

Abstract
The nuclear xenobiotic receptor CAR is a phenobarbital (PB)-activated transcription factor. Using a mouse model of two-step liver tumorigenesis, in which tumor growth was initiated by diethyl nitrosamine (DEN) and promoted by chronic treatment with PB, we previously demonstrated that tumors developed only in the presence of CAR. Here, we have identified the FAM84A (family with sequence similarity 84, member A) gene as a CAR-regulated gene that is over-expressed during development of phenobarbital-promoted mouse liver tumors. FAM84A mRNA was induced in the liver of DEN/PB-treated mice prior to the development of liver tumors and this induction continued in the non-tumor as well as tumor tissues of a tumor-bearing liver. Western blotting demonstated that FAM84A protein expression increased in mouse liver after PB treatment; however, the FAM84A protein in liver and liver tumors was not phosphorylated at the serine 38 residue, which has been reported to correlate with morphological changes in cells. Immunohistochemistry analysis revealed the cytoplasmic localization of FAM84A protein and its expression during tumor development in normal tissues (especially in hepatocytes around the central vein), eosinophilic foci, adenomas and carcinomas. HepG2 cell-based reporter assays indicated that CAR activated the FAM84A promoter. Exogenous over-expression of FAM84A in HepG2 cells resulted in increased cell migration. The physiological function of FAM84A remains unknown, but our results suggest that FAM84A is up-regulated by CAR during the development of liver tumors, and may play an important role in the progression of liver cancer by increasing cell migration.
Keywords: CAR, nuclear receptor, FAM84A, phenobarbital, liver cancer, non-genotoxic carcinogenesis
Introduction
CAR (NR1I3) is a member of the thyroid/steroid hormone nuclear receptor superfamily that was first characterized as the phenobarbital (PB)-activated nuclear receptor, inducing the cytochrome P450 2B (CYP2B) gene in the liver (1-3). Soon after, a large group of xenobiotics and therapeutics were characterized as CAR activators, establishing CAR as a nuclear receptor that plays a central role in regulating the hepatic capability of xenobiotic metabolism and excretion (4). The first set of CAR-regulated genes identified were those which encode xenobiotic metabolizing enzymes and transporters, such as numerous CYPs and MRPs (5,6). Our cDNA microarray studies and other experiments using Car-/- mice after PB treatment has extended the role of CAR beyond xenobiotic metabolism to the regulation of hepatic energy metabolism (7,8). For instance, the expression of phosphoenolpyruvate carboxykinase 1 (PEPCK1) which is known as one of gluconeogenic enzyme repressed by PB treatment in Car+/+ mice but not in Car-/- mice (8). In addition to regulating these normal liver functions, CAR can be a factor that causes various liver diseases, such as developing steatohepatitis, cholestasis and liver tumors (9).
Hepatocellular carcinoma (HCC) incidence is very frequent in the world. Formerly, the occurrence of HCC was reported frequently in Asia; however it is now spreading all over the world including Europe and North America (10). Several environmental factors such as infection with hepatitis B virus, hepatitis C virus or exogenous chemical exposure causing chronic inflammation of the liver, can lead to liver diseases, which often develop into HCC. However, the detailed molecular mechanism(s) of the development of HCC remains unknown.
It has been reported that primary exposure to genotoxic carcinogens and subsequent chronic treatment with non-genotoxic agents such as PB produces HCC in rodents, and this rodent model has been used to understand the development and promotion of HCC that does not involve genetic mutations (11,12). Given the fact that PB activates CAR, the two-step mouse model for HCC development was utilized with Car-/- mice in order to determine whether or not CAR plays a role in HCC development. Mice were initiated by a single injection of DEN and then liver tumors were promoted by chronic PB treatment over 34 weeks. The results obtained clearly revealed that the Car-/- mice did not develop liver tumors including HCC (13). Based on this observation, over 90 genes were identified as being CAR-regulated from a cDNA microarray study (unpublished data), and the FAM84A gene was further characterized as a gene that may be directly involved in liver tumor development after PB treatment.
Human FAM84A, based on its amino acid sequence identity with human collagen, type X and α1, is a novel collagen-type protein (14). The only study which investigated the biological function of FAM84A found that FAM84A was over-expressed in various colon cancer cell lines and also in many, but not all, human colon tumor tissues. This study also found that over-expression of ectopic FAM84A altered cell morphology and increased cell migration, thus suggesting that FAM84A may be a pro-tumor factor in colon cancers (14). However, FAM84A has not yet been studied in the liver. Here we have utilized real-time PCR, Western blot and immunohistochemistry to examine the expression of FAM84A in mouse livers, with respect to PB induction, CAR regulation, sex- and strain-dependency and expression in tumors. Furthermore, we showed that ectopic over-expression of FAM84A enhances cell migration, but not cell growth of hepatocellular carcinoma cells. The mouse FAM84A gene appears to be a novel marker for PB-inducible and CAR-regulated liver tumor development.
Materials and methods
Animals
Car+/+ and Car-/- mice were previously generated in a C3H/HeNCrlBR (C3He) background (13). C3H/HeNCrlBR (C3He) and C57BL/6 mice (5-7 week-old) were obtained from Charles River Laboratories International Inc. (MA, USA), housed in a specific pathogen-free rodent facility under a standard 12-h light/12-h dark cycle and were fed a standard rodent chow and water ad libitum. PB (100 mg/kg) was administered intraperitoneally. All animal procedures were approved by the Animal Ethics Committee of National Institute of Environmental Health Sciences.
Reagent
TCPOBOP was purchased from Sigma-Aldrich (MO, USA); androstenol from Steraloids (RI, USA); pGL3-basic from Promega Corp. (WI, USA); pcDNA3.1-V5-His-TOPO from Invitrogen (CA, USA).
Antibodies
To produce mouse anti-FAM84A serum, the C-terminal peptide CRVLQELEDFVDDKE was synthesized (Sigma Genosys, TX, USA) and was used to immunize rabbits. To produce antibodies to phosphorylated Ser38 of mouse FAM84A, the phosphopeptide AYFF(pS)DEEEDL DERGQC was synthesized (ANASPEC, CA, USA) and was used to immunize rabbits. The specificity of this phospho-(Ser38)peptide was verified by Western blots showing its binding to the phospho- but not to non-phospho-peptides.
Real-time PCR
Total RNAs were extracted from mouse livers using TRIzol reagent (Invitrogen), and used to synthesize cDNAs using the High Capacity cDNA reverse transcription kit (Applied Biosystems Inc., CA, USA). Real-time PCR was performed with the 7900HT fast real-time PCR System (Applied Biosystems Inc.). Forward and reverse primers for mouse FAM84A cDNA were: 5′-CCTCGCCGCGTCATTG-3′ and 5′-CAGTGGGCAACTCGCTGTAG-3′. PCR amplification was performed with SYBR-Green Master Mix (Applied Biosystems Inc.). The TaqMan rodent GAPDH (Applied Biosystems Inc.) was used as the reference house-keeping gene, and the expression level of FAM84A mRNA was normalized to that of GAPDH mRNA.
Western blot analysis
Mouse livers were homogenized in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% DOC (deoxycholate-Na), 0.1% SDS and 50 mM Tris-HCl) containing complete protease inhibitor cocktail (Roche Diagnostics, IN, USA). Liver homogenates were centrifuged at 15,000 rpm for 60 min to prepared cytosols. Cytosolic proteins were separated on a 12% SDS-polyacrylamide gel and were transferred onto an Immobilon-P membrane (Millipore, MA, USA). The membrane was then incubated overnight at 4°C with anti-FAM84A serum or anti-phospho-FAM84A (1:5000 dilution) in Tris-buffered saline, 0.05% (v/v) Tween-20 (TBS-T). After incubation with HRP-conjugated anti-rabbit IgG (1:10000 dilution; Cell Signaling Technology, MA, USA), protein bands on the membrane were visualized using ECL Plus Western blotting detection reagent (GE Healthcare Bio-Science Corp. WI, USA). Mouse FAM84A and its S38A mutant proteins were produced using the in vitro transcription/translation system: TNT T7 quick-coupled system (Promega Corp.) and used as markers.
DNA cloning and generation of FAM84A mutants
A full-length cDNA of mouse FAM84A (sequence reference no.; NCBI: NM_029007, Ensembl: ENSMUSG00000020607) was amplified using a set of primers, 5′-ACCATGGGCAACCAACTGGA-3′ and 5′-GCTACTCCTTGTCGTCCACA-3′, and was subsequently cloned into pcDNA3.1-His-V5-TOPO plasmid. We used the mouse brain cDNA for amplification because the expression level of FAM84A was very low in mouse liver. Serine 38 of mouse FAM84A was mutated to alanine using the Quickchange site directed mutagenesis kit (Stratagene, CA, USA) and primers, 5′-GGTTGCCTACTTCTTCGCGGATGAGGAGGAGGA-3′ and 5′-TCCTCCTCCTCATCCGCGAAGAAGTAGGCAACC-3′. A 9.0-kb promoter of the FAM84A gene was amplified using primers 5′-CTACCCACGTGGTCACTAATCCACAGTAGC-3′ and 5′-CTGGCTCCACTCCGCTTTCCTACAGC-3′ and was cloned into pGL3-basic. A 1.8-kb CYP2B6 promoter was previously cloned into pGL3-basic (15).
Reporter gene assay
HepG2 (human hepatocellular carcinoma cell line) cells were seeded on 24-well plates at a density of 4×104 cells per well in 10% MEM (minimal essential medium supplement with 10% (v/v) fetal bovine serum, antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) and 2 mM glutamine). About 24 h later, cells were transfected with a given luciferase reporter plasmid (100 ng/well), expression plasmid (100 ng/well) and phRL-TK plasmid (10 ng/well) using FuGENE6 (Roche Diagnostics,) for 24 h. Cells were then treated with TCPOBOP (250 nM), androstenol (8 μM) or DMSO for 48 h, and then whole extracts were prepared for luciferase assays using the Dual-luciferase reporter assay system (Promega Corp.).
Immunohistochemistry
C3H/HeNCrlBR (C3He) male mice were chronically treated with PB (500 ppm) in drinking water for 6 weeks, and then liver sections were prepared, fixed 10% formalin and embedded in paraffin. Sections of tumors were prepared from the C3He mice used in our previous study (13). Immunohistochemistry using a 1:1000 dilution of anti-FAM84A serum and pre-bleed serum was performed at the Histology Core Laboratory at NIEHS.
Laser capture microdissection
Frozen liver tissue sections were stained with 1% crystal violet acetate, and periportal or centrilobular areas were captured by laser capture microdissection (LCM) using an MMI CellCut, laser capture microscope (The Molecular Machines & Industries). Individual areas from four serial sections were dissected from the slides and captured by the caps of microcentrifuge tubes for RNA extraction. LCM was verified by light microscopic analysis. RNAs were extracted from each samples using the Arcturus PicoPure Kit (MDS analytical Technolog, CA, USA), and used for cDNA synthesis.
Recombinant adenoviruses
Based on the wild-type FAM84A expression plasmid, Ad-FAM84A expression adenovirus vector was constructed with the AdEasy Vector System kit (Q-BIO gene, Montreal, Canada) as described in the manufacturer's protocol. Adenoviruses were propagated in HEK293 cells. Ad-ß-gal encoding ß-galactosidase previously constructed in our laboratory was used as control. Virus titers were measured and calculated using the 50% tissue culture infectious dose assay (TCID50 method) as described in the manufacturer's application manual.
Colony formation assay
Human hepatocellular carcinoma cell line, Huh7 cells were seeded in 10-cm culture dishes 24 h before transfection. Each expression vector (pcDNA3.1-His-V5-Empty and FAM84A WT plasmid) was transfected using FuGENE 6 transfection reagent (Roche Diagnostics); 24 h after transfection, the harvested cells were diluted and reseeded on 10-cm culture dishes in triplicate. The transfected cells were allowed to grow in the presence of 0.8 mg/ml Geneticin (Invitrogen) for 2 weeks, after which the colonies formed from each cell were fixed with 10% formaldehyde, stained with 0.125% crystal violet and counted.
Cell migration assay
Transwell chambers (8-μm pore size; Corning, MA, USA) were used for the in vitro cell migration assay. Twenty-four hours post-seeding, HepG2 cells were infected with Ad-ß-gal or Ad-FAM84A-WT at 10 MOI (multiplicity of infection). Adenovirus-infected cells were seeded into the upper part of chamber at 5.0×104 with 300 μl of 10% MEM medium, and the lower compartment was filled with 600 μl of 10% MEM medium. After incubation for 72 h at 37°C in 5% CO2, cells were fixed with 10% formaldehyde Non-migrating cells were gently removed from the upper chamber with cotton, and cells on the underside of the membrane were stained with 0.1% crystal violet. Migrating cells were counted in four microscopic fields at ×100 magnification.
Statistical analysis
The statistical significance between two groups of data sets was determined by using the Student's t-test (two-tailed) with P<0.05 regarded as statistically significant.
Results
CAR-regulated induction of the FAM84A gene by PB
cDNA microarray analysis was first employed to identify the genes that are induced prior to the development of liver tumors and are then continuously expressed in the tumors of DEN/PB treated mice (unpublished data). From a number of genes that fitted this expression pattern, we focused on the FAM84A gene for further investigation since the human homologue has been reported to be over-expressed in human colon tumors (14). Furthermore, there have been no reports on FAM84A in liver. Our previous tumor study showed that the livers of the DEN/6 weeks PB-treated Car+/+ C3He mice did not develop lesions and tumors; DEN/23 weeks PB-treated Car+/+ C3He mice developed only a small number of small lesions (>2 mm) or no tumors, while DEN/32 weeks PB-treated Car+/+ C3He mice had multiple large-size tumors (13). Accordingly, RNA was prepared from the livers of these mice for subsequent real-time PCR to measure the expression level of FAM84A mRNA. In addition, RNA was prepared from the livers of the corresponding Car-/- C3He mice and also from the livers of mice treated only with DEN. The FAM84A mRNA was already induced in the livers of the DEN/6 weeks PB-treated Car+/+ but not in the Car-/- C3He mice and continued to be induced in the DEN/23 weeks PB-treated Car+/+ C3He mice (Fig. 1). The non-tumor and tumor tissues were separated from the livers of the DEN/32 weeks PB-treated Car+/+ C3He mice for subsequent analysis. Real-time PCR revealed that FAM84A mRNA was up-regulated in the tumor tissues as well as in the non-tumor tissues (Fig. 1). These results confirmed that the FAM84A gene undergoes CAR-mediated induction in the liver prior to tumor development and is continually induced in tumor tissues.
Figure 1.
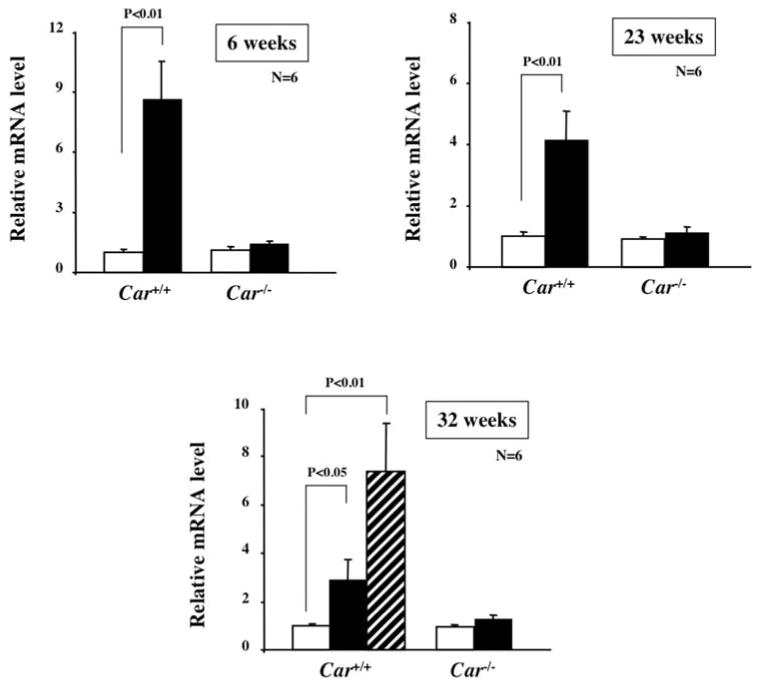
FAM84A mRNA induction in the livers of DEN/PB treated Car+/+ C3He and Car-/- C3He mice by PB. Total liver RNA was prepared from Car+/+ C3He and Car-/- C3He mice and the expression of FAM84A mRNA was evaluated by real-time PCR. Open and closed bars show the levels of FAM84A mRNA in the non-tumor tissues from the DEN-treated and DEN/PB-treated C3He mice, respectively. The hatched bar shows the FAM84a mRNA in tumor tissues. The 6, 23 and 32 weeks indicate the number of weeks that the mice were treated by PB. The relative levels of FAM84A mRNA were calculated by setting the levels in the livers of DEN-treated Car+/+ C3He mice as one. The data are reported as the mean ± SD, n=6.
Strain- and sex-dependent expressions
In mice, males are known to be more susceptible to HCC than females and C3He mice are more susceptible to HCC than C57BL6 mice (16,17). Mice were treated with PB for 24 h and liver RNA was prepared for real-time PCR to measure FAM84A mRNA. Consistent with the observation in mice chronically treated with PB (Fig. 1), the 24-h PB treatment induced FAM84A mRNA in male mice (Fig. 2A). With respect to the strain dependency, compared with an over 4-fold increase of FAM84A mRNA in the PB-treated C3He males, the corresponding PB-treated C57BL6 did not have a significant increase in FAM84A mRNA compared to controls (Fig. 2A). Thus, PB induction of the FAM84A mRNA was specific to the C3He males. This result suggests that the induction of FAM84A mRNA by PB in liver correlates with the strain-dependent HCC susceptibility. On the other hand, the level of FAM84A mRNA was not increased by PB treatment in C3He female mice, although the basal level of FAM84A mRNA was higher in females than in males (Fig. 2B). The mechanism of FAM84A gene regulation in liver may differ between male and female mice. This result suggests that the FAM84A gene may not be involved in sex-dependent HCC susceptibility caused by long-term treatment with PB.
Figure 2.
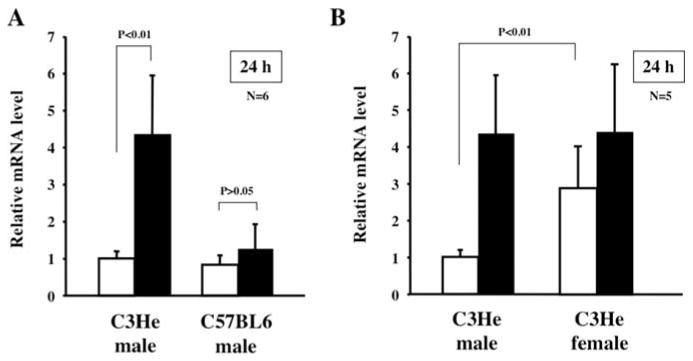
Mouse genetics and sex dependency of FAM84A induction by PB. (A) Total liver RNA was prepared from C3He and C57BL6 males treated with PBS or PB for 24 h. These RNAs were subsequently used for real-time PCR to measure FAM84A mRNA levels in the PBS- or PB-treated samples as shown by open and closed bars, respectively. The relative levels of FAM84A mRNA were calculated by setting the levels in the livers of PBS-treated C3He male mice as one. The data are reported as the mean ± SD, n=6. (B) Male-specific induction. Total RNA was prepared from the livers of five male and female of C3He mice treated with PBS or PB for 24 h. The levels of FAM84A mRNA were determined and are presented as described in (A).
Phosphorylation of FAM84A protein
For further experiments, we produced mouse FAM84A specific anti-serum using the C-terminal peptide of FAM84A. We also produced an anti-serum to detect the phosphorylation of serine 38 of mouse FAM84A, since it has been reported that serine 38 is a target site for phosphorylation of human FAM84A, which is involved in changes in cell morphology (14). We first measured FAM84A protein and phosphorylation of serine 38 by specific anti-sera using samples derived from Ad-FAM84A infected Huh7 cells or in vitro translation of FAM84A-WT or FAM84A-S38A expression plasmid. Two forms of FAM84A protein were detected in in vitro samples (Fig. 3A); the upper band is the serine 38-phosphorylated form and the lower band is the non-phosphorylated form corresponding to the S38A mutant protein (Fig. 3A). Over-expressed FAM84A protein in Huh7 cells by adenovirus infection was not phosphorylated (Fig. 3A) and the same result was observed in HepG2 cells (data not shown).
Figure 3.
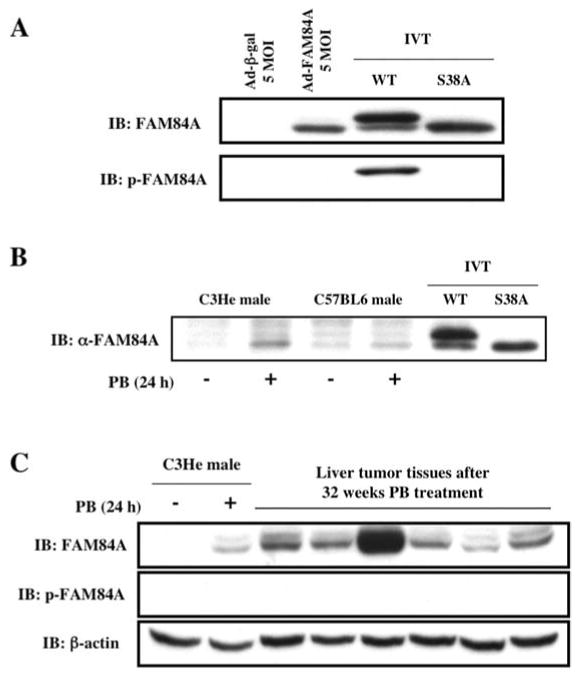
Phosphorylation of FAM84A at serine 38 and characterization of FAM84A protein in mouse liver. (A) Wild-type FAM84A protein was ectopically over-expressed in Huh7 cells that were infected with Ad-FAM84A at 5 MOI. Wild-type FAM84A (WT) and its serine 38 to alanine mutant protein (S38A) were in vitro translated using pcDNA3.1-His-V5 expression plasmids. Western blot analysis was performed using anti-FAM84A and anti-p-FAM84A serum. The in vitro-translated sample (IVT) of WT showed 2 bands, and the upper band was confirmed as phosphorylated FAM84A at serine 38. On the other hand, over-expressed FAM84A protein with Ad-FAM84A was not phosphorylated. (B) Cytosolic fractions were individually prepared from C3He male or C57BL6 male livers with or without PB treatment for 24 h, and were subjected to Western blot analysis using anti-FAM84A serum. IVT samples were run for comparison. The induction of non-phosphorylated FAM84A protein was detected in C3He male mouse liver after PB treatment, but not in C57BL6 mouse liver. (C) Cytosolic fractions were independently prepared from six different tumors derived from mice on chronic PB treatment for 32 weeks. The controls included the cytosolic fractions prepared from the livers of Car+/+ or Car-/- C3He mice treated with PB or PBS for 24 h. FAM84A was induced in PB treated Car+/+ mouse liver or liver tumor samples, but it was not phosphorylated.
Expression of FAM84A protein in mouse liver and tumor tissues
Cytosolic fractions were prepared from the livers of male C3He and C57BL6 mice, which were treated with PB for 24 h. While a slight induction of the FAM84A protein was observed in C57BL6 male mouse liver, an obvious induction of this protein was detected in C3He male mouse liver (Fig. 3B). This result was consistent with the results of real-time PCR (Fig. 2). However, the FAM84A protein that was expressed in each strain was apparently not phosphorylated (Fig. 3B). We also examined the expression of FAM84A using cytosolic fractions from different liver tumor tissues, showing that all 6 tumors expressed the FAM84A protein, but it was not phosphorylated at serine 38 (Fig. 3C). These results indicate that the FAM84A protein is induced by PB treatment in C3He male mouse liver, and also in liver tumors. However, the expressed FAM84A protein in mouse liver was not phosphorylated.
Activation of the mouse FAM84A promoter by CAR
A 9.0-kb FAM84A promoter was cloned into pGL3-basic, producing pGL3-FAM84A -9.0-kb luciferase reporter plasmid, which was subsequently co-transfected with the CAR expression plasmid (cloned into pcDNA3.1-His-V5-TOPO vector) and phRL-TK plasmid into HepG2 cells. These cells were treated with TCPOBOP (mouse CAR activator), androstenol (mouse CAR repressor) or DMSO, and cell extracts were prepared for luciferase assays. We also used the -1.8-kb CYP2B6 promoter, a known CAR-activated promoter, in transfection experiments as a positive control for the effects of CAR in the assay system (15). TCPOBOP treatment up-regulated the transcriptional activation of the -1.8-kb CYP2B6 promoter strongly, and this activation was attenuated by androstenol treatment (Fig. 4). Due to its constitutive activity, CAR activated the -9.0-kb FAM84A promoter; this activation was increased by TCPOBOP treatment while it was attenuated by androstenol treatment (Fig. 4). Compared to the control promoter, the -9.0-kb FAM84A promoter showed an up-regulation of transcriptional activity by CAR over-expression without TCPOBOP treatment. This -9.0-kb FAM84A promoter may be regulated by other transcriptional factors that are influenced by CAR over-expression, and not activation of CAR. However, statistically significant up-regulation of transcriptional activity of the -9.0-kb FAM84A promoter was observed with CAR activation, so these results indicate that CAR activates the FAM84A gene.
Figure 4.
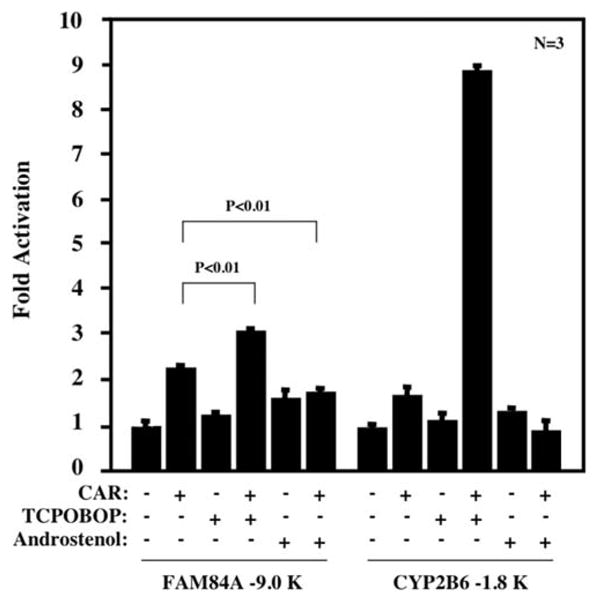
Activation of FAM84A promoter by CAR. The -9.0-kb FAM84A-Luc reporter plasmid was co-transfected with CAR expression plasmid into HepG2 cells for 24 h. Subsequently, these cells were treated with DMSO, CAR activator (250 nM TCPOBOP) or CAR repressor (8 μM androstenol) for an additional 48 h. Then whole cell extracts were prepared and used to measure luciferase activities, which were normalized against the values of Renilla luciferase activities. The activities of the FAM84A promoter are reported as the mean ± SD, n=3 with the activity of DMSO-treated cells as one. The -1.8-kp CYP2B6 reporter plasmid was used in a similar experiment as a positive control for CAR-mediated activation.
Intra-hepatic localization of FAM84A protein
Immunohistochemistry was performed using liver sections prepared from the DEN/6 weeks PB-treated Car+/+ C3He mice using anti-FAM84A serum. The anti-FAM84A serum immunostained the cytoplasm of the hepatocytes found around the central veins, but not those around the portal veins (Fig. 5A). Out of 31 central vein areas examined in DEN-treated and DEN/6 weeks PB-treated Car+/+ C3He males, 26 were positive for FAM84A staining, while all 27 of the portal vein regions were negative (Table I). As expected from the real-time PCR data (Fig. 2A), most of the hepatic central and the portal veins of C57BL6 mice were not stained by an anti-FAM84A serum (Table I). We also collected cells around the central veins and portal veins of 24 h PBS or PB treated Car+/+ C3He males using laser capture microdissection, and measured the expression of FAM84A mRNA by real-time PCR. The RNA for FAM84A was only induced in samples from the central veins following PB treatment (Fig. 5B). Thus, FAM84A was predominantly induced by PB in the central vein regions in C3He mice.
Figure 5.
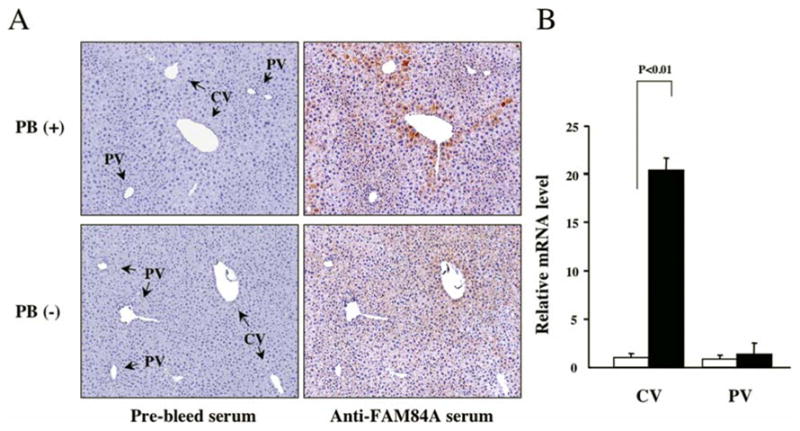
Intra-hepatic localization of FAM84A protein in PB-treated mice. (A) Liver sections were prepared the DEN/6 weeks PB-treated or DEN-treated Car+/+ C3He mice and subjected to immunohistochemical analysis using an anti-FAM84A serum and pre-bleed serum as control. CV and PV denote central and portal veins, respectively. FAM84A protein was induced in the PB-treated mouse liver around the CV regions, not the PV regions. (B) Using laser capture microdissection, cells around CV and PV were collected in the liver sections of C3He male mice treated with PBS or PB for 24 h. These RNAs were subsequently used for real-time PCR to measure FAM84A mRNA levels in the PBS- or PB-treated samples as shown by open and closed bars, respectively. The relative levels of FAM84A mRNA were calculated by setting the levels in the CV of PBS-treated C3He mice as one. The data are reported as the mean ± SD, n=3.
Table I.
FAM84A expression in central versus portal regions.
| Central vein | Portal vein | |||||
|---|---|---|---|---|---|---|
| Anti-serum | Positive | Total count | Positive | Total count | ||
| C3He Male | Non-PB | Pre-bleed | 0 | 25 | 0 | 20 |
| FAM84A | 2 | 25 | 0 | 20 | ||
| PB | Pre-bleed | 0 | 31 | 0 | 27 | |
| FAM84A | 26 | 31 | 0 | 27 | ||
| C57BL6 Male | Non-PB | Pre-bleed | 0 | 20 | 0 | 13 |
| FAM84A | 0 | 20 | 0 | 13 | ||
| PB | Pre-bleed | 0 | 20 | 0 | 18 | |
| FAM84A | 1 | 20 | 1 | 18 | ||
Liver sections were prepared from DEN/6 weeks PB-treated C3He male mice and C57BL6 male mice and were subjected to immunohistochemical analysis using an anti-FAM84A serum and a pre-bleed serum as control.
Expression of FAM84A during the development of liver tumors
Sections were prepared from the tumor-bearing livers of DEN/32 weeks PB-treated C3He mice for immunohistochemical analysis. A PB-promoted tumor-bearing liver characteristically develops eosinophilic foci that can progress into adenoma and eventually to carcinoma (18). As expected, out of a total of 47 foci observed in one of these tumor-bearing livers by H&E staining, 42 were eosinophilic and only 5 were basophilic (Table II). The eosinophilic foci were immunostained with anti-FAM84A serum (Fig. 6) and 69% of them were positive (Table II). Two of five basophilic foci that may have developed spontaneously showed a blush and diffuse staining, which was clearly different from the staining observed with eosinophilic foci. The reason for this result is not clear, but FAM84A may be involved with the promotion of either eosinophilic or basophilic foci. Twelve tumors were classified as adenoma, and nine of these cases were strongly stained by the anti-FAM84A serum (Fig. 6 and Table II). The strongest staining was observed in the peripheral region of hepatocellular carcinoma, although only one case was classified as carcinoma (Fig. 6). These results unequivocally indicate that FAM84A is expressed throughout the development of HCC after PB treatment, from normal hepatocytes to foci and carcinoma.
Table II.
FAM84A expression during stages of liver tumor development.
| FAM84A positive | FAM84A negative | Total no. | |
|---|---|---|---|
| Eosinophilic foci | 29 | 13 | 42 |
| Basophilic foci | 2 | 3 | 5 |
| Adenoma | 9 | 3 | 12 |
| Carcinoma | 1 | 0 | 1 |
After immunohistochemical staining, 12 liver sections prepared from DEN/32 weeks PB-treated Car+/+ C3He mice, groups of FAHs (foci of altered hepatocytes) were identified as eosinophilic and basophilic foci, adenoma and carcinoma.
Figure 6.
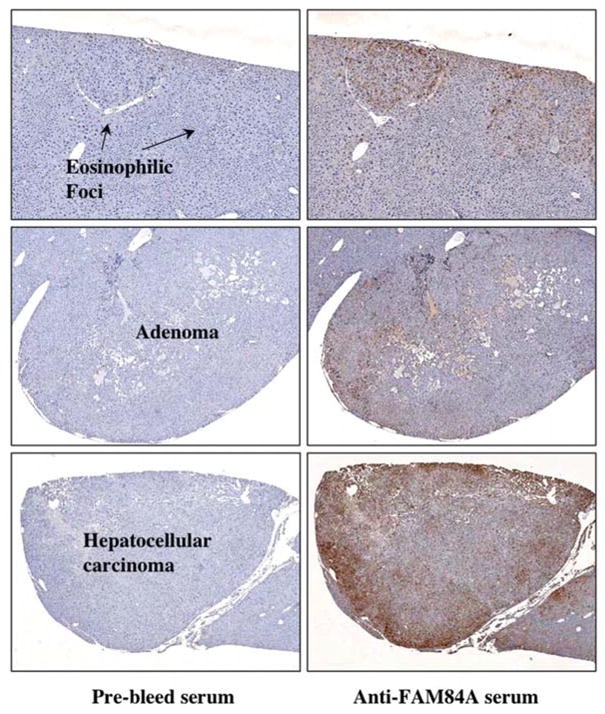
Expression and localization of FAM84A protein in mouse liver tumors. Sections were prepared from the tumor-bearing livers derived from DEN/32 weeks PB-treated Car+/+ C3He mice and subjected to immunohistochemical analysis with pre-bleed serum and anti-FAM84A serum. Higher FAM84A expression was detected in eosinophilic foci, adenoma and carcinoma compared to the non-tumor regions.
Effect of FAM84A on migration of liver cancer cells
Since human FAM84A was suggested to up-regulate the motility of cancer cells (14), we examined the putative function of mouse FAM84A for the development and promotion of liver cancer using human hepatocellular carcinoma cell lines. A colony formation assay using Huh7 cell was first performed to examine the role of FAM84A in cell growth. There were 38±4.9 (mean ± SD) colonies with empty-vector plasmid-transfected cells and 37±5.0 colonies for the FAM84A expression plasmid-transfected cells (Fig. 7A). We also performed the colony formation assay using other cancer cell lines, with no significant difference found (data not shown). This suggests that FAM84A over-expression may not affect cell growth and proliferation. Next we examined the effects of FAM84A on cell migration. Transient expression of FAM84A was performed via adenovirus-based over-expression in HepG2 cells. The migration rates in cells over-expressing FAM84A were significantly higher (P<0.01) than in control cells (187±20.9 and 120±17.3, respectively, Fig. 7B). We also used Huh7 cells for the migration experiment, but a clear difference was not observed (data not shown). These data indicate that FAM84A plays an important role in acceleration of cell migration, but this depends on the cell type.
Figure 7.
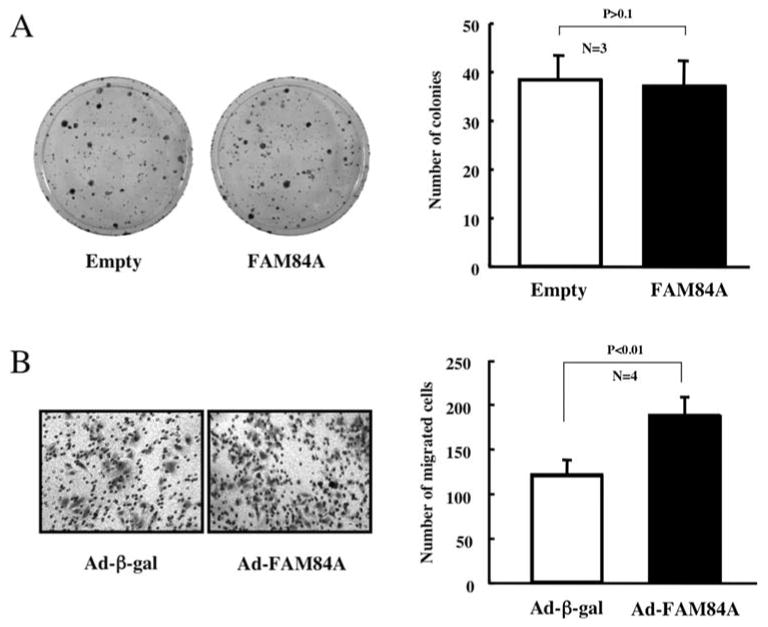
Effects of FAM84A over-expression on cell growth and migration. (A) Huh7 cells were transfected with pcDNA3.1-His-V5-Empty or pcDNA3.1-His-V5-FAM84A (WT) and grown on triplicate culture dishes in the presence of 0.8 mg/ml geneticin for 2 weeks. Geneticin-resistant colonies were fixed with 10% formaldehyde and stained with 0.125% crystal violet. The data are reported as the mean ± SD of three independent experiments. (B) HepG2 cells were infected with Ad-ß-gal or Ad-FAM84A for 24 h at 10 MOI and seeded on transwell chambers. After 72 h incubation, the migrated cells were fixed and stained with 0.1% crystal violet. The number of migrated cells was counted in four fields for each group. The results are reported as the mean ± SD of three independent experiments.
Discussion
Here, we identified FAM84A, which is expressed at a low level in normal mouse liver, as a gene which is activated by PB treatment in a CAR-dependent manner. The results from Western blot and immunohistochemistry analysis indicated that the expression of FAM84A continues in normal liver, eosinophilic foci, adenoma and carcinoma by single or chronic treatment of PB. This suggests that the induction of FAM84A by CAR activation may have an important role in liver tumor promotion. PB is well-known as a non-genotoxic carcinogen, and is also known as an activator of nuclear receptor CAR in rodent liver. In general, non-genotoxic carcinogens do not bind to DNA directly, but cause epigenetic alterations such as the regulation of gene expression by DNA methylation or protein function by post-translational modification. Although the molecular mechanism of liver tumorigenesis in rodents remains unknown, activation of CAR by PB resulting in FAM84A gene induction may support the changes leading to liver tumor development. This includes regulating the expression of enzymes, receptors or proteins involving in cell proliferation and/or apoptosis as well as xenobiotic metabolism or hepatic energy metabolism.
Oxidative stress caused by the overproduction of reactive oxygen species (ROS) is a consideration in many diseases including cancer (19). The ROS is a by-product in the process of monooxigenase reaction by cytochrome P450, and ROS causes several biological effects such as the damage of DNA, proteins or lipids (20). Previous studies have shown the relationship between PB treatment and oxidative stress in rat liver. The accumulation of 8-hydroxy-2′-deoxyguanosine (8-OhdG), which is the marker for oxidative DNA damage, and the induction of P450 isoenzymes CYP3A2 and CYP2B1/2 were observed in rat liver after PB treatment (21). The following report demonstrated that ketoconazole, known as a P450 inhibitor, reduced the induction of 8-OhdG by PB treatment in rat liver, suggesting that the induction of DNA oxidation is produced by P450s including CYP2B1 and CYP3A2 (22). Moreover, it is well-known that nuclear receptor CAR induced CYP2B1/2 expression following PB treatment in rat liver, suggesting that CAR could regulate the rate of oxidative stress via CYP2B1/2 induction. A similar mechanism also exists in mouse liver, and the frequent occurrence of lesions following chronic PB treatment and the up-regulation of oxidative stress may occur by the induction of P450s regulated by CAR. Here we showed that FAM84A is a candidate target gene of CAR. The function of this gene remains unknown, but recent findings suggested that Nse1 (an alias of FAM84A) contributes to genomic stability (23). Nse1 is one of subunit of the Smc5-Smc6 holocomplex, which facilitates DNA repair, and Nse1 contains a RING-like motif that often confers ubiquitin E3 ligase activity. These authors suggest that the RING-like motif is important for the integrity of the Smc5-Smc6 holocomplex and for its recruitment/retention at DNA lesions. The results of these yeast-based experiments require further molecular studies of the mechanism regulating such interacting proteins and FAM84A to prove our hypothesis. FAM84A may have a role in genomic stability through a similar mechanism in rodent liver, reducing the lesions resulting from oxidative stress and preventing tumor development at the early stage of PB treatment.
As one possible physiological function of FAM84A, we found the ectopic over-expression of mouse FAM84A enhanced cell migration in HepG2 cells. This implies that FAM84A may be an enhancer of tumor metastasis as well as of genomic stability. These functions appear to be inconsistent, but there may be equal and opposite reactions of FAM84A in the development and promotion of liver tumors. A previous report indicated that exogenous FAM84A expression affected the morphological changes of NIH3T3 cells (14), while we could not detect any morphological changes in FAM84A over-expressed cancer cells (data not shown). These authors reported that the phosphorylation of serine 38 was associated with changes in cell morphology. However, we did not detect serine 38 phosphorylation in over-expressed FAM84A protein in cancer cells, or induced FAM84A protein in mouse liver by PB treatment, neither in the FAM84A protein expressed in liver tumors. The regulation of FAM84A phosphorylation may be altered in normal liver tissue, in liver tumor and as well as in cultured hepatocellular carcinoma cells under the conditions of our studies. In conclusion, our results indicate that the over-expression of FAM84A, but not the phosphorylation of mouse FAM84A, at serine 38 after activation of CAR by chronic PB treatment may play an important role in liver tumorigenesis.
PB has been used as a drug to treat patients with epilepsy; however epidemiological studies have not shown PB-related tumors in humans, so humans may have a low sensitivity to toxic effects of PB in liver. However, we cannot conclude that PB does not have a link to human liver cancer development. The role of CAR in human liver tumorigenesis is not clear at present, but we can present an interesting suggestion on the correlation between CAR and liver cancer. The C3He mice are more susceptible to the promotion of liver tumors by PB than C57BL6 mice and recent genetic studies suggested that this susceptibility is linked to the region of marker D1Mi33 of mouse chromosome 1 (24). This marker maps to the human chromosome 1q21-23 region, and CAR also maps to this region. The amplification of the long arm of human chromosome 1 including 1q21-23 has been reported in hepatocellular carcinoma (25). In our experiments of comparative genomic hybridization (CGH) using human liver cancer tissue and normal tissue of the same liver, the amplification of two chromosome regions (1q23 and 8q24) were detected (unpublished data), suggesting that the CAR gene may be amplified and the expression of genes regulated by CAR may be increased or decreased. We also examined the expression of FAM84A mRNA in human hepatocellular carcinoma tissues by real-time PCR, and 11 cases out of a total 31 cases showed higher expression of FAM84A in tumor tissues compared to normal tissues derived from same liver (data not shown). Although the levels were not associated with the sex, age, etiology, multiplicity or stage of tumors, these findings suggest that CAR and the CAR target gene FAM84A may be linked to the development or promotion of human liver cancer. In addition, we demonstrated by immunohistochemistry experiments that the FAM84A protein was induced in mouse liver tumors ranging from eosinophilic foci to hepatocellular carcinoma. Although the biological process and detailed mechanism in which FAM84A is involved in human liver tumor development remains to be determined, our findings suggests the possibility that FAM84A may be a good diagnostic and prognosis marker of liver tumors including hepatocellular carcinomas.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, and National Institute of Environmental Health Sciences, Z01ES1005-01. We would like to acknowledge the efforts of all the staff of the NIEHS DNA Sequencing Core and Immunohistochemistry Core. We especially thank for Dr Todd Painter for valuable immunohistological discussion.
References
- 1.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6. J Biol Chem. 1999;274:6043–6046. doi: 10.1074/jbc.274.10.6043. [DOI] [PubMed] [Google Scholar]
- 3.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 4.Sueyoshi T, Negishi M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol. 2001;41:123–143. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- 5.Kodama S, Negishi M. Phenobarbital confers its diverse effects by activating the orphan nuclear receptor CAR. Drug Metab Rev. 2006;38:75–87. doi: 10.1080/03602530600569851. [DOI] [PubMed] [Google Scholar]
- 6.Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for inter-individual variability in response to drugs. J Clin Pharmacol. 2007;47:566–578. doi: 10.1177/0091270007299930. [DOI] [PubMed] [Google Scholar]
- 7.Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, Lehmann JM, Negishi M. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FoxO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kakizaki S, Yamazaki Y, Takizawa D, Negishi M. New insights on the xenobiotic-sensing nuclear receptors in liver diseases -CAR and RXR. Curr Drug Metab. 2008;9:614–621. doi: 10.2174/138920008785821666. [DOI] [PubMed] [Google Scholar]
- 10.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 11.Preat V, de Gerlache J, Lans M, Taper H, Roberfroid M. Comparative analysis of the effect of phenobarbital, dichloro-diphenyltrichloroethane, butylated hydroxytoluene and nafenopin on rat hepatocarcinogenesis. Carcinogenesis. 1986;7:1025–1028. doi: 10.1093/carcin/7.6.1025. [DOI] [PubMed] [Google Scholar]
- 12.Dragon YP, Pitot HC. The role of the stages of initiation and promotion in phenotypic diversity during hepatocarcinogenesis in the rat. Carcinogenesis. 1992;13:739–750. doi: 10.1093/carcin/13.5.739. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 2004;64:7197–7200. doi: 10.1158/0008-5472.CAN-04-1459. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi T, Masaki T, Sugiyama M, Atomi Y, Furukawa Y, Nakamura Y. A gene encoding a family with sequence similarity 84, member A (FAM84A) enhanced migration of human colon cancer cells. Int J Oncol. 2006;29:341–347. [PubMed] [Google Scholar]
- 15.Inoue K, Negishi M. Nuclear receptor CAR requires early growth response 1 to activate the human cytochrome P450 2B6 gene. J Biol Chem. 2008;283:10425–10432. doi: 10.1074/jbc.M800729200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poole TM, Drinkwater NR. Strain dependent effects of sex hormones on hepatocarcinogenesis in mice. Carcinogenesis. 1996;17:191–196. doi: 10.1093/carcin/17.2.191. [DOI] [PubMed] [Google Scholar]
- 17.Ito A, Takahashi T, Watanabe H, Ogundigie PO, Okamoto T. Significance of strain and sex differences in the development of 252Cf neutron-induced liver tumors in mice. Jpn J Cancer Res. 1992;83:1052–1056. doi: 10.1111/j.1349-7006.1992.tb02721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans JG, Collins MA, Savage SA, Lake BG, Butler WH. The histology and development of hepatic nodules in C3H/He mice following chronic administration of phenobarbitone. Carcinogenesis. 1986;7:627–631. doi: 10.1093/carcin/7.4.627. [DOI] [PubMed] [Google Scholar]
- 19.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Zangar RC, Davydov DR, Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Kinoshita A, Wanibuchi H, Imaoka S, Ogawa M, Masuda C, Morimura K, Funae Y, Fukushima S. Formation of 8-hydro-xydeoxyguanosine and cell-cycle arrest in the rat liver via generation of oxidative stress by phenobarbital: association with expression profiles of p21WAF/Cip1, cyclin D1 and Ogg1. Carcinogenesis. 2002;23:341–349. doi: 10.1093/carcin/23.2.341. [DOI] [PubMed] [Google Scholar]
- 22.Imaoka S, Osada M, Minamiyama Y, Yukimura T, Toyokuni S, Takemura S, Hiroi T, Funae Y. Role of phenobarbital-inducible cytochrome P450s as a source of active oxygen species in DNA-oxidation. Cancer Lett. 2004;203:117–125. doi: 10.1016/j.canlet.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Pebernard S, Perry JJ, Tainer JA, Boddy MN. Nse1 RING-like domain supports functions of the Smc5-Smc6 holocomplex in genome stability. Mol Biol Cell. 2008;19:4099–4109. doi: 10.1091/mbc.E08-02-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bilger A, Bennett LM, Carabeo RA, Chiaverotti TA, Dvorak C, Liss KM, Schadewald SA, Pitot HC, Drinkwater NR. A potent modifier of liver cancer risk on distal mouse chromosome 1: linkage analysis and characterization of congenic lines. Genetics. 2004;167:859–866. doi: 10.1534/genetics.103.024521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau SH, Guan XY. Cytogenetic and molecular genetic alterations in hepatocellular carcinoma. Acta Pharmacol Sin. 2005;26:659–665. doi: 10.1111/j.1745-7254.2005.00126.x. [DOI] [PubMed] [Google Scholar]