

Abstract
We have developed a heterobifunctional all-small molecule PROTAC (PROteolysis TArgeting Chimera) capable of inducing proteasomal degradation of the androgen receptor. This cell-permeable PROTAC consists of a non-steroidal androgen receptor ligand (SARM) and the MDM2 ligand known as nutlin, connected by a PEG-based linker. The SARM-nutlin PROTAC recruits the androgen receptor to MDM2, which functions as an E3 ubiquitin ligase. This leads to the ubiquitination of the androgen receptor, and its subsequent degradation by the proteasome. Upon treatment of HeLa cells with 10 μM PROTAC for 7h, we were able to observe a decrease in androgen receptor levels. This degradation is proteasome dependent, as it is mitigated in cells pre-treated with 10 μM epoxomicin, a specific protease inhibitor. These results have implications for the potential study and treatment of various cancers with increased androgen receptor levels.
The rational design of small molecules to probe biological systems is one of the primary goals of chemical biology. These bioorganic strategies are now providing the opportunity to explore one of the central challenges in biology: deciphering protein function within the cell.1-4 This task has traditionally been addressed by the removal or inhibition of the protein of interest in order to observe the biological consequences of its loss. The most common techniques for achieving this operate at the DNA or RNA level (i.e genetic knockout or RNAi, respectively). Though well-established, these methods can be cumbersome, often lack temporal control, and generally require some level of genetic manipulation. We are interested in developing small molecule probes to add to the chemical biology toolbox, which would allow improved control over protein expression levels without the need for genetic intervention.5 In recent years, we have developed a method to accomplish this by inducing selective post-translational protein degradation in vivo via heterobifunctional PROteolysis TArgeting Chimerae (PROTACs).6,7 Herein we describe a significant improvement to the PROTAC strategy, utilizing an all-small molecule-based design.
The PROTAC strategy employs the protein degradation machinery of the cell: the ubiquitin-proteasome pathway. Proteins removed via this pathway are targeted for degradation by the attachment of a polyubiquitin chain, leading to their recognition and subsequent degradation by the 26S proteasome.8 The selectivity of this process is governed by the recognition of the targeted protein by an E3 ubiquitin ligase associated with an E2 conjugating enzyme coupled to ubiquitin. Upon binding of the target protein, ubiquitin is transferred to an accessible lysine residue, thus labeling the protein for proteasome-mediated degradation. The PROTAC strategy seeks to exploit this degradation pathway by promoting the non-natural ubiquitination of a targeted protein. The PROTAC molecule consists of a ligand, which binds an E3 ubiquitin ligase, connected by a linker to another ligand that binds the target protein. The association between a protein and an E3 ligase, as induced by a PROTAC molecule, will lead to the transfer of ubiquitin and degradation of the targeted protein (Figure 1).
Figure 1.

PROTAC mechanism of action: The heterobifunctional PROTAC molecule contains two binding motifs: one recruits an E3 ligase, and another recruits the target protein. Following binding to the PROTAC, the E3 ligase catalyzes the synthesis of a polyubiquitin chain on the target, leading to its recognition by the 26S proteasome, and subsequent target protein degradation.
We have previously shown that a heterobifunctional PROTAC can actively induce selective intracellular protein degradation.6, 7 However, this first generation of PROTAC molecules contained a peptide-based ligand for the E3 ligase, which limited its permeability. The subsequent addition of a polyarginine chain mimicking the HIV viral TAT 9 protein overcame cell permeability issues in the next generation of PROTACs. While significant biological activity was seen using these early PROTACs, the synthesis, purification and stability issues associated with their high molecular weight and vulnerable peptide bonds, limited their broad applicability. We are now able to report an all-small molecule-based PROTAC that can efficiently induce intracellular protein degradation while bypassing the drawbacks associated with the peptide moiety in the previous generation of compounds.
The PROTAC molecule itself contains three distinct portions: a ligand for binding to the target protein, a ligand for binding to an E3 ligase, and a linker joining these two ligands. For this proof of concept all-small molecule PROTAC we chose to target the androgen receptor (AR), as we have had success degrading this protein previously using a peptide-based PROTAC.7 In addition, AR is an attractive target because it has been shown to promote the growth of prostate tumor cells, while the inhibition of AR has been shown to repress tumor cell growth.10 Moreover, there are a variety of known small molecule ligands for AR. For this PROTAC we opted to use a selective androgen receptor modulator (SARM), which binds AR with a Ki of 4 nM.11
We chose to recruit targeted the androgen receptor via this class of PROTACs to the E3 ligase MDM2, which is a 90 kDa protein whose natural substrate is p53.12 Recently a new class of imidazoline derivatives that bind MDM2 has been identified.13 These compounds, called nutlins, have been shown to disrupt the binding of MDM2 and its natural ligand p53 with IC50 values in the nano- to micromolar range, leading to the stabilization of p53 protein levels. For use in our all-small molecule PROTAC, a nutlin derivative (racemic nutlin-3, MDM2 binding affinity: enantiomer a IC50=13.6 μM, enantiomer b IC50=0.09 μM)13 was prepared utilizing an efficient one-pot condensation/oxidation strategy (Scheme 1).14,15 The triaryl imidazoline substrate was easily accessed via condensation of diamine 2 with substituted benzaldehyde 1. The intermediate 2,4,5 triarylimidazolidine was oxidized in situ by addition of NBS to the reaction mixture to provide the cis-imidazoline 3 in good yield. 5 and 6 were combined in the presence of base to give ester 10, which was then treated under standard hydrogenolysis conditions to give amine 8. Subsequent urea formation was performed as previously reported, to produce the acid 4.16
Scheme 1.
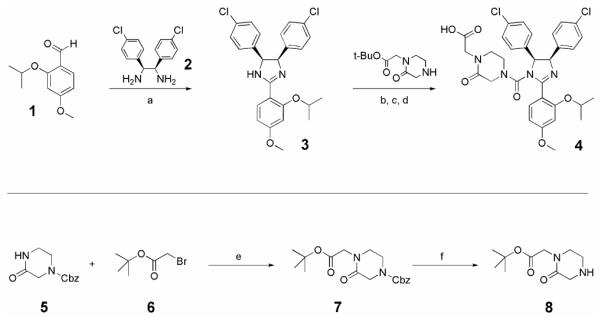
Synthesis of nutlin derivative 4
Reagents and conditions: (a) 2, CH2Cl2, 0°C, 2h, then NBS, 0°C-rt, 16h, 88%; (b) Triphosgene, Et3N, THF, 0°C, 2.5h; (c) 8, CH2Cl2, 0°C, 1.5h, 96%; (d) TFA, CH2Cl2, 96%; (e) 5, NaH, DMF, 0°C, 30min, then 6, rt, 4h; (f) H2, 10% Pd/C, MeOH, rt, 16h
A short soluble PEG linker was chosen to bridge the two terminal protein ligands; synthesis of this linker is shown in Scheme 2.17 First, azido alcohol 9 was treated with sodium iodoacetate in the presence of base to give acid 10, which was subsequently treated with 4-aminophenol hydrochloride under standard peptide coupling conditions (EDCI, HOBt) to afford phenol 11 in good yield. Having developed an efficient strategy for the preparation of nutlin derivatives, the synthesis of the SARM-nutlin PROTAC 14 proceeded smoothly as shown in Scheme 3.18 Epoxide 12 was prepared as previously reported using D-proline as a chiral auxiliary.11 Epoxide opening of 12 with phenol 11 was followed by a Staudinger reduction of the azide to provide amine 13. Coupling of 13 to the acid 4 afforded a diastereomeric mixture of the SARM-nutlin PROTAC, 14.
Scheme 2.
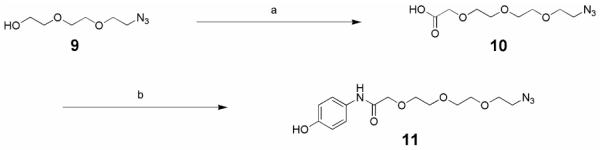
Synthesis of derivatized PEG linker 11
Reagents and conditions: (a) NaH, DMF, 0°C, 30 min., then NaIAc, rt, 32h; (b) CHCl2, 4-aminophenol hydrochloride, HOBt, DIPEA, rt, then 0°C, EDCI, then rt, 18h
Scheme 3.
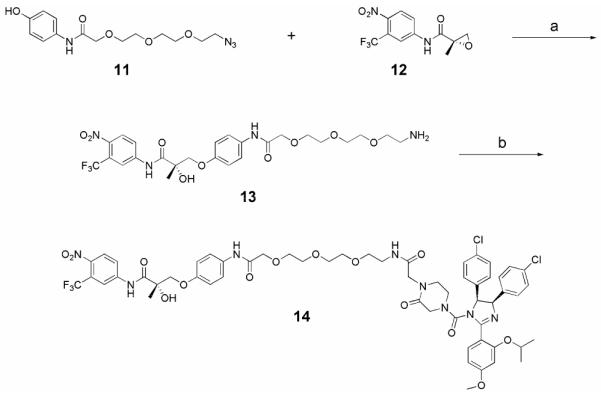
Synthesis of SARM-nutlin PROTAC 14
Reagents and conditions: (a) (i) K2CO3, 2-PrOH, 85°C; (ii) PPh3, H2O, THF, rt; (b) 4, HATU, DIPEA, DMF, rt
The SARM-nutlin PROTAC was tested for induced intracellular protein degradation using HeLa (human cervical carcinoma) cells transiently expressing the androgen receptor. Cells were treated with 10 μM PROTAC or vehicle alone, and incubated at 37°C. Cell lysates were prepared after 7h, separated by SDS-PAGE, and transferred to nitrocellulose membrane. Western blots were assessed using an anti-AR antibody to determine AR protein levels.19 We were able to observe a reproducible decrease in total AR in cells treated with the PROTAC as compared to the vehicle-treated cells (Figure 2). To verify that androgen receptor degradation induced by the PROTAC is proteasome dependent, we pre-treated cells with the specific proteasome inhibitor epoxomicin, at 10 μM concentration for 1h.20 These cells were then treated with either vehicle or 10 μM PROTAC. Again, cells were incubated for 7h, harvested, and analyzed by Western blot with anti-AR antibody.21 These results, shown in Figure 2, clearly show that PROTAC-mediated degradation is proteasome dependent, as AR degradation is inhibited in the presence of epoxomicin. A slight accumulation of AR is noticeable in epoxomicin-treated cells, reflecting the loss of normal protein turnover due to proteasome inhibition.
Figure 2.
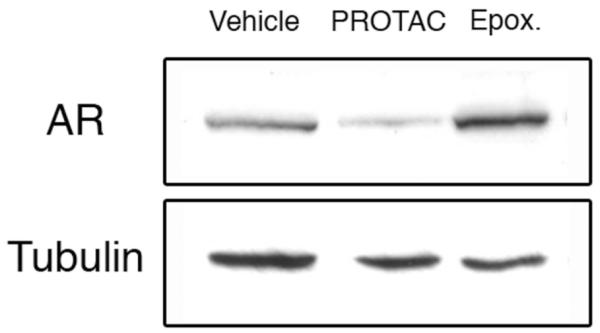
The SARM-nutlin PROTAC effectively degrades the androgen receptor (AR) in vivo. HeLa cells transiently expressing AR were treated with either vehicle, 10 μM PROTAC 14, or pre-treated with 10 μM epoxomicin and then 10 μM PROTAC, for 7h. Cells were then lysed and separated by SDS-PAGE. The total level of AR was detected by Western blotting using an anti-AR antibody (top). As a loading control, the same membrane was probed using an anti-α-tubulin antibody (bottom).
In summary, we report here the first synthesis of an all-small molecule PROTAC. We have shown this compound to be both cell permeable and capable of promoting selective interaction between an E3 ligase and a target protein, leading to specific intracellular protein degradation. We have also shown this degradation to be proteasome dependent. These results are an important step in the pursuit of small molecule PROTAC libraries, which could potentially provide new insights into a number of important biological pathways through loss-of-function phenotypes. In addition, these results lend support for potential PROTAC-based therapeutic approaches in the treatment of various diseases.
Acknowledgements
A.R.S. thanks the NSF for a predoctoral fellowship. M.P. thanks the Human Frontier Science Program Organization for a cross-disciplinary post-doctoral fellowship. This work was partially supported by the Korea Research Foundation Grant (KRF-2005-214-C00218) to H.S.T. This work was partially supported by the National Institutes of Health (CA118631). We gratefully acknowledge John Schneekloth and Nicholas Aberle for their thorough review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Mitchison TJ. Chem. Biol. 1994;1:3. doi: 10.1016/1074-5521(94)90034-5. [DOI] [PubMed] [Google Scholar]
- (2).Crews CM, Splittgerber U. Trends Biochem. Sci. 1999;24:317. doi: 10.1016/s0968-0004(99)01425-5. [DOI] [PubMed] [Google Scholar]
- (3).Sieber SA, Cravatt BF. Chem. Commun. 2006:2311. doi: 10.1039/b600653c. [DOI] [PubMed] [Google Scholar]
- (4).Alaimo PJ, Shogren-Knaak MA, Shokat K. Curr. Opin. Chem. Biol. 2001;5:360. doi: 10.1016/s1367-5931(00)00215-5. [DOI] [PubMed] [Google Scholar]
- (5).Schneekloth JS, Jr., Crews CM. ChemBioChem. 2005;6:40. doi: 10.1002/cbic.200400274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Proc. Natl. Acad. Sci. U.S.A. 2001;98:8554. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Schneekloth JS, Jr., Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. J. Am. Chem. Soc. 2004;126:3748. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- (8).Ciechanover A, Orian A, Schwartz AL. BioEssays. 2000;22:442. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- (9).Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13003. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Debes JD, Schmidt LJ, Huang H, Tindall DJ. Cancer Res. 2002;62:5632. [PubMed] [Google Scholar]
- (11).Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl C, Dalton JT, Miller DD. J. Med. Chem. 2004;47:993. doi: 10.1021/jm030336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Piette J, Neel H, Marechal V. Oncogene. 1997;15:1001. doi: 10.1038/sj.onc.1201432. [DOI] [PubMed] [Google Scholar]
- (13).Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- (14).Fujioka H, Murai K, Ohba Y, Hiramatsu A, Kita Y. Tetrahedron Lett. 2005;46:2197. [Google Scholar]
- (15).Experimental: Ester 7. To a solution of 1-benzyloxycarbonyl-3-oxopiperazine (234 mg, 1.0 mmol) in DMF (3 mL) at 0°C was added NaH (60%, 48 mg, 1.2 mmol). After stirring at 0 °C for 30 min, tert-butyl bromoacetate (160 μL, 1.1 mmol) was added to the mixture, which was then stirred at room temperature (rt) for 3.0 h. The resulting mixture was quenched with H2O (10 mL) and the aqueous solution was extracted twice with ethyl acetate. The extracts were dried over Na2SO4, filtered, and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:19 methanol-dichloromethane) to furnish 338 mg (97%) of ester 7. 1H NMR (400 MHz, CD3OD) δ 7.37-7.35 (m, 3H), 7.33-7.30 (m, 2H), 5.15 (s, 2H), 4.14 (s, 2H), 4.05 (s, 2H), 3.75 (s, 2H), 3.45 (t, J = 4.8 Hz, 2H), 1.45 (s, 9H). 13C NMR (100 MHz, CD3OD) δ 169.3, 169.2, 156.1, 137.7, 129.6, 129.2, 129.0, 83.3 (2C), 68.7 (2C), 50.1, 48.2, 28.2. TLC (10% CH3OH in CH2Cl2), Rf 0.58 (UV, CAM). Amine 8. To a solution of ester 7 (300 mg, 0.861 mmol) in MeOH (8.5 mL) at rt was added 10% Pd/C (26 mg, 30 mg/mmol). After introducing H2, the reaction mixture was stirred vigorously for 16 h. The resulting mixture was filtered through celite and the filtrate was concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:19 methanol-dichloromethane) to provide 172 mg (93%) of amine 8. 1H NMR (400 MHz, CDCl3) δ 4.02 (s, 2H), 3.55 (s, 2H), 3.37 (d, J = 5.4 Hz, 2H), 3.11 (d, J = 5.4 Hz, 2H), 1.72 (s, 1H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 168.4, 167.9, 82.0, 50.0, 49.2, 48.7, 43.0, 28.0. HRMS (ES+) calculated for C10H17N2O3Na [M+Na]+ 237.1209, found 237.1204. TLC (10% CH3OH in CH2Cl2), Rf 0.47 (Ninhydrin). Imidazoline 3. To a solution of 2-isopropoxy 4-methoxybenzaldehyde 1 (2.5 g, 12.88 mmol) in CH2Cl2 (130 mL) was added 1,2-bis(4-chlorophenyl)-1,2-ethanediamine (3.65 g, 12.98 mmol). After stirring at 0 °C for 2.0 h, NBS (2.5 g, 14.04 mmol) was added to the mixture. The reaction mixture was warmed to rt and further stirred at rt for 16 h. The resulting mixture was quenched with 10% KOH aqueous solution and the mixture was extracted three times with CH2Cl2. The extracts were dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:19 methanol-dichloromethane) to give 5.14 g (88%) of imidazoline 3. 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 8.7 Hz, 1H), 7.75 and 7.42 (d, J = 8.4 Hz, 1H), 7.04 (d, J = 8.4 Hz, 4H), 6.90 (d, J = 8.4 Hz, 4H), 6.61 (dd, J = 8.8, 2.3 Hz, 1H), 6.54 (d, J = 2.3 Hz, 1H), 5.36 (s, 2H), 4.74-4.68 (m, 1H), 3.86 (s, 3H), 1.37 (d, J = 6.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 164.1, 163.0, 157.3, 137.9, 133.0, 129.1, 128.8, 127.8, 127.4, 111.9, 105.5, 100.6, 77.2, 71.3, 55.6, 22.2. LRMS (ES+) [M+H]+ 455.4, [M+Na]+ 477.4. HRMS (ES+) calculated for C25H24N2O2Cl2 [M+H]+ 455.1287, found 455.1286. TLC (10% CH3OH in CHCl2), Rf 0.34 (UV). Nutlin tert-butyl ester. To a solution of 4,5-bis-(4-chloro-phenyl)-2-(2-isopropoxy-4-methoxy-phenyl)-4,5-dihydro-1H-imidazole 3 (15 mg, 0.033 mmol) dissolved in THF (1 mL) at 0 °C were added Et3N (24 uL, 0.174 mmol) and triphosgene (76 mg, 0.257 mmol). After stirring at 0 °C for 2.5 h, the mixture was evaporated and evacuated for 0.5 h. To the residue dissolved in CH2Cl2 (0.5 mL) at 0 °C was added dropwise a solution of amine 8 (110 mg, 0.515 mmol) in CH2Cl2 (0.5 mL) via cannula. The resulting mixture was stirred at 0 °C for 1.5 h. The mixture was quenched with saturated NaHCO3 aqueous solution (5 mL). The mixture was extracted three times with CH2Cl2 and the extracts were washed with saturated NaCl, dried over Na2SO4, filtered, and concentrated. The residue was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:19 methanol-dichloromethane) to give 22 mg (96%) of Nutlin ester. 1H NMR (500 MHz, CDCl3) δ 7.58 (d, J = 8.5 Hz, 1H), 7.07 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 6.54 (dd, J = 8.4, 2.2 Hz, 1H), 6.47 (d, J = 2.2 Hz, 1H), 5.55 (d, J = 9.7 Hz, 1H), 5.47 (d, J = 9.7 Hz, 1H), 4.64-4.57 (m, 1H), 4.02 (d, J = 10.0 Hz, 1H), 3.94 (d, J = 10.0 Hz, 1H), 3.84 (s, 3H), 3.76 (d, J = 9.1 Hz, 1H), 3.71 (d, J = 10.9 Hz, 1H), 3.52-3.45 (m, 1H), 3.33-3.28 (m, 1H), 3.07-3.05 (m, 2H), 1.42 (s, 9H), 1.38 (d, J = 6.0 Hz, 3H), 1.34 (d, J = 6.0 Hz, 3H). LRMS (ES+) [M+H]+ 695.39, [M+Na]+ 717.14. TLC (10% CH3OH in CHCl3), Rf 0.54 (UV, CAM). Nutlin acid 4. To a solution of Nutlin ester (14 mg, 0.020 mmol) dissolved in CH2Cl2 (1 mL) at 0 °C was added TFA (0.2 mL). After stirring at rt for 17 h, the resulting mixture was diluted with toluene (1 mL) and evaporated. The residue was purified by flash column chromatography (3:99 methanol-dichloromethane initially, grading to 1:9 methanol-dichloromethane) to give 12.3 mg (96%) of Nutlin acid 4. 1H NMR (400 MHz, CD3OD) δ 7.58 (d, J = 8.3 Hz, 1H), 7.13 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.6 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.3 Hz, 2H), 6.68-6.65 (m, 2H), 5.77 (d, J = 10.0 Hz, 1H), 5.57 (d, J = 10.0 Hz, 1H), 4.76-4.70 (m, 1H), 3.87 (s, 3H), 3.83-3.72 (m, 4H), 3.55-3.48 (m, 1H), 3.41-3.35 (m, 1H), 3.08-3.04 (m, 2H), 1.38 (d, J = 6.0 Hz, 3H), 1.35 (d, J = 6.0 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 167.0, 165.1, 163.4, 158.6, 155.6, 137.3, 136.2, 134.2, 133.1, 130.7, 129.9, 129.1, 129.0, 113.4, 106.5, 101.2, 72.5, 71.7, 70.1, 56.1, 50.4, 47.8, 43.1, 42.9, 22.5, 22.4, 13.4. HRMS (ES+) calculated for C32H32N4O6Cl2 [M+H]+ 639.1771, found 639.1779. [α]D20 + 6.8° (c = 0.365, MeOH). TLC (10% CH3OH in CHCl2), Rf 0.09 (UV, CAM).
- (16).Kong N, Liu EA, Vu BT. Preparation of cis-2,4,5-triphenylimidazolines and their use in the treatment of tumors. WO2003051359 Patent. 2003
- (17).Experimental: Azido alcohol 9. To a solution of triethylene glycol (1.185 g, 7.892 mmol) in CH2Cl2 (20 mL) at 0 °C were added Ag2O (2.01 g, 8.681 mmol) and MsCl (0.73 mL, 9.470 mmol). The black suspension was stirred at 0 °C for 10 min, allowed to warm to rt, and further stirred at rt for 26 h. The resulting mixture was filtered through celite with CH2Cl2. The filtrate was concentrated in vacuo and the residue was purified by short column chromatography to give 1.8 g of mono-mesylated alcohol. To a solution of the alcohol (1.8 g, 7.892 mmol) in DMF (25 mL) at rt was added NaN3 (770 mg, 11.838 mmol). The reaction mixture was heated to 110 °C, stirred at 110 °C for 5.5 h, and cooled to 0 °C. The resulting mixture was quenched with water (50 mL) and the mixture was extracted three times with ethyl acetate. The extracts were washed with saturated NaCl solution, dried over Na2SO4, filtered, and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:19 methanol-dichloromethane) to give 1.285 g (93%) of azido alcohol 9. 1H NMR (400 MHz, CDCl3) δ 3.73-3.71 (m, 2H), 3.67-3.66 (m, 6H), 3.60 (t, J = 4.2 Hz, 2H), 3.39 (t, J = 4.8 Hz, 2H), 2.32 (t, J = 6.2 Hz, 1H), 13C NMR (100 MHz, CDCl3) δ 72.5, 70.6, 70.4, 70.1, 61.8, 50.6. HRMS (ES+) calculated for C6H12N3O3Na [M+Na]+ 198.0849, found 198.0846. TLC (10% CH3OH in EtOAc), Rf 0.58 (CAM). Acid 10. To a solution of azido alcohol 9 (780 mg, 4.454 mmol) in DMF (4.5 mL) at 0 °C was added NaH (60%, 178 mg, 4.454 mmol). After stirring at 0 °C for 30 min, sodium iodoacetate (926 mg, 4.454 mmol) was added to the mixture. The reaction mixture was stirred at rt for 32 h and quenched with 1N-HCl in an ice bath. The mixture was extracted three times with ethyl acetate. The extracts were washed with saturated NaCl solution, dried over Na2SO4, filtered, and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:9 methanol-dichloromethane) to provide 860 mg (83%) of acid 10. 1H NMR (500 MHz, CDCl3) δ 8.02 (s, 1H), 4.16 (s, 2H), 3.77-3.75 (m, 2H), 3.71-3.70 (m, 4H), 3.68-3.66 (m, 4H), 3.39 (t, J = 4.9 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 172.5, 71.4, 70.7, 70.4, 70.2, 70.0, 68.7, 50.6. HRMS (ES+) calculated for C8H14N3O5Na [M+Na]+ 256.0904, found 256.0902. TLC (10% CH3OH in EtOAc), Rf 0.12 (CAM). Phenol 11. To a solution of acid 10 (250 mg, 1.072 mmol) and 4-aminophenol hydrochloride (156 mg, 1.072 mmol) in CH2Cl2 (4 mL) at rt were added HOBt (174 mg, 1.286 mmol) and DIPEA (0.47 mL, 2.680 mmol). The mixture was cooled to 0 °C and EDCI (226 mg, 1.179 mmol) was added to the mixture. The reaction mixture was allowed to room temperature and further stirred at rt for 18 h. The resulting mixture was quenched with H2O in an ice bath. The mixture was extracted three times with ethyl acetate. The extracts were washed with saturated NaCl solution, dried over Na2SO4, filtered, and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:9 methanol-dichloromethane) to afford 302 mg (87%) of phenol 11. 1H NMR (400 MHz, CDCl3) δ 8.69 (s, 1H), 7.36 (d, J = 8.8 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H), 4.10 (s, 2H), 3.77-3.71 (m, 6H), 3.67-3.64 (m, 2H), 3.58 (t, J = 5.2 Hz, 2H), 3.29 (t, J = 5.2 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 168.5, 153.3, 129.7, 122.5, 115.7, 71.1, 70.7, 70.5, 70.4, 70.3, 70.0, 50.6. HRMS (ES+) calculated for C14H19N4O5Na [M+Na]+ 347.1325, found 347.1324. TLC (10% CH3OH in EtOAc), Rf 0.51 (UV, CAM).
- (18).Experimental: Amine 13. To a solution of phenol 11 (31 mg, 0.094 mmol) and epoxide 12 (19 mg, 0.065 mmol) in 2-PrOH (1 mL) at rt was added K2CO3 (14 mg, 0.097 mmol). After refluxing for 2.0 h, the resulting mixture was cooled to room temperature and 2-PrOH was removed in vacuo. The residue was diluted with H2O (3 mL) and the mixture was extracted three times with ethylacetate and the extracts were washed with saturated NaCl, dried over Na2SO4, filtered, and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:19 methanol-dichloromethane) to give 48.5 mg (84%) of azide. 1H NMR (500 MHz, CDCl3) δ 9.29 (s, 1H), 8.73 (s, 1H), 8.11 (d, J = 2.1 Hz, 1H), 8.01 (dd, J = 8.9, 2.2 Hz, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.46 (d, J = 8.9 Hz, 2H), 6.83 (d, J = 8.9 Hz, 2H), 4.39 (d, J = 9.1 Hz, 1H), 4.09 (s, 2H), 3.94 (d, J = 9.1 Hz, 1H), 3.76-3.75 (m, 2H), 3.73-3.70 (m, 4H), 3.65 (dd, J = 4.9, 2.5 Hz, 2H), 3.59 (t, J = 5.4 Hz, 2H), 3.30 (t, J = 5.1 Hz, 2H), 1.78 (s, 1H), 1.56 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 172.7, 168.1, 154.5, 143.3, 141.5, 131.8, 127.1, 122.0, 121.8, 118.3, 115.2, 75.8, 72.7, 71.2, 70.7, 70.5, 70.4, 70.3, 70.0, 50.6, 23.0. [α]D20 + 24.7° (c = 0.230, MeOH). LRMS (ES+) [M+Na]+ 637.35. HRMS (ES+) calculated for C25H28N6O9F3 [M+Na]+ 637.1840, found 637.1838. TLC (5% CH3OH in CHCl2), Rf 0.38 (UV, CAM). To a solution of azide (20 mg, 0.0325 mmol) in THF (0.5 mL) at rt were added PPh3 (11 mg, 0.0423 mmol) and H2O (1 drop). The reaction mixture was stirred at rt for 34 h and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:9 methanol-dichloromethane) to afford 12.6 mg (65%) of amine 13. 1H NMR (500 MHz, CD3OD) δ 8.36 (d, J = 2.1 Hz, 1H), 8.16 (dd, J = 8.9, 2.2 Hz, 1H), 8.02 (d, J = 8.9 Hz, 1H), 7.46 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 4.30 (d, J = 9.5 Hz, 1H), 4.09 (s, 2H), 4.01 (d, J = 9.5 Hz, 1H), 3.75-3.73 (m, 2H), 3.72-3.71 (m, 2H), 3.69-3.68 (m, 2H), 3.62-3.61 (m, 2H), 3.47 (d, J = 5.2 Hz, 2H), 2.74 (d, J = 5.2 Hz, 2H), 1.52 (s, 3H). 13C NMR (125 MHz,CD3OD) δ 176.2, 170.7, 157.2, 144.2, 143.9, 132.3, 127.9, 124.2, 123.4 (2C), 119.4, 116.0 (2C), 76.5, 75.1, 72.9, 72.0, 71.5, 71.4, 71.3, 71.1, 41.9, 23.1. TLC (20% CH3OH in CHCl2), Rf 0.06 (UV, CAM, and Ninhydrin). PROTAC 14. To a solution of Nutlin acid 4 (11 mg, 0.017 mmol) in DMF (0.5 mL) at rt were added HATU (8 mg, 0.020 mmol) and DIPEA (10 uL, 0.057 mmol). After stirring at rt for 30 min, a solution of amine 13 (10 mg, 0.017 mmol) in DMF (0.5 mL) was added to the mixture and the reaction mixture was stirred at rt for 16.5 h. After the resulting mixture was quenched with H2O (5 mL) in an ice bath, the mixture was extracted four times with ethyl acetate and the extracts were washed with saturated NaCl, dried over Na2SO4, filtered, and concentrated. The concentrate was purified by flash column chromatography (1:99 methanol-dichloromethane initially, grading to 1:9 methanol-dichloromethane) to afford 12.6 mg (61%) of PROTAC 14 as a diastereomeric mixture. 1H NMR (500 MHz, CD3OD) δ 8.34 (d, J = 1.9 Hz, 1H), 8.15 (dt, J = 8.9, 2.5 Hz, 1H), 8.01 (dd, J = 8.9, 4.1 Hz, 1H), 7.56 (d, J = 8.1 Hz, 1H), 7.46 (d, J = 9.0 Hz, 2H), 7.12 (dd, J = 8.4, 1.5 Hz, 2H), 7.06 (dd, J = 8.6, 3.0 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.1 Hz, 2H), 6.90 (dd, J = 9.0, 1.5 Hz, 2H), 6.63 (dd, J = 8.5, 2.1 Hz, 1H), 6.62 (s, 1H), 5.73 (d, J = 10.1 Hz, 1H), 5.54 (d, J = 10.1 Hz, 1H), 4.73-4.68 (m, 1H), 4.30 (d, J = 9.5 Hz, 1H), 4.10 (s, 2H), 3.99 (dd, J = 9.5, 3.1 Hz, 1H), 3.87-3.80 (m, 2H), 3.84 (s, 3H), 3.78-3.74 (m, 2H), 3.73-3.70 (m, 2H), 3.70 (t, J = 3.6 Hz, 2H), 3.67-3.65 (m, 2H), 3.60-3.58 (m, 2H), 3.47 (t, J = 5.3 Hz, 2H), 3.50-3.45 (m, 2H), 3.38-3.30 (m, 2H), 3.01-2.92 (m, 2H), 1.51 (d, J = 3.7 Hz, 3H), 1.36 (d, J = 6.0 Hz, 3H), 1.33 (d, J = 6.0 Hz, 3H). 13C NMR (125 MHz, CD3OD) δ 176.1, 170.7, 169.8, 167.3, 165.0, 163.1, 158.5, 157.2, 155.8, 144.2, 143.9, 137.5, 136.4, 134.2, 133.0, 132.3, 130.7, 129.9, 129.1, 129.0, 127.9, 124.2, 123.5, 119.7, 116.1, 106.4, 101.2, 76.5, 75.1, 72.4, 72.1, 72.0, 71.5, 71.4, 71.1, 70.4, 70.1, 56.1, 54.8, 50.1, 48.0, 43.2, 40.3, 23.2, 22.5, 22.4. FTIR (neat, cm−1) 3240 (br), 2957, 2923, 1722, 1700, 1653, 1635, 1576, 1559, 1506, 1457, 1399, 1316, 1224, 1083, 1027, 822. LRMS (ES+) [M+H]+ 1209.74. HRMS (ES+) calculated for C57H61N8O14Cl2F3 [M+H]+ 1209.3709, found 1209.3694. [α]D20 + 32.5° (c = 0.135, MeOH). TLC (10% CH3OH in CHCl2), Rf 0.41 (UV, CAM).
- (19).HeLa cells were grown in DMEM containing 10% FBS and pen/strep at 37°C in a 7.5% CO2 humidified atmosphere. A 10 cm plate of HeLa cells (approximately 50-60% confluent) was transfected using Lipofectamine2000 with 25 ng of a plasmid expressing the androgen receptor (AR). After incubation for 8-10 h, transfected cells were collected and distributed 1:10 into the wells of a 6-well plate. Cells were allowed to adhere overnight. Fresh media was combined with the appropriate concentration of drug or vehicle in an eppendorf tube (2 μL of a 5 mM stock of PROTAC in DMSO/CremephorEL (50/50, v/v) into 1 mL of media (10 μM final concentration), or 2 μL of 50/50 DMSO/CremephorEL alone into 1 mL) and vortexed to mix. The media was removed from the wells of the 6-well plate, and the drug-treated media was carefully added so as not to disrupt the cells. Cells were incubated at 37°C for 7 h, then lysed in RIPA buffer with protease inhibitors. Samples were run on an 8% SDS-PAGE gel and transferred to nitrocellulose membrane. The membrane was probed with a mouse monoclonal anti-androgen receptor antibody (abcam, ab9474) at a 1:2000 dilution to determine total levels of AR. The membrane was then stripped and re-probed with a monoclonal anti-α-tubulin antibody (Sigma, T5168) at a 1:80,000 dilution as a loading control.
- (20).Meng L, Mohan R, Kwok BHB, Elofsson M, Sin N, Crews CM. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10403. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Preparation of 6-well plates same as described in reference 19. For the epoxomicin-treated cells, a 10 μM solution of epoxomicin in growth medium was mixed in an eppendorf tube and added to the cells. Cells were incubated for 1h. Following epoxomicin pre-treatment, media was removed from all wells, and drug-treated media + PROTAC was added (10 μM PROTAC, 10 μM PROTAC + 10 μM epoxomicin (1 μL of a 10 mM stock in DMSO), and vehicle). Following a 7h incubation, cells were harvested and lysates were analyzed as described in reference 19.