

Abstract
Escherichia coli MacAB-TolC is a tri-partite macrolide efflux transporter driven by hydrolysis of ATP. In this complex, MacA is the periplasmic membrane fusion protein that stimulates the activity of MacB transporter and establishes the link with the outer membrane channel TolC. The molecular mechanism by which MacA stimulates MacB remains unknown. Here, we report that the periplasmic membrane proximal domain of MacA plays a critical role in functional MacA-MacB interactions and stimulation of MacB ATPase activity. Binding of MacA to MacB stabilizes the ATP-bound conformation of MacB, whereas interactions with both MacB and TolC affect the conformation of MacA. A single G353A substitution in the C-terminus of MacA inactivates MacAB-TolC function by changing the conformation of the membrane proximal domain of MacA and disrupting the proper assembly of the MacA-MacB complex. We propose that MacA acts in transport by promoting MacB transition into the closed ATP-bound conformation and in this respect, is similar to the periplasmic solute-binding proteins.
INTRODUCTION
Periplasmic Membrane Fusion Proteins (MFPs) are a large family of proteins that associate with a variety of transporters in both gram-positive and gram-negative bacteria (Zgurskaya et al., 2009). In contrast to the well-characterized periplasmic solute-binding proteins, which preferentially associate with ATP-binding cassette (ABC) uptake transporters (Davidson et al., 2008), MFPs function with exporters belonging to the ABC, the Major Facilitator (MF) and the Resistance Nodulation Division (RND) Superfamilies of proteins. These transporters depend on MFPs to export from cells a variety of toxic molecules ranging in size from small organic solvents to large proteins such as hemolysin.
The MFP-dependent transporters vary dramatically in their structures and biochemical mechanisms (Zgurskaya, 2009). The RND and MF transporters are driven by a proton motive force, whereas ABC transporters are ATPases. In contrast to monomeric MF and dimeric ABC transporters devoid of large periplasmic extensions, RND pumps are trimers with large periplasmic domains. Yet MFPs that function with such diverse transporters are structurally conserved. Among others, the structures of E. coli AcrA and MacA that associate with the RND drug efflux pump AcrB and the ABC macrolide exporter MacB, respectively, are currently available (Mikolosko et al., 2006, Yum et al., 2009). The two proteins share a modular structure comprising the α-helical hairpin, lipoyl-binding, α–β-barrel and membrane proximal (MP) domains. Both AcrA and MacA are believed to function as hexamers but their interfaces with respective transporters are expected to be different (Yum et al., 2009, Tikhonova et al., 2011). MacA contains a single N-terminal transmembrane segment, which is likely to interface with MacB within the membrane (Tikhonova et al., 2007). In contrast, the signal peptide of AcrA is cleaved after lipid modification limiting the interface between AcrA and AcrB to their periplasmic domains (Zgurskaya & Nikaido, 1999a). Despite mechanistic diversity of transporters, all MFPs are thought to function in transport by stimulating the activity of transporters (Aires & Nikaido, 2005, Tikhonova et al., 2007, Zgurskaya & Nikaido, 1999b, Lin et al., 2009). However, the molecular mechanism of such stimulation remains unknown.
This study is focused on the mechanism of MacAB, a macrolide transporter from E. coli. The macrolide efflux activity of MacAB was identified by complementation studies of the drug susceptible phenotype of E. coli cells lacking the major multidrug efflux pump AcrAB (Kobayashi et al., 2001). In these cells, overproduction of MacAB leads to decreased susceptibility to 14- and 15- membered macrolide antibiotics, such as erythromycin and oleandomycin. As with other MFP-dependent transporters from gram-negative bacteria, the activity of MacAB requires a third component, the outer membrane channel TolC. Located in two different membranes, the MacAB complex and TolC interact with each other and form a large protein conduit across the two-membrane envelope of E. coli (Tikhonova et al., 2007). Such assembly is believed to enable macrolide efflux across the outer membrane into the external medium.
MacB and its homologs are categorized into a distinct subfamily of ABC transporters, the Macrolide Exporter family, because of their unusual topology. They contain only four transmembrane segments (TMSs) with the nucleotide-binding domains (NBDs) located at their N-termini (Kobayashi et al., 2003). In contrast, a typical prokaryotic ABC transporter contains at least six TMSs with the NBD attached to the C-terminus (Davidson & Chen, 2004). However, similar to other ABC transporters, MacB functions as a dimer, a feature dictated by the structure of the NBD domains. NBDs of all ABC systems display a series of highly conserved sequence motifs and upon dimerization form two composite catalytic sites. Previous studies from this and other groups showed that MacB possesses an intrinsic ATPase activity, which is not stimulated by its drug substrates (Lin et al., 2009, Tikhonova et al., 2007). Although MacB reconstituted into proteoliposomes is a slow ATPase, it is strongly stimulated in the presence of MacA. The kinetic studies indicated that the stimulation involves the ATP binding step but its molecular mechanism remains unclear.
Membrane topology and protein analyses suggested that on the cytosolic side and within the membrane, MacA interacts with MacB by its N-terminal TMS (Tikhonova et al., 2007). MacA deletion mutant lacking this domain is soluble in aqueous solutions and fails to support MacB function in vivo, as well as to stimulate its ATPase activity in vitro. But this mutant can still bind MacB. On the periplasmic side of the membrane, the membrane proximal (MP) domain of MacA is likely to interact with the large periplasmic loop between TMS1 and TMS2 of MacB (Kobayashi et al., 2003). The structure of the MP domain of MacA is unknown. However, in MexA, an MFP component of the RND efflux pump MexAB-OprM, the MP domain is composed of residues from both the N-terminal and C-terminal regions, which together form a compact β-roll extending contiguously from the β-barrel domain (Symmons et al., 2009). MacA lacking the C-terminal region of the MP domain does not bind MacB and as a result, is non-functional in both in vitro and in vivo assays (Tikhonova et al., 2007). The importance of the MP domains of MFPs for interaction with transporters is further supported by the recent studies of AcrAB transporter, which demonstrated that the MP domain of AcrA is critical for the assembly of AcrAB-TolC complex as well (Ge et al., 2009, Tikhonova et al., 2011). The ~70 C-terminal residues of the MP domain of MacA are the most conserved residues among MFPs associated with various transporters (Zgurskaya et al., 2009). The functional significance of this sequence conservation remains unknown.
In this study, we investigated the role of MacA in the assembly and function of MacAB-TolC pump. We report that the MP domain of MacA plays a critical role in the stimulation of MacB ATPase by promoting the closed ATP-bound state of MacB. The association with MacB and TolC changes the conformation of MacA. Point mutations in the MP domain of MacA prevent these conformational changes and disrupt the function of MacAB-TolC.
Results
G353 is important for macrolide efflux function of MacA
We previously found that MacA lacking the C-terminal 90 amino acid residues lost its ability to protect against macrolide antibiotics in vivo and stimulate the ATPase activity of MacB in vitro (Tikhonova et al., 2007). To identify the specific residues important for MacA activity we targeted the C-terminal G353 and G357, which are highly conserved among various MFPs (Ge et al., 2009). A cysteine substitution in the G353 position (MacAG353C) but not in G357 notably decreased minimal inhibitory concentrations (MICs) of erythromycin and oleandomycin, the two macrolide substrates of MacAB-TolC (Table 1). The susceptibility to macrolides also increased when G353 in MacA was substituted with either an alanine or a serine residue. In addition, the alanine substitution in G357 (MacAG357A) increased the susceptibility to macrolides by two folds.
Table 1.
Minimal inhibitory concentrations of macrolides in E. coli W4680AD cells carrying plasmids that produce the wild type and mutant MacA
| Plasmids | MIC (μg/ml)a |
|
|---|---|---|
| Erythromycin | Oleandomycin | |
| pUC18 | 2 | 2.44–4.88 |
| pUMacAB | 32–64 | 78–156 |
| pUMacAB-G353C | 8 | 19.5 |
| pUMacAB-G357C | 32 | 78 |
| pUMacAB-G353A | 16 | 39 |
| pUMacAB-G357A | 16 | 39 |
| pUMacAB-G353S | 16 | 39 |
| pUMacAB-G357S | 32 | 78 |
All MIC measurements were done in triplicate
MacA mutants containing cysteine and alanine substitutions in positions G353 and G357 were selected for further biochemical analysis. All mutant MacA proteins were produced from plasmids in similar amounts (data not shown). Furthermore, as determined by co-purification experiments, neither cysteine nor alanine substitutions in G353 and G357 positions disrupted interactions between components of the MacAB-TolC complex (Fig. 1). Taken together, these results suggest that G353 and G357 mutants do not have large scale structural abnormalities that would reduce their functionality.
Fig. 1. MacA mutants with substitutions in the C-terminal domain assemble into MacABTolC complexes.
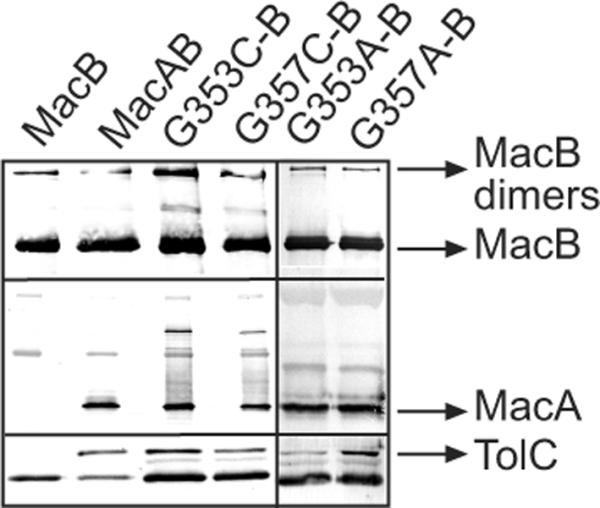
The 6His-tagged MacB was purified from E. coli TolC+ strain W4680AD (ΔacrAB, ΔacrD) carrying plasmids that co-express MacB along with MacAWT or MacA G353C, G357C, G353A and G357A mutants. The co-purification of MacAWT, MacA mutants and TolC with MacB was analyzed by 12% SDS-PAGE and immunoblotting with anti-MacB, anti-MacA and anti-TolC polyclonal antibodies.
Mutations in G353 and G357 change structure of the MP domain of MacA
To investigate possible reasons for the reduced functionality of MacAG353C/A and MacAG357C/A mutants, we next compared the proteolytic patterns of MacAwt and mutants. For this purpose, all proteins were purified and treated with increasing concentrations of trypsin. Consistent with previous studies (Tikhonova et al., 2007), a characteristic ~26 kDa fragment of MacAwt accumulates during the trypsin digest (Fig. 2B). The N-terminal sequencing and mass spectrometry showed that this fragment corresponds to the C-terminal half of the protein, which is produced by cleavage at K139 located in the loop of the α-helical hairpin of MacA (A140-H377 peptide) (Fig. 2A).
Fig. 2. Mutations in positions G353 and G357 affect the MP domain without disrupting oligomerization and the large scale structure of MacA. A.
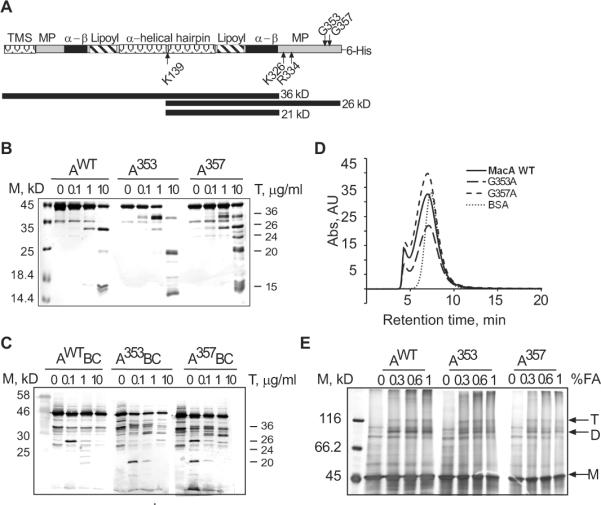
Schematic representation of the secondary structure of MacA. The transmembrane segment (TMS), membrane proximal (MP), α-β barrel (α-β), lipoyl, and α-helical hairpin domains of MacA are indicated. Sites of trypsin cleavage at K139 of the α-helical hairpin and K324, K326 and R334 residues in the MP domain are indicated by arrows. MacAWT proteolysis products 26 kD (A140-Q371+6His) and MacAG353A products 36 kD (M1- K324/326 or R334) and 20 kD (A140-K324/326 or R334) are also indicated. B. Trypsin (T) generated proteolytic patterns of the purified MacAWT and its G353A and G357A variants. 1.25 μM of each of the purified proteins were treated with increasing concentrations of trypsin (0, 0.1, 1.0, 10 μg/mL) and incubated for 30 min at 37°C. The reactions were terminated by boiling in SDS sample buffer for 5 min, resolved on 12% SDS-PAGE and visualized by silver staining. The molecular weight marker (M) is indicated on the left. Numbers on the right are molecular weights of proteolytic fragments in kD determined by mass spectrometry and mobilities in SDS-PAGE as compared to the marker. C. In vivo proteolytic patterns of MacAWT and its G353A and G357A mutants generated by trypsin. E. coli W4680AD (ΔacrAB, ΔacrD) carrying plasmids producing indicated MacA variants along with MacB (2.7×107 cells) were treated with increasing concentration of the trypsin (0.1, 1.0, 10.0 μg/mL) at 37°C for 30 min. The reactions were terminated by adding SDS sample buffer and boiled for 5 min. The whole cell lysates were resolved on 12% SDS-PAGE and visualized by polyclonal rabbit anti-MacA immunoblotting. D. Size-exclusion HPLC of purified MacAWT and mutants using Sephadex 300 (S-300) column. 0.1 mL of ~0.5 mg/mL of each protein was injected with buffer containing 20 mM HEPES-KOH (pH 7.7), 200 mM NaCl, 0.03% DDM, 15 mM MgCl2 at a flow rate of 1 ml/min. Protein elution was monitored by measuring absorbance at 280 nm. BSA (66 kD), was used as a molecular weight standard. E. Formaldehyde (FA) cross-linking of purified MacAWT and its variants. 1–2 μM purified proteins were treated with increasing concentrations of FA (0, 0.3, 0.6, 1.0 % v/v) and incubated in 37°C for 30 min. Reactions were terminated by boiling for 5 min in SDS sample buffer. Proteins were separated by 8% SDS-PAGE and stained with silver nitrate. MacA trimers (T), dimers (D) and monomers (M) are indicated.
The proteolytic pattern of MacAG353A mutant differed significantly from that of the wild type. MacAG353A was readily digested by trypsin with accumulation of two closely sized ~36 kD fragments (Fig. 2A and 2B). In contrast, the 26 kD band characteristic for MacAwt was only a minor band in the MacAG353A pattern. The N-terminal sequence of ~36 kD bands matched that of the whole protein indicating that the cleavage occurs in the C-terminal domain of MacAG353A. Based on the mass spectrometry analysis, the ~36 kD fragments are generated by cleavage at R334 and K326/K324 positions located in the MP domain of MacAG353A (Fig. 2A and Table S1).
Interestingly, MacAG357A mutant adopted an intermediate between the wild type and G353A state. This protein was readily cleaved at both K139 and R334/K326/K324 residues with accumulation of roughly the same amounts of 36 kD and 26 kD fragments (Fig. 2B). In addition, the patterns of both MacAG353A and MacAG357A contained the large amounts of ~20 kD bands, which likely correspond to A140–R334 and A140–K324/326 peptides with the calculated molecular weights 20 and 20.5 kD, respectively (Fig. 2B and Table S1). Taken together, these results suggest that in contrast to MacAwt, which is mainly cleaved by trypsin at K139, MacAG353A mutant is cleaved at the C-terminal R334 and K326/K324 first, followed by cleavage at K139 with the formation of ~20 kD fragment. In addition, the patterns of all three MacA variants contained ~15 kD band, which could correspond to the N-terminal half of the protein (Table 1S).
The trypsin digestion of MacA with cysteine substitutions in G353 and G357 positions produced patterns identical to those containing alanines suggesting that either mutation has the same effect on MacA structure (data not shown). Thus, mutations in the C-terminal region destabilize the MP domain of MacA and make it more accessible to cleavage by trypsin at amino acid residues R334 and K326/K324.
Previous structural and functional analyses suggested that MacA and other MFPs function in transport complexes as oligomers, in which the MP domains of neighboring protomers interact with each other (Symmons et al., 2009, Tikhonova et al., 2009, Yum et al., 2009). Therefore, the reduced functionality and increased accessibility to trypsin could be caused by defects in oligomerization of MacA mutants. Since the oxidative environment of the periplasm and the metal affinity purification promote disulfide bonding in MacA mutants with cysteine substitutions (data not shown), the subsequent experiments were carried out with G353A and G357A mutants.
The size exclusion chromatography of purified proteins showed that elution profiles of MacA mutants match that of the wild type protein (Fig. 2D). In agreement, treatment with formaldehyde showed that all three MacA variants are cross-linked into dimers and trimers (Fig. 2E). Thus, substitutions in G353 and G357 do not affect MacA oligomerization and the increased sensitivity to proteases is due to structural changes in the MP domains.
To determine whether the structural changes in the MP domains of MacA mutants persist in vivo, we treated the whole E. coli cells with proteases under conditions of a mild osmotic shock. Previous studies showed that such conditions permeabilize the outer membrane and make the periplasmic proteins accessible to exogenous proteases (Ge et al., 2009). Fig. 2C shows the anti-MacA immunoblotting analysis of trypsin treated TolC+ cells producing the plasmid-encoded MacA and MacB. As with the purified proteins, the proteolytic pattern of MacAG353A differed significantly from that of MacAwt, whereas MacAG357A showed the intermediate pattern. The proteolytic patterns in vivo were surprisingly similar to those in vitro. The most prominent band of MacAwt pattern was the 26 kD fragment (Fig. 2C). This fragment is also seen in the MacAG357A pattern but not in MacAG353A. In contrast, the characteristic 36 and 20 kDa fragments can be readily detected in mutant patterns but these fragments were not accumulated in MacAwt. This result shows that structural changes identified in the purified MacA mutants (Fig. 2B) persist in vivo. Furthermore, a significant fraction of MacA is accessible to trypsin at the C-terminal R334 and K326/K324 as well as K139 residue in the loop of the α-helical hairpin in the presence of MacB and TolC. This fraction of MacA is either not assembled into the complex or these residues are exposed to the periplasm even in the MacAB-TolC complex.
TolC dramatically sensitizes MacA to proteolysis
To investigate whether interactions with MacB and TolC affect the proteolytic patterns of MacA variants, MacA alone or in combination with MacB was produced in cells lacking the chromosomal copies of acrAB and/or tolC genes. Cells were treated with either trypsin or proteinase K (PK) and MacA proteolytic profiles were visualized by immunoblotting (Fig. 3 and Fig. S1). The PK proteolysis was more revealing than trypsin about MacA interactions with MacB and TolC in vivo. When produced alone, MacAwt was digested by PK into three major ~40, 38 and 24 kD products (Fig. 3A and Fig. S2). In support of the direct MacA-MacB interactions, a new protected 26 kD fragment of MacAwt was detected only in the presence of MacB (Fig. 3A, AWTB panel). The PK pattern of MacAwt was also altered by TolC (Fig. 3A, AWTC panel). However, in TolC+ cells the whole length MacAwt was digested almost completely into a stable 40 kD intermediate (marked by an asterisk) even at the lowest 0.1 μg/ml concentrations of PK. Since in cells only the periplasmic portion of MacAwt is accessible to PK, this fragment is generated by cleavage within the MP domain. Thus, the bipartite association with TolC sensitizes the MP domain of MacAwt to PK.
Fig. 3. In vivo proteolytic profiles of MacAWT and mutants produced in the presence and absence of MacB and TolC.
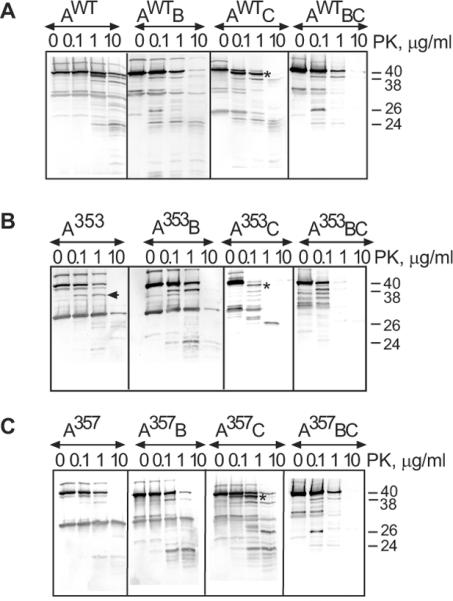
E. coli W4680AD (ΔacrAB, ΔacrD) and ECM2115 (MC4100 ΔacrAB ΔtolC) cells carrying plasmids producing MacA variants alone (AWT, A353, A357) or in the presence of MacB (AWTB, A353B, A357B) were treated with PK at indicated concentrations and analyzed by anti-MacA immunoblotting as described in Fig. 2C. The composition of complexes is shown above the patterns with TolC indicated by “C” when present in cells.
The assembly of MacAB-TolC complex protected the MP domain from cleavage as seen from the lack of the 40 kD product (Fig 3A, AwtBC panel). In addition, the 26 kD MacAwt band characteristic of the complex with MacB can be clearly seen on immunoblots. This result suggests that MacB protects the MP domain of MacAwt from PK, possibly by forming an interface with this domain. Proteolytic patterns of MacAWT in cells carrying different combinations of MacB and TolC were highly reproducible (Fig. S2).
The differences between MacAwt and its mutants identified by tryptic digests (Fig. 2C and Fig. S1) were also seen in PK profiles (Fig. 3). MacA variants were especially distinct in TolC+ cells. In the presence of TolC, the MP domain of MacAG353A was notably more vulnerable to PK than that of MacAwt (Fig. 3B, A353C panel). In particular, the stable 40 kD product of MacAwt generated by PK in the presence of TolC was readily digested in MacAG353A producing a ladder of fragments. The TolC-induced sensitization of the MP domain of MacAG353A was also seen in the tryptic digests (Fig. S1, panel A353C in B).
The presence of MacB protected MacAG353A from the TolC-induced digestion suggesting that this mutant binds MacB as well (Fig. 3B, A353C and A353BC panels). In addition, proteolytic patterns of MacAG353A in the presence of MacB differed from that of MacAG353A alone or in the complex with TolC. Specifically, a 36 kD fragment was generated in cells producing MacAG353A alone (indicated by an arrow), whereas a larger 38 kD product of MacAG353A emerged in the presence of MacB (Fig. 3B, A353 and A353B panels). Thus, in agreement with co-purification results (Fig. 1), the PK proteolysis indicates that MacAG353A binds MacB.
Proteolytic profiles of MacAG357A generated by PK contained features of both MacAwt and MacAG353A proteins (Fig. 3C). The presence of either TolC or MacB altered the proteolytic patterns of MacAG357A supporting the above conclusion that this protein is assembled into complexes. Although proteolytic patterns of MacAG357A in bi-partite complexes with MacB and TolC differed from those of MacAwt, profiles of these two MacA variants were similar when assembled into the tri-partite MacAB-TolC complexes (Fig. 3C, AWTBC and A357BC panels). Thus, in agreement with functional assays, the G357A substitution only mildly affected the MP domain of MacA.
Previous studies suggested that the assembly of the MFP-dependent exporters that secrete peptide toxins is modulated by the presence of substrates (Letoffe et al., 1994, Thanabalu et al., 1998). However, addition of increasing concentrations of oleandomycin did not affect the proteolytic patterns of MacA and its mutants whether expressed alone or assembled into the complexes with MacB and TolC (data not shown).
Thus, co-purification and proteolysis results are consistent with each other and indicate that both MacAG353A and MacAG357A interact with MacB and TolC and assemble into the tri-partite MacAB-TolC complexes. Association with TolC sensitizes the MP domain of all three MacA variants to proteolysis, whereas MacB protects MacA from proteases. The assembly into the complex however, does not rescue the structural defect in the MP domain of MacAG353A.
The MacAB-TolC complex assembled with MacAG353A is defective
We next investigated how MacB is affected by interactions with MacA and its mutants. For this purpose, MacB was produced alone or in the presence of MacA and/or TolC and the in vivo proteolysis was carried out as described above for MacA. The tryptic patterns of MacB alone were identical to those produced in the presence of either MacAwt or its mutants (Fig. S1). Therefore, we used the more robust PK to assess MacA-MacB interactions in vivo. As shown on Fig. 4A, when produced alone, MacB is cleaved by 1 μg/ml PK into three fragments with molecular weights ~42, 41 and 31 kD. The presence of MacAwt changed MacB pattern by appearance of an additional 33 kD fragment at 1 μg/ml of PK and a 29 kD fragment at 10 μg/ml of PK. Thus, interactions with MacA either directly protect MacB from PK cleavage or alter the MacB conformation in such way that at least two cleavage sites on MacB are no longer accessible to PK.
Fig. 4. In vivo proteolytic profiles of MacB produced in the presence and absence of MacAwt, its G353A and G357A mutants and TolC. A.
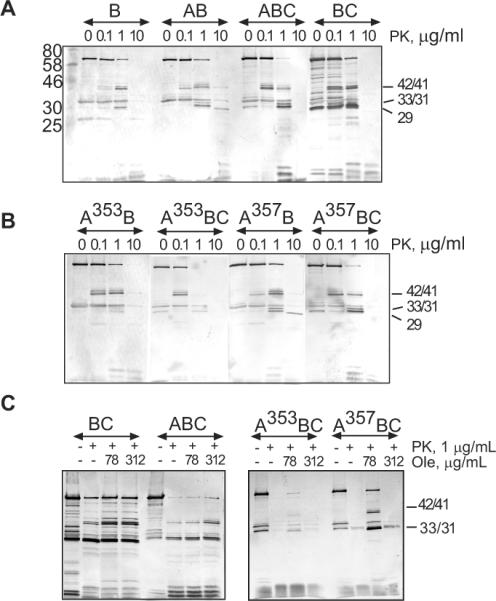
E. coli W4680AD (ΔacrAB, ΔacrD) and ECM2115 (MC4100 ΔacrAB ΔtolC) cells carrying plasmids producing MacB alone or in the presence of MacAwt were treated with PK at indicated concentrations and analyzed by anti-MacB immunoblotting as described in Fig. 2C. The composition of complexes is shown above the patterns with TolC indicated by “C” when present in cells. B. The same as A but MacB was co-expressed with MacAG353A and MacAG357A variants (A353B, A357B). C. Effect of oleandomycin on proteolysis of MacB in vivo. E. coli W4680AD (ΔacrAB, ΔacrD) cells producing MacB alone or in the presence of indicated MacA variants were pretreated with increasing concentrations of oleandomycin (Ole) (0, 78, 312 μg/mL), subjected to osmotic shock and then treated with PK (1 μg/mL) at 37°C for 30 min. The reactions were terminated by boiling in SDS sample buffer and MacB patterns visualized by anti-MacB immunoblotting.
Association with MacAG353A notably changed MacB accessibility to PK. The 42 and 41 kD fragments of MacB appeared at lower 0.1 μg/ml concentration of PK suggesting that these sites of MacB are more accessible to PK when bound to MacAG353A (Fig. 4B, panel A353B). Furthermore, when in the complex with MacAG353A, the proteolytic pattern of MacB contained only traces of the 33 and 31 kD bands characteristic for the MacAwt-MacB complex, whereas the 29 kD band was not detected at all. This result shows that MacAG353A binds MacB in vivo but the assembled complex differs from MacAwt-MacB. The proteolysis pattern of MacB in the complex with MacAG357A mutant was similar to that in the complex with MacAwt (Fig. 4B, panel A357B).
The association of MacAwt-MacB with TolC notably sensitized MacB to PK (Fig. 4A, panel ABC). In TolC+ cells, the 42 and 41 kDa fragments of MacB were detectable even at 0.1 μg/ml of PK, whereas the whole length MacB was almost completely digested by 1 μg/ml of PK. A similar trend was observed with G353A and G357A mutants suggesting that all MacB complexes interact with TolC (Fig. 4B, panels A353BC and A357BC). The TolC-induced vulnerability of MacB to intracellular proteases was further exaggerated in the absence of MacA (Fig. 4A, panel BC). This result strongly suggests that TolC promotes the proteolytic instability of MacAB complex by acting directly on MacB. Although previous co-purification experiments did not identify MacB-TolC interactions (Tikhonova et al., 2007), the TolC-dependent sensitization of MacB to proteases is consistent with direct binding of TolC to MacB. The proteolytic patterns of MacB were the same in the tri-partite complex assembled with TolC and either MacAwt or mutants suggesting that all three MacA variants protect MacB from TolC-induced cleavage by proteases.
Finally, the proteolysis patterns of MacB were studied in cells pre-treated with increasing concentrations of its substrate oleandomycin. No significant differences in MacB were found in TolC− cells suggesting that oleandomycin does not induce stable changes in MacB or MacAB complex (data not shown). Interestingly, in the presence of TolC, oleandomycin protected MacB and its 42, 41 and 31 kDa fragments from cleavage by PK (Fig. 4C, panel ABC). Similar protection was observed in TolC+ cells producing either MacB alone (BC) or the compromised MacAG353A-MacB and MacAG357A-MacB complexes. We conclude that treatment with oleandomycin counter-acts the destabilizing effect of TolC on MacB, perhaps by engaging TolC into other efflux complexes.
Taken together these results suggest that the association of MacB with MacAG353A fails to bring MacB into the same state as in the complex with either MacAwt or MacAG357A. The assembly of the tri-partite MacAB-TolC complex changes the structure of both MacA and MacB and makes them more accessible to PK cleavage. Oleandomycin offsets the effect of TolC on the proteolytic patterns of MacB but does not induce notable changes in MacA or MacB proteins.
Reconstituted MacAG353A mutant inhibits MacB ATPase
The experiments described above established that mutations in position G353 of MacA affect its MP domain and the mode of interaction with MacB. To investigate how these changes in MacA influence MacB activity, we purified MacA and its G353 and G357 mutants and reconstituted the respective MacAB complexes into proteoliposomes. As shown on Fig. 5A, MacAwt and mutants were reconstituted into proteoliposomes with the same efficiencies.
Fig. 5. Reconstituted MacAG353A mutant does not stimulate MacB ATPase. A.

Reconstitution of MacB and its complexes with MacA variants into proteoliposomes. Reconstituted proteoliposomes (1 μL each) were resolved on 10 % SDS-PAGE and were visualized by CBB staining. B. Dependence of ATP hydrolysis by MacB and MacAB proteoliposomes on the concentration of Mg-ATP. All reactions contained 0.42 μM MacB and 0.84–1.68 μM MacA. Filled symbols are observed values and the lines indicate predicted values calculated by Enzfitter software. Error bars are SDs (n = 3).
In agreement with previous studies, MacB alone is a slow ATPase, which hydrolyzes one ATP in about 30 sec (Fig. 5B, Table 2). Co-reconstitution with MacAwt increased the MacB turnover by more than ten times. Although MacAG353C improved the MacB turnover by two fold, MacAG353A mutant completely failed to stimulate MacB. Furthermore, association with MacAG353A inhibited the intrinsic ATPase activity of MacB. Changes in both Vmax and Km contributed to decrease in kcat/Km suggesting that structural changes in MacAG353A affect both ATP binding and hydrolysis by MacB. Thus, the impaired activity of G353 mutants in vivo, their altered proteolytic patterns and defects in the assembly of MacAB-TolC complex correlate directly with the lack of stimulation of the MacB ATPase.
Table 2.
Kinetic parameters of the reconstituted MacB and MacAB proteoliposomes
| Proteoliposomes | Vmax(μM/min) | Km(mM) | kcat(s−1) | kcat/Km(M−1s−1) |
|---|---|---|---|---|
| MacB | 0.82 ± 0.05 | 1.81 ± 0.11 | 0.03 ± 0.002 | 17.5 ± 2 |
| MacAB | 8.0 ± 0.06 | 0.37 ± 0.12 | 0.32 ± 0.002 | 849 ± 33 |
| MacAG353C-B | 1.45 ± 0.3 | 1.6 ± 0.6 | 0.056 ± 0.01 | 34 ± 5 |
| MacAG353A-B | 0.3 ± 0.016 | 1.0 ± 0.2 | 0.01 ± 0.006 | 12 ± 2 |
| MacAG357C-B | 7.6 ± 1.0 | 0.3 ± 0.04 | 0.29 ± 0.04 | 969 ± 16 |
| MacAG357A-B | 1.8 ± 0.3 | 0.55 ± 0.2 | 0.07 ± 0.01 | 131 ± 40 |
Interestingly, the G357 mutants that demonstrated the intermediate phenotype in the macrolide susceptibility and the proteolysis experiments also had intermediate activities in the ATPase assay. The activity of MacAG357C mutant was almost identical to that of MacAwt but kcat of MacB in the complex with MacAG357A decreased by 4.6 fold.
MacA but not its G353 mutant increases MacB affinity to ATP
To gain further insight into the mechanism of MacA, we used photoaffinity labeling with 8-azido[α-32P]ATP ((α32P)8N3ATP). MacB and MacAB proteoliposomes were incubated with increasing concentrations of (α32P)8N3ATP on ice, conditions which allow ATP binding but not hydrolysis, followed by UV irradiation. Autoradiography of proteins separated by SDS-PAGE showed that (α32P)8N3ATP was specifically cross-linked to MacB in both MacB and MacAwtB proteoliposomes (Fig. 6A). Binding and subsequent photocross-linking of (α32P)8N3ATP increased proportionally with the amount of (α32P)8N3ATP present in reaction. However, depending on concentration of (α32P)8N3ATP, three- to six fold more (α32P)8N3ATP was cross-linked to MacB in the presence of MacAwt than to MacB alone. The cross-linking was highly specific to MacB with less than 6% of total radioactivity non-specifically cross-linked to MacAwt. This result strongly suggests that MacAwt increases MacB affinity to ATP.
Fig. 6. MacA increases MacB affinity to ATP. A.
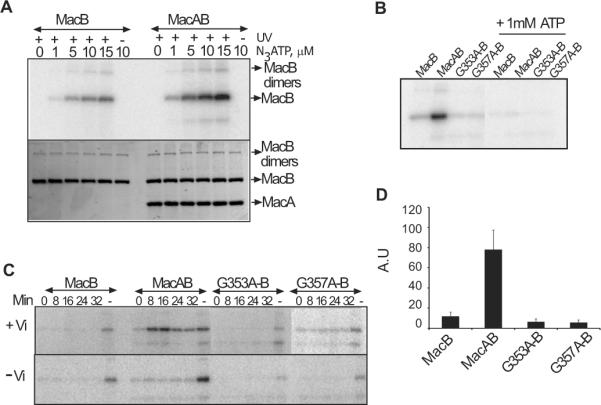
(α-32P)8N3ATP concentration dependent photoaffinity labeling of MacB and MacAB PLs. Proteoliposomes containing 1.1 μM MacB were mixed with increasing concentrations of (α-32P)8N3ATP and incubated on ice for 8 min in subdued light to mediate binding. The photocross-linking was induced by irradiating with 254 nm UV light for 8 min. In the control reaction, UV exposure was omitted. Samples were mixed with SDS sample buffer containing 10 mM DTT, resolved on 12 % SDS-PAGE and labeling was visualized by autoradiography (top panel). Protein amounts were determined by staining with CBB (bottom panel). B. Photoaffinity labeling of MacB and MacAB containing proteoliposomes in the presence or absence of 1 mM cold ATP. All reactions were performed as described above with 1.1 μM MacB, 2 mM MgCl2 and 2.5 μM (α-32P)8N3ATP and in the presence or absence of 1 mM cold ATP. C. Vanadate (Vi) mediated trapping of MacB in post hydrolysis transition state (top panel). All reactions contained 1.1 μM MacB, 2mM MgCl2 and 2.5 (α-32P)8N3ATP and 1 mM vanadate. Control reactions were carried out without vanadate (bottom panel). Reactions were incubated on ice for 8 min to allow nucleotide binding and then transferred to 37°C for indicated time to allow (α-32P)8N3ATP hydrolysis. In the binding only control (lane “-“), reactions were exposed to UV light immediately after incubation on ice and 1 mM cold ATP was omitted. Cross-linking was carried out by exposing the reactions to UV light at 254 nm for 8 min. Samples were mixed with SDS sample buffer containing 10 mM DTT and resolved on 12% SDS-PAGE. Labeling was analyzed by autoradiography and CBB staining to ensure loading of equivalent amounts of MacB. D. Quantification of extend of MacB labeling alone and when co-reconstituted with MacAwt and its G353A, G357A mutants. ImageQuant (Molecular dynamics) software was used to measure the intensity of the bands developed after autoradiography. Error bars are SDs (n = 3).
The incorporation of (α32P)8N3ATP was significantly diminished when MacB was co-reconstituted with either MacAG353A or MacAG357A mutants (Fig. 6B). Furthermore, binding and photocross-linking of (α32P)8N3ATP to MacB in complexes with MacA mutants was at least twice lower than that to MacB alone (Fig. 6D). Thus, MacA mutants not only fail to stimulate but in fact inhibit ATP binding to MacB.
To investigate the action of MacA in ATP hydrolysis, we used vanadate (Vi) trapping. In contrast to binding reactions, which were carried out on ice, the Vi trapping reactions were incubated at 37°C to promote ATP hydrolysis. Previous studies showed that under these conditions Vi replaces the released Pi and captures ADP in the NBD site forming a long lived intermediate that can be visualized by (α32P)8N3ATP labeling (Urbatsch et al., 1995). In agreement with binding data, significant trapping of the nucleotide was detected for MacAwtB complex (Fig. 6C). This trapping was dependent on the presence of Vi indicating that photolabeling is due to trapping of hydrolyzed nucleotide, as opposed to simple binding of the label to MacB. Thus, MacAwt-dependent increase in ATP affinity (Fig. 6B) correlates with the efficient ATP hydrolysis. In contrast, nucleotide trapping was very low in MacB alone or when co-reconstituted with MacA mutants (Fig. 6C and Table 2). Thus, the failure of MacAG353A or MacAG357A mutants to stimulate the ATPase activity of MacB is due to the loss of ability to increase the affinity of MacB to ATP.
MacA stabilizes the closed ATP-bound conformation of MacB
To investigate interactions between MacA and MacB during ATP hydrolysis, proteoliposomes containing MacB and MacAB were treated with PK in the presence or absence of ATP. In the absence of nucleotides, the presence of either MacAwt or its MacAG353A variant led to protection of MacB from PK, as seen from the increased amounts of proteolytic intermediates in MacAB proteoliposomes (Fig. 7B and 7C) as compared to MacB alone (Fig. 7A). The presence of ATP further enhanced the protective action of MacA on the proteolytic patterns of MacB (Fig. 7A, 7B and 7C). It appears that ATP and MacA act synergistically to induce conformational changes in MacB leading to accumulation of 35, 42, 55 and 65 kD fragments. The ATP induced protection of MacB sites targeted by PK was also clearly detected in proteoliposomes containing MacB alone (Fig. 7A) suggesting ATP binds to MacB even in the absence of MacA but its affinity is higher to MacAB complex.
Fig. 7. Proteolytic patterns of MacB proteoliposomes in the presence or absence of nucleotides.

Reconstituted proteoliposomes were pre-loaded when indicated with 1 mM Mg-ATP, 1mM Mg-AMPPNP or 1mM Mg-ATP plus 1 mM vanadate mixture. Reactions were incubated on ice for 8 min to allow nucleotide binding. Proteolysis was carried out with increasing concentrations (0, 0.1, 1.0, 10.0 μg/mL) of PK at 37°C for 30 min (conditions that allow ATP hydrolysis) and terminated by boiling with SDS sample buffer for 5 min. Samples were resolved on 12% SDS-PAGE and visualized by anti-MacB immunoblotting. A. Proteoliposomes containing 0.2 μM MacB alone. B. MacB co-reconstituted with MacAWT (AWTB). C. MacB co-reconstituted with MacAG353A mutant (A353AB) D. The same as in B and C but immunoblotting was carried with anti-MacA antibody. Molecular marker is indicated on the left and proteolytic products are indicated on the right.
On the other hand, ATP notably diminished differences in MacB patterns associated with different MacA variants. In the presence of ATP the pattern for MacAwt-B complex was very similar to that of MacAG353A-MacB without ATP (Figs. 7B and 7C). This result suggests that association with MacAG353A brings MacB into the same state as a combination of MacAwt and ATP together. In agreement, MacAwt co-reconstituted into proteoliposomes with MacB was visibly protected from PK in the presence of ATP, whereas the proteolytic pattern of MacAG353A was insensitive to ATP (Fig. 7D).
To further characterize the MacB state in the complex with MacA, reconstituted proteoliposomes were incubated with AMPPNP or ATP plus vanadate mixture and then subjected to proteolysis as described above. MacB is expected to bind AMPPNP but not hydrolyze this ATP analog. On the other hand, incubation with ATP-vanadate mixture is expected to trap MacB in the closed post-ATP hydrolysis state. We found however, that all three conditions ATP alone, AMPPNP and ATP plus vanadate lead to protection of MacB from PK with accumulation of 35, 42, 55 and 65 kD fragments (Fig. 7A, 7B and 7C). This protection is further enhanced by MacA suggesting that all these conditions induce the same conformational change in MacB, which is the closed nucleotide-bound conformation. In contrast, MacAG353A brings MacB into this conformation even in the absence of nucleotides.
Taken together, the photoaffinity labeling and proteolysis experiments show that MacB alone binds ATP and AMPPNP, whereas MacA promotes conformational transitions in MacB that increase ATP affinity. The MP domain of MacA plays a critical role in these conformational transitions. The similarity of proteolytic profiles of MacB in the complex with MacAG353A and in the complex with MacAwt and ATP together suggest that association with MacAG353A brings MacB into a closed conformation, which precludes ATP binding and inhibits the activity of the complex.
Discussion
MFP-dependent transporters are broadly represented in both gram-negative and gram-positive bacteria (Zgurskaya et al., 2009). However, how MFPs function in transport of various substrates remain unknown. In this study, we used partially functional mutants of MacA to gain insight into the molecular mechanism of MFPs. Our results suggest that the mechanism of MFPs involves stabilization of the closed ATP-bound conformation of MacB ATPase and in this respect, is similar to that of the periplasmic solute-binding proteins (BPs) (Orelle et al., 2008).
The main features of the catalytic cycles of ABC transporters are believed to be highly conserved (Davidson et al., 2008). These transporters cycle through at least four conformational states (Fig. 8): (i) binding of substrate and ATP, (ii) closing of NBD dimers and transport of substrate, (iii) hydrolysis of two ATP molecules, (iv) release of ADP and Pi and transition into the resting state (Higgins & Linton, 2004). Numerous studies of ABC importers and exporters have merged into a view that ABC transporters adopt an outward-facing conformation in the ATP-hydrolysis transition state and an inward-facing conformation in the resting state (the “alternate access model”) (Oldham et al., 2008, Dawson et al., 2007). In the case of BP-dependent transporters, BPs and substrates are believed to promote the closure of the nucleotide-binding interface of NBD dimers and by this means stimulate ATP hydrolysis (Orelle et al., 2008). Our results suggest that MFPs are functional analogs of BPs with an additional role in coupling ATP hydrolysis in the cytoplasm to transport of substrates across the outer membrane (Fig. 8).
Fig. 8. Adaptation of the alternate access model to MacAB-TolC complex.
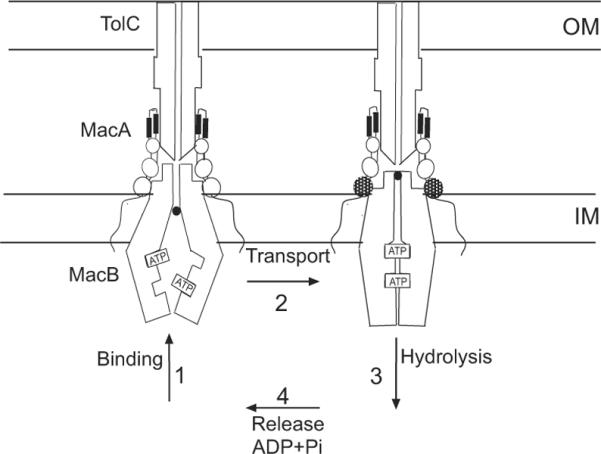
Since the intracellular ATP concentration is at least 30 times the Km of MacAB (for the glucose-fed exponentially growing E. coli, it is 9.6 mM ATP (Bennett et al., 2009)), the nucleotide-binding sites of MacB are probably saturated with ATP in the resting state in vivo. We propose that closure of the NBD interface requires not only the binding of ATP and substrates but also conformational changes in the MP domain of MacA. Association with TolC could drive conformational changes in the MP domain of MacA. OM, outer membrane; IM, inner membrane.
We previously found that MacB reconstituted into proteoliposomes alone is a very slow ATPase but that its activity is strongly stimulated in the presence of MacA (Tikhonova et al., 2007). The photoaffinity labeling showed that the dramatic difference between MacB and MacAB in ATP hydrolysis is mostly due to the difference in ATP affinity. Very little photocross-linking of (α32P)8N3ATP was detected in MacB, in contrast this ATP analog was readily photocross-linked to MacAB (Fig. 6). This result suggested that MacA could either increase the affinity of MacB to ATP in the binding step (1) or could promote closing of NBD dimers (2) (Fig. 8). In the proteolysis experiments, the presence of ATP altered the proteolytic pattern of MacB even when the transporter was reconstituted into proteoliposomes alone (Fig. 7A). Thus, MacB binds ATP but its affinity is much lower than that of MacAB. Surprisingly, the presence of either MacA or ATP resulted in protection of the same set of MacB fragments and the combination of two further enhanced the protection from the protease (Fig. 7A and 7B). This result implies that two ligands act synergistically and induce the same conformational change in MacB. Since this conformational change is the same in the presence of non-hydrolyzable AMPPNP and ATP-vanadate mixture, we propose that this conformational change is the closure of the NBD dimers stimulated by ATP and MacA.
Our results further indicate that the MP domain of MacA located in the periplasm plays an important role in the stimulation of ATP hydrolysis by MacB. A single G353A substitution in the MP domain renders MacA inactive in the in vitro stimulation of the ATPase activity of MacB (Fig. 5) and partially reduces the in vivo resistance to oleadomycin provided by MacAB-TolC complex (Table 1). Biochemical in vivo and in vitro analyses showed that this mutation changes the structure of the MP domain of MacAG353A without affecting its oligomerization and ability to interact with MacB and TolC (Fig. 1 and 2). Analogous results were obtained with the multidrug efflux pump AcrAB-TolC. A single G363 substitution in the C-terminus of AcrA significantly impaired multidrug efflux and affected the assembly of AcrAB-TolC complex (Ge et al., 2009, Tikhonova et al., 2011). Thus, despite dramatic structural and functional differences between MacB and AcrB efflux transporters, the MP domains of MacA and AcrA are very similar.
We propose that the MP domain of MFPs act as a switch in coupling energy consumption by transporter to efflux of substrates across the outer membrane. Our results show that interaction with TolC induces a conformational change in the MP domain of MacA that can be detected by TolC-dependent sensitization of MacA to proteases (Fig. 3 and Fig. S1). On the other hand, the MP domain of MacA interfaces with MacB. The in vivo proteolytic profiles of MacA generated by PK are especially revealing (Fig. 3). The bi-partite interactions with TolC made the MP domains of MacAwt and its mutants highly vulnerable to PK digest. However, the presence of MacB protected all three MacA variants from PK. Reciprocally all three MacA variants protected MacB from proteases (Fig. 4). Thus, MacA and MacB mutually protect and stabilize each other when assembled into the tri-partite complex with TolC. These results further imply that the conformation of the MP domain of MacA in the tri-partite MacAB-TolC complex differs from that in the bi-partite MacAB complex.
In the reconstituted proteoliposomes, the protective effect of MacA is also obvious and it is further enhanced by ATP (Fig. 7). Surprisingly, the strongest protection was seen with MacAG353A, which inhibits MacB ATPase and binding of ATP to MacB (Figs. 5C and 6D). Thus, the lack of stimulation of MacB ATPase could be because the association with MacAG353A stabilizes NBDs of MacB in their closed state preventing the access of ATP to the binding sites (Fig. 8). This result further suggests that conformational transitions in the MP domain of MacA modulate the ATPase activity of MacB.
Interestingly, the conformation of purified MacA is similar to that in the complex with TolC in vivo (Fig. 2B and 2C). It is tempting to propose that the stimulation of MacB ATPase is linked to the conformational changes in the MP domain of MacA induced by TolC. If this is case, the closure of NBDs and ATP hydrolysis are coupled to assembly of the tri-partite MacAB-TolC complex, whereas its disassembly is needed to prevent the futile ATP hydrolysis in the absence of transport. Perhaps extraction with detergents during purification leads to destabilization of the MP domain of MacA and its activation. This would explain why reconstituted MacA stimulates the ATPase activity of MacB even in the absence of TolC (Fig. 5).
Surprisingly, the presence of TolC also affected proteolytic profiles of MacB (Fig. 4), the result consistent with direct interactions between MacB and TolC. However, in contrast to MacA-TolC interactions, which are well-characterized in both in vitro and in vivo studies (Tikhonova et al., 2009, Tikhonova et al., 2007), the existence of a direct interaction between MacB and TolC was not shown. Recent studies of AcrAB-TolC pump showed that AcrB transporter directly binds TolC and that the assembly of the tri-partite complex is completed by interactions between AcrA and TolC bound to AcrB (Tikhonova et al., 2011). Although the periplasmic loops of MacB are smaller than those of AcrB, the periplasmic tunnel of TolC is large enough to directly bind MacB (Koronakis et al., 2000).
The role of substrates in MacA-dependent stimulation of MacB is still obscure. The ligand-free MacA stimulates the rates of ATP hydrolysis by up to 45 folds and the rates decrease in the presence of oleandomycin (Tikhonova et al., 2007). Oleandomycin did not affect the proteolytic patterns of MacA but protected MacB in vivo from TolC-stimulated proteolysis, likely by disassembling TolC from the complex (Fig. 4C). The MFP-dependent type-I secretion complexes engage TolC channel only in the presence of their substrates (Thanabalu et al., 1998). Perhaps, in vivo MacAB is presented with a physiological substrate, which promotes assembly of the complex with TolC, whereas resistance to macrolides is only an inadvertent effect of MacAB-TolC function.
EXPERIMENTAL PROCEDURES
Bacterial growth media and growth conditions
All bacterial cultures were grown at 30°C or 37°C in Luria-Bertani (LB) broth or LB agar (10 g of Bacto-tryptone, 5 g of yeast extract and 5 g of NaCl per 1 liter). Ampicillin (100 μg/ mL) was used for selection where indicated.
Strains and plasmids
E.coli strains W4680AD (K-12 but ΔacrAB ΔacrD), ET103 (BW25113 but ompT∷kan), ECM2115 (MC4100 ΔacrAB ΔtolC) (Tikhonova et al., 2007) were used in protein purification and functional assays. The pUMacAB (pUC18 carrying macAB, Ampr) plasmid (Tikhonova et al., 2007) and its derivatives producing MacA mutants were used for in vivo functional studies. MacA and MacB proteins with the six-His tags were over-expressed and purified from ET103 cells carrying the previously described pBAHis, pBABHis and pBBHis, the pBAD/MycHis-C derivatives carrying macA, macAB and macB, respectively (Tikhonova et al., 2007).
Site-directed mutagenesis
Mutations at positions G353 and G357 in macA gene were introduced by QuickChange XL site-directed mutagenesis kit (Stratagene). The PCR reactions were set up using pUMacAB, pBAHis, pBABHis as templates. The resulting constructs were verified by DNA sequencing (Oklahoma Medical Research Foundation).
Minimal Inhibitory Concentrations (MICs)
MICs of macrolide antibiotics erythromycin and oleandomycin were measured using two-fold micro-dilution method in a 96-well microtiter plate. Plasmids harboring wild type macAB and their derivatives were transformed into E. coli W4680AD (ΔacrAB ΔacrD) strain. Exponentially growing cells were inoculated at a density of 105 cells per well into LB medium supplemented with two-fold increments of the antibiotics under investigation. Cell growth was determined visually after overnight incubation at 30°C. The MICs of the drugs were determined as the lowest concentration of drugs that completely inhibits cell growth.
Protein purification and assays
All purified proteins in this study contain C-terminal 6His tags. Bacterial cultures were grown at 30°C and proteins were purified using metal affinity chromatography as described before (Tikhonova et al., 2007). Concentrations of purified proteins were determined by Bradford protein assay (Bio-Rad) with bovine serum albumin (BSA) as a standard. Concentrations of proteins in proteoliposomes were measured by quantitative SDS-PAGE with BSA as a standard as described before (Tikhonova et al., 2007).
Limited proteolysis
For proteolysis in vitro, MacAwt and its variants (1.2 μM final concentration each) or MacB (0.42 μM final concentration) were incubated with increasing concentrations of trypsin (0.0043 μM, 0.043 μM, 0.43 μM) and/or proteinase K (PK) (0.0035 μM, 0.035 μM, 0.35 μM) for 30 min at 37°C. Reactions were terminated by adding sodium dodecyl sulfate (SDS) sample buffer and boiling for 5 min. Samples were analyzed by 12% SDS-PAGE and protein fragments visualized by silver staining or by immunoblotting with corresponding polyclonal rabbit antibodies.
For proteolysis in vivo, 1 ml of exponentially growing cells (OD600 nm ~ 0.5–0.6) were harvested and washed with equal volumes of buffer I containing 20 mM Tris-HCl (pH 7.5), 100 mM NaCl followed by buffer II containing 20 mM Tris-HCl (pH 7.5), 5 mM EDTA. Finally the cells were resuspended in 640 μL of buffer III containing 20 mM Tris-HCl (pH 7.5), 5 mM EDTA, 20% sucrose and incubated on ice for 30 min. Digestion reactions were set up by adding increasing concentration of trypsin or PK to 45 μL of concentrated cells (2.7 × 107) and were incubated at 37°C for 30–60 min. Reactions were terminated by adding SDS sample buffer and boiling for 5 min. The whole cell lysates were resolved on 12% SDS-PAGE. MacA and MacB proteins and their proteolytic fragments were visualized by immunoblotting with polyclonal anti-MacA and anti-MacB antibodies.
For proteolysis in vivo in the presence of oleandomycin, buffers I and II were additionally supplemented with 1% glucose. Prior to resuspension of cells in buffer III, increasing concentrations of oleandomycin were added and cells were incubated for 30 min at room temperature. Cells were then resuspended in buffer III and treated with proteases as described above.
N-terminal peptide sequencing
Proteolytic fragments were resolved on 12% SDS-PAGE and transferred onto PVDF membrane in buffer containing 10 mM CAPS (pH 11.0) and 10% (v/v) methanol. Protein bands stained with Amido Black were excised from the membrane and analyzed using the Edman degradation method at the Iowa State University Protein Facility.
Size-Exclusion HPLC
Purified native MacA and MacA variants were analyzed by HPLC (Shimadzu) using Sephadex 300 size exclusion column. Proteins (0.05 mg) were injected in buffer containing 20 mM HEPES-KOH (pH 7.7), 200 mM NaCl, 0.03% DDM, 15 mM MgCl2 at a flow rate of 1 ml/min. Protein elution was monitored at 280 nm. BSA (66 kD) was used as a molecular weight standard.
Mass Spectrometry
Proteolysis of purified MacA and its G353A derivative was carried out as described above, except the reactions were terminated by addition of acetic acid to final concentration 15 % (v/v). The samples were desalted using C4 ZipTips (Millipore) and eluted with 50% (v/v) acetonitrile and 0.1% (v/v) trifluoroacetic acid. Matrix-assisted desorption ionization-time of flight (MALDI-TOF) analysis was done to determine masses of the peptide fragments generated by tryptic digest. Samples were spotted on sample grids and covered with matrix (α-cyano-4-hydroxycinnmic acid). MALDI-TOF mass spectra were collected by using a linear positive ion mode. Multiple attempts to identify the MacB proteolytic fragments failed because the fragments did not ionize.
Co-purification of MacAB-TolC complex
E. coli W4680AD cells were transformed with pBBHis producing MacBHis alone or pBABHis derivatives producing MacBHis along with MacA variants. Cells were grown in 250 mL of LB broth supplemented with 100 μg/mL ampicillin at 30°C till OD600 nm ~ 0.8 and induced with 0.1% arabinose for 3 hours. Cells were harvested, lysed and membrane fractions were collected as described before (Tikhonova et al., 2007). MacBHis-containing protein complexes were purified using Cu2+-charged NTA column chromatography.
ATP hydrolysis assay
Purified MacA and MacB proteins in 0.2% TX-100 were reconstituted into proteoliposomes as described before (Tikhonova et al., 2007). The rate of ATP hydrolysis by freshly reconstituted proteoliposomes was measured in a reaction buffer containing 20 mM HEPES-KOH (pH 7.0), 5 mM DTT, 50 mM KCl, 2 mM MgCl2. The final concentration of MacB was 0.42 μM. The total reaction volume was 10 μL. The 32P-γ-phosphate labeled ATP (3000 Ci mmol–1, Amersham) was mixed with unlabelled Mg-ATP at concentrations indicated in Figures. ATP was loaded into proteoliposomes by three rounds of freezing in liquid N2 followed by thawing in bath sonicator. Reactions were carried out at 37°C and the amounts of released Pi were analyzed by thin-layer chromatography as described before (Tikhonova et al., 2007).
Photoaffinity labeling of MacB with (α32P)8N3ATP
Purified MacBhis was co-reconstituted into proteoliposomes along with MacAwt, MacAG353A or MacAG357A in buffer containing 20 mM HEPES-KOH (pH 7.0) and 50 mM KCl. Reactions containing proteoliposomes (1.1 μM MacB final concentration), 2.5 μM (α32P)8N3ATP, 2 mM MgCl2 in a total volume of 10 μL were subjected to three cycles of freezing in liquid N2 and thawing in bath sonicator to facilitate the entry of ATP analogues into the lumen of proteoliposomes. Then the reaction mixture was incubated for 8 min on ice under subdued light to facilitate binding of ATP analog to MacB. Photocross-linking was carried out with UV light (wavelength 254 nm) for 8 min on ice. Post UV irradiation, reactions were mixed with 3.3 μL of SDS sample buffer and analyzed by 12% SDS-PAGE. Radioactivity was detected using Storm PhosphorImager (Molecular Dynamics). For competition experiments, 1 mM cold ATP was added to the reaction tubes prior to the addition of (α32P)8N3ATP, followed by incubation for ~ 5 min on ice.
For vanadate induced trapping of MacB in post ATP hydrolysis state, 100 mM Na3VO4 stock solution (pH ~10.0) was prepared according to the standard methods (Goodno, 1982) and boiled immediately before use. Reactions containing proteoliposomes (1.1 μM MacB final concentration), 4 μM (α32P)8N3ATP, 2 mM MgCl2 and 1 mM vanadate in a total volume of 10 μL were pre-loaded with ATP analogue and vanadate by three cycles of freezing/thawing. The reaction mixture was incubated for 8 min on ice under subdued light to enable binding of ATP analog to MacB. The reactions were then placed at 37°C to facilitate ATP hydrolysis and at different time intervals aliquots were taken, transferred on ice and 1 mM cold ATP was added to displace any bound but unhydrolyzed or non-specifically bound (α32P)8N3ATP. After ~10 min, the reactions were subjected to UV irradiation for azido cross-linking and analyzed as described above. The control reactions were carried out as described above but Vi or the hydrolysis step were omitted from the reaction mixtures.
Supplementary Material
Acknowledgement
This work was supported by National Institutes of Health Grant AI052293 to H.I.Z. We thank Drs. Shaorong Liu and Joann J. Lu for help with MALDI-TOF analyses of peptides.
References
- Aires JR, Nikaido H. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli. J Bacteriol. 2005;187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- Davidson AL, Dassa E, Orelle C, Chen J. Structure, Function, and Evolution of Bacterial ATP-Binding Cassette Systems. Microbiol. Mol. Biol. Rev. 2008;72:317–364. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson RJP, Hollenstein K, Locher KP. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Molecular Microbiology. 2007;65:250–257. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- Ge Q, Yamada Y, Zgurskaya H. The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB-TolC. J Bacteriol. 2009;191:4365–4371. doi: 10.1128/JB.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodno CC. Myosin active-site trapping with vanadate ion. Methods Enzymol. 1982;85(Pt B):116–123. doi: 10.1016/0076-6879(82)85014-3. [DOI] [PubMed] [Google Scholar]
- Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, Hirata T, Yamaguchi A. Membrane topology of ABC-type macrolide antibiotic exporter MacB in Escherichia coli. FEBS Lett. 2003;546:241–246. doi: 10.1016/s0014-5793(03)00579-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, Yamaguchi A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol. 2001;183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Letoffe S, Ghigo JM, Wandersman C. Secretion of the Serratia marcescens HasA protein by an ABC transporter. J Bacteriol. 1994;176:5372–5377. doi: 10.1128/jb.176.17.5372-5377.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HT, Bavro VN, Barrera NP, Frankish HM, Velamakanni S, van Veen HW, Robinson CV, Borges-Walmsley MI, Walmsley AR. MacB ABC transporter is a dimer whose ATPase activity and macrolide-binding capacity are regulated by the membrane fusion protein MacA. J Biol Chem. 2009;284:1145–1154. doi: 10.1074/jbc.M806964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational Flexibility in the Multidrug Efflux System Protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Curr Opin Struct Biol. 2008;18:726–733. doi: 10.1016/j.sbi.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C, Ayvaz T, Everly RM, Klug CS, Davidson AL. Both maltose-binding protein and ATP are required for nucleotide-binding domain closure in the intact maltose ABC transporter. Proc Natl Acad Sci U S A. 2008;105:12837–12842. doi: 10.1073/pnas.0803799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci U S A. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanabalu T, Koronakis E, Hughes C, Koronakis V. Substrate-induced assembly of a contiguous channel for protein export from E.coli: reversible bridging of an inner-membrane translocase to an outer membrane exit pore. Embo J. 1998;17:6487–6496. doi: 10.1093/emboj/17.22.6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Dastidar V, Rybenkov VV, Zgurskaya HI. Kinetic control of TolC recruitment by multidrug efflux complexes. Proc Natl Acad Sci U S A. 2009;106:16416–16421. doi: 10.1073/pnas.0906601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Devroy VK, Lau SY, Zgurskaya HI. Reconstitution of the Escherichia coli macrolide transporter: the periplasmic membrane fusion protein MacA stimulates the ATPase activity of MacB. Mol Microbiol. 2007;63:895–910. doi: 10.1111/j.1365-2958.2006.05549.x. [DOI] [PubMed] [Google Scholar]
- Tikhonova EB, Yamada Y, Zgurskaya HI. Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem Biol. 2011;18 doi: 10.1016/j.chembiol.2011.02.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbatsch IL, Sankaran B, Weber J, Senior AE. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J Biol Chem. 1995;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- Yum S, xu Y, Piao S, Sim SH, Kim HM, Jo WS, Kim KJ, Kweon HS, Jeong MH, Jeon H, Lee K, Ha NC. Crystal structure of the periplasmic component of a tripartite macrolide-specific efflux pump. J Mol Biol. 2009;387:1286–1297. doi: 10.1016/j.jmb.2009.02.048. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI. Multicomponent drug efflux complexes: architecture and mechanism of assembly. Future Microbiol. 2009;4:919–932. doi: 10.2217/fmb.09.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. AcrA is a highly asymmetric protein capable of spanning the periplasm. J. Mol. Biol. 1999a;285:409–420. doi: 10.1006/jmbi.1998.2313. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1999b;96:7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Yamada Y, Tikhonova EB, Ge Q, Krishnamoorthy G. Structural and functional diversity of bacterial membrane fusion proteins. Biochim Biophys Acta. 2009;1794:794–807. doi: 10.1016/j.bbapap.2008.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.