nature02373. [DOI] [PubMed] [Google Scholar]
- 39.Kleima F.J., Hofmann E., van Amerongen H. Förster excitation energy transfer in peridinin-chlorophyll-a-protein. Biophys. J. 2000;78:344–353. doi: 10.1016/S0006-3495(00)76597-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDermott G., Prince S.M., Isaacs N.W. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 1995;374:517–521. [Google Scholar]
- 41.Prince S.M., Papiz M.Z., Isaacs N.W. Apoprotein structure in the LH2 complex from Rhodopseudomonas acidophila strain 10050: modular assembly and protein pigment interactions. J. Mol. Biol. 1997;268:412–423. doi: 10.1006/jmbi.1997.0966. [DOI] [PubMed] [Google Scholar]
- 42.Lampoura S.S., Barzda V., van Amerongen H. Aggregation of LHCII leads to a redistribution of the triplets over the central xanthophylls in LHCII. Biochemistry. 2002;41:9139–9144. doi: 10.1021/bi025724x. [DOI] [PubMed] [Google Scholar]
- 43.Croce R., Mozzo M., Bassi R. Singlet and triplet state transitions of carotenoids in the antenna complexes of higher-plant photosystem I. Biochemistry. 2007;46:3846–3855. doi: 10.1021/bi602531k. [DOI] [PubMed] [Google Scholar]
- 44.Peterman E.J., Gradinaru C.C., van Amerongen H. Xanthophylls in light-harvesting complex II of higher plants: light harvesting and triplet quenching. Biochemistry. 1997;36:12208–12215. doi: 10.1021/bi9711689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.