

Abstract
Long-timescale molecular dynamics simulations (300 ns) are performed on both the apo- (i.e., camphor-free) and camphor-bound cytochrome P450cam (CYP101). Water diffusion into and out of the protein active site is observed without biased sampling methods. During the course of the molecular dynamics simulation, an average of 6.4 water molecules is observed in the camphor-binding site of the apo form, compared to zero water molecules in the binding site of the substrate-bound form, in agreement with the number of water molecules observed in crystal structures of the same species. However, as many as 12 water molecules can be present at a given time in the camphor-binding region of the active site in the case of apo-P450cam, revealing a highly dynamic process for hydration of the protein active site, with water molecules exchanging rapidly with the bulk solvent. Water molecules are also found to exchange locations frequently inside the active site, preferentially clustering in regions surrounding the water molecules observed in the crystal structure. Potential-of-mean-force calculations identify thermodynamically favored trans-protein pathways for the diffusion of water molecules between the protein active site and the bulk solvent. Binding of camphor in the active site modifies the free-energy landscape of P450cam channels toward favoring the diffusion of water molecules out of the protein active site.
Introduction
Cytochrome P450s are essential hemoprotein monooxygenases that catalyze a variety of biochemical processes, including drug metabolism, lipid and steroid biosynthesis, and degradation of pollutants (1,2). The active site of the enzyme contains a heme moiety. There is considerable interest in identifying and characterizing possible channels that connect the active site to the protein exterior, facilitating substrate entry into the active site and product release. Identification and characterization of the channels connecting the active site of P450s to the protein exterior provides important structural and functional information about P450s. The pioneering work of the Wade group (3–5) has described several potential channels involved in substrate/product access/release, and also potentially involved in substrate/product diffusion into and out of the protein, which have been confirmed to be important for P450 structure-function relationships and specificity (6,7).
Water molecules play an important role in enzymatic activity and must be able to enter and exit the active site of P450cam. Substrate binding is known to result in displacement of water molecules from the active site, favoring the ferrous Fe(II) state (8,9). It is proposed that in addition to their role in spin transition of the iron, water molecules are involved in a proton shuttle mechanism that allows the proton transfer needed for cleaving the Fe-O bond in the Fe-oxo intermediate (10–13). After the enzymatic reaction is complete, the product leaves the active site and water molecules must diffuse again into the active site for enzyme turnover.
Crystal structures of P450s provide atomic coordinates for cocrystallized water molecules in the active site, both in the apo and camphor-bound forms (Fig. 1 a). Observation of both the number and location of water molecules is fundamental for understanding the protein function (8–13) described above. Previous free-energy calculations performed on P450cam suggested that the number of water molecules present in the active site of P450cam was not higher than the number of water molecules observed in the crystal structures (8,14). However, the time length of these simulations was relatively short, and the structure of the active site stayed very close to that of the crystal structures. More recently, Rydberg et al. (15) followed the number of active-site water molecules exchanging with the bulk through several channels over 4-ns molecular dynamics (MD) simulations in several mammalian apo-P450s. In their landmark study, they showed that in mammalian P450s, the number of water molecules present in the active site could be significantly higher than that observed in the crystal structures, contrary to what has been suggested for bacterial P450s by others (8,14). Rydberg et al. also showed that several channels may exist for water diffusion, and that they differ among the various P450 species, as well as between mammalian and bacterial P450s.
Figure 1.
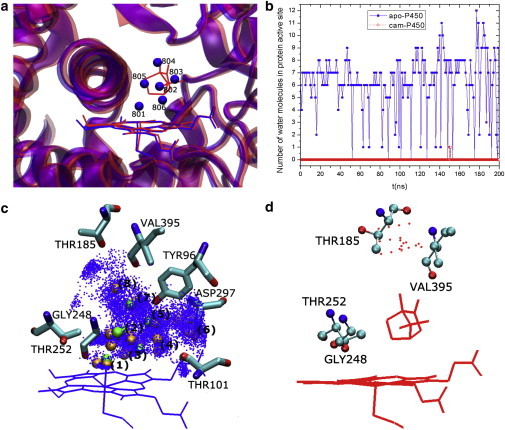
(a) Six water molecules (spheres) observed in active-site cavity of the x-ray crystal structure of apo-P450cam and none in camphor-bound P450cam. Protein backbone is shown in ribbons, and the heme group and camphor molecule are shown as lines. (b) Number of water molecules found in the protein-active-site cavity of apo-P450cam versus the camphor-bound P450cam during MD simulation (100–300-ns time window). (c and d) Distribution of water molecules (points) in apo-P450cam (c) and camphor-bound P450cam (d) obtained from the MD trajectories up to 5 Å from the camphor-binding region: eight water clusters (number labels 1–8) (light gray/orange spheres) were identified surrounding the crystal water locations (dark gray/green spheres) in apo-P450cam and none in camphor-bound P450cam.
Here, we address the structural and thermodynamic aspects of water diffusion between the bulk solvent and the protein active site in cytochrome P450cam from Pseudomonas putida. P450cam catalyzes the regio- and stereospecific hydroxylation of camphor and has long served as a model system for studying P450s. In this work, we perform 300-ns MD simulations, i.e., two orders of magnitude longer than those in the earlier study of water hydration in mammalian P450s (15). The length of these simulations allows calculation of the potential of mean force for unbiased water diffusion in protein channels, comparison of the thermodynamics of water diffusion in apo- and substrate-bound forms of P450cam, and characterization of the dynamics and energetic of P450 channels that connect the active site with the protein exterior.
Materials and Methods
Structures of cytochrome P450cam
The x-ray crystal structures of cytochrome P450cam were obtained from the Protein Data Bank (16) (ID codes 1PHC for apo-P450cam solved at 1.60 Å resolution (17) and 3L63 for camphor-bound P450cam at 1.50 Å resolution (18)). The 3L63 structure was solved at a higher resolution than the 2CPP camphor-bound structure (1.63 Å resolution) (19) and explicitly includes a K+ ion surrounded by protein residues Glu-84, Gly-93, Glu-94 and Tyr-96 (Fig. S1 b). Accordingly, water 515 in the 1PHC structure was replaced by a K+ ion in this simulation (17,19). The nine N-terminal residues missing in crystal structures were added as a random coil extending above Phe-26 and Leu-45 (Fig. S1 a) using program MOE (20) and further equilibrated following the simulation protocol described below. The two species (apo- and camphor-bound) of P450cam are superimposable (Fig. S1 a) with channels closed for substrate access (5). The psfgen plugin in VMD (21) was used to build the protein structural topology with protein residues set to the standard CHARMM protonation states at neutral pH, with the following exceptions following pKa calculations of ref (22): Asp-297 and Glu-366 are protonated, His-62 is protonated on δ-nitrogen, and seven other histidines (17, 21, 80, 176, 308, 337, and 355) are doubly protonated (Fig. S1 c). The heme iron atom was coordinated to the sulfur atom of Cys-357, providing a five-coordinate high-spin state in the camphor-bound P450cam, and it was also liganded to water 801 in the active site of apo-P450cam in a six-coordinated low-spin state (10,17,19). Crystallographic water molecules were kept, and each structure was further solvated in a rectangular box of TIP3P water molecules (14), extending at least 10 Å from the protein surface to the edge of the box using the Solvate plugin in VMD (21). K+ and Cl– ions were added at random locations in the bulk solvent to neutralize the system, achieving an ionic strength of 0.1 M using the Autoionize plugin in VMD (21). A schematic representation of the resulting model system for camphor-bound P450cam is shown in Fig. S1 d. The final systems contain 47,588 atoms (13,653 water molecules) for apo-P450cam and 46,568 atoms (13,305 water molecules) for substrate-bound P450cam.
Molecular-dynamics simulations
MD simulations of cytochrome P450cam were performed using the NAMD2.7 simulation engine (23). The CHARMM22 force field (24,25) was used for the protein, using the TIP3P model (26) for water molecules. Standard CHARMM force-field parameters (25) were used for the heme group (toppar_all22_prot_heme.str) including an explicit Fe-S bond to Cys-357. The partial charge of the SG atom in Cys-357 was adjusted to –0.07e to account for the resting state of P450cam in both apo and camphor-bound forms (22). CHARMM parameters for camphor were obtained from Schöneboom et al. (28). An integration timestep of 2 fs was used for the MD simulations presented here. The system was initially energy-minimized for 1000 steps with the location of atoms given in the crystal structures fixed and another 1000 steps with all atoms free to move, using the conjugate-gradient algorithm. The system was then gradually heated to 300 K at a rate of 60 K/ps and equilibrated at 300 K for 5 ns in constant volume (i.e., NVT ensemble), with the crystallographically identified atoms fixed. The system was further equilibrated for 5 ns by using the NPT (300 K and 1 atm with Nosé-Hoover Langevin piston-pressure control) ensemble with all atoms free to move and a 300-ns NPT product run was performed. A multiple-time-stepping algorithm (23) was used in which bonded and short-range, nonbonded interactions were computed every time step and long-range electrostatic interactions every 2 timesteps. A cutoff distance of 12 Å was used for van der Waals and short-range electrostatic interactions, and the long-range electrostatic interactions were computed using the particle-mesh Ewald summation method with a grid point density of 1/Å. The SHAKE (29) algorithm was applied to hydrogen-oxygen bonds in water molecules and hydrogen-containing bonds in the protein were treated as flexible. MD simulations were run on the University of Tennessee's Kraken supercomputer.
Water molecules in the protein active site and water passages through the protein
Bulk water molecules were defined as being at least 3 Å from the protein and within 2.5 Å of other bulk water molecules. Three subsequent layers of water molecules were then defined as surface water, deep surface water, and channel water, and contain water molecules in contact with the adjacent layer and not belonging to an already-assigned layer. The remaining water molecules are defined as cavity water molecules. In this work, we consider water molecules within 5 Å of camphor location, as found in the 3L63 camphor-bound crystal structure, as protein-active-site hydration water. This was preferred to considering all water molecules within 13 Å of the heme iron, as in Rydberg et al. (15), as it would have included water molecules located outside the active-site region (Fig. S3). The number and time of water passages between the protein active site and the bulk solvent were collected for both apo and camphor-bound P450cam by tracking the trajectory of each protein-active-site water molecule through the MD simulation (Fig. S4).
Identification of CAVER protein channels for water diffusion
To characterize possible water-diffusion pathways in cytochrome P450cam, CAVER (30) was used to identify channels that extend from the protein active site to the protein exterior in the protein snapshots of MD trajectories using 0.5-Å grid spacing. Protein snapshots were taken every 1 ns from the 100–300-ns part of the MD trajectories, resulting in 200 snapshots for both apo- and camphor-bound P450cam. The position of atom C1 in camphor from the x-ray crystal structure of camphor-bound P450cam was used as the starting point for the channel calculations. Each channel has a maximum probe radius (rmax) for spheres that can pass through the channel. It has been proposed that channels with rmax ≥ 1.2 Å can be a priori considered open for water diffusion or substrate access (4). Twenty channels with the lowest CAVER cost-function values (30) were identified for each snapshot. Fig. S9 shows front and top views of resulting channels for the apo- and camphor-bound P450cam.
To cluster the CAVER output channels, MD trajectory average structures were calculated for both apo- and camphor-bound P450cam (Fig. S8), and 21 unique channels were identified from their CAVER calculations (50 channels with the lowest cost-function values for each trajectory average structure were inspected using VMD (21)). The channels were labeled according to the nomenclature of earlier studies, based on their spatial location and the secondary-structure elements lining them at the protein surface (4,5) and used to cluster the CAVER output channels of MD trajectory snapshots, for which the exit mouth radius of each cluster was set as half of the minimum distance between the exit of the belonging channel and its neighboring ones, obtained from the trajectory average structure. Channels with the lowest CAVER cost-function values were selected from each cluster for the analysis described below.
A water molecule that has its oxygen atom located inside a given CAVER channel was counted as belonging to that channel. A water passage with water occasionally present in more than one CAVER channel (e.g., near the camphor binding site) at different simulation time and diffusing only partially through any CAVER channel (Fig. S11) was assigned to the CAVER channel in which the water molecule appears most often during its passage. The percentage of diffusing water molecules included in a given CAVER channel during passage was calculated by dividing the population of this water molecule in the channel by the total number of trajectory snapshots of the entire water passage (e.g., 282 snapshots for the 2.82-ns water passage shown in Fig. S4 b by taking one snapshot every 10 ps).
Potential-of-mean-force calculations
The potential of mean force (PMF) for water hydration in the protein was calculated as
| (1) |
where R is the gas constant, T is the temperature, is the density of water molecules in a given bin, and is the maximum density of water molecules. The protein active site was represented by a three-dimensional grid at 1-Å resolution and the water molecules' oxygen atoms were binned to the resulting grid for all 20,000 MD snapshots from the MD trajectories (i.e., one snapshot every 10 ps from the 100- to 300-ns MD trajectories) for both apo- and camphor-bound P450cam. Water hydration clusters were identified in the protein active site. The Volmap plugin in VMD (21) was used to create the water-density map and visualize free-energy isosurfaces to examine the diffusion pathways of protein-active-site water molecules.
PMF profiles were also calculated for water hydration inside the CAVER channels using Eq. 1. The CAVER protein channels have their reaction coordinates (i.e., center line) and the radii of comprising spheres explicitly defined (see Fig. 4). A water molecule with its oxygen atom located in a CAVER channel was assigned to one bin of the channel whose center is the closest to the water oxygen atom. All the water molecules appearing in the protein were included in the CAVER channel PMFs, i.e., not only the water molecules identified above as being present in the protein active site during simulation, which provides sufficient sampling and precludes the need for biased simulations. Five different bin sizes ranging from 1.0 to 3.0 Å were used for the PMF calculations in the CAVER channels to estimate the precision of the results (31).
Figure 4.
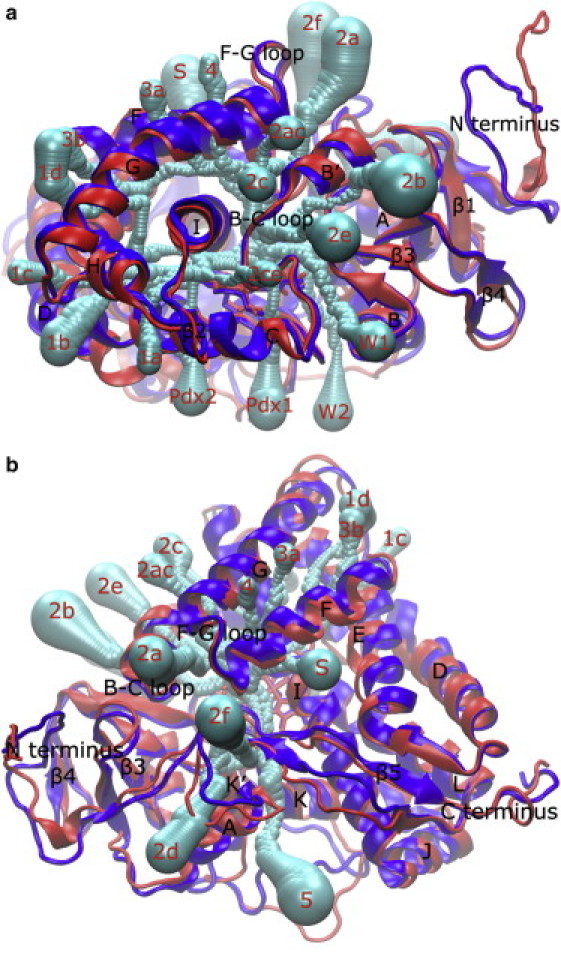
Schematic representation of P450cam channels identified with CAVER as shown on the MD trajectory average structures of apo-P450cam (dark gray/blue) and camphor-bound P450cam (light gray/red). (a) Front view looking down the I helix. (b) Top view looking down on the heme plane from the distal side. Protein secondary structures and channel exiting positions on the protein surface are labeled. Channel spheres are represented with half of the radii for clarity.
Results and Discussion
Hydration of the active site
Analysis of the MD trajectories indicates that the root mean-square deviations (RMSDs) between trajectory snapshots and the system dihedral energies converge after ∼100 ns for both apo- and substrate-bound P450cam (Fig. S2), and the last 200 ns (i.e., 100–300-ns) of the MD trajectories were used for subsequent analysis.
A total of 192 water molecules are present in the x-ray structure of apo-P450cam, including six in the active site and one close to the heme-7-propionate group (17). A total of 204 water molecules are observed in the camphor-bound form, including one close to the heme-7-propionate group and none in the active site (19) (Fig. 1 a). The number of water molecules in the active-site cavity of apo- and camphor-bound P450cam during 100–300-ns MD trajectories was calculated as described in Materials and Methods. As discussed in Rydberg et al. (15), it is difficult to define precisely the location and volume of the active site. Here, we focus on the active-site region that extends 5 Å away from the position of camphor in the x-ray structure (19).
As shown in Fig. 1 b, the instantaneous number of water molecules found in the active site of apo-P450cam varies between 0 and 12, exhibiting large fluctuations of active-site hydration over the course of the simulation. This fluctuation shows a significant exchange of water molecules between the protein-active-site cavity and the protein exterior, as previously described in the case of mammalian P450s (15). Water molecules spend only a limited amount of time in the active site and frequently exchange with the bulk through water-conducting channels (see Fig. S4 for an illustration of transient locations of water molecules diffusing in the protein). During the 100–300-ns simulation, the average number of water molecules present in the active site is calculated to be 6.4 for the apo form and zero for the substrate-bound form. In the latter case, only one water molecule is observed entering the active-site cavity briefly at ∼150 ns (Fig. S4, c and d). Although the number of water molecules can be significantly more than the number of crystallographic waters at a given time, the average number of water molecules in the active site is in very good agreement both with the number observed in the crystal structures (6 and 0 in the apo- and camphor-bound forms, respectively) and with previous free-energy calculations by Helms and Wade (14) and Oprea et al. (8). A difference of 6.7 water molecules was also found between apo- and camphor-bound P450cam from MD and grand Monte Carlo simulations by Deng and Roux (32). However, hydrostatic pressure experiments suggested that up to 19 water molecules may be present at a time in the active site or in close proximity, which is significantly larger than the six well-defined crystallographic water molecules (8,33). The results of Rydberg et al. (15) also suggest that more water molecules can be found in the protein-active-site cavity than those identified in the x-ray crystal structures of mammalian P450s. The simulations on P450cam presented here are in agreement with the Rydberg results, showing that the number of water molecules in the active site of P450cam may be up to twice the number of water molecules observed in the crystal structures.
The potential of mean force for water hydration sites in the protein-active-site cavity was calculated as described in Materials and Methods. For apo-P450cam, eight well-defined water clusters were found in the active site with free energy <1.2 kcal/mol (i.e., 2 kBT) above that of the most populated water cluster. This suggests that although as many as 12 water molecules may be present at one time in the active site of apo-P450cam (Fig. 1 b), not all the water hydration sites are equally thermodynamically stable. It has been suggested that the crystallization conditions could favor only the most stable locations for a water molecule to be in the active site (15), making them stable enough to be experimentally observed. These results suggest indeed that only a limited number of hydration sites have a free energy within 2 kBT of the most hydrated water site, and that these sites are collectively preferred to accommodate the average number of 6.4 water molecules in the vicinity of the camphor-binding region in the active site.
As shown in Fig. 1 c, five water clusters (numbered 1, 2, 3, 5, and 7) of the eight identified share closely similar locations to crystallographic water molecules 801, 805, 806, 803, and 804, respectively. Water 801 was linked to the heme iron in the simulation of apo-P450cam described here and as such stayed close to the heme iron. In comparison, the study of five mammalian P450s with five-coordinate, high-spin Fe(III) (15) did not identify a water molecule staying close to the heme's iron. These results indicate that the active-site hydration of apo-P450cam can be appropriately described with a six-coordinate, low-spin Fe(III) state corresponding to the configuration of apo-P450cam x-ray crystal structure (17).
In the case of the substrate-bound P450cam, no water cluster was found in the camphor-binding region (Fig. 1 d), although water density appears at several locations between residues Thr-185 and Val-395 due to the presence of a water molecule at ∼150 ns in the simulation (Fig. S4). Water molecules can only approach the distal region of the protein-active-site cavity with a minimum Fe-O (water) distance of 10.43 Å. This agrees with the fact that there is no water molecule observed in the active-site cavity of the camphor-bound P450cam crystal structure (18,19). Camphor was found to maintain its crystallographic orientation in the protein active site throughout the MD simulation except for a 14-ns time period at ∼50 ns during the 100–300-ns trajectory (Fig. S5).
Thermodynamics for diffusion of protein-active-site water molecules
In the simulation of apo-P450cam, 41 unique water molecules were identified in the protein-active-site cavity during the 100–300-ns MD trajectory (including the six water molecules present in the crystal structure). The water molecule liganded to heme iron stayed in its liganded location, and four other water molecules did not diffuse outside of the protein-active-site cavity (but occasionally left the camphor-binding region for parts of the simulation), the other 36 water molecules were found to exchange dynamically between the protein active site and the bulk solvent during MD simulation (Fig. S4 a). For camphor-bound P450cam, only one water molecule was observed to diffuse into and out of the distal region of the protein-active-site cavity at ∼150 ns (Fig. S4, c and d). As one water molecule can diffuse into and out of the protein active site multiple times during the 100–300-ns simulation (Fig. S4 a), the total number of complete water passages between the protein active site and the bulk solvent was counted as 51 for apo-P450cam and 2 for camphor-bound P450cam.
Study of water diffusion from 4 ns MD simulation of six mammalian P450s (15) indicated there the number of water molecules exchanging between the protein active site and the bulk varied greatly, between 0 and 125 molecules, depending on the P450 species and on the size of the protein active site cavity. These results indicate that up to 36 water molecules diffuse through P450cam in a 200-ns MD trajectory, i.e., 50 times longer than that of Rydberg et al. (15). The apparent lower number of water passages observed in P450cam could be due to its relatively small protein-active-site volume, which contains fewer water molecules susceptible to diffusion than in the case of mammalian P450s, with their larger protein-active-site cavity. There is an average of 6.4 water molecules present in the camphor-free P450cam active site, whereas on average, the mammalian P450s include 41–58 water molecules in their active-site cavity, though there are only two for CYP2A6, in which no water diffusion was observed during the simulation reported by Rydberg et al. (15).
Three-dimensional PMF profiles were calculated using protein-active-site water molecules of all 20,000 snapshots (i.e., one snapshot every 10 ps from the 100–300-ns MD trajectories) for both apo- and camphor-bound P450cam, as described in Materials and Methods. The most populated bin corresponds to the water molecule liganded to heme iron in apo-P450cam and all free-energy isosurfaces here are given relative to this bin, which sets the zero free energy. The free-energy isosurfaces shown in Figs. 2 and 3 indicate the most thermodynamically favorable pathways for water molecules exchanging between the protein-active-site cavity and the bulk solvent.
Figure 2.
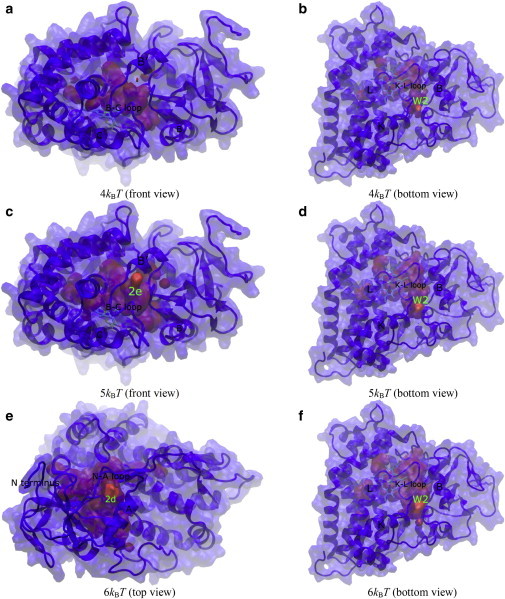
Free-energy isosurfaces of hydration in apo-P450cam. Relevant protein secondary structures and water diffusion exits on the protein surface are labeled. Front, top, and bottom views of the protein are looking down the I helix, looking down on the heme plane from the distal side, and looking up from below the heme plane, respectively.
Figure 3.
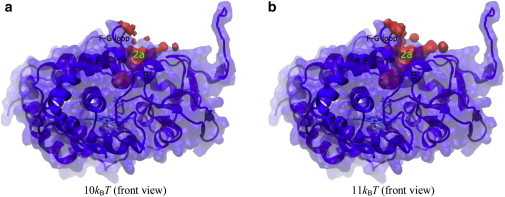
Free-energy isosurfaces of hydration in substrate-bound P450cam. Same representations are used as in Fig. 2.
For apo-P450cam, the free-energy isosurfaces below 4 kBT stay confined in the protein-active-site cavity and do not extend to the protein surface (Fig. S7, a and b). The free-energy isosurface connects to the protein surface at 4 kBT only between the B helix and the K-L loop on the protein surface below the heme plane (Fig. 2, a and b, and Fig. S7 c), which corresponds to the exit of CAVER protein channel W2, as described below. As free energy is increased to 5 kBT, the free-energy isosurface also connects to the bulk between the B′ helix and the B-C loop that corresponds to CAVER protein channel 2e (Fig. 2 c)(4). The free-energy isosurface patch originating from pathway W2 enlarges on the protein surface at 5 kBT (Fig. 2 d) and becomes even larger at 6 kBT (Fig. 2 f). From 5 kBT to 6 kBT, the free-energy isosurface was also found to protrude the protein surface represented by the loop between the N-terminus and the A helix that corresponds to the exit of CAVER channel 2d (Fig. 2 e) (4). These results suggest that the pathways preferentially conduct water diffusion in the order 2e at 4 kBT, W2 at 4–6 kBT, 2d at 5–6 kBT, and then the rest. For camphor-bound P450cam, the free-energy isosurface displays a gap along the pathway corresponding to the proposed substrate access channel, 2a, between the B′ helix and the F-G loop at 10 kBT (Fig. 3 a). The gap disappears and the isosurface connects the protein-active-site cavity and the bulk completely at 11 kBT (Fig. 3 b). This suggests that the diffusion of water molecules through pathway 2a exhibits a free-energy barrier of ∼10–11 kBT.
CAVER protein channels and observed water passages
Channels connecting the protein active site to the bulk solvent were calculated using CAVER and clustered, and the observed water passages during MD simulations were assigned to the protein channels as described in Materials and Methods for both apo- and camphor-bound P450cam. The resulting channel rmax, length, and CAVER cost-function value, the number of water passages and water-passage times, and the percentage of finding water molecules in the associated protein channel during each passage, are listed in Table 1. Schematic representations of the protein channels are shown in Fig. 4. The channels are labeled using the nomenclature established by the Wade group (4,5). Channel W splits into W1 and W2; channel 1 splits into 1a, 1b, 1c and 1d; and channel 3 splits into 3a and 3b, as observed in the MD simulations presented here. In addition, two previously unreported channels, labeled Pdx1 and Pdx2, are identified that connect the active site to the Pdx protein electron partner in the P450cam-Pdx complex (4,34,35).
Table 1.
List of channels identified in simulations of cytochrome P450cam
| Channel | X ray∗ | Occur.† | rmax ≥ 1.2 Ň | rmax (Å)§ | l (Å) § | Cost§ | Nwp¶ | twp (ns)‖ | pwp-channel(%)∗∗ |
|---|---|---|---|---|---|---|---|---|---|
| Apo-P450cam | |||||||||
| W1 | Y | 48 | 1 | 1.26 | 36.5 | 31.49 | 3 | 3.59 ± 2.52 | 55.7 ± 19.6 |
| W2 | Y | 40 | 1 | 1.23 | 42 | 32.23 | 11 | 12.69 ± 8.22 | 44.6 ± 33.2 |
| S | Y | 140 | 1 | 1.23 | 31.5 | 30.23 | 1 | 3.48 | 16.0 |
| 1a | Y | 17 | 0 | 1.06 | 37 | 45.38 | 0 | — | — |
| 1b | Y | 0 | 0 | — | — | — | — | — | — |
| 1c | N | 3 | 0 | 0.81 | 37.5 | 53.38 | 0 | — | — |
| 1d | N | 1 | 0 | 0.9 | 47 | 55.33 | 0 | — | — |
| 2a | Y | 180 | 2 | 1.27 | 29 | 19.73 | 0 | — | — |
| 2ac | Y | 74 | 0 | 0.99 | 28.5 | 35.66 | 0 | — | — |
| 2b | Y | 10 | 0 | 0.81 | 35.5 | 46.01 | 0 | — | — |
| 2c | N | 29 | 0 | 0.99 | 30.5 | 40.83 | 0 | — | — |
| 2ce | N | 18 | 0 | 0.71 | 33.5 | 52.87 | 0 | — | — |
| 2d | N | 73 | 5 | 1.32 | 53 | 38.16 | 7 | 9.98 ± 5.58 | 54.6 ± 4.5 |
| 2e | Y | 124 | 11 | 1.3 | 23.5 | 18.06 | 27 | 3.25 ± 2.08 | 48.7 ± 23.6 |
| 2f | N | 13 | 2 | 1.39 | 51 | 39.46 | 2 | 4.65 ± 2.60 | 69.5 ± 0.7 |
| 3a | Y | 29 | 0 | 1.01 | 28.5 | 48.65 | 0 | — | — |
| 3b | N | 23 | 0 | 1.09 | 46 | 44.19 | 0 | — | — |
| 4 | N | 33 | 0 | 1.06 | 26 | 37.24 | 0 | — | — |
| 5 | N | 91 | 0 | 1.10 | 46 | 38.66 | 0 | — | — |
| Pdx1 | Y | 27 | 0 | 0.79 | 27.5 | 41.44 | 0 | — | — |
| Pdx2 | Y | 26 | 0 | 1.07 | 32 | 45.8 | 0 | — | — |
| Camphor-bound P450cam | |||||||||
| W1 | Y | 71 | 0 | 0.9 | 35 | 32.06 | 0 | — | — |
| W2 | Y | 74 | 0 | 1.04 | 31.5 | 26.3 | 0 | — | — |
| S | Y | 183 | 0 | 1.07 | 36 | 29.29 | 0 | — | — |
| 1a | Y | 9 | 0 | 0.85 | 38.5 | 48.98 | 0 | — | — |
| 1b | Y | 22 | 0 | 0.83 | 36.5 | 44.32 | 0 | — | — |
| 1c | N | 3 | 0 | 0.89 | 40 | 51.7 | 0 | — | — |
| 1d | N | 0 | 0 | — | — | — | — | — | - |
| 2a | Y | 146 | 5 | 1.29 | 32 | 17.84 | 2 | 0.62 ± 0.37 | 42.5 ± 13.4 |
| 2ac | Y | 63 | 0 | 1.08 | 28.5 | 28.96 | 0 | - | — |
| 2b | Y | 13 | 0 | 1.06 | 39.5 | 46.07 | 0 | — | — |
| 2c | N | 1 | 0 | 0.71 | 24.5 | 48.44 | 0 | — | — |
| 2ce | N | 1 | 0 | 1.06 | 37 | 53.82 | 0 | — | — |
| 2d | N | 0 | 0 | — | — | — | — | — | — |
| 2e | Y | 113 | 0 | 0.96 | 24.5 | 26.75 | 0 | — | — |
| 2f | N | 0 | 0 | — | — | — | 0 | — | — |
| 3a | Y | 116 | 0 | 1 | 31 | 38.03 | 0 | — | — |
| 3b | N | 2 | 0 | 1.11 | 42 | 50.53 | 0 | — | — |
| 4 | N | 4 | 0 | 0.76 | 26.5 | 55.14 | 0 | — | — |
| 5 | N | 36 | 0 | 1.11 | 48 | 43.46 | 0 | — | — |
| Pdx1 | Y | 25 | 0 | 0.93 | 28.5 | 39.82 | 0 | — | — |
| Pdx2 | Y | 160 | 2 | 1.29 | 34.5 | 32.01 | 0 | — | — |
Y, channel appears in the x-ray crystal structure; N, channel is not observed in the crystal structure.
Number of channel-appearing occurrences in the CAVER output calculated for the 200 MD trajectory snapshots (i.e., the 100–300-ns time window with one snapshot taken every 1 ns).
Number of appearing occurrences with rmax ≥ 1.2 Å.
Maximum probe radius, rmax, and length, l, for channel with the lowest CAVER cost-function value.
Number of water passages.
Water passage time.
Percentage probability of finding water in the corresponding CAVER channel during passage.
The maximum probe radius (rmax) of the channels identified in the crystal structures of apo- and camphor-bound P450cam (i.e., W1, W2, S, 1a, 1b, 2a, 2ac, 2b, 2e, 3a, Pdx1, and Pdx2) were found to range from 0.6 Å to 1.0 Å. In previous studies (4), a protein channel with rmax ≥ 1.2 Å was considered a priori as open for water diffusion or substrate access. As channels in the crystal structures of P450cam have rmax values much smaller than 1.2 Å, they have been considered to be closed to water or substrate/product diffusion (4,5). In the MD simulations presented here, the radii of these protein channels were found to undergo significant fluctuation. Seven channels exhibit rmax ≥ 1.2 Å during simulation of apo-P450cam, i.e., W1 (water channel 1), W2 (water channel 2), S (solvent channel), 2a (proposed substrate access channel), 2d, 2e, and 2f, and six of the seven channels (except channel 2a) are used for conducting water passages (Table 1). For camphor-bound P450cam, two channels, 2a and Pdx2, exhibit rmax ≥ 1.2 Å, but only channel 2a is able to conduct water passage between the protein-active-site cavity and the bulk solvent. The protein residues lining these channels that conduct water passages are listed in Table S1, Table S2, Table S3, Table S4, Table S5, Table S6, and Table S7. It is worthy of note that channels 2a in apo-P450cam and Pdx2 in camphor-bound P450cam do not appear to conduct water passages between the protein-active-site cavity and the bulk during MD simulations (Table 1) even though they are observed in the crystal structure and exhibit occasional rmax ≥ 1.2 Å. Therefore, rmax ≥ 1.2 Å is a factor for defining a channel as being able to conduct water molecules, but should not be used as the only criterion.
Among the above-identified channels that conduct water passages in apo-P450cam, channel 2e, which exhibits the largest number of rmax ≥ 1.2 Å occurrences (i.e., 11; see Fig. S10), the shortest length (23.5 Å), and the lowest CAVER cost-function value (i.e., 18.06), appears to be the most frequently used pathway for water exchange (i.e., 27 passages). It is followed by the other five channels: W2 (11 water passages), 2d (7 passages), W1 (3 passages), 2f (2 passages), and S (1 passage). Correspondingly, channels 2d and 2f have significant numbers of rmax ≥ 1.2 Å occurrences (5 and 2, respectively), even though W1, W2, and S have only one occurrence each (Table 1 and Fig. S10). In a similar way, channel 2a has the largest number of rmax ≥ 1.2 Å occurrences (i.e., 5) and the lowest CAVER cost-function value (i.e., 17.84) in camphor-bound P450cam and is involved in the only two water passages observed in the simulation, although its length (28.5 Å) is not the shortest compared with that of channel 2e (24.5 Å). The K+ ion described in the crystal structures was observed to diffuse dynamically into and out of its binding site near the C-terminus of the protein B′ helix in apo- and camphor-bound P450cam (see Fig. S6), apparently keeping the B′ helix ordered (Fig. S8) and closing channel 2ac, with no water passage observed in either structure (Table 1). These results suggest that the number of water passages associated with each protein channel correlate with the channel-opening size (e.g., rmax).
For water passages between the protein active cavity and the bulk solvent, the average passage time ranges from 3.25 ns (channel 2e) to 12.69 ns (channel W2) in apo-P450cam, with comparatively large variations found between water passages associated with different channels (Table 1). Two individual water passages that are typically fast and slow are illustrated in Fig. S4 b. The time of water passages associated with one protein channel also displays significant fluctuation, with fairly large standard deviations (Table 1). The only water molecule found to diffuse into the distal region of protein active site cavity in the camphor-bound P450cam exhibits a passage time of 0.62 ns (Fig. S4, c and d).
It is important to note that water molecules diffusing into and out of the protein active site were found to fit only partially into a CAVER protein channel during passage (Fig. S11). The percentage of water molecules found for a given pathway (Figs. 2 and 3) in the associated CAVER channel ranges from 43% to 70% during the water passages, except for the pathway associated with channel S in apo-P450cam, where water was found to diffuse inside the CAVER channel for only 16% of the passage time. Hence, water molecules that undergo complete passages through the protein diffuse for only about half of their passage time inside the CAVER protein channels.
Thermodynamics of water hydration in CAVER protein channels
The PMF was calculated in apo- and camphor-bound P450cam for CAVER protein channels W1, W2, S, 2a, 2d, 2e, and 2f, which allow water diffusion (see Materials and Methods). The resulting PMF profiles are shown in Fig. 5, and the residues corresponding to their respective reaction coordinate values are listed in the tables in the Supporting Material. Five bin sizes, ranging from 1.0 Å to 3.0 Å, were used to calculate PMF profiles of the CAVER channels to estimate their precision, i.e., the dependence of the free-energy profiles on the bin size (31). Variations in the PMF profiles can originate from poor sampling of water molecules in the channels with small bin sizes of 1.0 Å and 1.5 Å, especially for channels with a small number of water passages (e.g., S, W1, and 2a). Free-energy barriers obtained from the channel PMF profiles decrease by 0.37 kcal/mol (channel 2e) to 1.36 kcal/mol (channel S) when the bin size is increased from 1.5 Å to 3.0 Å, i.e., up to ∼30% of the free-energy barrier at the 1.5-Å bin size.
Figure 5.
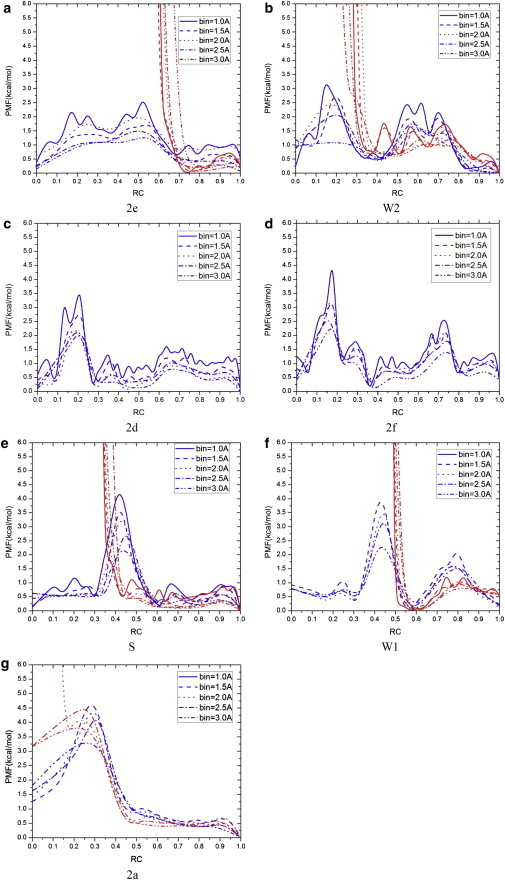
PMF for hydration in CAVER channels 2e (a), W2 (b), 2d (c), 2f (d), S (e), W1 (f), and 2a (g) for apo-P450cam (dark gray/blue) and camphor-bound P450cam (light gray/red). Five different reaction-coordinate bin sizes (1.0–3.0 Å) were used and PMF profiles were smoothed with B-spline curves.
The PMF profiles obtained with the 1.5-Å bin size (close to a typical 1.4-Å water probe radius) are analyzed in the following discussion. For apo-P450cam, channel 2e (Fig. 5 a) displays a rather flat PMF profile, with the lowest free-energy barrier found among the channels, i.e., 1.84 kcal/mol (∼3 kBT). The free-energy barrier increases for the other channel PMF profiles in the order W2 (2.84 kcal/mol), 2d (3.01 kcal/mol, i.e., ∼5 kBT), 2f (3.59 kcal/mol, i.e., ∼6 kBT), S (4.18 kcal/mol, i.e., ∼7 kBT), W1 (4.31 kcal/mol), and 2a (4.84 kcal/mol, i.e., ∼8 kBT). The PMF barriers correlate with the number of water passages with the protein channels (shown in Table 1), i.e., a low number of water passages leads to a higher PMF barrier.
In the case of camphor-bound P450cam, the thermodynamic equilibrium is shifted in favor of water molecules diffusing out of the protein active site when comparing its channel PMF profiles with those of apo-P450cam (Fig. 5). Note that channels 2d and 2f do not appear in the CAVER calculation of the camphor-bound P450cam MD trajectory (Fig. S9 d and Table 1). All the channel PMF profiles exhibit infinite free-energy values (i.e., absence of water molecules) for reaction coordinates corresponding to the camphor-binding region (see Fig. 5 and the tables in the Supporting Material), except for channel 2a which conducts water diffusion to the distal region of the protein active site as described above. Although the PMF profile for channel 2a is divergent at 1.0-Å and 1.5-Å bin sizes, it exhibits a free-energy barrier of 4.17 kcal/mol (i.e., ∼7 kBT) near the protein active site at the 2.0-Å bin size. Outside of the protein active site, the PMF profile of channel 2a is essentially similar to that calculated for the apo form (Fig. 5 g), unlike the case of channels 2e, W2, S, and W1.
The results of these MD simulations suggest that channels 2e and W2 provide the most thermodynamically favorable pathways for water diffusing into and out of the protein-active-site cavity in apo-P450cam, followed by 2d, 2f, S, and then W1. For the camphor-bound P450cam, although channel 2a can be used for water to reach the distal region of the protein-active-site cavity, all other channels appear to prevent water diffusion to the active site because of the presence of camphor.
Conclusions
The molecular dynamics simulations performed and analyzed here, which are two orders of magnitude longer than previous simulations, allow observation of water diffusion between the protein-active-site cavity and the bulk water by means of which to quantify P450cam's hydration thermodynamics without the need for biased sampling methods. The hydration of the camphor-free species shows that a large number of water molecules, up to 12 in the simulations presented here, can be present in the protein-active site cavity at a given time, but also that only some regions in the active site are preferred for hydration. In the apo form, an average of 6.4 water molecules is found to be present in the camphor-binding region, in very good agreement with the hydration observed in the crystal structure. In the camphor-bound form, no water molecules were found on average over the simulation time, also in agreement with crystallographic data. These results suggest that there are only a limited number of thermodynamically stable hydration sites in the protein active site, potentially leading to a limited number of water molecules being resolved in the crystal structure. The hydration dynamics of P450cam's active site is very dynamic, with water molecules frequently exchanging positions inside the active site, preferentially clustering in some regions, but able to explore a larger fraction of the accessible volume in solution at room temperature.
Channels that allow such bulk water diffusion to the active site and back to the protein exterior can be approximated according to geometrical criteria, but the energetics of water diffusion in these channels varies greatly between the channels and between the camphor-bound and apo forms, and water molecules can be located in protein regions that are not described by CAVER protein channels. Free-energy profiles for water hydration in the protein, from either CAVER channels or the diffusion of protein-active-site water molecules through the entire protein, converge on pathways 2e and W2, exhibiting the lowest free-energy barriers for water diffusion in apo-P450cam, followed by a number of others: 2d, 2f, S, and W1. They are thermodynamically preferred for water molecules diffusing into and out of the protein active site. In camphor-bound P450, nearly all the channels are thermodynamically favorable for driving water out of the protein active site, although pathway 2a (i.e., the proposed substrate access channel) could occasionally allow water diffusion to the distal region of the protein-active-site cavity. Directed mutations of the residues involved in the barriers and valleys of the water diffusion channels described here could potentially lead to modified active-site hydration dynamics and thus to modified reaction dynamics.
Acknowledgments
We thank Jerry Parks for proofreading the manuscript and members of the University of Tennessee/Oak Ridge National Laboratory Center for Molecular Biophysics for scientific discussions.
Y.M. was supported in part by the Department of Energy EpsCOR implementation award (grant DE-FG02-08ER46528), and by the University of Tennessee. J.B. was supported by the University of Tennessee. Computing time on the Kraken supercomputer was supported by a National Science Foundation TeraGrid award (grant TG-MCA08X032).
Supporting Material
References
- 1.Lynch T., Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician. 2007;76:391–396. [PubMed] [Google Scholar]
- 2.Poulos T.L. Cytochrome P450 flexibility. Proc. Natl. Acad. Sci. USA. 2003;100:13121–13122. doi: 10.1073/pnas.2336095100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wade R.C., Winn P.J., Schlichting E., Sudarko A survey of active site access channels in cytochromes P450. J. Inorg. Biochem. 2004;98:1175–1182. doi: 10.1016/j.jinorgbio.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Cojocaru V., Winn P.J., Wade R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Lüdemann S.K., Lounnas V., Wade R.C. How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. J. Mol. Biol. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 6.Li X.C., Baudry J., Schuler M.A. Structural and functional divergence of insect CYP6B proteins: from specialist to generalist cytochrome P450. Proc. Natl. Acad. Sci. USA. 2004;101:2939–2944. doi: 10.1073/pnas.0308691101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen Z.M., Baudry J., Schuler M.A. Ile115Leu mutation in the SRS1 region of an insect cytochrome P450 (CYP6B1) compromises substrate turnover via changes in a predicted product release channel. Protein Eng. Des. Sel. 2005;18:191–199. doi: 10.1093/protein/gzi023. [DOI] [PubMed] [Google Scholar]
- 8.Oprea T.I., Hummer G., Garcia A.E. Identification of a functional water channel in cytochrome P450 enzymes. Proc. Natl. Acad. Sci. USA. 1997;94:2133–2138. doi: 10.1073/pnas.94.6.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sligar S.G., Gunsalus I.C. Proton coupling in the cytochrome P-450 spin and redox equilibria. Biochemistry. 1979;18:2290–2295. doi: 10.1021/bi00578a024. [DOI] [PubMed] [Google Scholar]
- 10.Schlichting I., Berendzen J., Sligar S.G. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 11.Vidakovic M., Sligar S.G., Poulos T.L. Understanding the role of the essential Asp251 in cytochrome p450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- 12.Benson D.E., Suslick K.S., Sligar S.G. Reduced oxy intermediate observed in D251N cytochrome P450cam. Biochemistry. 1997;36:5104–5107. doi: 10.1021/bi963170q. [DOI] [PubMed] [Google Scholar]
- 13.Loida P.J., Sligar S.G. Molecular recognition in cytochrome P-450: mechanism for the control of uncoupling reactions. Biochemistry. 1993;32:11530–11538. doi: 10.1021/bi00094a009. [DOI] [PubMed] [Google Scholar]
- 14.Helms V., Wade R.C. Hydration energy landscape of the active site cavity in cytochrome P450cam. Proteins. 1998;32:381–396. [PubMed] [Google Scholar]
- 15.Rydberg P., Rod T.H., Ryde U. Dynamics of water molecules in the active-site cavity of human cytochromes P450. J. Phys. Chem. B. 2007;111:5445–5457. doi: 10.1021/jp070390c. [DOI] [PubMed] [Google Scholar]
- 16.Berman H.M., Westbrook J., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poulos T.L., Finzel B.C., Howard A.J. Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y.T., Wilson R.F., Goodin D.B. P450cam visits an open conformation in the absence of substrate. Biochemistry. 2010;49:3412–3419. doi: 10.1021/bi100183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulos T.L., Finzel B.C., Howard A.J. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 20.Chemical Computing Group. 2010. Molecular Operating Environment, version 2010. Chemical Computing Group, Montréal, Canada.
- 21.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 22.Zheng J.J., Altun A., Thiel W. Common system setup for the entire catalytic cycle of cytochrome P450(cam) in quantum mechanical/molecular mechanical studies. J. Comput. Chem. 2007;28:2147–2158. doi: 10.1002/jcc.20701. [DOI] [PubMed] [Google Scholar]
- 23.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mackerell A.D., Bashford D., Karplus M. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB J. 1992;6:A143. [Google Scholar]
- 25.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 27.Reference deleted in proof.
- 28.Schöneboom J.C., Lin H., Shaik S. The elusive oxidant species of cytochrome P450 enzymes: characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 2002;124:8142–8151. doi: 10.1021/ja026279w. [DOI] [PubMed] [Google Scholar]
- 29.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 30.Petrek M., Otyepka M., Damborský J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trzesniak D., Kunz A.P.E., van Gunsteren W.F. A comparison of methods to compute the potential of mean force. ChemPhysChem. 2007;8:162–169. doi: 10.1002/cphc.200600527. [DOI] [PubMed] [Google Scholar]
- 32.Deng Y.Q., Roux B. Computation of binding free energy with molecular dynamics and grand canonical Monte Carlo simulations. J. Chem. Phys. 2008;128:115103. doi: 10.1063/1.2842080. [DOI] [PubMed] [Google Scholar]
- 33.Di Primo C., Hui Bon Hoa G., Sligar S. Effect of the tyrosine 96 hydrogen bond on the inactivation of cytochrome P-450cam induced by hydrostatic pressure. Eur. J. Biochem. 1990;193:383–386. doi: 10.1111/j.1432-1033.1990.tb19350.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhang W., Pochapsky S.S., Jain N.U. Solution NMR structure of putidaredoxin-cytochrome P450cam complex via a combined residual dipolar coupling-spin labeling approach suggests a role for Trp106 of putidaredoxin in complex formation. J. Mol. Biol. 2008;384:349–363. doi: 10.1016/j.jmb.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis D.F.V., Hlavica P. Interactions between redox partners in various cytochrome P450 systems: functional and structural aspects. Biochim. Biophys. Acta. 2000;1460:353–374. doi: 10.1016/s0005-2728(00)00202-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.