

Abstract
Cardiac hypertrophy entails complex structural remodeling involving rearrangement of muscle fibers, interstitial fibrosis, accumulation of extracellular matrix, and angiogenesis. Many of the processes underlying cardiac remodeling have features in common with chronic inflammatory processes. During these processes, nonmyocytes, such as endothelial cells, fibroblasts, and immune cells, residing in or infiltrating into the myocardial interstitium play active roles. This paper mainly addresses the functional roles of nonmyocytes during cardiac remodeling. In particular, we focus on the communication between cardiomyocytes and nonmyocytes through direct cell-cell interactions and autocrine/paracrine-mediated pathways.
1. Introduction
Cardiac hypertrophy is an essential adaptive process, through which the heart responds to various mechanophysical, metabolic, and genetic stresses. On the other hand, the hypertrophy induced by sustained overload eventually leads to contractile dysfunction and heart failure. Cardiac hypertrophy involves cellular and molecular events within both cardiomyocytes and nonmyocytes. Cardiomyocytes show phenotypic modification that results in cellular hypertrophy accompanied by reexpression of several fetal genes, abnormal Ca2+ handling, oxidative stress and mitochondrial DNA damage, and cardiomyocyte death due to necrosis or apoptosis [1]. In addition to cardiomyocytes, the myocardium contains a variety of nonmyocytes, including vascular endothelial, and smooth muscle cells, fibroblasts and immune cells, which all appear to be crucially involved in the myocardial response to external and internal stress [2, 3]. During cardiac hypertrophy and the progression to heart failure, the myocardium exhibits complex structural remodeling involving rearrangement of muscle fibers, fibrosis, accumulation of extracellular matrix (ECM), cellular death, and angiogenesis [4]. Many of the processes underlying these phenomena are also seen in chronic inflammatory diseases and are mediated by cellular interactions among cardiomyocytes and nonmyocytes. In this paper, we will focus on the functional roles of nonmyocytes and the cellular communication ongoing during the development of cardiac hypertrophy and heart failure under noninfectious and noninfarction conditions, such as pressure overload.
2. Fibroblasts
Cardiac fibroblasts are critically involved in the development of cardiac fibrosis [4, 5]. They can produce a wide variety of ECM proteins, including interstitial collagens, proteoglycans, glycoproteins, and proteases [6]. Morphologically, fibroblasts are flat, spindle-shaped cells with multiple processes sprouted from the cell body, which lacks a basement membrane [7]. Fibroblasts play central roles in two types of fibrosis: reparative and reactive. Reparative (replacement) fibrosis or scarring accompanies cardiomyocyte death. Reactive fibrosis appears as “interstitial” or “perivascular” fibrosis and does not directly associate with cardiomyocyte death [8, 9].
Increases in fibrosis result in mechanical stiffness and cardiac diastolic dysfunction [10]. In addition, by forming a barrier between cardiomyocytes, fibrosis can impair the electrical coupling of cardiomyocytes, leading to cardiac systolic dysfunction [11]. Moreover, perivascular fibrosis can increase oxygen and nutrient diffusion distances, leading to pathological remodeling [12]. Thus, fibrosis profoundly affects cardiomyocyte metabolism and performance, and ultimately ventricular function [13]. However, the functions of fibroblasts are not limited to producing ECM. Cardiac fibroblasts interact with other cell types, most notably cardiomyocytes. This interaction may be direct via physical contacts or indirect via paracrine factors. Thus fibroblasts are involved in much more than deposition of collagen [4, 7, 14].
In response to external stress, fibroblasts change their phenotype and become myofibroblasts [15, 16], which express several smooth muscle (SM) markers, including SM α-actin, SM22α, SMemb/nonmuscle myosin heavy chain-B, and tropomyosin [16–18]. However, more stringent SM markers (e.g., SM myosin heavy chains) are not expressed in myofibroblasts [19]. With the exception of heart valve leaflets, myofibroblasts are not found in normal cardiac tissue [4].
So far, no common definitive fibroblast-specific marker that could be used to identify fibroblasts in different tissues has been determined. In fact, fibroblasts in different tissues likely differ with respect to their cellular origins and functions. Several markers have been used to identify cardiac fibroblasts. Discoidin domain receptor 2 (DDR2) is specifically expressed by fibroblasts within the heart [20, 21], and, in a recent report, was successfully used as a marker in a flow cytometric analysis of cardiac cells [22]. This study found that fibroblasts represent a substantial portion of the cells in the mammalian heart. For example, the adult murine heart consists of approximately 55% cardiomyocytes and 45% nonmyocytes (~27% fibroblasts), and the adult rat heart consists of 30% cardiomyocytes and 70% nonmyocytes (~67% fibroblasts). Periostin [23] and thymus cell antigen-1 (Thy1/CD90) [14, 24] also have been used as markers of fibroblasts in developing and adult hearts. However, because fibroblasts can acquire heterogeneous phenotypes [25–27], those markers may not be capable of identifying all fibroblasts under all physiological and pathological conditions [5, 28]. Commonly used fibroblasts markers are reviewed elsewhere [28].
2.1. Origin and Phenotype of Cardiac Fibroblasts
The majority of resident cardiac fibroblasts are thought to arise from embryonic proepicardial organ (Figure 1) [23, 29]. Proepicardial and primitive epicardial cells undergo epithelial-mesenchymal transition (EMT) and then migrate into the myocardium, where they progressively differentiate into interstitial fibroblasts, perivascular fibroblasts, and coronary SMCs [30, 31]. These resident fibroblasts have traditionally been thought to be the sole source of cardiac fibroblasts, but more recently other cellular origins of cardiac fibroblasts have been proposed [23]. Endothelial cells and pericytes may also contribute to cardiac fibroblasts via endothelial-to-mesenchymal transition (EndMT) and EMT, respectively [32, 33]. In addition, bone marrow-derived cells may acquire fibroblast-like phenotypes [32, 34–37], and fibrocytes, circulating mesenchymal progenitor cells of bone-marrow origin, have been shown to be recruited to ischemic hearts and to express SM α-actin, collagen I, vimentin, and DDR2 [38]. Finally, it has been suggested that monocytes/macrophages represent another potential source of myofibroblasts in ischemic hearts [39]. How these various bone-marrow-derived cells are related to one another remains unclear, as does their precise lineage origins, in part, because of a lack of definitive markers for fibroblasts and the different myeloid cell subsets. Macrophages may also promote fibrosis by producing cytokines, such as TGF-β. It is therefore likely that bone-marrow-derived cells play multiple roles in cardiac fibrosis. Further studies will be needed to clarify precisely how these cells contribute to cardiac fibrosis.
Figure 1.
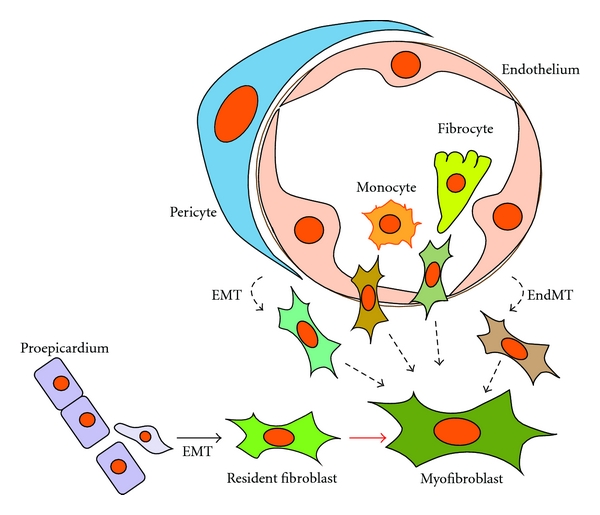
Diverse origins of cardiac fibroblasts. Resident cardiac fibroblasts are thought to arise from the proepicardium and embryonic epicardium during development. In response to fibrogenic stimuli, however, many other cell types, including bone marrow-derived cells, pericytes, and endothelial cells, may also acquire myofibroblast-like phenotypes. This scheme depicts the possible origins of cardiac fibroblasts. EMT: epithelial-to-mesenchymal transition; EndMT: endothelial-to-mesenchymal transition.
2.2. Mediators of Intercellular Communications in Fibrogenic and Cardiomyocyte Hypertrophic Responses
Cardiomyocyte-specific deletion of genes has been shown to affect not only cardiomyocyte functionality but also the phenotypes and functions of fibroblasts [4]. Conversely, recent studies have shown that cardiac fibroblasts control cardiomyocyte proliferation in the developing ventricles during embryogenesis and that fibroblasts promote cardiomyocyte hypertrophy through paracrine factors and ECM [14, 24, 40]. These findings are indicative of the communication between cardiomyocytes and fibroblasts. Here we describe factors that mediate intercellular communication within the myocardium (Figure 2).
Figure 2.
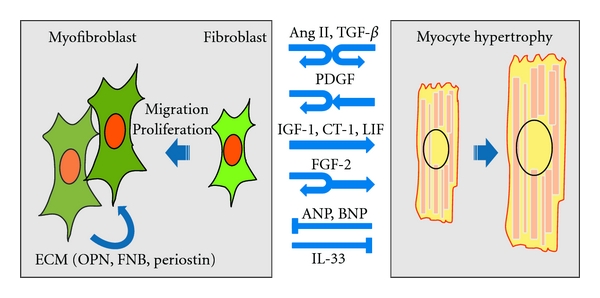
Reciprocal interactions between fibroblasts and cardiomyocytes. Many growth factors and cytokines have been shown to act in autocrine and/or paracrine fashion to induce hypertrophic responses in cardiomyocytes and activate fibroblasts. This scheme depicts only some of the factors identified. Ang II: angiotensin II; TGF-β: transforming growth factor-β; PDGF: platelet-derived growth factors; IGF-1: insulin-like growth factor-1; CT-1: cardiotrophin-1; LIF: leukemia inhibitory factor; FGF-2: fibroblast growth factor 2; ANP: atrial natriuretic peptide; BNP: brain natriuretic peptide; IL-33: interleukin-33; ECM: extracellular matrix; OPN: osteopontin; FBN: fibronectin.
2.2.1. Angiotensin II
Ang II is a pleiotropic vasoactive peptide that plays key roles in the development of cardiac fibrosis and remodeling. Although most of the cardiovascular effects of Ang II are mediated via the Ang II type 1 receptor (AT1), the Ang II type 2 receptor (AT2) may be also important, as expression of both receptors is upregulated in various cardiac diseases [41–43]. Continuous infusion of Ang II into mice induces cardiac hypertrophy and fibrosis [44, 45]. Under these conditions, most of the proliferating fibroblasts were found to be surrounding cardiomyocytes carrying the AT1a receptor [46], suggesting that activation of cardiomyocytes via AT1a receptors also affects fibroblasts. Ang II also stimulates paracrine release of growth factors and cytokines, including TGF-β1 and endothelin-1 (ET-1) from cardiomyocytes [47–49]. On the other hand, AT1 receptor expression is greater in fibroblasts than cardiomyocytes [47], and Ang II directly stimulates fibroblast proliferation, collagen and ECM synthesis, and expression of fibroblast growth factor 2 (FGF2) [50]. It therefore seems likely that reciprocal interactions between cardiomyocytes and fibroblasts via paracrine factors are important for myocardial responses to Ang II. Consistent with these in vitro and in vivo findings in animal models are recent clinical studies demonstrating that blockade of the rennin-angiotensin system in patients, using a direct renin inhibitor, an angiotensin converting enzyme inhibitor, or an angiotensin receptor blocker, effectively reduces cardiac fibrosis and remodeling in addition to reducing blood pressure [51].
AT2 receptor expression is also upregulated in failing human hearts, mainly in cardiac fibroblasts [43, 52]; however, the function of AT2 remains controversial. Initially, AT2 was reported to mediate effects opposing the growth-promoting signals mediated by AT1 [53], but since then there have been several reports that AT2 also stimulates prohypertrophic signaling [54, 55]. The function of AT2 may depend on the adaptor proteins recruited to the receptor and the pathophysiological conditions [56, 57].
2.2.2. Transforming Growth Factor-β
TGF-β exists in three isoforms (TGF-β1, TGF-β2, and TGFβ-3) that have distinct but overlapping functions in immunity, inflammation, and tissue repair, and TGF-β also has a central role in fibroblast activation and differentiation into myofibroblasts [58]. TGF-β is initially produced as a latent complex bound to latent TGF-β binding protein (LTBP) within the interstitium. It is activated physiochemically by altered pH, a large group of proteases and enzymes, high-energy ionizing radiation, or integrin-mediated mechanisms [59, 60]. Activated TGF-β binds to heterodimers comprised of TGF-β type 1 receptor (TGF-βR1) and type 2 receptor (TGF-βR2) on both cardiomyocytes and fibroblasts [61]. TGF-βR1 (ALK5; activin-linked kinase 5) then phosphorylates receptor-regulated Smads (R-Smads: Smad2 and Smad3), which in turn associate with a common-mediator Smad (co-Smad: Smad4) and are translocated into the nucleus, where they act as transcription factors [62]. Smad3 is required for TGF-β to induce expression of collagen, fibronectin, and other ECM genes [63–66].
TGF-β promotes myofibroblast differentiation and ECM production by fibroblasts, and Ang II-induced cardiac hypertrophy is also mediated in part through TGF-β secreted from AT1-expressing fibroblasts [47]. TGF-β1-deficient mice subjected to chronic subpressor doses of Ang II showed no significant cardiac hypertrophy or fibrosis [67], which suggests that strategies to block TGF-β signaling may be useful for treating fibrogenic cardiac remodeling. Indeed, a TGF-β neutralizing antibody inhibited fibroblast activation and proliferation, and diastolic dysfunction in pressure-overloaded rats [68]. Similarly, an ALK5 inhibitor attenuated fibroblast activation and systolic dysfunction in an experimental rat model of myocardial infarction [69]. However, fibrosis was attenuated in Smad3-dificient mice subjected to experimental cardiac pressure overload, cardiac hypertrophy and heart failure were aggravated [70]. Moreover, TGF-β neutralizing antibody increased mortality and worsened cardiac remodeling, which correlated with reduction of ECM in a rat MI model [71]. These results indicate that the consequences of inhibiting TGF-β signaling can vary depending on the disease model and the timing of the inhibition, presumably because TGF-β signaling has an essential adaptive role in the myocardium under stress. Seemingly maladaptive functions, such as fibrosis, might also be essential for adaptation in other contexts. It will, therefore, be important to clarify the spatiotemporal functions of TGF-β signaling in different disease contexts if we are to develop effective therapeutic approaches involving TGF-β.
2.2.3. Fibroblast Growth Factor-2
FGF-2 is alternatively translated as a high molecular weight (Hi-FGF-2) and a low molecular weight (Lo-FGF-2) isoform from the single Fgf2 gene [72]. The Hi-FGF-2 isoform contains nuclear localization sequences and is found predominantly within the nucleus, while the Lo-FGF-2 isoform is localized in the ECM and cytoplasm [72]. Cardiac fibroblasts predominantly express Hi-FGF-2, which acts in a paracrine fashion to promote cardiomyocyte hypertrophy [49]. Hi-FGF-2 also acts in an autocrine fashion on the fibroblasts themselves to stimulate release of other pro-hypertrophic factors, such as cardiotrophin-1 (CT-1) [49, 73, 74]. In addition, Lo-FGF-2 elicits cardioprotective effects against postischemic cardiac dysfunction [72].
2.2.4. Interleukins
The IL-6 family of cytokines, including leukemia inhibitory factor (LIF) and CT-1, are expressed by cardiac fibroblasts and cardiomyocytes. LIF and CT-1 secreted from fibroblasts mediate Ang II-induced cardiomyocyte hypertrophy [75, 76]. LIF was also shown to induce fibroblast hypertrophy but to inhibit myofibroblast transition and collagen deposition [77].
IL-33 is produced primarily by cardiac fibroblasts, and its expression is upregulated by cyclic strain [78]. IL-33 binds to a transmembrane form of the ST2 receptor (ST2L) on cardiomyocytes and inhibits the hypertrophic response of cultured cardiomyocytes to pro-hypertrophic stimuli. In vivo, IL-33 inhibits cardiomyocyte hypertrophy as well as fibrosis induced by pressure overload [40, 79].
2.2.5. Serotonin
Serotonin (5-hydroxytryptamine [5-HT]) acts via its 5-HT2B receptor (5-HT2BR) to contribute to cardiac hypertrophy. Indeed, plasma serotonin levels and cardiac 5-HT2BR expression are both elevated in human heart failure. 5-HT2BRs mainly colocalize with AT1 receptors in fibroblasts [80, 81]. Isoproterenol (ISO) and Ang II-induced cardiac hypertrophy is suppressed in 5-HT2BR-deficient mice, and this effect is accompanied by reduced production of cytokines (IL-6, IL-1β, TGF-β, and TNF-α) and reactive oxygen species in cardiac fibroblasts [82, 83]. Similarly, pharmacological blockade of 5-HT2BRs prevents ISO-induced murine cardiac hypertrophy [82]. Moreover, mice in which expression of 5-HT2BR is limited to their cardiomyocytes are also resistant to ISO-induced cardiac hypertrophy and dysfunction, as well as to ISO-induced upregulation of the cytokines. This suggests that signaling through 5-HT2BRs on fibroblasts stimulates production of cytokines that promote cardiomyocyte hypertrophy [81, 84].
2.2.6. Platelet-Derived Growth Factors
PDGF-A and -B are secreted from cardiomyocytes and fibroblasts and play critical roles in cardiac fibrosis and angiogenesis through their interactions with the protein tyrosine kinase receptors PDGF receptor (PDGFR)-α and -β [85, 86]. PDGF signaling activates fibroblast proliferation and migration and ECM deposition. PDGF expression is significantly increased in cardiac hypertrophy and fibrosis [87], atrial fibrillation [88], and chronic rejection of cardiac allografts [89]. PDGF-C and -D may also contribute to cardiac fibrosis and remodeling. Transgenic mice exhibiting cardiomyocyte-specific expression of PDGF-C and -D develop hyperproliferation of myocardial interstitial cells, resulting in progressive fibrosis leading to dilated cardiomyopathy and heart failure [90–92].
PDGF signaling has been assessed as a therapeutic target for cardiac remodeling. The synthetic retinoid Am80 inhibited upregulation of PDGF-A by inhibiting the transcription factor Krüppel-like factor 5 (KLF5), thereby suppressing Ang II-induced cardiac fibrosis (Figure 3) [87, 93]. In addition, a neutralizing PDGFRα-specific antibody attenuated induction of pressure overload-induced atrial fibrosis and fibrillation [88]. However, several inhibitors of receptor tyrosine kinases, including PDGFRs, have been linked to the development of cardiomyopathy in some treated patients [94–97]. PDGFR-β on cardiomyocytes is indispensable for the cardiac response to pressure overload and may regulate angiogenesis [98]. For therapies targeting PDGF signaling to cardiac remodeling, it will be important to further clarify the precise roles played by PDGFs and PDGFRs under various pathological and physiological conditions.
Figure 3.
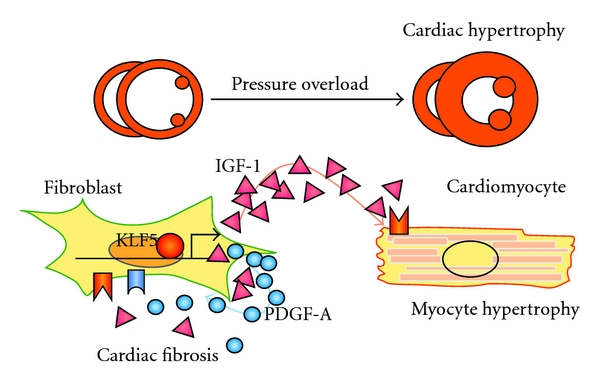
A model for the regulation of fibroblasts by KLF5 during cardiac hypertrophy. The transcription factor KLF5 controls Igf1 and Pdgfa expression in cardiac fibroblasts. IGF-1 is a major cardiotrophic factor secreted from fibroblasts, and PDGF-A is primarily involved in mediating the migration and proliferation of fibroblasts.
2.2.7. Insulin-Like Growth Factor-1
IGF-1 exerts adaptive and cardioprotective effects in response to stress. The majority of serum IGF-1 is liver-derived and plays a critical role during normal body development. However, postnatal body growth is preserved, even in the complete absence of IGF-1 expression by hepatocytes; autocrine/paracrine IGF-1 appears to have important regulatory functions under these conditions [99]. In the heart, IGF-1 is mainly expressed in cardiac fibroblasts [14] and activates downstream signal transducers, such as phosphoinositide 3-kinase (PI3K), leading to cardiomyocyte hypertrophy [100]. We recently found that cardiac IGF-1 is transactivated by KLF5 (Figure 3) [14]. Although cardiac fibroblast-specific deletion of Klf5 ameliorated the cardiac hypertrophy and fibrosis elicited by moderate-intensity pressure overload, it resulted in severe heart failure in high-intensity pressure overload. Similarly, administration of a peptide inhibitor of IGF-1 severely exacerbated heart failure induced by high-intensity pressure overload. These findings indicate that induction of IGF-1 is an essential cardioprotective response; that cardiac fibroblasts play a pivotal role in the myocardial adaptive response to pressure overload; that KLF5 controls IGF-1 expression in cardiac fibroblasts in response to stress [14].
2.2.8. Connective Tissue Growth Factor
CTGF (also known as CCN2) is expressed in fibroblasts and cardiomyocytes and regulates ECM deposition and wound healing [101, 102]. CTGF is induced by TGF-β, Ang II, and ET-1. By itself, CTGF only weakly promotes fibrosis and cardiomyocyte hypertrophy, but it appears that it may promote a more robust effect by acting as a cofactor for TGF-β [103, 104]. Transgenic mice exhibiting cardiomyocyte-specific expression of CTGF did not develop cardiac hypertrophy or fibrosis under baseline conditions but showed significantly increased fibrosis and contractile dysfunction in response to pressure overload [105]. Another group of transgenic mice developed age-dependent cardiac hypertrophy and dysfunction, though Ang II did not increase fibrosis in young transgenic mice [106]. Thus, the cardiac actions of CTGF will require further study.
2.2.9. Natriuretic Peptides
Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are usually synthesized in the atria and ventricles, respectively [107]. Plasma levels of these peptide hormones are widely used as biomarkers when making a diagnosis or determining risk stratification in a variety of cardiac disease states. In addition, infusion of synthetic ANP or BNP is useful for treating cardiac heart failure and remodeling, mainly by optimizing intravascular volume and arterial pressure [108]. ANP and BNP also exert anti-hypertrophic and antifibrogenic effects on the heart, and knockout mice deficient in their common receptor, guanylyl cyclase-A (GC-A), showed cardiac hypertrophy and extensive interstitial fibrosis that was at least partially independent of blood pressure [109–111]. In fibroblasts, BNP inhibits TGF-β-regulated genes related to fibrosis (collagen I, fibronectin, and CTGF), proliferation (PDGF-A and IGF-1), and inflammation (COX2, IL-6, and TNF) [112], while ANP suppresses ET-1 expression and cell proliferation [113]. This suggests ANP and BNP secreted from cardiomyocytes suppress the fibrogenic activity of fibroblasts.
2.2.10. ECM Molecules
ECM serves as an important intermediary for intercellular communication by transducing intracellular signals via its receptor molecules (integrins and CD44) on myocardial cells [24, 114–116]. Production, degradation and modification of ECM components are dynamically regulated under both physiological and pathological conditions. Fibroblasts are a major source of nonbasement membrane collagen and other ECM proteins, and other cells in the myocardium, including cardiomyocytes, endothelial cells, and SMCs, also produce sets of ECM components [117]. Cardiac fibroblasts and macrophages are major producers of matrix metalloproteases (MMPs), which degrade ECM proteins.
The integrin family consists of 18 α and 8 β subunits, which form 24 known α-β-heterodimers. Integrins serve as cell-ECM and cell-cell adhesion molecules and also function as signal-transducing receptors for ECM proteins, including collagen, laminin, fibronectin (FBN), and osteopontin (OPN) [116, 118]. Cardiac-specific integrin signaling in genetically engineered animal models are reviewed elsewhere [116].
CD44 was originally described as a receptor for hyaluronan (HA), a ubiquitous constituent of the ECM, but is now known to interact with collagen, laminin, FBN, and OPN [119]. CD44-HA interactions play an important role in regulating leukocyte extravasation into sites of inflammation and in mediating efficient phagocytosis. CD44 also contributes to the resolution of inflammation through removal of matrix breakdown products, clearance of apoptotic neutrophils, and fibroblast migration [120]. In injured hearts, CD44 is upregulated in fibroblasts, leukocytes, and endothelial cells, particularly those cells surrounding and within the coronary arteries [120, 121]. Cd44-deficient mice subjected to myocardial infarction show increased myocardial infiltration by leukocytes and expression of proinflammatory cytokines, followed by decreased fibroblast infiltration and fibrosis and enhanced dilative cardiac remodeling [120]. Finally, Cd44-deficient cardiac fibroblasts exhibit diminished proliferation and collagen synthesis in response to TGF-β. This suggests CD44 is important for resolution of postinfarction inflammatory processes and for regulation of fibroblast function.
FBN and OPN are upregulated in cardiac hypertrophy and by Ang II. They contain the arginine-glycine-aspartate (RGD) tripeptide integrin binding motif and activate integrin-mediated proliferation, survival, adhesion, differentiation, and migration of myocardial cells [122–124]. FBN is mainly expressed in fibroblasts and acts in a paracrine fashion to regulate cardiomyocyte proliferation through β1 integrin signaling during embryonic heart development [24].
OPN is strongly expressed in chronic inflammatory and autoimmune diseases and promotes the recruitment and retention of macrophages and T cells at inflamed sites [125]. Cardiomyocyte-specific overexpression of OPN results in dilated cardiomyopathy and severe fibrosis, with recruitment of activated T cells showing Th1 polarization [125]. Moreover, OPN (Spp1)-deficient fibroblasts are less proliferative and less adherent to ECM substrates, while Spp1-deficient mice exhibit less Ang II-induced cardiac fibrosis [123, 126]. Recently, OPN has emerged as a novel biomarker of various cardiac diseases [127–129].
Periostin is primarily expressed in myocardial fibroblasts, and its expression is upregulated by pressure overload and myocardial infarction. Periostin can serve as a ligand for αvβ3, αvβ5, and α4β6 integrins [23] and can also directly interact with other ECM proteins, including FBN, tenascin-C, collagen I/V, and heparin [130]. Collagen fibrils from periostin (Postn)-null mice are reduced in size, somewhat disorganized, and less efficiently cross-linked, indicating that periostin facilitates proper organization of the ECM [23, 131, 132]. In addition, periostin is induced via TGF-β signaling and may then enable collagen realignment in response to TGF-β [130, 133, 134]. Postn-deficient hearts subjected to pressure overload or ischemic insult exhibited less fibrosis but more frequent rupture of the ventricular wall [132, 135].
3. Endothelial Cells
Vascular endothelial cells are also crucially involved in the development of cardiac hypertrophy, remodeling, and failure. Endothelial cells are capable of producing a wide variety of functional agonists and antagonists, including vasodilators and vasoconstrictors, procoagulants and anticoagulants, and inflammatory and anti-inflammatory factors. Endothelial cells maintain homeostasis by controlling the balance of these various mediators [136]; endothelial dysfunction disturbs that balance and leads to pathological inflammatory processes. For instance, activated endothelial cells express the adhesion molecules, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which recruit and promote the infiltration of immune cells into the myocardium in response to various stimuli.
Endothelial cell-derived nitric oxide (NO), produced by endothelial NO synthase (eNOS), is a key regulator of vasodilation. NO also reduces vascular permeability and thrombogenesis, and it inhibits inflammation by suppressing signaling by adhesion molecules, proinflammatory cytokines, and NF-κB [136]. Under pathological conditions, the bioavailability of NO is diminished [137, 138]. For example, sustained pressure overload triggers eNOS uncoupling, which reduces NO signaling, increases levels of eNOS-derived reactive oxygen species, and promotes endothelial dysfunction. NO is also known to influence myocardial excitation-contraction coupling, substrate metabolism, and hypertrophy, as well as cell survival, which are at least in part dependent on eNOS and nNOS expression in cardiomyocytes [139]. For example, eNOS (Nos3) knockout mice develop concentric left ventricular hypertrophy and fibrosis [140], indicating the importance of the autocrine and paracrine effects of NO in cardiac remodeling.
Accumulating evidence indicates that impaired angiogenesis contributes to the transition of cardiac hypertrophy to heart failure. Hypertrophic stimuli induce expression of the angiogenic growth factors, vascular endothelial growth factor (VEGF), and angiopoietin 2 [141], which promote angiogenesis and blood flow in response to reductions in coronary perfusion pressure or ischemia. Blockade of VEGF action using an adenoviral vector encoding a decoy VEGF receptor or an anti-VEGF antibody promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload in mice [142, 143]. Likewise, TNP-470, an inhibitor of angiogenesis, also induced cardiac dysfunction [144]. Conversely, VEGF treatment during prolonged pressure overload preserved contractile function [144, 145].
Within myocardium subjected to pressure overload, hypoxia-inducible factor-1-(HIF-1-) mediated transactivation of VEGF in cardiomyocytes plays an important role during induction of angiogenesis. Furthermore, it has been proposed that, in response to sustained pressure overload, p53 accumulates in cardiomyocytes and inhibits HIF-1 activity, thereby impairing cardiac angiogenesis and contractile function [144]. However, there are also conflicting data showing that ventricular deletion of HIF-1α prevents hypertrophy-induced activation of peroxisome proliferator-activated receptor-(PPAR-) γ and contractile dysfunction [146].
Endothelin-1 (ET-1) is a major growth factor secreted from endothelial cells that contributes to cardiac hypertrophy and fibrosis. ET-1 was originally identified as an endothelium-derived vasoconstrictor [147], but it is also expressed in various nonendothelial cells, including fibroblasts and cardiomyocytes, and it exerts both autocrine and paracrine effects that appear to be important for cardiomyocyte hypertrophy [47, 48, 148]. Although cardiomyocytes predominantly express ETA ET-1 receptors, the hypertrophic response to Ang II and ISO is unaltered in cardiomyocyte-specific ETA receptor-(Ednra-) deficient mice, suggesting the possible involvement of ETA receptors on nonmyocytes and ETB receptors in cardiomyocyte hypertrophy [48, 149]. Consistent with that idea, the combined ETA/ETB receptor antagonist bosentan inhibited Ang II-induced cardiac hypertrophy [150]. It was also shown that endothelium-derived ET-1 promotes cardiac fibrosis in diabetic heart [151].
4. Immune Cells
A variety of immune cells, including macrophages, T cells, and mast cells, reside in the myocardium under physiological conditions. They are also induced to infiltrate the myocardium under pathogenic conditions and to promote cardiac remodeling, in part by releasing cytokines, growth factors, and MMPs [152, 153].
Macrophages are essential effector cells involved in tissue remodeling and fibrosis. It is becoming increasingly clear that macrophages can have diverse phenotypes and functions [154]. In vitro studies have shown that Th1 cytokines, alone or in contact with microbial products, elicit classical M1 activation of macrophages, which then release proinflammatory cytokines and reactive oxygen species. Th2 cytokines (IL-4 and IL-13) elicit an alternative form of macrophage activation designated M2. M2 macrophages are thought to suppress immune responses and promote fibrosis and tissue remodeling, though M2 activation is a rather generic term often used to describe any form of macrophage activation other than classical M1. Previous studies have shown that macrophages are involved in cardiac hypertrophy and remodeling. For instance, Ang II-induced cardiac hypertrophy and fibrosis were diminished in macrophage-specific mineralocorticoid receptor-(MR-) deficient mice [155]. The MR-deficient macrophages exhibited M2-type activation and reduced expression of proinflammatory cytokines, suggesting it is M1-type macrophages that are involved in the cardiac hypertrophy and fibrosis induced by Ang II. Similarly, a monoclonal neutralizing anti-MCP-1 antibody attenuated not only macrophage accumulation, but also fibroblast proliferation and fibrosis, resulting in amelioration of cardiac diastolic dysfunction [156]. These results demonstrate the pathological involvement of macrophages in cardiac hypertrophy and fibrosis. By contrast, macrophage depletion using liposomal clodronate induces abundant infiltration of inflammatory cells, predominantly CD4+ lymphocytes, and aggravates cardiac dysfunction in hypertensive rats harboring the mouse renin gene (Ren2) [157]. This suggests macrophages exert a protective effect against cardiac dysfunction induced by hypertension. Clearly further studies are needed to clarify the seemingly diverse functions of macrophages in cardiac hypertrophy and heart failure. It is very likely that macrophage function changes with time and in different pathological contexts [158]. It will therefore be important to characterize the different functions and phenotypes of macrophages at different times during the processes of cardiac hypertrophy and heart failure and to elucidate the underlying molecular mechanisms. These studies will be essential for the development of novel therapeutic interventions affecting macrophages.
Mast cells reside in the myocardium, and their numbers are increased in hypertrophied and failing hearts. Mast cells are an important source of an array of cytokines, growth factors, chemokines, and other mediators. Histamine is a major mediator released upon mast cell degranulation in the heart and may be involved in heart failure. Consistent with that idea, inhibition of histamine using the histamine H2 receptor antagonist famotidine reportedly ameliorates heart failure [159, 160]. In addition, mast cells secrete the proteases renin and chymase, which, respectively, catalyze the conversion of angiotensinogen to angiotensin I and angiotensin I to Ang II and may thus activate the local rennin-angiotensin system in the heart [161]. Mast cell degranulation also releases preformed TGF-β, PDGF-A, and TNF-α [88, 161, 162], and inhibition of mast cells suppresses cardiac dysfunction and atrial fibrillation induced by pressure and volume overload [88, 163]. These results strongly suggest that mast cells are involved in inflammatory processes that contribute to remodeling in the heart.
T cells also reside in the myocardium. Although little is known about their function in cardiac pathology, Kvakan et al. recently reported that adoptive transfer of CD4+ CD25+ regulatory T cells suppresses cardiac hypertrophy and fibrosis induced by Ang II in mice, and this effect was accompanied by a marked reduction in infiltration of inflammatory cells [164].
5. Cardiac Progenitor Cells
In addition to fibroblasts and immune cells, the cardiac stroma contains a number of mesenchymal cell types. Previous studies have demonstrated that a fraction of these mesenchymal cells, referred to as cardiac progenitor cells (CPCs), have the potential to differentiate into cardiomyocytes under certain conditions [165]. There is also some evidence of cellular interactions between CPCs and their surrounding cells, including cardiomyocytes and fibroblasts. For instance, IGF-1 and Ang II produced by surrounding cells affect the survival of CPCs [166, 167]. In addition, human CPCs were shown to be connected to cardiomyocytes and fibroblasts via gap junctions and adherens junctions [168, 169], which may enable cellular communication via mediating molecules, including miRNA [170]. Communication between CPCs and surrounding cells through Notch-Notch ligand and Eph-ephrin signaling has also been shown [171, 172]. As such, cellular communication between mesenchymal cells, including CPCs, and their surrounding cells, including cardiomyocytes and fibroblasts, may contribute to the myocardial response to injury.
6. Conclusions
Both cardiomyocytes and nonmyocytes play essential roles in the processes involved in the development of cardiac hypertrophy, remodeling, and failure. The cellular and molecular processes that contribute to cardiac remodeling and failure share many features with chronic inflammatory processes in other tissues. Thus sterile stresses such as pressure overload and Ang II appear to activate pathways that are commonly used in inflammatory processes, including those involving immune cells in the myocardium. As we have seen here, dynamic cellular interactions among cardiomyocytes and nonmyocytes are a driving force for these inflammatory processes. In that regard, it will be important to further clarify the functional involvement of the different cell types residing in the myocardium and the underlying molecular control mechanisms. The function of a particular cell type may change with time and in response to different insults. As discussed, the functions of some myocardial cells, such as fibroblasts and macrophages, appear maladaptive under certain conditions but are in fact essential for adaptive responses at different times and in different disease models. Elucidation of these complex processes could lead to identification of novel therapeutic targets for the treatment of cardiac hypertrophy and heart failure.
Acknowledgments
This work was supported in part by a Grant-in-Aid from JSPS (to N. Takeda and I. Manabe); Grant-in-Aid for Scientific Research on Innovative Areas “Homeostatic Inflammation” from MEXT, Japan (to I. Manabe); research grants from the Sumitomo Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, Takeda Science Foundation (to I. Manabe), and Mitsubishi Pharma Research Foundation (to N. Takeda).
References
- 1.Oka T, Komuro I. Molecular mechanisms underlying the transition of cardiac hypertrophy to heart failure. Circulation Journal. 2008;72(supplement A):13–16. doi: 10.1253/circj.cj-08-0481. [DOI] [PubMed] [Google Scholar]
- 2.Zak R. Cell proliferation during cardiac growth. The American Journal of Cardiology. 1973;31(2):211–219. doi: 10.1016/0002-9149(73)91034-5. [DOI] [PubMed] [Google Scholar]
- 3.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28(109):41–61. [PubMed] [Google Scholar]
- 4.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis involvement in cardiac hypertrophy. Circulation Research. 2002;91(12):1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 5.Souders CA, Bowers SLK, Baudino TA. Cardiac fibroblast: the renaissance cell. Circulation Research. 2009;105(12):1164–1176. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanekar S, Hirozanne T, Terracio L, Borg TK. Cardiac fibroblasts: form and function. Cardiovascular Pathology. 1998;7(3):127–133. doi: 10.1016/s1054-8807(97)00119-1. [DOI] [PubMed] [Google Scholar]
- 7.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? American Journal of Physiology. 2006;291(3):H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 8.Anderson KR, Sutton MG, Lie JT. Histopathological types of cardiac fibrosis in myocardial disease. Journal of Pathology. 1979;128(2):79–85. doi: 10.1002/path.1711280205. [DOI] [PubMed] [Google Scholar]
- 9.Weber KT, Pick R, Jalil JE, Janicki JS, Carroll EP. Patterns of myocardial fibrosis. Journal of Molecular and Cellular Cardiology. 1989;21(supplement 5):121–131. doi: 10.1016/0022-2828(89)90778-5. [DOI] [PubMed] [Google Scholar]
- 10.Chaturvedi RR, Herron T, Simmons R, et al. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation. 2010;121(8):979–988. doi: 10.1161/CIRCULATIONAHA.109.850677. [DOI] [PubMed] [Google Scholar]
- 11.Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side- to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing and Clinical Electrophysiology. 1997;20(2):397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 12.Sabbah HN, Sharov VG, Lesch M, Goldstein S. Progression of heart failure: a role for interstitial fibrosis. Molecular and Cellular Biochemistry. 1995;147(1-2):29–34. doi: 10.1007/BF00944780. [DOI] [PubMed] [Google Scholar]
- 13.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacology and Therapeutics. 2009;123(2):255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Takeda N, Manabe I, Uchino Y, et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. Journal of Clinical Investigation. 2010;120(1):254–265. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. American Journal of Physiology. 1999;277(1):C1–C19. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 16.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano: regulation of connective tissue remodelling. Nature Reviews Molecular Cell Biology. 2002;3(5):349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 17.Shiojima I, Aikawa M, Suzuki JI, Yazaki Y, Nagai R. Embryonic smooth muscle myosin heavy chain SMemb is expressed in pressure-overloaded cardiac fibroblasts. Japanese Heart Journal. 1999;40(6):803–818. doi: 10.1536/jhj.40.803. [DOI] [PubMed] [Google Scholar]
- 18.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovascular Research. 2000;48(1):89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 19.Hautmann MB, Adam PJ, Owens GK. Similarities and differences in smooth muscle alpha-actin induction by TGF-beta in smooth muscle versus non-smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(9):2049–2058. doi: 10.1161/01.atv.19.9.2049. [DOI] [PubMed] [Google Scholar]
- 20.Goldsmith EC, Hoffman A, Morales MO, et al. Organization of fibroblasts in the heart. Developmental Dynamics. 2004;230(4):787–794. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- 21.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovascular Research. 2005;65(1):40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 22.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. American Journal of Physiology. 2007;293(3):H1883–H1891. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 23.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circulation Research. 2009;105(10):934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ieda M, Tsuchihashi T, Ivey KN, et al. Cardiac fibroblasts regulate myocardial proliferation through β1 integrin signaling. Developmental Cell. 2009;16(2):233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fries KM, Blieden T, Looney RJ, et al. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clinical Immunology and Immunopathology. 1994;72(3):283–292. doi: 10.1006/clin.1994.1144. [DOI] [PubMed] [Google Scholar]
- 26.Jelaska A, Strehlow D, Korn JH. Fibroblast heterogeneity in physiological conditions and fibrotic disease. Springer Seminars in Immunopathology. 1999;21(4):385–395. [PubMed] [Google Scholar]
- 27.Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biology and Therapy. 2006;5(12):1640–1646. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 28.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. Journal of Cellular Physiology. 2010;225(3):631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carmona R, Guadix JA, Cano E, et al. The embryonic epicardium: an essential element of cardiac development. Journal of Cellular and Molecular Medicine. 2010;14(8):2066–2072. doi: 10.1111/j.1582-4934.2010.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olivey HE, Mundell NA, Austin AF, Barnett JV. Transforming growth factor-β stimulates epithelial-mesenchymal transformation in the proepicardium. Developmental Dynamics. 2006;235(1):50–59. doi: 10.1002/dvdy.20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Epstein JA. Cardiac development and implications for heart disease. New England Journal of Medicine. 2010;363(17):1638–1647. doi: 10.1056/NEJMra1003941. [DOI] [PubMed] [Google Scholar]
- 32.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nature Medicine. 2007;13(8):952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 33.Sartore S, Chiavegato A, Faggin E, et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circulation Research. 2001;89(12):1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 34.van Amerongen MJ, Bou-Gharios G, Popa ER, et al. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. Journal of Pathology. 2008;214(3):377–386. doi: 10.1002/path.2281. [DOI] [PubMed] [Google Scholar]
- 35.Kania G, Blyszczuk P, Stein S, et al. Heart-infiltrating prominin-1+/cd133+ progenitor cells represent the cellular source of transforming growth factor β-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circulation Research. 2009;105(5):462–470. doi: 10.1161/CIRCRESAHA.109.196287. [DOI] [PubMed] [Google Scholar]
- 36.Möllmann H, Nef HM, Kostin S, et al. Bone marrow-derived cells contribute to infarct remodelling. Cardiovascular Research. 2006;71(4):661–671. doi: 10.1016/j.cardiores.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-β and peroxisome proliferator-activated receptor. Journal of Biological Chemistry. 2007;282(31):22910–22920. doi: 10.1074/jbc.M703597200. [DOI] [PubMed] [Google Scholar]
- 38.Haudek SB, Xia Y, Huebener P, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18284–18289. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujita J, Mori M, Kawada H, et al. Administration of granulocyte colony-stimulating factor after myocardial infarction enhances the recruitment of hematopoietic stem cell-derived myofibroblasts and contributes to cardiac repair. Stem Cells. 2007;25(11):2750–2759. doi: 10.1634/stemcells.2007-0275. [DOI] [PubMed] [Google Scholar]
- 40.Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circulation Research. 2010;106(1):47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez JJ, Lorell BH, Ingelfinger JR, et al. Distribution and function of cardiac angiotensin AT1-and AT2-receptor subtypes in hypertrophied rat hearts. American Journal of Physiology. 1994;267(2):H844–H852. doi: 10.1152/ajpheart.1994.267.2.H844. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki J, Matsubara H, Urakami M, Inada M. Rat angiotensin II (type 1A) receptor mRNA regulation and subtype expression in myocardial growth and hypertrophy. Circulation Research. 1993;73(3):439–447. doi: 10.1161/01.res.73.3.439. [DOI] [PubMed] [Google Scholar]
- 43.Tsutsumi Y, Matsubara H, Ohkubo N, et al. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Circulation Research. 1998;83(10):1035–1046. doi: 10.1161/01.res.83.10.1035. [DOI] [PubMed] [Google Scholar]
- 44.Harada K, Komuro I, Zou Y, et al. Acute pressure overload could induce hypertrophic responses in the heart of angiotensin II type 1a knockout mice. Circulation Research. 1998;82(7):779–785. doi: 10.1161/01.res.82.7.779. [DOI] [PubMed] [Google Scholar]
- 45.Harada K. Pressure overload induces cardiac hypertrophy in angiotensin II type 1a receptor knockout mice. Circulation. 1998;97(19):1952–1959. doi: 10.1161/01.cir.97.19.1952. [DOI] [PubMed] [Google Scholar]
- 46.Matsusaka T, Katori H, Inagami T, Fogo A, Ichikawa I. Communication between myocytes and fibroblasts in cardiac remodeling in angiotensin chimeric mice. Journal of Clinical Investigation. 1999;103(10):1451–1458. doi: 10.1172/JCI5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-β1 and endothelin-1 from fibroblasts. Cardiovascular Research. 1998;40(2):352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 48.Harada M, Itoh H, Nakagawa O, et al. Significance of ventricular myocytes and nonmyocytes interaction during cardiocyte hypertrophy: evidence for endothelin-1 as a paracrine hypertrophic factor from cardiac nonmyocytes. Circulation. 1997;96(10):3737–3744. doi: 10.1161/01.cir.96.10.3737. [DOI] [PubMed] [Google Scholar]
- 49.Pellieux C, Foletti A, Peduto G, et al. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. Journal of Clinical Investigation. 2001;108(12):1843–1851. doi: 10.1172/JCI13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang ZS, Jeyaraman M, Wen GB, et al. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. Journal of Molecular and Cellular Cardiology. 2007;42(1):222–233. doi: 10.1016/j.yjmcc.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 51.Hoogwerf BJ. Renin-Angiotensin System Blockade and Cardiovascular and Renal Protection. American Journal of Cardiology. 2010;105(1, supplement) doi: 10.1016/j.amjcard.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Saito Y, Kuwahara K, et al. Guanylyl cyclase-A inhibits angiotensin II type 2 receptor-mediated pro-hypertrophic signaling in the heart. Endocrinology. 2009;150(8):3759–3765. doi: 10.1210/en.2008-1353. [DOI] [PubMed] [Google Scholar]
- 53.Masaki H, Kurihara T, Yamaki A, et al. Cardiac-specific overexpression of angiotensin II AT2 receptor causes attenuated response to AT1 receptor-mediated pressor and chronotropic effects. Journal of Clinical Investigation. 1998;101(3):527–535. doi: 10.1172/JCI1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ichihara S, Senbonmatsu T, Price E, Jr., Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation. 2001;104(3):346–351. doi: 10.1161/01.cir.104.3.346. [DOI] [PubMed] [Google Scholar]
- 55.D’Amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension. 2005;46(6):1347–1354. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]
- 56.Opie LH, Sack MN. Enhanced angiotensin II activity in heart failure: reevaluation of the counterregulatory hypothesis of receptor subtypes. Circulation Research. 2001;88(7):654–658. doi: 10.1161/hh0701.089175. [DOI] [PubMed] [Google Scholar]
- 57.Funke-Kaiser H, Reinemund J, Steckelings UM, Unger T. Adapter proteins and promoter regulation of the angiotensin AT2 receptor—implications for cardiac pathophysiology. Journal of the Renin-Angiotensin-Aldosterone System. 2010;11(1):7–17. doi: 10.1177/1470320309343652. [DOI] [PubMed] [Google Scholar]
- 58.Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB Journal. 2004;18(7):816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 59.Munger JS, Huang X, Kawakatsu H, et al. The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 60.Hyytiäinen M, Penttinen C, Keski-Oja J. Latent TGF-β binding proteins: extracellular matrix association and roles in TGF-β activation. Critical Reviews in Clinical Laboratory Sciences. 2004;41(3):233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 61.Engelmann GL, Grutkoski PS. Cordinate TGF-β receptor gene expression during rat heart development. Cellular and Molecular Biology Research. 1994;40(2):93–104. [PubMed] [Google Scholar]
- 62.Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-β . Journal of Biochemistry. 2010;147(6):781–792. doi: 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang YC, Piek E, Zavadil J, et al. Hierarchical model of gene regulation by transforming growth factor β . Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10269–10274. doi: 10.1073/pnas.1834070100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-β/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. Journal of Biological Chemistry. 2001;276(20):17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- 65.Bujak M, Ren G, Kweon HJ, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116(19):2127–2138. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 66.Ryer EJ, Hom RP, Sakakibara K, et al. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arteriosclerosis Thrombosis and Vascular Biology. 2006;26(4):780–786. doi: 10.1161/01.ATV.0000209517.00220.cd. [DOI] [PubMed] [Google Scholar]
- 67.Schultz JEJ, Witt SA, Glascock BJ, et al. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. Journal of Clinical Investigation. 2002;109(6):787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuwahara F, Kai H, Tokuda K, et al. Transforming growth factor-β function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106(1):130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 69.Tan SM, Zhang Y, Connelly KA, Gilbert RE, Kelly DJ. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. American Journal of Physiology. 2010;298(5):H1415–H1425. doi: 10.1152/ajpheart.01048.2009. [DOI] [PubMed] [Google Scholar]
- 70.Divakaran V, Adrogue J, Ishiyama M, et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circulation. 2009;2(6):633–642. doi: 10.1161/CIRCHEARTFAILURE.108.823070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frantz S, Hu K, Adamek A, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Research in Cardiology. 2008;103(5):485–492. doi: 10.1007/s00395-008-0739-7. [DOI] [PubMed] [Google Scholar]
- 72.Liao S, Bodmer J, Pietras D, Azhar M, Doetschman T, Schultz JEJ. Biological functions of the low and high molecular weight protein isoforms of fibroblast growth factor-2 in cardiovascular development and disease. Developmental Dynamics. 2009;238(2):249–264. doi: 10.1002/dvdy.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aoyama T, Takimoto Y, Pennica D, et al. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. Journal of Molecular and Cellular Cardiology. 2000;32(10):1821–1830. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- 74.Freed DH, Moon MC, Borowiec AM, Jones SC, Zahradka P, Dixon IMC. Cardiotrophin-1: expression in experimental myocardial infarction and potential role in post-MI wound healing. Molecular and Cellular Biochemistry. 2003;254(1-2):247–256. doi: 10.1023/a:1027332504861. [DOI] [PubMed] [Google Scholar]
- 75.King KL, Winer J, Phillips DM, Quach J, Williams PM, Mather JP. Phenylephrine, endothelin, prostaglandin F(2α), and leukemia inhibitory factor induce different cardiac hypertrophy phenotypes in vitro. Endocrine Journal. 1998;9(1):45–55. doi: 10.1385/endo:9:1:45. [DOI] [PubMed] [Google Scholar]
- 76.Kuwahara K, Saito Y, Harada M, et al. Involvement of cardiotrophin-1 in cardiac myocyte-nonmyocyte interactions during hypertrophy of rat cardiac myocytes in vitro. Circulation. 1999;100(10):1116–1124. doi: 10.1161/01.cir.100.10.1116. [DOI] [PubMed] [Google Scholar]
- 77.Wang F, Trial J, Diwan A, et al. Regulation of cardiac fibroblast cellular function by leukemia inhibitory factor. Journal of Molecular and Cellular Cardiology. 2002;34(10):1309–1316. doi: 10.1006/jmcc.2002.2059. [DOI] [PubMed] [Google Scholar]
- 78.Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie ANJ, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. Journal of Clinical Investigation. 2007;117(6):1538–1549. doi: 10.1172/JCI30634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nature Reviews Drug Discovery. 2008;7(10):827–840. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liang YJ, Lai LP, Wang BW, et al. Mechanical stress enhances serotonin 2B receptor modulating brain natriuretic peptide through nuclear factor-κB in cardiomyocytes. Cardiovascular Research. 2006;72(2):303–312. doi: 10.1016/j.cardiores.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 81.Jaffré F, Bonnin P, Callebert J, et al. Serotonin and angiotensin receptors in cardiac fibroblasts coregulate adrenergic-dependent cardiac hypertrophy. Circulation Research. 2009;104(1):113–123. doi: 10.1161/CIRCRESAHA.108.180976. [DOI] [PubMed] [Google Scholar]
- 82.Jaffré F, Callebert J, Sarre A, et al. Involvement of the serotonin 5-HT2B receptor in cardiac hypertrophy linked to sympathetic stimulation: control of interleukin-6, interleukin-1β, and tumor necrosis factor-α cytokine production by ventricular fibroblasts. Circulation. 2004;110(8):969–974. doi: 10.1161/01.CIR.0000139856.20505.57. [DOI] [PubMed] [Google Scholar]
- 83.Monassier L, Laplante MA, Jaffré F, Bousquet P, Maroteaux L, de Champlain J. Serotonin 5-HT2B receptor blockade prevents reactive oxygen species-induced cardiac hypertrophy in mice. Hypertension. 2008;52(2):301–307. doi: 10.1161/HYPERTENSIONAHA.107.105551. [DOI] [PubMed] [Google Scholar]
- 84.Shyu KG. Editorial: serotonin 5-HT2B receptor in cardiac fibroblast contributes to cardiac hypertrophy: a new therapeutic target for heart failure? Circulation Research. 2009;104(1):1–3. doi: 10.1161/CIRCRESAHA.108.191122. [DOI] [PubMed] [Google Scholar]
- 85.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine and Growth Factor Reviews. 2004;15(4):255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 86.Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS Journal. 2005;272(22):5723–5741. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 87.Shindo T, Manabe I, Fukushima Y, et al. Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nature Medicine. 2002;8(8):856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 88.Liao CH, Akazawa H, Tamagawa M, et al. Cardiac mast cells cause atrial fibrillation through PDGF-A—mediated fibrosis in pressure-overloaded mouse hearts. Journal of Clinical Investigation. 2010;120(1):242–253. doi: 10.1172/JCI39942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tuuminen R, Nykanen AI, Krebs R, et al. PDGF-A, -C, and -D but not PDGF-B increase TGF-beta1 and chronic rejection in rat cardiac allografts. Arteriosclerosis Thrombosis and Vascular Biology. 2009;29(5):691–698. doi: 10.1161/ATVBAHA.108.178558. [DOI] [PubMed] [Google Scholar]
- 90.Li X, Pontén A, Aase K, et al. PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nature Cell Biology. 2000;2(5):302–307. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- 91.Pontén A, Li X, Thorén P, et al. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. American Journal of Pathology. 2003;163(2):673–682. doi: 10.1016/S0002-9440(10)63694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pontén A, Folestad EB, Pietras K, Eriksson U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circulation Research. 2005;97(10):1036–1045. doi: 10.1161/01.RES.0000190590.31545.d4. [DOI] [PubMed] [Google Scholar]
- 93.Fujiu K, Manabe I, Ishihara A, et al. Synthetic retinoid Am80 suppresses smooth muscle phenotypic modulation and in-stent neointima formation by inhibiting KLF5. Circulation Research. 2005;97(11):1132–1141. doi: 10.1161/01.RES.0000190613.22565.13. [DOI] [PubMed] [Google Scholar]
- 94.Chu TF, Rupnick MA, Kerkela R, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. The Lancet. 2007;370(9604):2011–2019. doi: 10.1016/S0140-6736(07)61865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmidinger M, Zielinski CC, Vogl UM, et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology. 2008;26(32):5204–5212. doi: 10.1200/JCO.2007.15.6331. [DOI] [PubMed] [Google Scholar]
- 96.Khakoo AY, Kassiotis CM, Tannir N, et al. Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer. 2008;112(11):2500–2508. doi: 10.1002/cncr.23460. [DOI] [PubMed] [Google Scholar]
- 97.Kerkelä R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nature Medicine. 2006;12(8):908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 98.Chintalgattu V, Ai D, Langley RR, et al. Cardiomyocyte PDGFR-β signaling is an essential component of the mouse cardiac response to load-induced stress. Journal of Clinical Investigation. 2010;120(2):472–484. doi: 10.1172/JCI39434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sjögren K, Liu JL, Blad K, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(12):7088–7092. doi: 10.1073/pnas.96.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McMullen JR. Role of insulin-like growth factor 1 and phosphoinositide 3-kinase in a setting of heart disease. Clinical and Experimental Pharmacology and Physiology. 2008;35(3):349–354. doi: 10.1111/j.1440-1681.2007.04873.x. [DOI] [PubMed] [Google Scholar]
- 101.Perbal B. CCN proteins: multifunctional signalling regulators. The Lancet. 2004;363(9402):62–64. doi: 10.1016/S0140-6736(03)15172-0. [DOI] [PubMed] [Google Scholar]
- 102.Daniels A, van Bilsen M, Goldschmeding R, van der Vusse GJ, van Nieuwenhoven FA. Connective tissue growth factor and cardiac fibrosis. Acta Physiologica. 2009;195(3):321–338. doi: 10.1111/j.1748-1716.2008.01936.x. [DOI] [PubMed] [Google Scholar]
- 103.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circulation Research. 2010;106(11):1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 104.Hayata N, Fujio Y, Yamamoto Y, et al. Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochemical and Biophysical Research Communications. 2008;370(2):274–278. doi: 10.1016/j.bbrc.2008.03.100. [DOI] [PubMed] [Google Scholar]
- 105.Yoon PO, Lee MA, Cha H, et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. Journal of Molecular and Cellular Cardiology. 2010;49(2):294–303. doi: 10.1016/j.yjmcc.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 106.Panek AN, Posch MG, Alenina N, et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS ONE. 2009;4(8) doi: 10.1371/journal.pone.0006743. Article ID e6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuwahara K, Nakao K. Regulation and significance of atrial and brain natriuretic peptides as cardiac hormones. Endocrine Journal. 2010;57(7):555–565. doi: 10.1507/endocrj.k10e-150. [DOI] [PubMed] [Google Scholar]
- 108.Saito Y. Roles of atrial natriuretic peptide and its therapeutic use. Journal of Cardiology. 2010;56(3):262–270. doi: 10.1016/j.jjcc.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 109.Oliver PM, Fox JE, Kim R, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kishimoto I, Rossi K, Garbers DL. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2703–2706. doi: 10.1073/pnas.051625598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Knowles JW, Esposito G, Mao L, et al. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. Journal of Clinical Investigation. 2001;107(8):975–984. doi: 10.1172/JCI11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kapoun AM, Liang F, O’Young G, et al. B-Type natriuretic peptide exerts broad functional opposition to transforming growth factor-β in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circulation Research. 2004;94(4):453–461. doi: 10.1161/01.RES.0000117070.86556.9F. [DOI] [PubMed] [Google Scholar]
- 113.Glenn DJ, Rahmutula D, Nishimoto M, Liang F, Gardner DG. Atrial natriuretic peptide suppresses endothelin gene expression and proliferation in cardiac fibroblasts through a GATA4-dependent mechanism. Cardiovascular Research. 2009;84(2):209–217. doi: 10.1093/cvr/cvp208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aikawa R, Nagai T, Kudoh S, et al. Integrins play a critical role in mechanical stress-induced p38 MAPK activation. Hypertension. 2002;39(2):233–238. doi: 10.1161/hy0202.102699. [DOI] [PubMed] [Google Scholar]
- 115.Ross RS, Borg TK. Integrins and the myocardium. Circulation Research. 2001;88(11):1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- 116.Lal H, Verma SK, Foster DM, et al. Integrins and proximal signaling mechanisms in cardiovascular disease. Frontiers in Bioscience. 2009;14:2307–2334. doi: 10.2741/3381. [DOI] [PubMed] [Google Scholar]
- 117.Bowers SLK, Banerjee I, Baudino TA. The extracellular matrix: at the center of it all. Journal of Molecular and Cellular Cardiology. 2010;48(3):474–482. doi: 10.1016/j.yjmcc.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hannigan GE, Coles JG, Dedhar S. Integrin-linked kinase at the heart of cardiac contractility, repair, and disease. Circulation Research. 2007;100(10):1408–1414. doi: 10.1161/01.RES.0000265233.40455.62. [DOI] [PubMed] [Google Scholar]
- 119.Orian-Rousseau V. CD44, a therapeutic target for metastasising tumours. European Journal of Cancer. 2010;46(7):1271–1277. doi: 10.1016/j.ejca.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 120.Huebener P, Abou-Khamis T, Zymek P, et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. Journal of Immunology. 2008;180(4):2625–2633. doi: 10.4049/jimmunol.180.4.2625. [DOI] [PubMed] [Google Scholar]
- 121.Hellman U, Hellström M, Mörner S, et al. Parallel up-regulation of FGF-2 and hyaluronan during development of cardiac hypertrophy in rat. Cell and Tissue Research. 2008;332(1):49–56. doi: 10.1007/s00441-007-0562-8. [DOI] [PubMed] [Google Scholar]
- 122.Crawford DC, Chobanian AV, Brecher P. Angiotensin II induces fibronectin expression associated with cardiac fibrosis in the rat. Circulation Research. 1994;74(4):727–739. doi: 10.1161/01.res.74.4.727. [DOI] [PubMed] [Google Scholar]
- 123.Collins AR, Schnee J, Wang W, et al. Osteopontin modulates angiotensin II- induced fibrosis in the intact murine heart. Journal of the American College of Cardiology. 2004;43(9):1698–1705. doi: 10.1016/j.jacc.2003.11.058. [DOI] [PubMed] [Google Scholar]
- 124.Chen H, Huang XN, Stewart AFR, Sepulveda JL. Gene expression changes associated with fibronectin-induced cardiac myocyte hypertrophy. Physiological Genomics. 2004;18(3):273–283. doi: 10.1152/physiolgenomics.00104.2004. [DOI] [PubMed] [Google Scholar]
- 125.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(11):2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 126.Matsui Y, Jia N, Okamoto H, et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension. 2004;43(6):1195–1201. doi: 10.1161/01.HYP.0000128621.68160.dd. [DOI] [PubMed] [Google Scholar]
- 127.Klingel K, Kandolf R. Osteopontin: a biomarker to predict the outcome of inflammatory heart disease. Seminars in Thrombosis and Hemostasis. 2010;36(2):195–202. doi: 10.1055/s-0030-1251504. [DOI] [PubMed] [Google Scholar]
- 128.Waller AH, Sanchez-Ross M, Kaluski E, Klapholz M. Osteopontin in cardiovascular disease: a potential therapeutic target. Cardiology in Review. 2010;18(3):125–131. doi: 10.1097/CRD.0b013e3181cfb646. [DOI] [PubMed] [Google Scholar]
- 129.Georgiadou P, Iliodromitis EK, Kolokathis F, et al. Osteopontin as a novel prognostic marker in stable ischaemic heart disease: a 3-year follow-up study. European Journal of Clinical Investigation. 2010;40(4):288–293. doi: 10.1111/j.1365-2362.2010.02257.x. [DOI] [PubMed] [Google Scholar]
- 130.Snider P, Hinton RB, Moreno-Rodriguez RA, et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circulation Research. 2008;102(7):752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Morris RA, Damon B, Mironov V, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. Journal of Cellular Biochemistry. 2007;101(3):695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shimazaki M, Nakamura K, Kii I, et al. Periostin is essential for cardiac healing after acute myocardial infarction. Journal of Experimental Medicine. 2008;205(2):295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen YF, Feng JA, Li P, et al. Dominant negative mutation of the TGF-beta receptor blocks hypoxia-induced pulmonary vascular remodeling. Journal of Applied Physiology. 2006;100(2):564–571. doi: 10.1152/japplphysiol.00595.2005. [DOI] [PubMed] [Google Scholar]
- 134.Iekushi K, Taniyama Y, Azuma J, et al. Novel mechanisms of valsartan on the treatment of acute myocardial infarction through inhibition of the antiadhesion molecule periostin. Hypertension. 2007;49(6):1409–1414. doi: 10.1161/HYPERTENSIONAHA.106.080994. [DOI] [PubMed] [Google Scholar]
- 135.Oka T, Xu J, Kaiser RA, et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circulation Research. 2007;101(3):313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Esper RJ, Nordaby RA, Vilariño JO, Paragano A, Cacharrón JL, Machado RA. Endothelial dysfunction: a comprehensive appraisal. Cardiovascular Diabetology. 2006;5, article 4 doi: 10.1186/1475-2840-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kai H, Kuwahara F, Tokuda K, Imaizumi T. Diastolic dysfunction in hypertensive hearts: roles of perivascular inflammation and reactive myocardial fibrosis. Hypertension Research. 2005;28(6):483–490. doi: 10.1291/hypres.28.483. [DOI] [PubMed] [Google Scholar]
- 138.Kai H, Mori T, Tokuda K, et al. Pressure overload-induced transient oxidative stress mediates perivascular inflammation and cardiac fibrosis through angiotensin II. Hypertension Research. 2006;29(9):711–718. doi: 10.1291/hypres.29.711. [DOI] [PubMed] [Google Scholar]
- 139.Seddon M, Shah AM, Casadei B. Cardiomyocytes as effectors of nitric oxide signalling. Cardiovascular Research. 2007;75(2):315–326. doi: 10.1016/j.cardiores.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 140.Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovascular Research. 2005;66(3):444–453. doi: 10.1016/j.cardiores.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 141.Shiojima I, Sato K, Izumiya Y, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. Journal of Clinical Investigation. 2005;115(8):2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47(5):887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jiang Y, Reynolds C, Xiao C, et al. Dietary copper supplementation reverses hypertrophic cardiomyopathy induced by chronic pressure overload in mice. Journal of Experimental Medicine. 2007;204(3):657–666. doi: 10.1084/jem.20061943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sano M, Minamino T, Toko H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 145.Friehs I, Barillas R, Vasilyev NV, Roy N, McGowan FX, del Nido PJ. Vascular endothelial growth factor prevents apoptosis and preserves contractile function in hypertrophied infant heart. Circulation. 2006;114(1, supplement):I290–I295. doi: 10.1161/CIRCULATIONAHA.105.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Krishnan J, Suter M, Windak R, et al. Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metabolism. 2009;9(6):512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 147.Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 148.Fareh J, Touyz RM, Schiffrin EL, Thibault G. Endothelin-1 and angiotensin II receptors in cells from rat hypertrophied heart: receptor regulation and intracellular Ca2+ modulation. Circulation Research. 1996;78(2):302–311. doi: 10.1161/01.res.78.2.302. [DOI] [PubMed] [Google Scholar]
- 149.Kedzierski RM, Grayburn PA, Kisanuki YY, et al. Cardiomyocyte-specific endothelin A receptor knockout mice have normal cardiac function and an unaltered hypertrophic response to angiotensin II and isoproterenol. Molecular and Cellular Biology. 2003;23(22):8226–8232. doi: 10.1128/MCB.23.22.8226-8232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Herizi A, Jover B, Bouriquet N, Mimran A. Prevention of the cardiovascular and renal effects of angiotensin II by endothelin blockade. Hypertension. 1998;31(1):10–14. doi: 10.1161/01.hyp.31.1.10. [DOI] [PubMed] [Google Scholar]
- 151.Widyantoro B, Emoto N, Nakayama K, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121(22):2407–2418. doi: 10.1161/CIRCULATIONAHA.110.938217. [DOI] [PubMed] [Google Scholar]
- 152.Hinglais N, Heudes D, Nicoletti A, et al. Colocalization of myocardial fibrosis and inflammatory cells in rats. Laboratory Investigation. 1994;70(2):286–294. [PubMed] [Google Scholar]
- 153.Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J, Michel JB. Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovascular Research. 1996;32(6):1096–1107. doi: 10.1016/s0008-6363(96)00158-7. [DOI] [PubMed] [Google Scholar]
- 154.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Usher MG, Duan SZ, Ivaschenko CY, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. Journal of Clinical Investigation. 2010;120(9):3350–3364. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kuwahara F, Kai H, Tokuda K, et al. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43(4):739–745. doi: 10.1161/01.HYP.0000118584.33350.7d. [DOI] [PubMed] [Google Scholar]
- 157.Zandbergen HR, Sharma UC, Gupta S, et al. Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy. Journal of Cardiovascular Pharmacology and Therapeutics. 2009;14(1):68–75. doi: 10.1177/1074248408329860. [DOI] [PubMed] [Google Scholar]
- 158.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kim J, Ogai A, Nakatani S, et al. Impact of blockade of histamine H2 receptors on chronic heart failure revealed by retrospective and prospective randomized studies. Journal of the American College of Cardiology. 2006;48(7):1378–1384. doi: 10.1016/j.jacc.2006.05.069. [DOI] [PubMed] [Google Scholar]
- 160.Takahama H, Asanuma H, Sanada S, et al. A histamine H receptor blocker ameliorates development of heart failure in dogs independently of beta-adrenergic receptor blockade. Basic Research in Cardiology. 2010;105(6):787–794. doi: 10.1007/s00395-010-0119-y. [DOI] [PubMed] [Google Scholar]
- 161.Balakumar P, Singh AP, Ganti SS, et al. Resident cardiac mast cells: are they the major culprit in the pathogenesis of cardiac hypertrophy? Basic & Clinical Pharmacology & Toxicology. 2008;102(1):5–9. doi: 10.1111/j.1742-7843.2007.00147.x. [DOI] [PubMed] [Google Scholar]
- 162.Gilles S, Zahler S, Welsch U, Sommerhoff CP, Becker BF. Release of TNF-α during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovascular Research. 2003;60(3):608–616. doi: 10.1016/j.cardiores.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 163.Brower GL, Janicki JS. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. Journal of Cardiac Failure. 2005;11(7):548–556. doi: 10.1016/j.cardfail.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 164.Kvakan H, Kleinewietfeld M, Qadri F, et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119(22):2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 165.Sturzu AC, Wu SM. Developmental and regenerative biology of multipotent cardiovascular progenitor cells. Circulation Research. 2011;108(3):353–364. doi: 10.1161/CIRCRESAHA.110.227066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Davis ME, Hsieh PCH, Takahashi T, et al. Local myocardial insulin-like growth factor 1 (IGF-1) delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(21):8155–8160. doi: 10.1073/pnas.0602877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gonzalez A, Rota M, Nurzynska D, et al. Activation of cardiac progenitor cells reverses the failing heart senescent phenotype and prolongs lifespan. Circulation Research. 2008;102(5):597–606. doi: 10.1161/CIRCRESAHA.107.165464. [DOI] [PubMed] [Google Scholar]
- 168.Urbanek K, Cesselli D, Rota M, et al. Stem cell niches in the adult mouse heart. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(24):9226–9231. doi: 10.1073/pnas.0600635103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bearzi C, Rota M, Hosoda T, et al. Human cardiac stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(35):14068–14073. doi: 10.1073/pnas.0706760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hosoda T, Zheng H, Cabral-da-Silva M, et al. Human cardiac stem cell differentiation is regulated by a mircrine mechanism. Circulation. 2011;123(12):1287–1296. doi: 10.1161/CIRCULATIONAHA.110.982918. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 171.Boni A, Urbanek K, Nascimbene A, et al. Notch1 regulates the fate of cardiac progenitor cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(40):15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 172.Goichberg P, Bai Y, D'Amario D, et al. The ephrin A1-EphA2 system promotes cardiac stem cell migration after infarction. Circulation Research. 2011;108(9):1071–1083. doi: 10.1161/CIRCRESAHA.110.239459. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]