

Abstract
Different types of human gene mutation may vary in size, from structural variants (SVs) to single base-pair substitutions, but what they all have in common is that their nature, size and location are often determined either by specific characteristics of the local DNA sequence environment or by higher-order features of the genomic architecture. The human genome is now recognized to contain ‘pervasive architectural flaws’ in that certain DNA sequences are inherently mutation-prone by virtue of their base composition, sequence repetitivity and/or epigenetic modification. Here we explore how the nature, location and frequency of different types of mutation causing inherited disease are shaped in large part, and often in remarkably predictable ways, by the local DNA sequence environment. The mutability of a given gene or genomic region may also be influenced indirectly by a variety of non-canonical (non-B) secondary structures whose formation is facilitated by the underlying DNA sequence. Since these non-B DNA structures can interfere with subsequent DNA replication and repair, and may serve to increase mutation frequencies in generalized fashion (i.e. both in the context of subtle mutations and SVs), they have the potential to serve as a unifying concept in studies of mutational mechanisms underlying human inherited disease.
Keywords: inherited disease, human genome, disease genes, mutational mechanisms, mutation hotspots, DNA sequence motifs, genome architecture, non-B DNA structures
Introduction
“Where, when, and in which individual a particular mutation will appear is unpredictable”.
Theodosius Dobzhansky (1970) Genetics of the Evolutionary Process
“A mutation is in itself a microscopic event, a quantum event, to which the principle of uncertainty consequently applies – an event which is hence by its very nature essentially unpredictable”.
Jacques Monod (1971) Chance and Necessity: An Essay on the Natural Philosophy of Modern Biology
Although mutation is still often casually described as a ‘random process’ [Gerrish, 2002; Ayala, 2007; Kondrashov and Kondrashov, 2010], there is now abundant evidence that the process of mutation is far from random. Indeed, over the last 20 years, it has become ever clearer that human gene mutation is frequently a highly sequence-specific process, irrespective of the type of lesion involved. Further, we have come to understand that certain DNA sequences are inherently mutation-prone by virtue of their base composition, sequence repetitivity, epigenetic modification, and/or characteristic secondary structures, and hence have a tendency to mutate in very specific ways. This inherent mutability pertains not only with respect to gross gene lesions but also to subtle mutations such as single base-pair (bp) substitutions. Thus, whereas highly prominent genomic structural features may act at a distance so as to induce gross genomic rearrangements, the nature, location and frequency of micro-lesions are often influenced by their immediate DNA sequence context. The recognition that certain DNA sequences are inherently hypermutable has been accompanied by an emerging understanding of how DNA sequence influences (and indeed often underpins) secondary structure formation, how certain local DNA structures can themselves be mutagenic, and how the type and frequency of the resulting mutations can in turn help to explain the nature and prevalence of specific human genetic diseases [Rogozin and Pavlov, 2003; Bacolla et al., 2008; Arnheim and Calabrese, 2009]. Studies of hypermutable sequences have also provided important insights into the endogenous nature of many of the known mechanisms of mutagenesis, for example CpG deamination or slipped mispairing at the DNA replication fork, that are responsible for quite different types of recurring micro-lesion.
Human mutational spectra are increasingly being ascertained on a genome-wide scale, as for example in sequenced cancer genomes that can constitute an intricate patchwork of clustered, or even overlapping, somatic lesions. Here, however, we have attempted to focus on those mutations that have occurred in the germline and which underlie human inherited disease. Many of these lesions have become explicable (albeit retrospectively) in terms of their underlying mutational mechanisms by reference to local genome structure and sub-structure. In this review, we explore how the nature, location and frequency of the many different types of human gene mutation causing inherited disease are shaped in large part, and often in remarkably predictable ways, by the local DNA sequence environment. The central hypothesis we aim to discuss herein is that sites of mutation leading to inherited disease often coincide with DNA sequences known to possess peculiar biochemical and/or structural features, ranging from the spontaneous deamination of single bases to the cooperative transition from the canonical right-handed double-helix to complex secondary structures, including triplexes, slipped-out bases and cruciforms (collectively termed non-B DNA) such that the root cause of the vulnerability of DNA to mutation often resides within its own sequence.
The text below is organized operationally into sections, allowing us to address sequentially the impact of DNA sequence architecture upon (i) single nucleotide substitutions, (ii) microdeletions, microinsertions and indels, (iii) structural variants (SVs) including copy number variations, (iv) microsatellite mutation and (v) mutations in or involving the mitochondrial genome. We then discuss the extent to which non-canonical (non-B) DNA structure-forming sequences have the potential to contribute to a generalizable and hence potentially unifying hypothesis in the field of mutagenesis, on the basis that non-B DNA structures appear to have the capacity to increase the mutation frequency not only with respect to SVs but also in the context of subtle mutations.
Single Nucleotide Substitutions
The CpG Dinucleotide Mutation Hotspot
“It is quite clear that the abnormal hemoglobins of man reveal a pattern of nucleotide replacements which is distinctly non-random. It is also clear that the major contributor.…is the G→A transition. Precisely the same conclusions are obtained from the data on the evolution of cytochrome c. This occurs in spite of the fact that in the case of hemoglobin we are probably looking almost exclusively at deleterious mutations, whereas in the case of cytochrome c we are looking only at mutations which have survived the rigors of selection”.
W. M. Fitch (1967) J. Mol. Biol. 26:499-507.
“Our observation of recurrent CG-TG mutations strongly supports the view that these dinucleotides are mutation hotspots”.
H. Youssoufian et al. (1986) Nature 324:380-382.
5-methylcytosine (5mC) is the most frequent post-synthetic (epigenetic) DNA modification in the human genome and is largely, but not exclusively, confined to the CpG dinucleotide. The first hint that the CpG dinucleotide might constitute a hotspot for pathological mutations in the human genome came 25 years ago with the finding that two different CGA>TGA (Arg>Term) nonsense mutations in the factor VIII gene (F8; MIM# 306700) had recurred quite independently in unrelated individuals causing hemophilia A [Youssoufian et al., 1986]. The potential generality of this phenomenon soon became evident with the finding that 12 of the 34 (35%) single base-pair substitutions then known to cause human inherited disease were C>T and G>A (on the other strand) transitions within CpG dinucleotides [Cooper and Youssoufian, 1988]. Further studies confirmed that the CpG dinucleotide was also a mutation hotspot in a number of other human disease genes including PAH [MIM# 612349; Abadie et al., 1989], SERPINC1 [MIM# 107300; Perry and Carrell, 1989], F9 [MIM# 300746; Koeberl et al., 1990], LDLR [MIM# 606945; Rideout et al., 1990], RB1 [MIM# 180200; Mancini et al., 1997], HPRT1 [MIM# 308000; O‘Neill and Finette, 1998] and DMD [MIM# 300377; Buzin et al., 2005]. As mutation data accumulated, CGA>TGA transitions were encountered disproportionately frequently as a cause of human genetic disease [Krawczak et al., 1998]. This was not simply due to the hypermutabilty of the CpG dinucleotide but also because such nonsense mutations are inherently more likely than missense mutations to come to clinical attention owing to their greater functional impact [Mort et al., 2008].
From the outset, it was realised that the hypermutability of the CpG dinucleotide was related to its role as the major site of cytosine methylation in the human genome. The reason traditionally put forward to explain this association has been that while cytosine spontaneously deaminates to uracil (which is efficiently recognized as a non-DNA base and removed by uracil-DNA glycosylase), the spontaneous deamination of 5mC yields thymine [Shen et al., 1994] thereby creating G•T mismatches whose removal by methyl-CpG binding domain protein 4 (MBD4) and/or thymine DNA glycosylase followed by base excision repair (BER) is inherently less efficient [Hendrich et al., 1999; Waters and Swann, 2000; Walsh and Xu, 2006; Cortázar et al., 2007; Boland and Christman, 2008]. This notwithstanding, it should be appreciated that CpG transitions do not originate exclusively via the spontaneous deamination of 5mC but may also arise through the action of other mechanisms and processes e.g. nucleotide misincorporation during replication [Shen et al., 1992; Zhang and Mathews, 1994; Pfeifer, 2006]. Irrespective of the precise nature of the underlying mutational mechanism, Krawczak et al. [1998] estimated that, in the context of inherited disease, the rate of CG>TG (and CG>CA on the other strand) transitions was five times that of the base mutation rate. Subsequent estimates of 5mC hypermutability, derived from various studies of polymorphism, pathological mutations or sequence divergence in an evolutionary context, have ranged from four- to fifteen-fold [Nachman and Crowell, 2000; Kondrashov 2003; Tomso and Bell, 2003; Jiang and Zhao, 2006a; Zhao and Zhang, 2006; Zhang et al., 2007; Elango et al., 2008; Misawa and Kikuno, 2009; Li et al., 2009]. Ultimately, the question of whether or not a given CpG dinucleotide is hypermutable in the context of inherited disease is determined by its methylation status in the germline. An added level of complexity is however likely to be introduced into the equation by site-specific differences in the efficiency of DNA methylation (by DNA methyltransferases) that are conferred by the immediate flanking sequence [Wienholz et al., 2010]. It would also appear that local DNA structure, specifically in the form of sequences capable of forming DNA structures other than the canonical right-handed double-helix (collectively called non-B DNA), can influence the efficiency of DNA methylation [Halder et al., 2010]. In passing, another potential source of 5mC-associated mutations is the genome-wide induction of single-strand breaks generated during the waves of demethylation and remethylation in the zygote [Wossidlo et al., 2010]; such a mechanism may account for the large deletions stimulated by knocked-in (CG•CG) tracts in the mouse [Wang et al., 2008].
Self evidently, since the CpG dinucleotide is a hotspot for mutation, the CpG mutation rate is considerably higher than the non-CpG mutation rate. However, it would appear that the non-CpG mutation rate is contingent to some extent upon the local CpG content [Walser et al., 2008]. This correlation between the CpG and non-CpG mutation rates seems to be independent of G+C content, recombination rate and chromosomal location but, intriguingly, approximates to a sigmoidal curve [Walser and Furano, 2010]. This is potentially explicable in terms of the effect of CpG content on the non-CpG mutation rate being subject to a certain threshold (~0.53%), with ‘saturation’ being attained when the CpG content rises above a particular level (~0.63%). In addition, the mutational spectrum (transition/transversion ratio) of non-CpG sites was noted to change with CpG content [Walser and Furano, 2010] supporting the authors’ contention that this ‘CpG effect’ could be an intrinsic property of the DNA sequence.
A CpHpG Trinucleotide Mutation Hotspot Associated with Human Inherited Disease?
It has been known for some time that cytosine methylation also occurs in the context of CpNpG sites (where N represents any nucleotide) in mammalian genomes [Woodcock et al., 1987; Clark et al., 1995; Ramsahoye et al., 2000] and in vitro [Pradhan et al., 1999]. Since the intrinsic symmetry of the CpNpG trinucleotide would support a semi-conservative model of replication of the methylation pattern (as with the CpG dinucleotide), it comes as no surprise that both maintenance and de novo methylation occurs at CpNpG sites in mammalian cells [Clark et al., 1995]. In their landmark paper on the human methylome, Lister et al. [2009] reported abundant DNA methylation in CpHpG trinucleotides (where H = A, C or T); more specifically, some 17.3% of 5mC in embryonic stem cells was found to occur within CpApG, CpCpG and CpTpG with a further 7.2% of 5mC occurring in CpHpH. Although Lister et al. [2009] suggested that non-CpG methylation is almost entirely lost upon differentiation (a conclusion based upon the analysis of fetal lung fibroblasts), others have noted CpNpG methylation within human genes in a variety of different somatic tissues [Lee et al., 2010; Laurent et al., 2010]. If we therefore assume not only that CpHpG methylation occurs in the germline but also that 5mC deamination can occur within a CpHpG context, then it follows that methylated CpHpG sites are very likely to constitute mutation hotspots causing human inherited disease. Initial indirect evidence that this might indeed be the case came from the observation that disproportionately high numbers of C>T and G>A transitions occur at CpNpG sites in studies of the human genes, NF1 [MIM# 613113; Rodenhiser et al., 1997] and BRCA1 [MIM# 113705; Cheung et al., 2007].
In the light of the above, Cooper et al. [2010] revisited the question of CpG dinucleotide hypermutability and explored the potential contribution that CpHpG transitions might make to human inherited disease. A total of 54,625 missense and nonsense mutations from 2,113 genes causing inherited disease were retrieved from the Human Gene Mutation Database [http://www.hgmd.org; Stenson et al., 2009]. Some 18.2% of these pathological lesions were found to be C>T and G>A transitions located within CpG dinucleotides (compatible with a model of methylation-mediated deamination of 5mC), a ~10-fold higher proportion than would have been expected by chance alone [Cooper et al., 2010]. The corresponding proportion for the CpHpG trinucleotide was 9.9%, a ~2-fold higher proportion than would have been expected by chance alone. Cooper et al. [2010] therefore estimated that ~5% of missense/nonsense mutations causing human inherited disease could be attributable to methylation-mediated deamination of 5mC within a CpHpG context. Irrespective of the functional role(s) of cytosine methylation in the human genome, it would appear that methylation of the CpHpG trinucleotide may leave a significant imprint on the spectrum of point mutations causing human genetic disease.
Other Sequence Specificities that Underlie the Local Context Dependency of Human Point Mutation
“A model for frameshift mutation can often be hypothesized from knowledge of the DNA sequence and contexts of the mutants and the sequence-specific behaviour of enzymes believed to be involved in mutation”.
L. S. Ripley (1990) Annu. Rev. Genet. 24:189-213.
In addition to the CpG and CpHpG effects discussed above, other types of nucleotide substitution also display context dependence in that substitution rates are dependent upon the identity of the neighbouring bases. For example, Krawczak et al. [1998] observed a subtle and locally confined influence of the surrounding DNA sequence on relative rates of single-base-pair substitutions causing human inherited disease. Most notably, T>(C,A), A>(C,G), and G>(T,C) appear to be biased by the nucleotide at position −1, whereas T>(C,G), C>(G,A), A>(T,G), and G>T are biased by the nucleotide at position +1. However, the nearest-neighbour influence decreases markedly with distance from the site of nucleotide substitution. A significant, albeit weak effect was also observed for position +2, but only for five specific substitutions viz. T>C, C>T, A>G and G>(T,C). Interestingly, the six substitutions significantly influenced by the −1 (5′) nucleotide can be matched with their complementary substitutions being significantly influenced by the +1 (3′) neighbour, and vice versa. When nearest-neighbour effects were analyzed in such a way as to allow for neighbouring dinucleotides rather than mononucleotides, more substitutions were found to exhibit a statistically significant (albeit weaker) rate dependency [Krawczak et al.. 1998]. Nevertheless, this effect was again found not to extend beyond positions −2 and +3. Hence, in most cases, the influence of the surrounding DNA sequence would appear to extend no further than ~2 bp from the site of nucleotide substitution. This notwithstanding, recent work suggests that the presence in the vicinity of sequences capable of forming non-B DNA may be capable of exerting an influence on nucleotide mutability [Bacolla et al., 2011].
One possible mechanism to account for an influence of the local DNA sequence environment on the nature and location of single base-pair substitutions is misalignment mutagenesis [Kunkel, 1990]. Transient misalignment of the primer template, caused by looping out of a single template base can give rise to nucleotide misincorporation during DNA replication [Kunkel, 1990]. If not promptly repaired, such misaligned structures can be bypassed and extended by low-fidelity DNA polymerases, ultimately giving rise to heritable mutations [Sutton, 2010]. Employing primer-template models in vitro, Chi and Lam [2008; 2009] have shown that the relative stabilities of misaligned DNA structures, and hence the likelihood of their templating mutations, are dependent upon the terminal base-pair at the replicating site, the identity of the templating base and the nature of the upstream and downstream nucleotides. If this were to play an important role in the generation of single-base-pair substitutions in human genes, then a substantial proportion of single base-pair substitutions should exhibit identity between the newly introduced base and one of the bases immediately flanking the site of mutation. Consistent with this prediction, Krawczak et al. [1998] previously showed that mutations causing human inherited disease display a degree of mutational bias that favours substitutions in the direction of the flanking bases, at least for certain codon positions. Mutation toward the 5′ flanking nucleotide was found to occur significantly more often than expected at the second position of the codon but not at the first or last position; mutation toward the 3′ flanking base was found to be favoured at the first position of a codon but was disfavoured at the second position. These findings were held to be suggestive of a mutational mechanism, involving positions 1 and 2 in the codon (both of which are critical for the specification of the encoded amino acid residue), that is biased toward the nucleotide at the other position. Inspection of the genetic code revealed that such a bias invariably serves to avoid the de novo introduction of termination codons [Krawczak et al., 1998]. Finally, although no specific preponderance of repeat-sequence motifs was noted in the vicinity of the nucleotide substitutions, a moderate correlation between the relative mutability and thermodynamic stability of DNA triplets emerged [Krawczak et al., 1998]. This was suggestive either of inefficient DNA replication in regions of high stability or the transient stabilization of misaligned intermediates. Not surprisingly, nearest neighbour effects are not confined to mutations causing inherited disease. Indeed, they are also evident in the spectrum of single nucleotide polymorphisms in the human genome [Zhao and Boerwinkle, 2002; Zhang and Zhao, 2004; Jiang and Zhao, 2006b] as well as in the context of evolutionary substitutions [Blake et al., 1992; Hess et al., 1994; Siepel and Haussler, 2004; Nevarez et al., 2010; Ma et al., 2010], findings that argue strongly for the ubiquity of the underlying mutational mechanisms.
The molecular basis of the sequence dependency of human mutation is clearly complex since the extensive inter- and intra-chromosomal variation in the mutation rate cannot be due entirely to neighbouring nucleotide effects [Hodgkinson et al., 2009]. Instead, a given mutational spectrum is likely to result from a combination of a number of different processes such as (i) the sequence specificity of both exogenous mutagens and endogenous mutational mechanisms, (ii) cellular attempts to repair the mutation in question followed by replication of the repaired DNA and (iii) chromatin composition (i.e. bulk vs. epigenetically modified nucleosomes [Tolstorykov et al., 2011]). Whilst imbalances in intracellular pools of dNTPs have long been known to exert a general mutagenic effect [Mathews, 2006], different exogenous mutagens can target specific sequence contexts [Pfeifer and Besaratinia, 2009]. Thus, both benzo[a]pyrene and UV light have been reported to display a target site specificity for CpG dinucleotides [Denissenko et al., 1997; You and Pfeifer, 2001], although both mutagens are rather more likely to be relevant to mutagenesis in the soma than in the germline. However, since the context-dependent pattern of (germline) mutations occurring during mammalian evolution correlates strongly with empirically determined patterns of oxidative damage [Stoltzfus, 2008; Sedelnikova et al., 2010], we may infer that oxidative damage probably plays a key role in germline mutation and that at least some of the context dependency of mutations is bound up with this mechanism of mutagenesis [Hsu et al., 2004]. This may be particularly relevant for sequences containing clustered guanine residues, since during repair these readily oxidized bases may give rise to opposing (or nearly opposing) single-strand breaks, which may then yield mutagenic double-strand breaks [Sedelnikova et al., 2010].
Different DNA polymerases and repair enzymes also exhibit their own characteristic sequence specificities and error signatures [Donigan and Sweasy, 2009; Mazurek et al., 2009; Korona et al., 2011; Lange et al., 2011]. In transcribed regions, transcription-coupled repair gives rise to inter-strand asymmetries in the mutation rate [Green et al., 2003; Polak and Arndt, 2008; Mugal et al., 2009] which are superimposed upon the intrinsic replication-associated mutational asymmetries that are thought to result from a combination of (i) the unequal rates of complementary base misincorporation by DNA polymerases and (ii) the different efficiencies of action of DNA mismatch repair enzymes on the leading and lagging DNA strands [Chen et al., 2011]. Different base mismatches, arising as a consequence of base misincorporation during DNA replication, display context dependency with respect to helix stability [SantaLucia and Hicks, 2004] and this strongly influences the local sequence bias exhibited by the resulting mutations [Nakken et al., 2010]. Local DNA flexibility is also known to be capable of modulating the efficiency of the enzymes involved in both base excision repair and mismatch repair [Seibert et al., 2002; Seibert et al., 2003; Wang et al., 2003; Isaacs and Spielmann, 2004] and this flexibility is itself sequence-dependent [Geggier and Vologodskii, 2010; Peters and Maher, 2010]. Finally, DNA repair efficiency may be influenced by nucleosome positioning [Ying et al., 2010] which is also DNA sequence-dependent [Chung and Vingron, 2009; Cui and Zhurkin, 2010; Wu et al., 2010]. Thus, a variety of different properties of a given DNA sequence and its structure are likely to impact on the inherent mutability of that sequence and the efficiency with which mutations arising are subsequently repaired.
Gene Conversion
Gene conversion occurs during homologous recombination and refers to the unidirectional transfer of genetic material from a ‘donor’ sequence to a highly homologous ‘acceptor’ [reviewed in Chen et al., 2007]. It can affect paralogous sequences (nonallelic homologous gene conversion) or different alleles at a given locus. Gene conversion appears to be most efficient when the sequences involved share homology over a range between 295 bp and 1 kb, but efficiency tails off rapidly if the length of the homologous stretch is less than 200 bp [Liskay et al., 1987]. The suggestion has also been made that gene conversion occurs optimally when the homology of the paralogous sequences involved exceeds 92% [Wolf et al., 2009].
A variety of DNA sequences, including direct repeats, inverted repeats, minisatellite repeats, the χ recombination hotspot and alternating purine–pyrimidine tracts with Z-DNA-forming potential have frequently been noted in association with gene conversion events in human genes indicative of the sequence-directed nature of this mutational mechanism [see Chuzhanova et al., 2009]. These somewhat anecdotal findings have recently been formalized by a methodical statistically-based analysis of 27 well-characterized human gene conversion mutations [Chuzhanova et al., 2009]. The lengths of the maximal converted tracts (MaxCTs) associated with these pathogenic gene conversions tended to be fairly short, rarely exceeding 1 kb. In silico analysis of the DNA sequence tracts involved in the 27 non-overlapping pathogenic gene conversion events in 19 different genes yielded several novel findings [Chuzhanova et al., 2009]. First, gene conversion events tended to occur preferentially within (C+G)- and CpG-rich regions. Second, sequences with the potential to form non-B DNA structures were found to occur disproportionately within MaxCTs and/or short flanking regions. Third, MaxCTs were enriched in several sequence motifs including a truncated version of the χ element (a TGGTGG motif) and the classical meiotic recombination hotspot, CCTCCCCT. Finally, there was a tendency for gene conversion to occur in genomic regions that had the potential to fold into stable hairpin conformations [Chuzhanova et al., 2009].
Another important aspect of this topic that is relevant to our brief relates to the phenomenon of biased gene conversion (BGC). Gene conversion is said to be biased if one of the two DNA molecules involved in the gene conversion event is more likely than the other to be the donor. In the case of allelic gene conversion, BGC leads to an excess of the ‘favoured’ allele in the pool of gametes and therefore tends to increase the frequency of this allele in the general population. Analysis of polymorphism and nucleotide substitution patterns in primate genomes has provided firm evidence for the action of BGC favouring GC alleles over AT alleles, i.e. the derived allele frequency of AT>GC mutations is higher than that of GC>AT mutations [Duret and Galtier, 2009; Clément and Arndt, 2011]. Recently, it has been shown that the spectrum of missense polymorphisms in human populations exhibits the footprints of GC-favoured BGC [Necşulea et al., 2011]. This pattern cannot be explained in terms of selection and is evident with all nonsynonymous mutations, including those implicated in human genetic disease. Necşulea et al. [2011] have speculated that the genes most likely to be influenced by this effect will be those that are AT-rich (i.e. those genes for which the opportunities for AT>GC mutations are maximized) and which coincide with recombination hotspots, “an additional argument for these hotspots being an Achilles’ heel of the human genome”.
Microdeletions, Microinsertions and Indels
“Just as the constant increase of entropy is the basic law of the universe, so it is the basic law of life to be ever more highly structured and to struggle against entropy”.
Václav Havel
The sequence context of microdeletions and microinsertions (<21 bp in length) causing human genetic disease was studied by Ball et al. [2005] who analysed a total of 3,767 microdeletions (from 426 genes) and 1,960 microinsertions (from 307 genes). Deletions of 1 bp were the most common type of microdeletion analyzed (48% of the total) while 2,815 microdeletions (75% of the total) were between 1 and 3 bp in length. Of the 3,144 microdeletions located within coding regions, 2,758 (88%) were of a length that was not a multiple of three and hence would be expected to alter the reading frame. Some 45% of microdeletions led to the removal of a repeated sequence, an event termed “deduplication” by Kondrashov and Rogozin [2004] in order to highlight the identity of the deleted sequence and the sequence abutting the site of deletion; Kondrashov and Rogozin [2004] observed a deduplication frequency of 66%. In the study of Ball et al. [2005], the proportion of deduplications decreased with increasing length of the deletion. For deletions of 2–5 bp, 38% were found to be deduplications whereas for deletions of ≥6 bp it was only 3%. By contrast, some 85% of microinsertions represented duplications of sequence bordering the site of mutation, comparable to the 81% reported by Kondrashov and Rogozin [2004]; this proportion was independent of the length of the insertion. Ball et al. [2005] reported that 1 bp constituted by far the most common length of microinsertion, with 66% of the total being of this size. As with microdeletions, the distribution was somewhat skewed, with some 1,571 microinsertions (80%) being between 1 and 3 bp in length. Of the 1,660 microinsertions located within gene coding regions, 1,556 (94%) were of a length that was not a multiple of three, and which would therefore be expected to alter the reading frame. Comparable results have been reported from extensive surveys of microinsertion/microdeletion polymorphisms in the human genome [Mills et al., 2006; Tan and Li, 2006].
Ball et al. [2005] found that many of the lesions of >1 bp were potentially explicable in terms of slippage mutagenesis, and involved the addition or removal of one copy of a mono-, di-, or tri-nucleotide repeat. Various sequence motifs were found to be over-represented in the vicinity of both microinsertions and microdeletions, including the heptanucleotide CCCCCTG that shares homology with the complement of the 8-bp human minisatellite conserved sequence/χ-like element (GCWGGWGG) [Ball et al., 2005]. The previously reported indel hotspot GTAAGT [Chuzhanova et al., 2003a] and its complement ACTTAC were also found to be overrepresented in the vicinity of both microinsertions and microdeletions, thereby providing a first example of a mutational hotspot that is common to different types of gene lesion. Other motifs overrepresented in the vicinity of microdeletions and microinsertions included DNA polymerase pause sites and topoisomerase cleavage sites [Ball et al., 2005]. Analysis of DNA sequence complexity also demonstrated that a combination of slipped mispairing mediated by direct repeats, and secondary structure formation promoted by symmetric elements, can account for the majority of documented microdeletions and microinsertions [Ball et al., 2005]. Thus, microinsertions and microdeletions exhibit strong similarities in terms of the characteristics of their flanking DNA sequences, implying that they are generated by very similar underlying mechanisms.
Once again, replication slippage is the key to understanding the genesis of microdeletions and microinsertions. Replication slippage involves DNA polymerase pausing at a direct repeat sequence, enzyme dissociation, reannealing of the polymerase to a second direct repeat copy in the vicinity to generate a misaligned intermediate, followed by resumption of DNA replication [Kunkel, 2004; Garcia-Diaz et al., 2006]. In vitro studies have shown that the fidelity of DNA replication is strongly dependent upon both the local DNA sequence environment and the type of DNA polymerase involved [Kunkel, 2004; Loeb and Monat, 2008]. Further, different DNA polymerases appear to be characterized by subtly different types of misalignment mutagenesis during DNA replication/repair, giving rise to different types of lesion [Eckert et al., 2002; Wolfle et al., 2003; Tippin et al., 2004; Zhang and Dianov, 2005; Arana et al., 2007; Lyons and O’Brien, 2010]. The considerable explanatory value of these studies for slippage-mediated mutagenesis in vivo is evidenced by the concordance noted between in vitro and in vivo mutational spectra [Muniappan and Thilly, 2002]. Further, it has long been recognized that mononucleotide tracts are hotspots for microinsertions and microdeletions causing human genetic disease [Kondrashov and Rogozin, 2004; Truong et al., 2010; Ivanov et al., 2011] while Ball et al. [2005] noted that oligonucleotides of 5–7 bp that were overrepresented in the vicinity of both microdeletions and microinsertions frequently contain A, C, or G mononucleotide tracts of 4–7 bp.
In his study of inherited mutations in a total of 20 human genes, Kondrashov [2003] reported a strong correlation between the rates of microdeletion and microinsertion. Such a correlation was also evident for the much larger number of genes examined by Ball et al. [2005]. The observation that the propensity of a given gene to undergo microdeletion is related to its propensity to undergo microinsertion could be a consequence of the presence of certain DNA sequences that are prone to both types of lesion [Truong et al., 2010]. Consistent with this view, Ball et al. [2005] reported strong similarities between microinsertions and microdeletions in terms of the sequence characteristics and repetitivity of the flanking DNA sequence, the overrepresentation of motifs known to play a role in recombination, mutation, cleavage, and rearrangement, and the likely involvement of various types of repetitive sequence element in the mutational mechanism. Similar conclusions have been drawn from studies of microdeletions and microinsertions identified in an evolutionary context [Zhang and Gerstein, 2003; Taylor et al., 2004; Messer and Arndt, 2007; Tanay and Siggia, 2008; Kvikstad et al., 2009; Sjödin et al., 2010]. Taken together, these results are consistent with the view that microdeletions and microinsertions are generated by very similar sequence-directed molecular mechanisms. The observation, noted above, that a GTAAGT hotspot of indel formation [Chuzhanova et al., 2003a] is significantly overrepresented in the vicinity of both microdeletions and microinsertions [Ball et al., 2005; Ivanov et al., 2011] suggests that some sequence motifs may represent hotspots for different types of mutation. Any such model, perhaps involving the repeat-mediated formation and resolution of secondary structure intermediates, would be most satisfying in that it could serve to mechanistically unify the various different types of microrearrangement described in human genes.
Structural Variation Including Copy Number Variants
“The distribution of break points in human chromosomes….is non-random with seemingly preferential breakages in negative band areas in terms of Giemsa banding. The determination of ‘hot-spots’ for breakage in the human genome may help us in….investigating the cause or causes which give rise to some of these abnormalities”.
C.W. Yu, D.S. Borgaonkar & D.R. Bolling (1978) Hum. Hered.28:210-225.
Structural variation of the human genome is characterized by a variety of different types of gross rearrangement including deletions, duplications, insertions (termed Copy Number Variants, CNVs) as well as inversions and translocations. Four major mutational mechanisms account for these structural variants (SVs): nonallelic homologous recombination, non-homologous end joining, replication-based mechanisms and L1-retrotransposition (Fig. 1) [Conrad et al., 2010; Kidd et al., 2010; Mills et al., 2011]. In what follows, we shall describe some well-studied examples of structural variation in the human genome, with an emphasis on disease-associated SVs as well as gross chromosomal aberrations such as translocations and isochromosomes that illustrate the sequence-directed nature of the above mentioned mutational mechanisms.
Figure 1.

Mutational mechanisms leading to gross genomic rearrangements (structural variants) including copy number variations. Non-homologous end joining (NHEJ) comprises two sub-pathways, classical or canonical NHEJ (C-NHEJ) and alternative NHEJ (A-NHEJ). In practice, some clinically observed mutations represent the end result of an untraceable in vivo mutational process and can often be explained by two or even three different mechanisms. Hence, it is often not possible to distinguish a break-induced replication (BIR) event from a non-allelic homologous recombination (NAHR) event, a microhomology-mediated BIR (MMBIR) event from a serial replication slippage (SRS) or fork stalling and template switch (FoSTeS) event, and a NHEJ event (where microhomology-mediated) from a ‘microhomology-mediated replication-dependent recombination (MMRDR) event. By contrast, mutations resulting from telomere healing or L1 retrotransposition can usually be unequivocally attributed owing to the presence of signature sequences. DSB, double-strand break; GC, gene conversion; RC, replication fork; RS, replication slippage; SRS, serial replication slippage; SSA, single-strand annealing.
Nonallelic Homologous Recombination (NAHR)
Sequence analysis of the breakpoints of 1,054 SVs identified in the genomes of 17 healthy human individuals of different geographical origins indicated that NAHR accounts for 22.5% of insertions and deletions, as well as 69.1% of the inversions identified [Kidd et al., 2010; Fig. 2]. The majority of SVs identified in this study are likely to represent more or less neutral polymorphisms but at least 1% are estimated to be disease-associated. Interestingly, some of the SVs that segregate as polymorphisms within the normal population predispose to further structural changes such as disease-associated deletions and duplications [Antonacci et al., 2010; Ciccone et al., 2006; Giglio et al., 2002; Gimelli et al., 2003; Hobart et al., 2010; Osborne et al., 2001; Visser et al., 2005]. Thus, for example, heterozygosity for a ~970 kb inversion polymorphism of the MAPT locus [MIM# 157140] at 17q21.3 predisposes to the NAHR events that underlie the 17q21.31 microdeletion syndrome [MIM# 610443; Antonacci et al., 2009; Koolen et al., 2006; Koolen et al., 2008; Rao et al., 2010; Shaw-Smith et al., 2006]. The most likely explanation for this phenomenon is that inversion heterozygosity perturbs the pairing of homologous chromosomes during meiosis, which then promotes interchromosomal NAHR between the inversion-flanking low copy repeats (LCRs) thereby giving rise to the 17q21.3 microdeletion.
Figure 2.
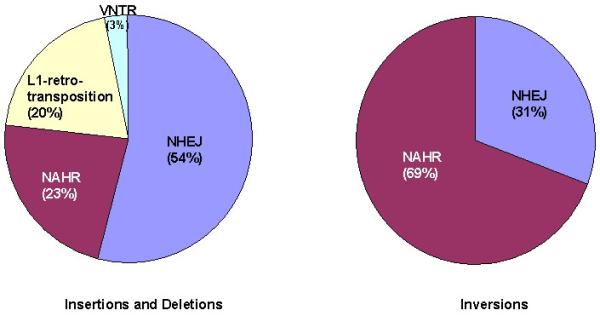
Contribution of mutational mechanisms to the formation of structural variants (SVs) <5kb according to Kidd et al. [2010]. The contraction or expansion of variable number of tandem repeats (VNTRs) accounts for ~3% of the detected SVs.
NAHR-mediated SVs are not randomly distributed across the human genome but rather are frequently located within complex regions that are enriched with segmental duplications. NAHR between segmental duplications not only causes submicroscopic CNVs giving rise to microdeletion and microduplication syndromes [reviewed by Guo et al., 2008; Stankiewicz and Lupski, 2010], but is also involved in the generation of cytogenetically visible chromosomal aberrations including isodicentric chromosomes and translocations. Thus, the isodicentric Xp11 chromosomes responsible for Turner syndrome do not simply occur at random but instead are mediated by NAHR between large inverted repeats comprising repetitive gene clusters and segmental duplications, which themselves correspond to regions of CNV [Scott et al., 2010; Koumbaris et al., 2011]. Recent findings also indicate that NAHR represents a major mechanism underlying unbalanced recurrent translocations, which are mediated either by interchromosomal LCRs or segmental duplications located on non-homologous chromosomes [Ou et al., 2011]. Regions containing highly redundant gene duplications such as those involving the olfactory receptor multigene family, located in the subtelomeric regions of human chromosomes, appear to be particularly prone to mediate interchromosomal NAHR causing recurrent translocations. These findings serve to emphasize the point that segmental duplications or LCRs are ubiquitous ‘soft spots’ in the human genome that have the potential to mediate SVs and other chromosomal rearrangements such as translocations. Clearly, not all LCRs are prone to undergo recurrent NAHR events; as deduced from LCRs that are known to be involved in recurrent pathogenic large deletions and duplications, the sequence requirements for LCRs to be frequently involved in mediating genomic instability include >95% sequence identity, >10 kb of LCR length and a distance between the LCRs of 50 kb-10 Mb [Bailey et al., 2002]. Based upon these criteria, a map of potential ‘rearrangement hotspots’ in the human genome has been defined and some of these predicted hotspots have already been found to be prone to recurrent disease-associated SVs [Mefford et al., 2007, 2008; Sharp et al., 2006, 2008; Shaw-Smith et al., 2006; Ou et al., 2011]. It should be kept in mind that LCRs are themselves non-randomly distributed at the chromosomal level [Bailey and Eichler, 2006; Marques-Bonet and Eichler, 2009].
Pathogenic NAHR and normal meiotic AHR (allelic homologous recombination) appear to have similar sequence requirements, as suggested by the spatial coincidence of AHR and meiotic NAHR hotspots [Lindsay et al., 2006; De Raedt et al., 2006]. This view is supported by the observation that the 13-bp sequence motif CCNCCNTNNCCNC, located within 40% of AHR hotspots, is also present in the NAHR hotspots that mediate CNVs [Myers et al., 2008]. PRDM9, a meiosis-specific protein which contains zinc finger arrays, binds to this motif and targets the initiation of recombination to specific locations (hotspots) in the genome [Baudat et al., 2010; Berg et al., 2010; Paranov et al., 2010]. Genetic variation at the PRDM9 locus has been shown to exert a powerful effect on recombination hotspot activity in sperm. Further, subtle changes within the zinc finger array serve to create hotspot-non-activating or -enhancing variants, suggesting that PRDM9 is a major regulator of AHR hotspot activity in the human genome [Berg et al., 2010]. Importantly, genetic variation at the PRDM9 locus [MIM# 609760] also influences NAHR activity as is evident in the context of the Charcot-Marie-Tooth type 1A-repeat (CMT1A-REP)-mediated duplications and deletions at 17p11.2 [MIM# 118220]; in the sperm of healthy donors homozygous for the A allele of PRDM9, de novo rearrangements between the CMT1A-REPs were observed >20-fold more frequently than in individuals homozygous for non-A alleles [Berg et al., 2010]. Taken together, these findings indicate that the locations of meiotic NAHR hotspots are not only determined by highly homologous target sequences but also by specific DNA sequence motifs and the proteins (such as PRDM9) which bind to them so as to perform their functions as trans-regulators of meiotic recombination.
Disease-associated NAHR also occurs in mitotic cells [reviewed by Moynahan and Jasin, 2010]. Although both meiotic NAHR and mitotic NAHR may be mediated by the same pairs of LCRs [Carvalho and Lupski, 2008; Messiaen et al., 2011], they are very likely to differ in terms of the underlying determinants for DSB formation, since SPO11 and other recombination initiating factors are expressed exclusively in meiotic cells [Shannon et al., 1999; Hayashi et al., 2005]. This is consistent with the observation that mitotic NAHR events causing large deletions of the NF1 gene region do not cluster in highly localized hotspots that are limited to a few hundred base-pairs, in contrast to the majority of NF1 deletions which are caused by meiotic NAHR [De Raedt et al., 2006; Roehl et al., 2010]. The observed properties of type-1 NF1 deletions are largely consistent with the finding that certain NAHR hotspots predominate during meiosis and are found only rarely (or not at all) during mitosis [Messiaen et al., 2011; Turner et al., 2008]. Breakpoint regions of structural variants generated by meiotic NAHR events have been previously found to be (i) biased toward GC-rich regions and (ii) to manifest higher DNA helix stability and lower DNA flexibility as compared with rearrangements caused by NHEJ [Lam et al., 2010; Lopez-Correa et al., 2001; Visser et al., 2005]. Interestingly, both the DNA stability and GC content have been found to be significantly higher in the PRS1 and PRS2 meiotic NAHR hotspots causing type-1 NF1 deletions than in the breakpoint regions of the mitotic type-2 NF1 deletions [Roehl et al., 2010]. However, in passing, we should point out that mitotic NAHR-mediated deletions also appear to be sequence-directed since short repeats capable of forming non-B DNA structures have been found to be over-represented within the breakpoint regions of mitotic type-2 NF1 deletions [Roehl et al., 2010].
Non-Homologous End Joining (NHEJ)
The defining characteristic of NHEJ (Fig. 1) is the ligation of DSB ends without the requirement for extensive homology, in stark contrast to the mechanism of homologous recombination. The presence of terminal microhomologies (typically 1-3 bp) facilitates canonical NHEJ (C-NHEJ) but this appears not to be an absolute requirement for it to occur. C-NHEJ of ends from simultaneous DSBs accounts for a diverse range of genomic rearrangements [Chen et al., 2010; Kidd et al., 2010].
Increasing evidence has emerged to support the view that when the core C-NHEJ factors (i.e. Ku and/or DNA ligase IV-XRCC4) are absent, DSB ends can still be repaired by NHEJ. This latter pathway, originally termed microhomology-mediated end joining (MMEJ) is now commonly known as alternative NHEJ (A-NHEJ) [Boboila et al., 2010; Fattah et al., 2010; Helmink et al., 2011; Lee-Theilen et al., 2011; Simsek and Jasin, 2010; Yan et al., 2007; Zhang and Jasin, 2011]. The process of A-NHEJ is presumed to involve a 5′ to 3′ end resection of DNA DSB(s), thereby exposing microhomologies between the resulting two 3′ single-strand DNA tails; subsequent annealing at the region of microhomology followed by 3′-flap removal and gap filling then gives rise to deletions or translocations [Lee-Theilen et al., 2011; Zhang and Jasin, 2011]. As compared with C-NHEJ, A-NHEJ is inherently more prone to generate large genomic rearrangements, particularly translocations [Boboila et al., 2010; Fan et al., 2010; Helmink et al., 2011; Simsek and Jasin, 2010; Yan et al., 2007]. Approximately 30-50% of all structural variants in the human genome have originated through microhomology–mediated NHEJ events [Conrad et al., 2010; Kidd et al., 2010].
Although some NHEJ events will have resulted from the repair of DSBs that originated quasi-randomly, there are also many well documented cases in which the location of the NHEJ-initiating DSBs appears to be highly dependent upon the local DNA sequence environment. The role of the local DNA sequence context in generating NHEJ-mediated germline mutations is exemplified by the constitutional t(11;22), the most common type of recurrent non-Robertsonian translocation in humans. The breakpoint sequences of both chromosomes are characterized by several hundred base-pairs of inverted AT-rich repeats; similar sequences have also been identified at the breakpoints of other non-recurrent translocations [Kehrer-Sawatzki et al., 1997; Kurahashi et al., 2010; Rhodes et al., 1997]. Evidently, NHEJ of two ends from different DSBs requires such ends to be physically located in the immediate vicinity. In mammalian cells, high-precision tracking of tagged broken chromosome ends indicates that these ends can only partially separate and, consequently, DSBs preferentially undergo translocations with those chromosomes with whom they share nuclear space [Soutoglou et al., 2007; Wijchers and de Laat, 2011]. This provides strong support for the ‘contact-first’ hypothesis, which proposes that interactions between different DSBs can only take place if they are colocalized at the time of DNA damage [Nikiforova et al., 2000]. Consistent with this hypothesis, close spatial proximity has been observed between several frequent translocation partners [reviewed by Meaburn et al., 2007; Wijchers and de Laat, 2011].
A meta-analysis of germ-line and somatic DNA breakpoint junction sequences derived from a total of 219 different rearrangements (most of which are likely to be NHEJ events) underlying human inherited disease and cancer allowed the first methodical examination of the local DNA sequence environment of translocation and deletion breakpoints across a wide variety of different gene loci [Abeysinghe et al., 2003; Chuzhanova et al., 2003b]. A number of recombination-predisposing motifs and non-B DNA-forming sequences were found to be overrepresented at these breakpoints as compared with randomly selected control sequences, indicative of the sequence-directed nature of many NHEJ mediated rearrangements.
It has been observed that at least one of the breakpoints of NHEJ-mediated rearrangements is often located within repetitive elements (such as LTRs, LINE or Alu elements) and sequence motifs capable of causing DSBs have been frequently identified in the vicinity of the breakpoints of these NHEJ-mediated rearrangemetns [Inoue et al., 2002; Kehrer-Sawatzki et al., 2005, 2008; Nobile et al., 2002; Oshima et al., 2009; Shaw and Lupski, 2005; Stankiewicz et al., 2003; Toffolatti et al., 2002; Vissers et al., 2009; Yatsenko et al., 2009]. Importantly, the breakpoints of many non-recurrent CNVs mediated by NHEJ map to LCRs [Carvalho et al., 2009; Stankiewicz et al., 2003; Kehrer-Sawatzki et al., 2005, 2008; Shaw and Lupski, 2005; Zhang et al., 2010] suggesting that LCRs can promote genomic instability by inducing certain chromatin secondary structures thereby alleviating NHEJ-mediated rearrangement.
Replication-based Mechanisms
Replication slippage or template switching during replication account for both small and large deletions and duplications with terminal microhomologies (Fig. 1). Recently, relevant replication-based models including serial replication slippage (SRS) [Chen et al., 2005a; Chen et al., 2005b; Chen et al., 2005c], fork stalling and template switching (FoSTes) [Lee et al., 2007] and microhomology-mediated break-induced replication (MMBIR) [Hastings et al., 2009], which were collectively termed microhomology-mediated replication-dependent recombination (MMRDR) by Chen et al. [2010], have been used to explain the generation of a diverse range of complex genomic rearrangements [Bauters et al., 2008; Carvalho et al., 2009; Chauvin et al., 2009; Collie et al., 2010; Koumbaris et al., 2011; Sheen et al., 2007; Vissers et al., 2009; Zhang et al., 2009, 2010].
For example, DNA replication stalling-induced chromosome breakage has turned out to be an important mechanism causing deletions at chromosomal ends. Different types of telomeric deletions have been described (Fig. 3) [Kulikowski et al., 2010]: type A terminal deletions are formed by chromosomal ends that are stabilized by the capture of a telomere from another source, whereas type B deletions are actually interstitial deletions towards the chromosomal ends. By contrast, type C deletions describe the process by which chromosomal ends are stabilized by telomere healing, namely the telomerase-dependent de novo addition of telomeres at non-telomeric sites. Terminal deletions associated with inverted duplications [Zuffardi et al., 2009] can be classified as either type A or type C. Recently, Hannes et al. [2010] succeeded in cloning the breakpoints of nine chromosome 4p terminal deletions. All nine cases were shown to be type C terminal deletions. Bioinformatics analysis of the breakpoint-flanking regions involved in these nine cases, together with 12 previously fully characterized type C terminal deletions, led to the realization that there is an enrichment in secondary structure-forming sequences and replication stalling site motifs in these regions as compared with a randomly selected sequence dataset [Hannes et al., 2010].
Figure 3.

Schematic representation of the different types of cytogenetically defined terminal deletions in terms of end stabilization. Whereas blocks indicate telomeres, filled circles indicate centromeres. In type A, the captured telomere and associated sequence are highlighted in blue. In type B, an internal portion of the chromosome (sequence highlighted in red in the normal chromosome) is deleted. In type C, the de novo telomere is highlighted in purple. In the box, two different mechanisms of end stabilization for terminal deletions associated with inverted duplications (indicated by facing arrows) are illustrated; the use of colour is consistent with the scheme used for types A and C.
Certain sequence features, such as microsatellites and transposon-rich regions, can serve to induce replication stalling, thereby acting as potential sources of genome instability [e.g. Cha and Kleckner, 2002; Pelletier et al., 2003]. On this basis, Koszul and colleagues [2004] proposed a two-step mechanism to account for the generation of large segmental duplications: “First, a replication fork pauses and collapses generating a chromosome breakage. Second, the double-strand break can be processed into a new replication fork either intra- or inter-molecularly by a break-induced replication-like mechanism that does not necessarily need a long sequence homology”. It was this ‘microhomology-dependent BIR’ model (Fig. 1) that was subsequently deployed to explain disease-causing copy number mutations. In MMBIR, replication ends with the engagement of a misaligned template instead of reannealing to its original template; the synthesis of the second strand then follows the synthesis of the first [reviewed in Chen et al., 2010]. In practice, mutations due to SRS/FoSTes are often indistinguishable from those due to MMBIR. Indeed, the two terms have sometimes been used interchangeably [e.g. Choi et al., 2011; Zhang et al., 2009].
All the replication-based models recently proposed to account for the formation of structural variants and/or mutations in the human genome stress the importance of genomic architectural elements such as palindromic DNA, stem-loop structures, repeats etc, features which may facilitate the initial stalling of the replication fork [Gu et al., 2008; Chen et al., 2010].
Process and effect of retrotransposition in relation to local sequence context and mutation L1 Retrotransposition
L1 elements comprise ~17% of the human reference genome sequence [Lander et al., 2001]. Retrotranspositionally competent L1 elements are typically ~6.0 kb in length and comprise a 5′-untranslated region (UTR), two non-overlapping open reading frames (ORF1 and ORF2), a short 3′-UTR, and a poly(A) tail. Whereas ORF1 encodes an RNA-binding protein, ORF2 encodes a protein with endonuclease (L1 EN) and reverse transcriptase (L1 RT) activities. L1 retrotransposition is thought to occur by target site-primed reverse transcription; briefly, it would appear that the L1 EN cleaves genomic DNA at a degenerate consensus target sequence (3′-A/TTTT-5′ and variants thereof), thereby freeing up a 3′-OH group that then serves as a primer for the reverse transcription of L1 RNA by L1 RT. The nascent L1 cDNA then recombines with genomic DNA, generating in the process the characteristic hallmarks of L1 retrotransposition such as 5′ truncations, a 3′ poly(A) tail and target site duplications (TSDs) of variable length [Cordaux and Batzer, 2009; Kazazian, 2004]. L1 retrotransposition requires a precise interplay between ORF1p, ORF2p, and L1 RNA [Doucet et al., 2010].
Of the >500,000 L1 copies in the reference human genome, only 80–100 are believed to be capable of active retrotransposition [Brouha et al., 2003]. Recent studies have however revealed that (i) the actual number of highly active or “hot” L1s in the human population is much higher than that identified in the reference human genome [Beck et al., 2010], and (ii) L1 retrotransposition has played a more important role in generating structural variation in the human genome than previously appreciated [Ewing and Kazazian, 2010; Huang et al., 2010; Iskow et al., 2010; Xing et al., 2009]. The rate of L1 retrotransposition in humans has been estimated by one study to be one insertion in every 108 births [Huang et al., 2010] and between 1/95 and 1/270 births by another [Ewing and Kazazian, 2010]. The number of dimorphic L1 elements in the human population with allele frequencies >0.05 is estimated to be between 3,000 and 10,000 [Ewing and Kazazian, 2010], far exceeding the ~400 human L1 retrotransposon insertion polymorphisms (RIPs) registered in dbRIP [Wang et al., 2006a].
L1 retrotransposition can affect the primary structure of the human genome in a variety of ways other than by simple self-insertion. For example, L1 elements are also able to mobilize non-autonomous sequences in trans, including repetitive Alu sequences, SVA (short interspersed nucleotide elements-R, variable-number-of-tandem-repeats, and Alu) elements, and processed pseudogenes [Cordaux and Batzer, 2009; Kazazian, 2004; Konkel and Batzer, 2010] (Fig. 1). In addition, L1 retrotransposition can give rise large genomic deletions [Callinan et al., 2005; Han et al., 2005; Xing et al., 2009]. L1 elements can also undergo retrotransposition in the germline [Ostertag et al., 2002], during early embryonic development [Garcia-Perez et al., 2007; Garcia-Perez et al., 2010; Kano et al., 2009; van den Hurk et al., 2007], in certain somatic cells [Coufal et al., 2009; Muotri et al., 2005] and in the human lung cancer genome [Iskow et al., 2010].
L1 retrotransposition can also give rise to human inherited disease. Since the first report of Kazazian et al. [1988], L1-mediated simple L1, Alu and SVA insertions have been increasingly reported to cause inherited disease [see Chen et al., 2005d for publications prior to 2005 and subsequently, Apoil et al., 2007; Bochukova et al., 2009; Bouchet et al., 2007; Chen et al., 2008; Gallus et al., 2010; Musova et al., 2006]. Following our own retrospective identification of pathogenic large genomic deletions caused by LI-mediated Alu insertions [Chen et al., 2005d], pathogenic large genomic deletions caused by L1-mediated L1 [Miné et al., 2007; Morisada et al., 2010], a number of Alu [Okubo et al., 2007; Schollen et al., 2007] and SVA [Takasu et al., 2007] insertions have been reported in prospective screens while the first cases of L1-driven pseudogene insertion causing human genetic disease have also been reported [Awano et al., 2010; Tabata et al., 2008].
The non-random insertion of L1-mediated retrotranspositional elements into the human genome can be considered at two distinct levels. First, consistent with the known target site specificity for L1 EN, the study of pre-insertion sites of de novo L1 insertions in cultured human cancer cells revealed an AT-rich bias in the 50 bp flanking the insertion sites [Gasior et al., 2007]. The genome-wide profiling of human L1(Ta) retrotransposons has also revealed a tendency for L1(Ta)s to accumulate within AT-rich regions [Huang et al., 2010]. L1(Ta) (transcribed L1, subset a) is the youngest L1 family that is currently capable of active retrotransposition, and hence the L1 family that is largely responsible for generating L1 insertion (presence/absence) polymorphisms in the human genome. In addition, the currently reported pathogenic L1-mediated events have almost invariably integrated at L1 EN consensus target sites. Second, in the abovementioned study of pre-insertion sites of de novo L1 insertion in cultured human cancer cells, a statistically significant cluster of such insertions was localized in the vicinity of the c-myc gene (MYC; MIM# 190080). This finding suggested that in addition to the local sequence determinants (i.e. L1 EN target sites), other features of the flanking genomic region may also influence the insertion preference of L1-mediated insertions [Gasior et al., 2007]. Apparent insertion clusters have also been observed in the context of pathogenic L1-mediated events. Thus, three independent Alu insertions have been found to be integrated into a 104 bp region of the FGFR2 gene [MIM# 176943; Bochukova et al., 2009; Oldridge et al., 1999] while two independent L1 insertions have been reported to have inserted into exon 44 of the dystrophin gene (DMD; MIM# 300377) within an 89 bp region [Musova et al., 2006; Narita et al., 1993].
The above notwithstanding, the most striking finding pertinent to the non-random nature of L1 retrotranspositional insertion is that independent L1 retrotransposition elements can integrate at precisely the same chromosomal sites [Chen et al., 2005d]. Thus, an L1 element and an Alu sequence are known to have become inserted at exactly the same location in the APC gene [MIM# 611731] in two unrelated individuals [Halling et al., 1999; Miki et al., 1992]; whilst the L1 element was a somatic insertion, the Alu sequence was a germline insertion. In addition, two markedly different Alu Ya5a2 elements have become integrated at precisely the same site in the F9 gene [MIM# 300746] causing severe hemophilia B [Vidaud et al., 1993; Wulff et al., 2000]. Finally, an SVA element and an Alu sequence have inserted at the same site within the coding region of the BTK gene [MIM# 300300; Conley et al., 2005]. These observations are consistent with some genomic locations being exquisitely prone to L1 retrotransposition [Chen et al., 2005d].
Alu-mediated Recombination (AMR)
A canonical Alu element is about 300 base-pairs long, comprising two related GC-rich monomers separated by an A-rich linker region and ending with a poly(A) tail [Cordaux and Batzer, 2009]. Owing to the high frequency (>1 million copies) of complete or partial Alu elements in the human reference genome (~10.6% of the genome sequence) [Lander et al., 2001], they serve as a huge reservoir of sequences for homology-based recombination. AMR between nonallelic sequences is also a frequent cause of human genetic disease as evidenced by the many recently described examples [e.g. Abo-Dalo et al., 2010; Champion et al., 2010; Cozar et al., 2011; Gentsch et al., 2010; Goldmann et al., 2010; Franke et al., 2009; Resta et al., 2010; Shlien et al., 2010; Tuohy et al., 2010; Yang et al., 2010; Zhang et al., 2010].
The importance of Alu elements in the context of mediating genomic deletions is unlikely owing simply to their sheer abundance in the human genome. In other words, Alu elements themselves must possess inherent recombination-predisposing properties [Rudiger et al., 1995]. A survey of a small subset (n = 36) of Alu-mediated rearrangements in several human genes identified a 26 bp core sequence that is often located at or close to the sites of recombination [Rudiger et al., 1995]. Importantly, this core sequence contains the pentanucleotide motif CCAGC, which represents a truncated version of the χ recombination hotspot (consensus sequence: 5′-GCTGGTGG-3′ or its complement, 5′-CCACCAGC) [Kenter and Birshtein, 1981; Smith, 1983]. This is likely to have had important implications with respect to many of the subsequently found AMR-mediated pathogenic deletions. In the absence of any meta-analysis or systematic review, we shall mention only two studies, to which some of us contributed. The first study reported a gross HFE [MIM# 613609] deletion consistent with AMR; the 17 bp crossover region contained two sequence motifs, CCACCA and CCAGC, both truncated versions of the χ recombination hotspot [Le Gac et al., 2008]. It should be noted that CCACCA has also been noted to be a mutational ‘super-hotspot’ common to microdeletions, microinsertions, and indels [Ball et al., 2005]. The second study reported, among others, a 2,769 bp SERPINC1 [MIM# 107300] deletion mediated by Alu elements. The 13 bp crossover region (i.e. GCCACCACGCCCG) was also found to contain the CCACCA mutational ‘super-hotspot’ [Picard et al., 2010]. In passing, it should also be appreciated that Alu elements are particularly prone to form non-B DNA structures (e.g. slipped structures) owing to their containing two related GC-rich monomers (Fig. 4).
Figure 4.

Non-B DNA. Types and ribbon models of the most common non-B DNA conformations formed by repetitive DNA motifs, followed by the general sequence requirements for each structure and examples of DNA sequences (redrawn from Bacolla et al., 2004). Y, pyrimidine (C or T); R, purine (A or G). It should however be noted that, for Z-DNA at least, one strand must contain alternating G residues; x and y, any intervening sequence separating the repeats by typically 0-5 nt [Cer et al., 2011].
Microsatellite Mutation
“The distribution of interspersed repeats close to and even within genes has brought the mechanism of their mutation into the arena of human molecular genetics. These sequences have a unique form of mutation: variation in copy number. The rate of the mutation is related to the copy number, and therefore, the mutability of the product of a change in copy number is different from that of its predecessor. For this reason, we have termed this mechanism dynamic mutation.”
R.I. Richards & G.R. Sutherland (1992) Cell 70:709-712.
Microsatellites, defined as the repetition of short (1-6 bp) DNA tandem motifs, display somewhat higher (yet individually distinct) mutation rates than the average nucleotide substitution rate genome-wide. Microsatellites comprise ~3% of the human genome [Lander et al., 2001] and the proportion of mononucleotide and dinucleotide repeat tracts that display length polymorphism in the human population (involving the generation of multiple alleles) has been found to increase almost exponentially above a length threshold of ~10 nt [Kelkar et al., 2010]. In similar vein, analyses of length polymorphisms of trinucleotide sequences in several human transcriptomes, assessed from the coding portions of RefSeq genes annotated in the human reference genome, have revealed the existence of multiple length difference alleles above ~25 bp, as opposed to essentially only two alleles for shorter (<~16 bp) tracts [Molla et al., 2009]. With an estimated average mutation rate of 10−5, microsatellites accumulate mutations at a rate three orders of magnitude higher than the average rate of nucleotide substitutions genome-wide (2 × 10−8) [Molla et al., 2009].
In addition to length changes, single base changes within microsatellites also occur at higher frequencies than the genome-wide average, not only within the microsatellite repeats but also in the bases adjacent to the repeats [Siddle et al., 2011]. From the analysis of 1000 Genomes Project pilot data, variability genome-wide has been noted to be at its maximum (accounting for 42.5% and 28%, respectively) at the dinucleotides (TG•CA) and (TA•TA) [McIver et al., 2011], which are the most abundant microsatellites, and for which mutation rates of up to 10−2 per locus per gamete per generation have been reported [Eckert and Hile, 2009]. Parent/child transmission studies have revealed that several of the most mutable loci also contain compound sequences comprising two or more different types of microsatellite repeat [Brinkmann et al., 1998; Dupuy et al., 2004; Eckert and Hile, 2009]. Thus, in addition to tract length, microsatellite sequence composition also exerts a powerful influence on the mutation rate. Systematic analyses, performed in human colorectal cancer cells defective in post-replicative mismatch repair (MMR), have provided evidence for heteroduplex DNA at (A•T)10, (G•C)10, (CA•TG)13 and (CA•TG)23 target microsatellites, with one strand containing the initial number of repeats and the complementary strand containing either +1 or −1 repeats. Hence, the lack of correction by MMR of bulges and unpaired/mispaired loops resulting from the misalignment of repetitive DNA during DNA synthesis appears to be the most plausible mechanism for the observed increase in mutation rates at microsatellite loci (Fig. 5A). Indeed, strand slippage of repetitive DNA motifs represents a significant cause of mutation genome-wide, as mentioned previously. Mutation rates are consequential to combined kinetic reactions involving strand slippage and their subsequent repair, both of which display complex dependence upon DNA sequence. For example, in MLH1-deficient (MMR) HCT116 cells, (A•T)10 repeats display 5- to 15-fold higher susceptibility to replication errors than (G•C)10 repeats [Campregher et al., 2010], whereas the longer (A•T)17 tract is 7- to 15-fold less prone to replication errors than (G•C)17 on the same genetic background [Boyer et al., 2002]. Conversely, in isogenic cells complemented for MMR function, (G•C)16 repeats (which exhibit a ~20- to 60-fold higher error rate during replication than (G•C)10 tracts in the absence of MLH1), are repaired ~10 times more efficiently than (A•T)10 repeats [Campregher et al., 2010]. However, repair efficiency varies by more than two orders of magnitude between different genetic backgrounds [Boyer et al., 2002]. In addition to sequence composition, the repair of slipped-out bases is dependent upon their size and densities along the DNA chain, decreasing sharply as a function of both loop size (1 – 30 bases) and local concentration [Panigrahi et al., 2010]. Finally, slippage-dependent mutation rates at microsatellites are highly sensitive to their flanking sequence. For example, (A•T)7 and (A•T)10 repeat tracts exhibit an ~3-fold higher mutation rate when inserted within exon 10 of the ACVR2A gene [MIM# 102581] than within exon 3 of the TGFBR2 gene [MIM# 190182] in MMR-deficient cells, whereas the converse is seen for (A•T)13 tracts [Chung et al., 2008]. In addition, −2 bp deletions resulting from multiple slippage events were only seen in the TGFBR2 exonic context. The DNA sequence features responsible for these complex patterns are largely unknown; however, both base stacking interactions [Bacolla et al., 2008; Yang, 2008] and energy coupling reactions between bases distally located within loops and/or the flanking duplex region [Völker et al., 2010] are likely to be involved.
Figure 5.

Models for microsatellite repeat expansion. Panel A. Strand slippage during DNA replication. A microsatellite repeat sequence (black and grey segments) and the associated DNA polymerase complexes (protein complexes are not shown for reasons of clarity) may transiently dissociate from the template strand and form an intra-strand hairpin stabilized by Watson-Crick and non-Watson-Crick (such as T:T) pairs with backward slippage between the repeats. The hairpin may be incorporated into genomic DNA during the next round of DNA synthesis (not shown), yielding an expansion. Panel B. Replication-independent expansion in quiescent oocytes. A base lesion (black bulge) is cleaved by base excision repair (see McMurray, 2010 for details) and the ensuing 3′-OH terminus is extended by DNA polymerase beta, displacing the DNA strand ahead of it. The displaced single-strand forms large hairpin structures, which are resistant to FEN1 cleavage and are stabilized by mismatch repair proteins (MSH2/MSH3). Further DNA ligation leaves long hairpin structures, which are subsequently incorporated into DNA (not shown) by cleavage and synthesis on the complementary strand. Panel C. Fork stalling and restart. A hairpin structure on the template for lagging strand DNA synthesis blocks replication. Replication restarts through fork-reversal (chicken-foot structure) which in the process yields a second hairpin on the newly synthesized leading strand that will subsequently be incorporated into chromosomal DNA, leading to expansion (see Mirkin, 2007 for details). Panel D. Fork stalling and template switching. After fork stalling (see Panel C), strand switching occurs, whereby the 3′-end of the growing leading strand copies microsatellite repeats from the newly synthesized lagging strand, yielding an expansion. It should be noted that large and small hairpins are not distinguished in the Figure. Panel E. Unequal crossing-over between normal alleles. Alleles a and b contain polymorphic alanine-encoding repeats (white squares, GCA; black squares, GCG; and grey squares, GCC) that can anneal in an out-of phase manner during crossover, resulting in an expanded allele and a smaller allele (not shown).
Some 30 human inherited diseases associated with neuromuscular and developmental disorders have now been linked to the expansion of a microsatellite repeat within the corresponding disease-associated gene [Brouwer et al., 2009; Lopez Castel et al., 2010; Wells and Ashizawa, 2006]. Expansions generally originate from ‘at-risk’ (premutation) alleles, from which the addition of up to thousands of repeat units, usually trinucleotides, takes place within parent-child transmissions. The number of repeats in normal alleles is highly variable between loci, but is generally limited to fewer than 40-45 repeats. Small expansions into the premutation range (~29-35 repeats within coding regions and ~55-200 repeats in non-coding regions) and/or loss of interruptions within the repeat tract act to destabilize the sequences, which then become increasingly prone to further expansion [Brouwer et al., 2009; McMurray, 2010; Orr and Zoghbi, 2007], triggering an escalating positive feed-back loop that creates pathogenic mutation alleles within a few generations [Wells and Ashizawa, 2006]. As the lengths of the microsatellites increase, the severity of the disease symptoms generally worsen and/or the age of onset decreases, a phenomenon termed ‘genetic anticipation’.
The dramatic intergenerational instability observed in microsatellite expansion diseases (MEDs) differs markedly from the population-based microsatellite instability described above. Indeed, several molecular mechanisms in addition to slippage are believed to occur. The microsatellite sequences involved in MEDs include the trinucleotide repeats (GAA•TTC) in intron 1 of the frataxin gene [FXN; MIM# 606829] in Friedreich ataxia; (CTG•CAG) in the 3′UTR of the DMPK gene [MIM# 605377] in myotonic dystrophy type 1 and the ataxin 8 opposite strand gene [ATXN8OS; MIM# 603680] in spinocerebellar ataxia type 8 (SCA8), in the 5′UTR of the serine/threonine-protein phosphatase 2A 55 kDa regulatory subunit B beta isoform gene [PPP2R2B; MIM# 604325] in SCA12 and within the coding regions of 9 polyglutamine expansion diseases; (CGG•CCG) in the 5′UTR of the fragile X mental retardation 1 gene [FMR1; MIM# 309550] in fragile X syndrome and fragile X-associated ataxia and tremor, the 5′UTR of the AF4/FMR2 family member 2 gene [AFF2; MIM# 300806] in FRAXE-associated mental retardation and the coding regions of 9 polyalanine expansion diseases; the tetranucleotide (CCTG•CAGG) in intron 1 of the CCHC-type zinc finger, nucleic acid binding protein gene [CNBP; MIM# 116955] in myotonic dystrophy type 2 and the pentanucleotide (ATTCT•AGAAT) in intron 9 of the ataxin 10 gene [ATXN10; MIM# 611150] in SCA10 [Lopez Castel et al., 2010; McMurray, 2010; Messaed and Rouleau, 2009]. A tenth polyalanine expansion disorder associated with the Zic family member 3 gene [ZIC3; MIM# 300265] and leading to X-linked heterotaxy with VACTERL association has recently been described [Wessels et al., 2010]. Most of these microsatellite sequences have been shown to be capable of adopting specific secondary structures (non-B DNA), including hairpin-loops, three-(triplex) and four-stranded (quadruplex) structures and left-handed Z-DNA [Lopez Castel et al., 2010; Mirkin, 2007; Renciuk et al., 2011; Wells and Ashizawa, 2006]. Below, we review some of the most compelling evidence in support of a role for DNA secondary structures in either microsatellite expansion and/or the process of pathogenesis.
Mechanisms Underlying Repeat Expansion
In fragile X syndrome, premutations expand to full mutation only upon maternal transmission, whereas full mutations invariably contract to premutations upon paternal transmission [Brouwer et al., 2009; Jin and Warren, 2000]. Expansion is believed to occur early in oogenesis, a stage when primary oocytes remain quiescent (i.e. do not divide) for years. Work both in the mouse and in vitro supports the view that following DNA damage (including oxidation-related DNA damage) within the (CGG•CCG) sequence, repair of the damaged bases (a process which involves both base excision and mismatch repair) entails the formation of stable hairpins on one DNA strand, which then direct DNA synthesis to the complementary strand in order to incorporate the looped-out structures into de novo DNA [Entezam et al., 2010; Lopez Castel et al., 2010; McMurray, 2010], resulting in expansion (Fig. 5B). In contrast to this replication-independent mechanism of repeat expansion, in the majority of the other MEDs in which large expansions also occur, similarly stable hairpins are thought to form, mainly on the lagging strand during DNA replication, thereby blocking DNA synthesis; further resolution of stalled replication forks and reinitiation of synthesis could then lead to expansion [Mirkin, 2007] (Fig. 5C). For the smaller expansions seen in polyglutamine diseases, slippage during DNA replication involving small hairpin-loops, as in the case of microsatellite length polymorphism (see above) remains the most likely mechanism [Lopez Castel et al., 2010; Mirkin, 2007; Wells and Ashizawa, 2006] (Fig. 5A). In polyalanine expansion diseases, in which the coding trinucleotide tracts are short and often interrupted, pedigree analyses support the occurrence of both fork stalling and template switching, triggered by secondary structure formation (Fig. 5D), as well as unequal crossing-over between two normal alleles [Arai et al., 2010; Cocquempot et al., 2009; Messaed and Rouleau, 2009; Warren, 1997] (Fig. 5E).
Pathogenesis Resulting From Repeat Expansion
At the time the molecular basis of Friedreich ataxia [MIM# 229300] was first reported [Campuzano et al., 1996], a substantial number of studies had already been performed on the biophysical properties of the (GAA•TTC) sequence. Indeed, the asymmetric purine•pyrimidine composition was known to enable the formation of three-stranded structures [Wells et al., 1988]. In triplex DNA, the purine-rich strand of duplex DNA binds a third strand through specific Hoogsteen hydrogen bonds, including A:A and G:G (purine-rich third strand) and A:T and G:C+ (pyrimidine-rich third strand) pairs. Thus, mirror symmetry within purine•pyrimidine sequences is required to yield stable triplex structures [Frank-Kamenetskii and Mirkin, 1995]. As expected, long (GAA•TTC) tracts cloned in plasmids were found to interact with each other and form stable intermolecular DNA structures that were interpreted as triplexes (sticky DNA) [Sakamoto et al., 1999]. However, their exceptionally high thermal stability, and the number of negative superhelical turns remaining in plasmids after DNA structure formation, suggest that other conformations, such as duplex-duplex interactions, are also feasible for long (GAA•TTC) repeats [Son et al., 2006]. In Friedreich ataxia, the FXN locus is silenced [Al-Mahdawi et al., 2008]. Within the FXN gene, local chromatin is characterized by hypoacetylation of histones H3 and H4 and methylation of histone H3 at Lys9 (H3K9), which are hallmarks of transcriptionally inactive heterochromatin [Punga and Buhler, 2010]. However, heterochromatin does not spread to the 5′ and 3′ sections of the gene and only transcriptional elongation (rather than initiation) appears to be impaired in patient-derived lymphoblastoid cell lines. Removal of H3K9 methylation marks is however ineffective in re-establishing transcriptional elongation [Punga and Buhler, 2010], strongly supporting a model in which the expanded (GAA•TTC) repeat itself or, more likely its folding into a secondary structure, imposes a direct block upon the transcriptional apparatus [Punga and Buhler, 2010].
Expansion of the (CGG•CCG) repeat in the FMR1 gene also leads to gene silencing in fragile X syndrome. A decrease in histone H3 and H4 acetylation is evident in pathological full mutation alleles, accompanied by de novo methylation of the repeat tract and the upstream CpG island in the promoter region [Jin and Warren, 2000]. Studies in intact ovaries of fetuses and chorionic villus samples harbouring full-mutations [reviewed in Jin and Warren, 2000] suggest that methylation is a dynamic process that takes place over an extended time period. However, the mechanisms by which expanded (CGG•CCG) repeats induce methylation remain unclear. (CGG•CCG) repeats have been shown to fold into hairpin-loops [Amrane and Mergny, 2006; Darlow and Leach, 1998], quadruplexes [Khateb et al., 2004; Usdin and Woodford, 1995], left-handed Z-DNA [Renciuk et al., 2011], to possess inherently high flexibility (bending) [Bacolla et al., 1997] and are predicted to sustain stable ‘bubbles’ despite their high CG content [Alexandrov et al., 2011]. In vitro, the ability of DNA methyltransferase 1 (DNMT1) to methylate (CGG•CCG) repeats increases with increasing negative supercoiling [Bacolla et al., 2001]. Hence, it is possible that the formation of alternative DNA structures and/or open (denatured) states favored by torsional stress at long repeat tracts, might nucleate unscheduled de novo methylation in the FMR1 gene, leading to gene silencing.
Microsatellite Polymorphisms and Susceptibility to Disease
In additions to the MEDs described above, length polymorphism at specific microsatellites within genes or their promoters has been associated with phenotypic trait variation and/or susceptibility to disease [Bacolla et al., 2008; Gemayel et al., 2010]. For example, a highly polymorphic (GT•CA)n repeat within the proximal SLC11A1 gene [MIM# 600266] promoter regulates variation in allele expression [Bayele et al., 2007] by directly modulating the recruitment of HIF-1α to the repeat sequence through its ability to interconvert from the canonical right-handed B- to left-handed Z-DNA. In addition, surrogate stimuli of the innate immune response (such as E. coli and S. typhimurium LPS, mannose- and phosphoinositide-capped lipidoarabinomannans from M. bovis and M. smegmatis, respectively), stimulate HIF-1α-dependent transactivation. Given the prominent role of HIF-1α in integrating innate immune responses to infection and inflammation, this SLC11A1 repeat polymorphism is believed to contribute to the heritable variation in susceptibility to infection and/or inflammation that is observed within and between populations [Bayele et al., 2007].
A recent study on the relationships between matrix metalloproteinase genetic polymorphisms and vulnerable plaques in a cerebrovascular disease patients cohort revealed a significant association between prognosis and the length of a polymorphic (CA•TG)13-26 microsatellite upstream of the MMP9 [MIM# 120361] transcriptional start site [Fiotti et al., 2011]. Specifically, carriers of ≥22 repeats displayed ~50% larger plaques and had a significantly higher risk of persistent angina and ischemic stroke than non-carriers. Consistent with this association, long (CA•TG)-containing alleles manifest increased MMP9 gene expression relative to shorter ones [Shimajiri et al., 1999].
Other examples on the involvement of polymorphic microsatellites in disease susceptibility include a (CA•TG) dinucleotide repeat in the EGFR [MIM # 131550] 5′UTR and gastrointestinal cancers [Baranovskaya et al., 2009], an (AAAT•ATTT) tetranucleotide repeat in intron 27b of the NF1 gene and mental retardation [Védrine et al., 2011], an (AAAG•CTTT) repeat in the estrogen receptor-related γ (ESRRG; MIM# 602969) 5′UTR and breast cancer [Galindo et al., 2011], a (GGGCGG•CCGCCC) hexanucleotide repeat in the arachidonate 5-lipoxygenase gene (ALOX5; MIM# 152390) and risk of carotid atherosclerosis and myocardial infarction [Vikman et al., 2009], and a (CATT•AATG) tetranucleotide repeat in the macrophage migration inhibitory factor gene (MIF; MIM# 153620) promoter and duodenal ulcer, rheumatoid arthritis and psoriasis [Shiroeda et al., 2010].
Mutations in or Involving the Mitochondrial Genome
The mitochondrial genome differs from the nuclear genome in a variety of different respects, most notably in terms of its high copy number (with the consequent potential for heteroplasmy), matrilineal inheritance, a 10 to 17-fold higher mutation rate despite having its own DNA repair system [Liu and Demple, 2010], active exposure to reactive oxygen species [Sedelnikova et al., 2010], a unique mode of DNA replication [Wanrooij and Falkenberg, 2010] and the virtual lack of any recombination [Krishnan and Turnbull, 2010]. A wealth of knowledge has now accumulated with respect to the spectrum of germline mitochondrial genome mutations that are responsible for heritable mitochondrial disease [Taylor and Turnbull, 2005; Neiman and Taylor, 2009; Wallace, 2010]. Despite these basic differences, the nature, location and frequency of the many different types of mutation in the mitochondrial genome are also strongly influenced by the local DNA sequence environment. Thus, as already reported for the nuclear genome, direct repeats have been frequently noted at mitochondrial DNA (mtDNA) breakpoints in mtDNA deletion syndromes [Samuels et al., 2004]. Indeed, mtDNA deletions may be separated into two types, type I (with a direct repeat) and type II (with an imperfect or no direct repeat), with respect to the sequences present at the two breakpoints. Sadikovic et al. [2010] have recently shown that, irrespective of the presence or absence of a direct repeat, most mtDNA deletions are characterized by an increase in sequence homology surrounding the breakpoints. This finding is consistent with sequence homology being a key determinant of breakpoint location in mtDNA deletion syndromes. In accord with an expectation that the longest direct repeats would be likely to demarcate the most dramatic mtDNA deletion hotspots, the most common mtDNA deletion (8470-13447), which is flanked by the longest (13 bp) direct repeat, has been noted in 37% of mtDNA deletion syndrome patients [Sadikovic et al., 2010]. The presence of sequence homologies at the deletion breakpoints is suggestive of a role for sequence homology not only in the generation of the initial break but also in the subsequent repair of the mtDNA damage. It has been suggested that direct repeats serve to promote breakpoint generation when there is an error in mtDNA replication due either to the illegitimate alignment of direct repeats [Holt et al., 2000] or to mtDNA damage [Krishnan et al., 2008]. Defects in mtDNA replication, resulting from the inappropriate alignment of direct repeats or mis-annealing of a single-stranded mtDNA molecule following the occurrence of a double strand break, both require the presence of direct repeats (or at the very least some sequence homology).
The mitochondrial genome is however also involved in a very different type of mutation. Numerous fragments of mitochondrial DNA are present throughout the human nuclear genome, these fragments having migrated from the mitochondrial genome over evolutionary time [Mishmar et al., 2004; Ricchetti et al., 2004]. An occasional consequence of these migrations in extant genomes is the de novo disruption of nuclear genes resulting in a heritable disease. Once again, the nature and location of these highly unusual lesions are both strongly influenced by the local DNA sequence environment. Probably the best characterized example of a pathogenic mitochondrial-nuclear DNA transfer is that described by Turner et al. [2003] in a sporadic case of Pallister-Hall syndrome [MIM# 146510], a condition usually inherited in an autosomal dominant fashion. The mutation involved a de novo nucleic acid transfer from the mitochondrial to the nuclear genome, more specifically the insertion of a 72-bp segment into exon 14 of the GLI3 gene [MIM# 165240] thereby creating a premature stop codon. The insertion site in the GLI3 gene was flanked by inverted repeat elements that could have facilitated hairpin-loop formation. Although no similarity of the 72-bp mitochondrial (mt) DNA insert and the GLI3 gene was apparent, Turner et al. [2003] noted significant sequence identity (~60%) of a 112-bp region (interrupted by a 31 bp inverted repeat) 5′ to the GLI3 gene insertion site and an 81 bp region of the mitochondrial genome immediately 5′ to the 72 bp insertion sequence. They therefore proposed that a mtDNA fragment, initially >72 bp in length, had interfered with the resolution of a transient GLI3 hairpin-loop structure, leading to the illegitimate insertion of a 72 bp mtDNA fragment during DNA repair.
A further example of this type of insertion was recently described in an isolated case of lissencephaly [MIM# 607432; Millar et al., 2010]: a de novo 130 bp mtDNA insertion into the 5′ untranslated region of the PAFAH1B1 gene [MIM# 601545], 7 bp upstream of the translational initiation site. The inserted DNA sequence was found to exhibit perfect homology to two non-contiguous regions of the mitochondrial genome [8,479 to 8,545 and 8,775 to 8,835, containing portions of two genes, MTATP8 (MIM# 516070) and ATP6 (MIM# 516060)]. Several other examples of mitochondrial-nuclear DNA transfer have been reported as a cause of human inherited disease. However, in the context of the mutation reported here, the mtDNA insertion polymorphism in intron 1 of the FOXO1A gene [MIM# 136533; Giampieri et al., 2004] is perhaps the most intriguing, since this 39 bp insertion was derived from the mtDNA sequence between nucleotides 8,531 and 8,569 containing the MTATP8 and MTATP6 genes. The mtDNA sequence inserted into the FOXO1A gene therefore overlaps with the 130 bp PAFAH1B1 gene insert reported by Millar et al. [2010] by 14 bases (8,532 to 8,545), raising the possibility of the preferential insertion (into the nuclear genome) of certain mtDNA fragments.
Non-B DNA: A Unifying Hypothesis?
In the preceding sections, numerous examples of mutations have been provided in which the formation of non-B DNA conformations (including cruciforms, looped-out bases, quadruplex, triplex and Z-DNA structures) [Figs. 4 and 5] has been postulated to account for intermediate (and transient) forms of DNA that generally serve to promote genetic instability while giving rise specifically to frameshift mutations, repeat expansions and other gross rearrangements. However, with the notable exception of heteroduplex formation by microsatellite repeats in MMR-deficient human cells, direct evidence for such structures having formed and being responsible for the reported mutations has been lacking, with most conclusions being drawn from experiments performed either in vitro or using episomal systems in bacteria and yeast [Mirkin, 2007]. Here, we review some of the work that has directly addressed the extent to which non-B DNA structures can induce human genomic rearrangements.
As already mentioned, the t(11;22)(q23;q11) is a recurrent balanced translocation and is the most frequent of non-Robertsonian translocations, i.e. those that do not involve the large heterochromatic regions of acrocentric chromosomes [Kurahashi et al., 2006]. Although carriers of t(11;22) are generally healthy or only mildly affected, their offspring may come to clinical attention as a consequence of severe mental retardation and morphologic anomalies, associated with the inheritance of the supernumerary der(22) chromosome (Emanuel syndrome, MIM# 609209) [Kurahashi et al., 2006]. Positional cloning permitted the identification of junction fragments in ~40 cases studied, which revealed the clustering of t(11;22) breakpoints at the centre of two large A+T-rich regions (~450 and ~590 bp, respectively), one on each chromosome, and each capable of forming a near-perfect cruciform due to the arrangement of the A+T-rich bases as an inverted repeat [Kurahashi and Emanuel, 2001]. These sequences were termed ‘palindromic AT-rich regions’, or PATRR11 and PATRR22. Most of the chromosomal breaks occurred within the predicted single-stranded loops that separated the two arms of each cruciform, one on chromosome 11 and the other on chromosome 22. Interestingly, despite the A+T-richness, no significant homology was apparent between PATRR11 and PATRR22, suggesting that t(11;22) events resulted from double-stranded break repair by a non-homologous end-joining mechanism.
Further support for this model has come from the analysis of two independent cases of neurofibromatosis type 1 (NF1) caused by a rare t(17;22)(q11;q11) translocation that disrupted the NF1 gene on chromosome 17. Molecular cloning identified PATRR22 as the region responsible for the rearrangements on chromosome 22, whereas an additional ~200 bp PATRR (PATRR17) within intron 31 of the NF1 gene was revealed to be the partner breakage site on chromosome 17 [Kehrer-Sawatzki et al., 1997; Kurahashi et al., 2006]. Thus, a mechanism similar to t(11;22) was apparent in both cases. More recently, analyses of at least 12 individuals with both balanced and unbalanced t(8;22)(q24.13;q11.2) translocations also showed the consistent involvement of PATRR22, as well as a predicted 129-145 bp long undisrupted cruciform structure at PATRR8 involving a sequence that is ~97% A+T [Sheridan et al., 2010]. Hence, in all these instances, a PATRR predicted to fold into a stable cruciform and hosting chromosomal breaks at the single-stranded centre loop, is believed to have been directly involved in the translocation process. Based on the PATRR-dependent cruciform model, Sheridan et al. made the prediction that if both t(11;22) and t(8;22) were recurrent events involving the common PATRR22 region, then t(8:11) might also occur at some frequency, even although carriers of such a rearrangement had not been reported in the literature [Sheridan et al., 2010]. The use of specific PCR primers on sperm samples from healthy males confirmed the occurrence of just such an event, which took place with an estimated frequency of <2.6 × 10−6. Additional sperm analyses aimed at detecting the frequencies of t(11;22) and t(8;22) in healthy males also provided strong support for the PATRR-dependent cruciform model for translocation. The t(11;22) was found to occur at a frequency of ~10−5, whereas the frequency of t(8;22) ranged from ~10−6 to 10−5. Importantly, these frequencies were found to vary by more than two orders of magnitude and correlated in a predictable manner with PATRR sequence length polymorphisms (i.e. the existence of multiple alleles of variable length in the general population). Specifically, homozygous males carrying long PATRRs with the inverted symmetry required to extrude cruciform structures from regular duplex DNA were associated with high translocation frequencies, whereas carriers of shorter alleles in which such inverted symmetry was either reduced or lost, manifested fewer, if any, translocation events [Kato et al., 2006; Sheridan et al., 2010]. These results therefore provide compelling support for the hypothesis that cruciform structures generated by PATRR sequences are responsible for recurrent non-Robertsonian translocations, by providing a substrate for the generation of structure-directed double-strand breaks.
In addition to these composite cases that share common recombination hotspots, a number of other studies have reported the occurrence of non-B DNA-forming sequences at breakpoints of rearrangements associated with inherited disease. For example, a common 1.1 Mb deletion on chromosome 14q32 has been identified in two unrelated patients diagnosed with uniparental disomy. An expanded (TGG)n repeat was identified on either side of the deletion, suggesting that either non-allelic homologous recombination between the two repeat tracts and/or the formation of non-B structures (such as quadruplexes) adjacent to the repeats, could have induced strand breakage thereby triggering the deletion [Bena et al., 2010]. Additional examples include the presence of triplex-, quadruplex- and hairpin-forming sequences at sites of subtelomeric rearrangements associated with mental retardation [Rooms et al., 2007] and other abnormalities (ear shape, scoliosis) [Bonaglia et al., 2009], short cruciform structures flanking a large (~30 kb) heterozygous deletion that removed the entire SPINK1 gene [MIM# 167790], associated with idiopathic pancreatitis [Masson et al., 2007] and similar structures formed by inverted Alu repeats flanking deletions in the OTC gene [MIM# 300461] leading to ornithine transcarbamylase deficiency [Quental et al., 2009]. Comprehensive meta-analyses that have aimed to determine whether non-B DNA-forming motifs are enriched at rearrangement breakpoints have also supported the involvement of DNA secondary structural features in promoting genetic instability [Bacolla et al., 2004; Wells, 2007; Bengesser et al., 2010; Quemener et al., 2010; Roehl et al., 2010].
The abovementioned studies, together with those cited in previous sections, raise the question as to how non-B DNA structures form on chromatin and how they induce genetic instability. A large number of studies, performed in vitro and on model organisms, now support the conclusion that non-B DNA conformations may arise through various mechanisms, including the folding of single-stranded DNA regions during replication [Mirkin, 2007], transcription [Belotserkovskii et al., 2007; Lin et al., 2010; Tornaletti, 2009] and repair [Wang and Vasquez, 2009], as well as through the generation of unrestrained negative supercoiling [Napierala et al., 2005] either via such processes as transcription and replication or upon nucleosome release. In the specific case of MEDs, a number of studies in bacteria, yeast, mammalian cell culture and mouse models support the conclusion that the extent of instability is intimately associated with replication fork dynamics, being generally greater when microsatellite repeats are close to, or part of, a replication origin and/or when more stable hairpins may form on the lagging, rather than on the leading, strands [Wells and Ashizawa, 2006; Potaman et al., 2003; Liu et al. 2010; Nichol Edamura et al., 2005; Yang et al., 2003; Tomé et al., 2011]. As already mentioned (Fig. 5), collapsed replication forks may lead to aberrant repair, including recombination, at non-B DNA conformations leading to instability. Hence, initiation of replication, recombination and mutagenesis probably constitute the three corners of a triangle associated with a number of human pathological conditions. On the other hand, while we believe that these processes are adequate to explain the transient formation of short non-B DNA regions, they appear insufficient to account for the formation of much larger structures, such as the cruciforms extruded from PATRR elements. Hence, it is possible that other as yet unidentified mechanisms of secondary structure formation also operate.
Regarding the mechanisms underlying non-B DNA induced genetic instability, studies in bacteria, yeast and mouse are consistent with the recognition and cleavage of non-B DNA structures by DNA repair enzyme pathways [Lopez Castel et al., 2010; Wang et al., 2008; Wang et al., 2006b; Wang and Vasquez, 2004; Wang and Vasquez, 2009] and the local induction of oxidative damage [Bacolla et al., 2011], followed by DSB repair via non-homologous end-joining. Nevertheless, a number of questions still remain to be addressed. For example, although the t(11;22), t(8;22) and t(8;11) translocations were detected in sperm samples, as mentioned above, they were not observed in somatic cells, despite the occurrence of such recombination events in episomal DNA systems in cell culture [Inagaki et al., 2009]. Thus, it appears that during the course of meiosis, ‘natural’ chromatin might offer a more favourable environment for the generation of non-B DNA conformations and the ensuing genomic instability than mitotic cells.
Finally, the survey presented here raises the question as to the overall impact that non-canonical DNA conformations might have in the context of the causation of human genetic disease. Since the number of pathological conditions specifically listed above is necessarily quite limited, it would at first sight appear as if the overall impact of non-B DNA structures in human inherited disease could be rather modest. However, the specific examples described above were for the most part confined to either repeat expansions or the close proximity observed between the location of chromosomal strand breaks and the presence of potential non-canonical DNA structures. A recent study using human osteosarcoma cell lines has shown that non-canonical DNA conformations are capable of increasing the overall spectrum of mutations (from single base substitutions to gross rearrangements) in a reporter gene in cis by exposing those distant DNA sequences to oxidative damage [Bacolla et al., 2011]. Further, in this study, the spectrum of single base substitutions was shown to be indistinguishable from that induced by other conditions known to lead to an hyperoxidative state (such as Werner deficiency and lung tumorigenesis), an observation which lends support to a model whereby DNA bases become oxidized, followed by the transfer of their oxidized state (‘hole migration’) to target neighbouring bases. If these observations are eventually found to be relevant in the context of ‘natural’ chromatin during meiosis, then the impact of non-canonical DNA conformations on human inherited disease, both with respect to gross rearrangements and single base substitutions, would be even greater than the current review already appears to suggest.
Concluding Remarks
“The human genome [is] riddled with structural and operational deficiencies ranging from the subtle to the egregious. These genetic defects register not only as deleterious mutational departures from some hypothetical genomic ideal but as universal architectural flaws in the standard genomes themselves”.
John C. Avise (2010) Proc. Natl. Acad. Sci.USA 107:8969-8976.
In the above discussion, we have seen that the most plausible explanations for many types of inherited mutation almost invariably invoke either the immediate DNA sequence environment or higher order (but nevertheless still comparatively local) features of genome structure and sub-structure. Different types of mutation may vary dramatically in size (from gross genomic rearrangements down to subtle gene lesions at the single base-pair level) but what they have in common is that their nature, location and extent are often determined by specific characteristics of the local DNA sequence environment. Thus, both the non-randomness and sequence directedness of human gene mutation are reflections of the influence of a number of different genomic features including base composition, epigenetic modification and sequence repetitivity. In addition, the presence of certain DNA sequence motifs may serve to induce mutations by initiating or modulating specific biological processes (e.g. recombination or DNA repair) associated with that motif. Together, such sequence features exert a profound influence over the likelihood of occurrence of specific types of mutation at specific sites or in particular genomic locations.
It has also come to be realised that the mutability of a given gene/genomic region can be mediated indirectly through a variety of non-standard secondary structures whose formation is facilitated by the underlying DNA sequence. These unusual secondary structures may be slipped mispairing intermediates or any one of a number of different non-B DNA structures that can interfere with subsequent DNA replication and repair. It is also becoming apparent that once formed, non-B DNA structures can serve to increase the mutation frequency in generalized fashion, inducing large deletions and other gross genomic rearrangements as well as subtle mutations such as single base-pair substitutions. For reasons that we do not yet fully understand, the single nucleotide substitution rate often covaries with the frequency of insertions, deletions and other rearrangements in the human genome [Longman-Jacobsen et al., 2003; Yang et al., 2004; Marques-Bonet et al., 2007; Tian et al., 2008]. One explanation could be that the single nucleotide substitution rate becomes elevated as a direct consequence of the low fidelity of the error-prone DNA polymerases used to repair regions that have been subject to structural alteration [De and Babu, 2010], an hypothesis not inconsistent with the concept of transient hypermutability [Chen et al., 2009].
Since the human genome is a product of molecular evolution rather than some form of ‘intelligent design’, it is scarcely surprising to find that it contains “pervasive architectural flaws” rendering it “the antithesis of thoughtful organic engineering” [Avise, 2010; Chapman, 2010]. Indeed, over evolutionary time, and as an integral part of its development, the extant human genome has acquired a variety of rearrangements including inversions, insertions and duplications [Cooper, 1999] that, by virtue of their structure and/or organization, now constitute mutation hotspots. This should not of course be held to imply that a relatively immutable primeval genome once existed which then proceeded to decay to an imperfect state over evolutionary time; genomes always were and always will be mutable, and it could not be otherwise since mutability constitutes the major driving force behind the evolution of all life forms. In yet another manifestation of the much vaunted ‘Goldilocks principle’, if somehow the genomes of our ancestors had been immutable, we would not now be around to register it. There is however a price that extant organisms, including humans, must pay for the inherent mutability of their genomes: genetic disease. There are numerous examples of benign (or relatively benign) genetic changes or rearrangements that occurred during the evolutionary history of our species, and which gave rise to particular types of genomic organization or even specific DNA sequences that are now inherently hypermutable and hence responsible for the recurrence of pathological mutations in extant humans [Laken et al., 1997; Huang et al., 2004; Mirkin, 2007; Bacolla et al., 2008; Kim et al., 2008; Wolf et al., 2009; Witherspoon et al., 2009; Fu et al., 2010]. In this review, we have come to appreciate, through perusal of some of the many published studies of molecular defects identified in individuals afflicted by inherited disease, that the structure of the human genome is inherently nemesistic in the sense that it contains buried within it the seeds of its own destruction, or at the very least its own decay. Our task is to come to understand the ground rules that characterize the different mechanisms of mutagenesis in order to apply this knowledge in the context not only of the analysis and diagnosis of genetic disease, but also eventually perhaps, in the cause of its therapeutic correction.
Acknowledgments
This work was supported in part by a National Cancer Institute/National Institutes of Health Contract HHSN261200800001E (to A.B.).
References
- Abadie V, Lyonnet S, Maurin N, Berthelon M, Caillaud C, Giraud F, Mattei JF, Rey J, Rey F, Munnich A. CpG dinucleotides are mutation hot spots in phenylketonuria. Genomics. 1989;5:936–939. doi: 10.1016/0888-7543(89)90137-7. [DOI] [PubMed] [Google Scholar]
- Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN. Translocation and gross deletion breakpoints in human inherited disease and cancer. I: Nucleotide composition and recombination-associated motifs. Hum Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- Abo-Dalo B, Kutsche K, Mautner V, Kluwe L. Large intragenic deletions of the NF2 gene: breakpoints and associated phenotypes. Genes Chrom Cancer. 2010;49:171–175. doi: 10.1002/gcc.20733. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, Trabzuni D, Pook M. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17:735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- Alexandrov BS, Valtchinov VI, Alexandrov LB, Gelev V, Dagon Y, Bock J, Kohane IS, Rasmussen KO, Bishop AR, Usheva A. DNA dynamics is likely to be a factor in the genomic nucleotide repeats expansions related to diseases. PLoS ONE. 2011;6:e19800. doi: 10.1371/journal.pone.0019800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrane S, Mergny JL. Length and pH-dependent energetics of (CCG)n and (CGG)n trinucleotide repeats. Biochimie. 2006;88:1125–1134. doi: 10.1016/j.biochi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Antonacci F, Kidd JM, Marques-Bonet T, Ventura M, Siswara P, Jiang Z, Eichler EE. Characterization of six human disease-associated inversion polymorphisms. Hum Mol Genet. 2009;18:2555–2566. doi: 10.1093/hmg/ddp187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonacci F, Kidd JM, Marques-Bonet T, Teague B, Ventura M, Girirajan S, Alkan C, Campbell CD, Vives L, Malig M, Rosenfeld JA, Ballif BC, Shaffer LG, Graves TA, Wilson RK, Schwartz DC, Eichler EE. A large and complex structural polymorphism at 16p12.1 underlies microdeletion disease risk. Nat Genet. 2010;42:745–750. doi: 10.1038/ng.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apoil PA, Kuhlein E, Robert A, Rubie H, Blancher A. HIGM syndrome caused by insertion of an AluYb8 element in exon 1 of the CD40LG gene. Immunogenetics. 2007;59:17–23. doi: 10.1007/s00251-006-0175-5. [DOI] [PubMed] [Google Scholar]
- Arai H, Otagiri T, Sasaki A, Umetsu K, Hayasaka K. Polyalanine expansion of PHOX2B in congenital central hypoventilation syndrome: rs17884724:A>C is associated with 7-alanine expansion. J Hum Genet. 2010;55:4–7. doi: 10.1038/jhg.2009.109. [DOI] [PubMed] [Google Scholar]
- Arana ME, Takata K, Garcia-Diaz M, Wood RD, Kunkel TA. A unique error signature for human DNA polymerase nu. DNA Repair. 2007;6:213–223. doi: 10.1016/j.dnarep.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnheim N, Calabrese P. Understanding what determines the frequency and pattern of human germline mutations. Nat Rev Genet. 2009;10:478–488. doi: 10.1038/nrg2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise JC. Footprints of nonsentient design inside the human genome. Proc Natl Acad Sci USA. 2010;107:8969–8976. doi: 10.1073/pnas.0914609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awano H, Malueka RG, Yagi M, Okizuka Y, Takeshima Y, Matsuo M. Contemporary retrotransposition of a novel non-coding gene induces exon-skipping in dystrophin mRNA. J Hum Genet. 2010;55:785–790. doi: 10.1038/jhg.2010.111. [DOI] [PubMed] [Google Scholar]
- Ayala FJ. Darwin’s greatest discovery: design without designer. Proc Natl Acad Sci USA. 2007;104(Suppl. 1):8567–8573. doi: 10.1073/pnas.0701072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Gellibolian R, Shimizu M, Amirhaeri S, Kang S, Ohshima K, Larson JE, Harvey SC, Stollar BD, Wells RD. Flexible DNA: genetically unstable CTG.CAG and CGG.CCG from human hereditary neuromuscular disease genes. J Biol Chem. 1997;272:16783–16792. doi: 10.1074/jbc.272.27.16783. [DOI] [PubMed] [Google Scholar]
- Bacolla A, Pradhan S, Larson JE, Roberts RJ, Wells RD. Recombinant human DNA (cytosine-5) methyltransferase. III. Allosteric control, reaction order, and influence of plasmid topology and triplet repeat length on methylation of the fragile X CGG.CCG sequence. J Biol Chem. 2001;276:18605–18613. doi: 10.1074/jbc.M100404200. [DOI] [PubMed] [Google Scholar]
- Bacolla A, Jaworski A, Larson JE, Jakupciak JP, Chuzhanova N, Abeysinghe SS, O’Connell CD, Cooper DN, Wells RD. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc Natl Acad Sci USA. 2004;101:14162–14167. doi: 10.1073/pnas.0405974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Wells RD. Non-B DNA conformations, genomic rearrangements, and human disease. J Biol Chem. 2004;279:47411–47414. doi: 10.1074/jbc.R400028200. [DOI] [PubMed] [Google Scholar]
- Bacolla A, Larson JE, Collins JR, Li J, Milosavljevic A, Stenson PD, Cooper DN, Wells RD. Abundance and length of simple repeats in vertebrate genomes are determined by their structural properties. Genome Res. 2008;18:1545–1553. doi: 10.1101/gr.078303.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Wang G, Jain A, Chuzhanova NA, Cer RZ, Collins JR, Cooper DN, Bohr VA, Vasquez KM. Non-B DNA-forming sequences and WRN deficiency independently increase the frequency of base substitution in human cells. J Biol Chem. 2011;286:10017–10026. doi: 10.1074/jbc.M110.176636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Ball EV, Stenson PD, Abeysinghe SS, Krawczak M, Cooper DN, Chuzhanova NA. Microdeletions and microinsertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat. 2005;26:205–213. doi: 10.1002/humu.20212. [DOI] [PubMed] [Google Scholar]
- Baranovskaya S, Martin Y, Alonso S, Pisarchuk KL, Falchetti M, Dai Y, Khaldoyanidi S, Krajewski S, Novikova I, Sidorenko YS, Perucho M, Malkhosyan SR. Down-regulation of epidermal growth factor receptor by selective expansion of a 5′-end regulatory dinucleotide repeat in colon cancer with microsatellite instability. Clin Cancer Res. 2009;15:4531–4537. doi: 10.1158/1078-0432.CCR-08-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauters M, Van Esch H, Friez MJ, Boespflug-Tanguy O, Zenker M, Vianna-Morgante AM, Rosenberg C, Ignatius J, Raynaud M, Hollanders K, Govaerts K, Vandenreijt K, Niel F, Blanc P, Stevenson RE, Fryns JP, Marynen P, Schwartz CE, Froyen G. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res. 2008;18:847–858. doi: 10.1101/gr.075903.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayele HK, Peyssonnaux C, Giatromanolaki A, Arrais-Silva WW, Mohamed HS, Collins H, Giorgio S, Koukourakis M, Johnson RS, Blackwell JM, Nizet V, Srai SK. HIF-1 regulates heritable variation and allele expression phenotypes of the macrophage immune response gene SLC11A1 from a Z-DNA forming microsatellite. Blood. 2007;110:3039–3048. doi: 10.1182/blood-2006-12-063289. [DOI] [PubMed] [Google Scholar]
- Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141:1159–1170. doi: 10.1016/j.cell.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem. 2007;282:32433–3241. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- Bena F, Gimelli S, Migliavacca E, Brun-Druc N, Buiting K, Antonarakis SE, Sharp AJ. A recurrent 14q32.2 microdeletion mediated by expanded TGG repeats. Hum Mol Genet. 2010;19:1967–1973. doi: 10.1093/hmg/ddq075. [DOI] [PubMed] [Google Scholar]
- Bengesser K, Cooper DN, Steinmann K, Kluwe L, Chuzhanova NA, Wimmer K, Tatagiba M, Tinschert S, Mautner VF, Kehrer-Sawatzki H. A novel third type of recurrent NF1 microdeletion mediated by nonallelic homologous recombination between LRRC37B-containing low-copy repeats in 17q11.2. Hum Mutat. 2010;31:742–751. doi: 10.1002/humu.21254. [DOI] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Lam KW, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake RD, Hess ST, Nicholson J. The influence of nearest neighbours on the rate and pattern of spontaneous point mutations. J Mol Evol. 1992;34:189–200. doi: 10.1007/BF00162968. [DOI] [PubMed] [Google Scholar]
- Boboila C, Jankovic M, Yan CT, Wang JH, Wesemann DR, Zhang T, Fazeli A, Feldman L, Nussenzweig A, Nussenzweig M, Alt FW. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc Natl Acad Sci USA. 2010;107:3034–3039. doi: 10.1073/pnas.0915067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochukova EG, Roscioli T, Hedges DJ, Taylor IB, Johnson D, David DJ, Deininger PL, Wilkie AO. Rare mutations of FGFR2 causing apert syndrome: identification of the first partial gene deletion, and an Alu element insertion from a new subfamily. Hum Mutat. 2009;30:204–211. doi: 10.1002/humu.20825. [DOI] [PubMed] [Google Scholar]
- Boland MJ, Christman JK. Characterization of Dnmt3b:thymine-DNA glycosylase interaction and stimulation of thymine glycosylase-mediated repair by DNA methyltransferase(s) and RNA. J Mol Biol. 2008;379:492–504. doi: 10.1016/j.jmb.2008.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Massagli A, Galluzzi R, Ciccone R, Zuffardi O. A familial inverted duplication/deletion of 2p25.1-25.3 provides new clues on the genesis of inverted duplications. Eur J Hum Genet. 2009;17:179–186. doi: 10.1038/ejhg.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet C, Vuillaumier-Barrot S, Gonzales M, Boukari S, Bizec CL, Fallet C, Delezoide AL, Moirot H, Laquerriere A, Encha-Razavi F, Durand G, Seta N. Detection of an Alu insertion in the POMT1 gene from three French Walker Warburg syndrome families. Mol Genet Metab. 2007;90:93–96. doi: 10.1016/j.ymgme.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Boyer JC, Yamada NA, Roques CN, Hatch SB, Riess K, Farber RA. Sequence dependent instability of mononucleotide microsatellites in cultured mismatch repair proficient and deficient mammalian cells. Hum Mol Genet. 2002;11:707–713. doi: 10.1093/hmg/11.6.707. [DOI] [PubMed] [Google Scholar]
- Brinkmann B, Klintschar M, Neuhuber F, Hühne J, Rolf B. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am J Hum Genet. 1998;62:1408–1415. doi: 10.1086/301869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr. Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Willemsen R, Oostra BA. Microsatellite repeat instability and neurological disease. Bioessays. 2009;31:71–83. doi: 10.1002/bies.080122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzin CH, Feng J, Yan J, Scaringe W, Liu Q, den Dunnen J, Mendell JR, Sommer SS. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum Mutat. 2005;25:177–188. doi: 10.1002/humu.20132. [DOI] [PubMed] [Google Scholar]
- Callinan PA, Wang J, Herke SW, Garber RK, Liang P, Batzer MA. Alu retrotransposition-mediated deletion. J Mol Biol. 2005;348:791–800. doi: 10.1016/j.jmb.2005.02.043. [DOI] [PubMed] [Google Scholar]
- Campregher C, Scharl T, Nemeth M, Honeder C, Jascur T, Boland CR, Gasche C. The nucleotide composition of microsatellites impacts both replication fidelity and mismatch repair in human colorectal cells. Hum Mol Genet. 2010;19:2648–2657. doi: 10.1093/hmg/ddq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. Copy number variation at the breakpoint region of isochromosome 17q. Genome Res. 2008;18:1724–1732. doi: 10.1101/gr.080697.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, Shaw C, Peacock S, Pursley A, Tavyev YJ, Ramocki MB, Nawara M, Obersztyn E, Vianna-Morgante AM, Stankiewicz P, Zoghbi HY, Cheung SW, Lupski JR. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cer RZ, Bruce KH, Mudunuri US, Yi M, Volfovsky N, Luke BT, Bacolla A, Collins JR, Stephens RM. Non-B DB: a database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res. 2011;39:D383–391. doi: 10.1093/nar/gkq1170. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–606. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- Champion KJ, Basehore MJ, Wood T, Destree A, Vannuffel P, Maystadt I. Identification and characterization of a novel homozygous deletion in the alpha-N-acetylglucosaminidase gene in a patient with Sanfilippo type B syndrome (mucopolysaccharidosis IIIB) Mol Genet Metab. 2010;100:51–56. doi: 10.1016/j.ymgme.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Chapman RW. The genome is the perfect imperfect machine. Proc Natl Acad Sci USA. 2010;107:E119. doi: 10.1073/pnas.1006896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvin A, Chen JM, Quemener S, Masson E, Kehrer-Sawatzki H, Ohmle B, Cooper DN, Le Maréchal C, Férec C. Elucidation of the complex structure and origin of the human trypsinogen locus triplication. Hum Mol Genet. 2009;18:3605–3614. doi: 10.1093/hmg/ddp308. [DOI] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Meta-analysis of gross insertions causing human genetic disease: novel mutational mechanisms and the role of replication slippage. Hum Mutat. 2005a;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Complex gene rearrangements caused by serial replication slippage. Hum Mutat. 2005b;26:125–134. doi: 10.1002/humu.20202. [DOI] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Intrachromosomal serial replication slippage in trans gives rise to diverse genomic rearrangements involving inversions. Hum Mutat. 2005c;26:362–373. doi: 10.1002/humu.20230. [DOI] [PubMed] [Google Scholar]
- Chen JM, Stenson PD, Cooper DN, Férec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum Genet. 2005d;117:411–427. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Chuzhanova N, Férec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet. 2007;8:762–775. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- Chen JM, Masson E, Macek M, Jr., Raguenes O, Piskackova T, Fercot B, Fila L, Cooper DN, Audrezet MP, Férec C. Detection of two Alu insertions in the CFTR gene. J Cyst Fibros. 2008;7:37–43. doi: 10.1016/j.jcf.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Chen JM, Férec C, Cooper DN. Closely spaced multiple mutations as potential signatures of transient hypermutability in human genes. Hum Mutat. 2009;30:1435–1448. doi: 10.1002/humu.21088. [DOI] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Férec C, Kehrer-Sawatzki H, Patrinos GP. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol. 2010;20:222–233. doi: 10.1016/j.semcancer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Chen CL, Duquenne L, Audit B, Guilbaud G, Rappailles A, Baker A, Huvet M, d’Aubenton-Carafa Y, Hyrien O, Arneodo A, Thermes C. Mol Biol Evol. 2011. Replication-associated mutational asymmetry in the human genome. In press. [DOI] [PubMed] [Google Scholar]
- Cheung LW, Lee YF, Ng TW, Ching WK, Khoo US, Ng MK, Wong AS. CpG/CpNpG motifs in the coding region are preferred sites for mutagenesis in the breast cancer susceptibility genes. FEBS Lett. 2007;581:4668–4674. doi: 10.1016/j.febslet.2007.08.061. [DOI] [PubMed] [Google Scholar]
- Chi LM, Lam SL. Nuclear magnetic resonance investigation of primer-template models: formation of a pyrimidine bulge upon misincorporation. Biochemistry. 2008;47:4469–4476. doi: 10.1021/bi8001456. [DOI] [PubMed] [Google Scholar]
- Chi LM, Lam SL. NMR investigation of DNA primer-template models: guanine templates are less prone to strand slippage upon misincorporation. Biochemistry. 2009;48:11478–11486. doi: 10.1021/bi9014049. [DOI] [PubMed] [Google Scholar]
- Choi BO, Kim NK, Park SW, Hyun YS, Jeon HJ, Hwang JH, Chung KW. Inheritance of Charcot-Marie-Tooth disease 1A with rare nonrecurrent genomic rearrangement. Neurogenetics. 2011;12:51–58. doi: 10.1007/s10048-010-0272-3. [DOI] [PubMed] [Google Scholar]
- Chung HR, Vingron M. Sequence-dependent nucleosome positioning. J Mol Biol. 2009;386:1411–1422. doi: 10.1016/j.jmb.2008.11.049. [DOI] [PubMed] [Google Scholar]
- Chung H, Young DJ, Lopez CG, Le TA, Lee JK, Ream-Robinson D, Huang SC, Carethers JM. Mutation rates of TGFBR2 and ACVR2 coding microsatellites in human cells with defective DNA mismatch repair. PLoS ONE. 2008;3:e3463. doi: 10.1371/journal.pone.0003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuzhanova NA, Anassis EJ, Ball EV, Krawczak M, Cooper DN. Meta-analysis of indels causing human genetic disease: mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat. 2003a;21:28–44. doi: 10.1002/humu.10146. [DOI] [PubMed] [Google Scholar]
- Chuzhanova N, Abeysinghe SS, Krawczak M, Cooper DN. Translocation and gross deletion breakpoints in human inherited disease and cancer II: Potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum Mutat. 2003b;22:245–251. doi: 10.1002/humu.10253. [DOI] [PubMed] [Google Scholar]
- Chuzhanova N, Chen JM, Bacolla A, Patrinos GP, Férec C, Wells RD, Cooper DN. Gene conversion causing human inherited disease: evidence for involvement of non-B-DNA-forming sequences and recombination-promoting motifs in DNA breakage and repair. Hum Mutat. 2009;30:1189–1198. doi: 10.1002/humu.21020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccone R, Mattina T, Giorda R, Bonaglia MC, Rocchi M, Pramparo T, Zuffardi O. Inversion polymorphisms and non-contiguous terminal deletions: the cause and the (unpredicted) effect of our genome architecture. J Med Genet. 2006;43:e19. doi: 10.1136/jmg.2005.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nat Genet. 1995;10:20–27. doi: 10.1038/ng0595-20. [DOI] [PubMed] [Google Scholar]
- Clément Y, Arndt PF. Substitution patterns are under different influences in primates and rodents. Genome Biol Evol. 2011;3:236–245. doi: 10.1093/gbe/evr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquempot O, Brault V, Babinet C, Herault Y. Fork stalling and template switching as a mechanism for polyalanine tract expansion affecting the DYC mutant of HOXD13, a new murine model of synpolydactyly. Genetics. 2009;183:23–30. doi: 10.1534/genetics.109.104695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collie AM, Landsverk ML, Ruzzo E, Mefford HC, Buysse K, Adkins JR, Knutzen DM, Barnett K, Brown RH, Jr., Parry GJ, Yum SW, Simpson DA, Olney RK, Chinnery PF, Eichler EE, Chance PF, Hannibal MC. Non-recurrent SEPT9 duplications cause hereditary neuralgic amyotrophy. J Med Genet. 2010;47:601–607. doi: 10.1136/jmg.2009.072348. [DOI] [PubMed] [Google Scholar]
- Conley ME, Partain JD, Norland SM, Shurtleff SA, Kazazian HH., Jr. Two independent retrotransposon insertions at the same site within the coding region of BTK. Hum Mutat. 2005;25:324–325. doi: 10.1002/humu.9321. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet. 2010;42:385–391. doi: 10.1038/ng.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Cooper DN. Human Gene Evolution. BIOS Scientific; Oxford: 1999. [Google Scholar]
- Cooper DN, Mort M, Stenson PD, Ball EV, Chuzhanova NA. Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides as well as in CpG dinucleotides. Hum Genomics. 2010;4:406–410. doi: 10.1186/1479-7364-4-6-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortázar D, Kunz C, Saito Y, Steinacher R, Schär P. The enigmatic thymine DNA glycosylase. DNA Repair. 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, Gage FH. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozar M, Bembi B, Dominissini S, Zampieri S, Vilageliu L, Grinberg D, Dardis A. Molecular characterization of a new deletion of the GBA1 gene due to an inter Alu recombination event. Mol Genet Metab. 2011;102:226–228. doi: 10.1016/j.ymgme.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Cui F, Zhurkin VB. Structure-based analysis of DNA sequence patterns guiding nucleosome positioning in vitro. J Biomol Struct Dyn. 2010;27:821–841. doi: 10.1080/073911010010524947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlow JM, Leach DR. Evidence for two preferred hairpin folding patterns in d(CGG).d(CCG) repeat tracts in vivo. J Mol Biol. 1998;275:17–23. doi: 10.1006/jmbi.1997.1452. [DOI] [PubMed] [Google Scholar]
- De S, Babu MM. A time-invariant principle of genome evolution. Proc Natl Acad Sci USA. 2010;107:13004–13009. doi: 10.1073/pnas.0914454107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Raedt T, Stephens M, Heyns I, Brems H, Thijs D, Messiaen L, Stephens K, Lazaro C, Wimmer K, Kehrer-Sawatzki H, Vidaud D, Kluwe L, Marynen P, Legius E. Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat Genet. 2006;38:1419–1423. doi: 10.1038/ng1920. [DOI] [PubMed] [Google Scholar]
- Denissenko MF, Chen JX, Tang MS, Pfeifer GP. Cytosine methylation determines hot spots of DNA damage in the human p53 gene. Proc Natl Acad Sci USA. 1997;94:3893–3898. doi: 10.1073/pnas.94.8.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donigan KA, Sweasy JB. Sequence context-specific mutagenesis and base excision repair. Mol Carcinogen. 2009;48:362–368. doi: 10.1002/mc.20497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet AJ, Hulme AE, Sahinovic E, Kulpa DA, Moldovan JB, Kopera HC, Athanikar JN, Hasnaoui M, Bucheton A, Moran JV, Gilbert N. Characterization of LINE-1 ribonucleoprotein particles. PLoS Genet. 2010;6:e1001150. doi: 10.1371/journal.pgen.1001150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy BM, Stenersen M, Egeland T, Olaisen B. Y-chromosomal microsatellite mutation rates: differences in mutation rate between and within loci. Hum Mutat. 2004;23:117–124. doi: 10.1002/humu.10294. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Eckert KA, Mowery A, Hile SE. Misalignment-mediated DNA polymerase beta mutations: comparison of microsatellite and frame-shift error rates using a forward mutation assay. Biochemistry. 2002;41:10490–10498. doi: 10.1021/bi025918c. [DOI] [PubMed] [Google Scholar]
- Eckert KA, Hile SE. Every microsatellite is different: Intrinsic DNA features dictate mutagenesis of common microsatellites present in the human genome. Mol Carcinog. 2009;48:379–388. doi: 10.1002/mc.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango N, Kim SH, Vigoda E, Yi SV. Mutations of different molecular origins exhibit contrasting patterns of regional substitution rate variation. PLoS Comput Biol. 2008;4:e1000015. doi: 10.1371/journal.pcbi.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Lokanga AR, Le W, Hoffman G, Usdin K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. 2010;31:611–616. doi: 10.1002/humu.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH., Jr. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010;20:1262–1270. doi: 10.1101/gr.106419.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116:5298–5305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiotti N, Moretti ME, Bussani R, Altamura N, Zamolo F, Gerloni R, Ukovich L, Ober E, Silvestri F, Grassi G, Adovasio R, Giansante C. Features of vulnerable plaques and clinical outcome of UA/NSTEMI: Relationship with matrix metalloproteinase functional polymorphisms. Atherosclerosis. 2011;215:153–159. doi: 10.1016/j.atherosclerosis.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- Franke G, Bausch B, Hoffmann MM, Cybulla M, Wilhelm C, Kohlhase J, Scherer G, Neumann HP. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: Molecular characterization and genotype-phenotype correlations in VHL patients. Hum Mutat. 2009;30:776–786. doi: 10.1002/humu.20948. [DOI] [PubMed] [Google Scholar]
- Fu W, Zhang F, Wang Y, Gu X, Jin L. Identification of copy number variation hotspots in human populations. Am J Hum Genet. 2010;87:494–504. doi: 10.1016/j.ajhg.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo CL, McCormick JF, Bubb VJ, Abid Alkadem DH, Li LS, McIver LJ, George AC, Boothman DA, Quinn JP, Skinner MA, Garner HR. A long AAAG repeat allele in the 5′ UTR of the ERR-γ gene is correlated with breast cancer predisposition and drives promoter activity in MCF-7 breast cancer cells. Breast Cancer Res Treat. 2011 doi: 10.1007/s10549-010-1237-9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallus GN, Cardaioli E, Rufa A, Da Pozzo P, Bianchi S, D’Eramo C, Collura M, Tumino M, Pavone L, Federico A. Alu-element insertion in an OPA1 intron sequence associated with autosomal dominant optic atrophy. Mol Vis. 2010;16:178–183. [PMC free article] [PubMed] [Google Scholar]
- Gallus GN, Cardaioli E, Rufa A, Da Pozzo P, Bianchi S, D’Eramo C, Collura M, Tumino M, Pavone L, Federico A. Alu-element insertion in an OPA1 intron sequence associated with autosomal dominant optic atrophy. Mol Vis. 2010;16:178–183. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell. 2006;124:331–342. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez JL, Marchetto MC, Muotri AR, Coufal NG, Gage FH, O’Shea KS, Moran JV. LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet. 2007;16:1569–1577. doi: 10.1093/hmg/ddm105. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, Hammer GD, Collins KL, O’Shea KS, Menendez P, Moran JV. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature. 2010;466:769–773. doi: 10.1038/nature09209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior SL, Preston G, Hedges DJ, Gilbert N, Moran JV, Deininger PL. Characterization of pre-insertion loci of de novo L1 insertions. Gene. 2007;390:190–198. doi: 10.1016/j.gene.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geggier S, Vologodskii A. Sequence dependence of DNA bending rigidity. Proc Natl Acad Sci USA. 2010;107:15421–15426. doi: 10.1073/pnas.1004809107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010;44:445–477. doi: 10.1146/annurev-genet-072610-155046. [DOI] [PubMed] [Google Scholar]
- Gentsch M, Kaczmarczyk A, van Leeuwen K, de Boer M, Kaus-Drobek M, Dagher MC, Kaiser P, Arkwright PD, Gahr M, Rösen-Wolff A, Bochtler M, Secord E, Britto-Williams P, Saifi GM, Maddalena A, Dbaibo G, Bustamante J, Casanova JL, Roos D, Roesler J. Alu-repeat-induced deletions within the NCF2 gene causing p67-phox-deficient chronic granulomatous disease (CGD) Hum Mutat. 2010;31:151–158. doi: 10.1002/humu.21156. [DOI] [PubMed] [Google Scholar]
- Gerrish P. Evolution plays dice. Nature. 2002;420:756–757. doi: 10.1038/420756a. [DOI] [PubMed] [Google Scholar]
- Giampieri C, Centurelli M, Bonafè M, Olivieri F, Cardelli M, Marchegiani F, Cavallone L, Giovagnetti S, Mugianesi E, Carrieri G, Lisa R, Cenerelli S, Testa R, Boemi M, Petropoulou C, Gonos ES, Franceschi C. A novel mitochondrial DNA-like sequence insertion polymorphism in intron I of the FOXO1A gene. Gene. 2004;327:215–219. doi: 10.1016/j.gene.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, Tonnies H, Ventura M, Zollino M, Neri G, Barber J, Wieczorek D, Rocchi M, Zuffardi O. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet. 2002;71:276–285. doi: 10.1086/341610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, Zuffardi O. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849–858. doi: 10.1093/hmg/ddg101. [DOI] [PubMed] [Google Scholar]
- Goldmann R, Tichy L, Freiberger T, Zapletalova P, Letocha O, Soska V, Fajkus J, Fajkusova L. Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med Genet. 2010;11:115. doi: 10.1186/1471-2350-11-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green P, Ewing B, Miller W, Thomas PJ, NISC Comparative Sequencing Program. Green ED. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514–517. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008:1–4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder R, Halder K, Sharma P, Garg G, Sengupta S, Chowdhury S. Guanine quadruplex DNA structure restricts methylation of CpG dinucleotides genome-wide. Mol Biosyst. 2010;6:2439–2447. doi: 10.1039/c0mb00009d. [DOI] [PubMed] [Google Scholar]
- Halling KC, Lazzaro CR, Honchel R, Bufill JA, Powell SM, Arndt CA, Lindor NM. Hereditary desmoid disease in a family with a germline Alu I repeat mutation of the APC gene. Hum Hered. 1999;49:97–102. doi: 10.1159/000022852. [DOI] [PubMed] [Google Scholar]
- Hannes F, Van Houdt J, Quarrell OW, Poot M, Hochstenbach R, Fryns JP, Vermeesch JR. Telomere healing following DNA polymerase arrest-induced breakages is likely the main mechanism generating chromosome 4p terminal deletions. Hum Mutat. 2010;31:1343–1351. doi: 10.1002/humu.21368. [DOI] [PubMed] [Google Scholar]
- Han K, Sen SK, Wang J, Callinan PA, Lee J, Cordaux R, Liang P, Batzer MA. Genomic rearrangements by LINE-1 insertion-mediated deletion in the human and chimpanzee lineages. Nucleic Acids Res. 2005;33:4040–4052. doi: 10.1093/nar/gki718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–378. doi: 10.1038/nature04112. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, Sleckman BP. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature. 2011;469:245–249. doi: 10.1038/nature09585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- Hess ST, Blake JD, Blake RD. Wide variations in neighbour-dependent substitution rates. J Mol Biol. 1994;236:1022–1033. doi: 10.1016/0022-2836(94)90009-4. [DOI] [PubMed] [Google Scholar]
- Hobart HH, Morris CA, Mervis CB, Pani AM, Kistler DJ, Rios CM, Kimberley KW, Gregg RG, Bray-Ward P. Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:220–228. doi: 10.1002/ajmg.c.30258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, Ladoukakis E, Eyre-Walker A. Cryptic variation in the human mutation rate. PLoS Biol. 2009;7:e1000027. doi: 10.1371/journal.pbio.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Lorimer HE, Jacobs HT. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell. 2000;100:515–524. doi: 10.1016/s0092-8674(00)80688-1. [DOI] [PubMed] [Google Scholar]
- Hsu GW, Ober M, Carell T, Beese LS. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004;431:217–221. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- Huang H, Winter EE, Wang H, Weinstock KG, Xing H, Goodstadt L, Stenson PD, Cooper DN, Smith D, Albà MM, Ponting CP, Fechtel K. Evolutionary conservation and selection of human disease gene orthologs in the rat and mouse genomes. Genome Biol. 2004;5:R47. doi: 10.1186/gb-2004-5-7-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, Wheelan SJ, Ji H, Boeke JD, Burns KH. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;141:1171–1182. doi: 10.1016/j.cell.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki H, Ohye T, Kogo H, Kato T, Bolor H, Taniguchi M, Shaikh TH, Emanuel BS, Kurahashi H. Chromosomal instability mediated by non-B DNA: cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2009;19:191–198. doi: 10.1101/gr.079244.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Thurston VC, Clarke JT, Yoneyama A, Rosenbarker L, Bird TD, Hodes ME, Shaffer LG, Lupski JR. Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet. 2002;71:838–853. doi: 10.1086/342728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs RJ, Spielmann HP. A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair. 2004;3:455–464. doi: 10.1016/j.dnarep.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov D, Hamby SE, Stenson PD, Phillips AD, Kehrer-Sawatzki H, Cooper DN, Chuzhanova N. Comparative analysis of germline and somatic microlesion mutational spectra in 17 human tumor suppressor genes. Hum Mutat. 2011;32:620–632. doi: 10.1002/humu.21483. [DOI] [PubMed] [Google Scholar]
- Jiang C, Zhao Z. Directionality of point mutation and 5-methylcytosine deamination rates in the chimpanzee genome. BMC Genomics. 2006a;7:316. doi: 10.1186/1471-2164-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Zhao Z. Mutational spectrum in the recent human genome inferred by single nucleotide polymorphisms. Genomics. 2006b;88:527–534. doi: 10.1016/j.ygeno.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet. 2000;9:901–908. doi: 10.1093/hmg/9.6.901. [DOI] [PubMed] [Google Scholar]
- Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH., Jr. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev. 2009;23:1303–1312. doi: 10.1101/gad.1803909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Inagaki H, Yamada K, Kogo H, Ohye T, Kowa H, Nagaoka K, Taniguchi M, Emanuel BS, Kurahashi H. Genetic variation affects de novo translocation frequency. Science. 2006;311:971. doi: 10.1126/science.1121452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH., Jr. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr., Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164–166. doi: 10.1038/332164a0. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Häussler J, Krone W, Bode H, Jenne DE, Mehnert KU, Tümmers U, Assum G. The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum Genet. 1997;99:237–247. doi: 10.1007/s004390050346. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Kluwe L, Fünsterer C, Mautner VF. Extensively high load of internal tumors determined by whole body MRI scanning in a patient with neurofibromatosis type 1 and a non-LCR-mediated 2-Mb deletion in 17q11.2. Hum Genet. 2005;116:466–475. doi: 10.1007/s00439-005-1265-4. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Schmid E, Fünsterer C, Kluwe L, Mautner VF. Absence of cutaneous neurofibromas in an NF1 patient with an atypical deletion partially overlapping the common 1.4 Mb microdeleted region. Am J Med Genet A. 2008;146A:691–699. doi: 10.1002/ajmg.a.32045. [DOI] [PubMed] [Google Scholar]
- Kelkar YD, Strubczewski N, Hile SE, Chiaromonte F, Eckert KA, Makova KD. What is a microsatellite: a computational and experimental definition based upon repeat mutational behavior at A/T and GT/AC repeats. Genome Biol Evol. 2010;2:620–635. doi: 10.1093/gbe/evq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenter AL, Birshtein BK. Chi, a promoter of generalized recombination in lambda phage, is present in immunoglobulin genes. Nature. 1981;293:402–404. doi: 10.1038/293402a0. [DOI] [PubMed] [Google Scholar]
- Khateb S, Weisman-Shomer P, Hershco I, Loeb LA, Fry M. Destabilization of tetraplex structures of the fragile X repeat sequence (CGG)n is mediated by homolog-conserved domains in three members of the hnRNP family. Nucleic Acids Res. 2004;32:4145–4154. doi: 10.1093/nar/gkh745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Lam HY, Urban AE, Korbel JO, Affourtit J, Grubert F, Chen X, Weissman S, Snyder M, Gerstein MB. Analysis of copy number variants and segmental duplications in the human genome: evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl DD, Bottema CD, Ketterling RP, Bridge PJ, Lillicrap DP, Sommer SS. Mutations causing hemophilia B: direct estimate of the underlying rates of spontaneous germ-line transitions, transversions, and deletions in a human gene. Am J Hum Genet. 1990;47:202–217. [PMC free article] [PubMed] [Google Scholar]
- Konkel MK, Batzer MA. A mobile threat to genome stability: The impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol. 2010;20:211–221. doi: 10.1016/j.semcancer.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 2003;21:12–27. doi: 10.1002/humu.10147. [DOI] [PubMed] [Google Scholar]
- Kondrashov FA, Kondrashov AS. Measurements of spontaneous rates of mutations in the recent past and the near future. Phil Trans R Soc B. 2010;365:1169–1176. doi: 10.1098/rstb.2009.0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS, Rogozin IB. Context of deletions and insertions in human coding sequences. Hum Mutat. 2004;23:177–185. doi: 10.1002/humu.10312. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, Knoers NV, van Kessel AG, Sistermans EA, Veltman JA, Brunner HG, de Vries BB. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destrée A, Grisart B, Rooms L, Van der Aa N, Field M, Hackett A, Bell K, Nowaczyk MJ, Mancini GM, Poddighe PJ, Schwartz CE, Rossi E, De Gregori M, Antonacci-Fulton LL, McLellan MD, 2nd, Garrett JM, Wiechert MA, Miner TL, Crosby S, Ciccone R, Willatt L, Rauch A, Zenker M, Aradhya S, Manning MA, Strom TM, Wagenstaller J, Krepischi-Santos AC, Vianna-Morgante AM, Rosenberg C, Price SM, Stewart H, Shaw-Smith C, Brunner HG, Wilkie AO, Veltman JA, Zuffardi O, Eichler EE, de Vries BB. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korona DA, LeCompte KG, Pursell ZF. The high fidelity and unique error signature of human DNA polymerase ε. Nucleic Acids Res. 2011;39:1763–1773. doi: 10.1093/nar/gkq1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koszul R, Caburet S, Dujon B, Fischer G. Eucaryotic genome evolution through the spontaneous duplication of large chromosomal segments. EMBO J. 2004;23:234–243. doi: 10.1038/sj.emboj.7600024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumbaris G, Hatzisevastou-Loukidou H, Alexandrou A, Ioannides M, Christodoulou C, Fitzgerald T, Rajan D, Clayton S, Kitsiou-Tzeli S, Vermeesch JR, Skordis N, Antoniou P, Kurg A, Georgiou I, Carter NP, Patsalis PC. FoSTeS, MMBIR and NAHR at the human proximal Xp region and the mechanisms of human Xq isochromosome formation. Hum Mol Genet. 2011;20:1925–1936. doi: 10.1093/hmg/ddr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Ball EV, Cooper DN. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet. 1998;63:474–488. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Turnbull DM. Mitochondrial DNA and genetic disease. Essays Biochem. 2010;47:139–151. doi: 10.1042/bse0470139. [DOI] [PubMed] [Google Scholar]
- Kulikowski LD, Yoshimoto M, da Silva Bellucco FT, Belangero SI, Christofolini DM, Pacanaro AN, Bortolai A, Cardozo Smith M de A, Squire JA, Melaragno MI. Cytogenetic molecular delineation of a terminal 18q deletion suggesting neo-telomere formation. Eur J Med Genet. 2010;53:404–407. doi: 10.1016/j.ejmg.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Misalignment-mediated DNA synthesis errors. Biochemistry. 1990;29:8003–8011. doi: 10.1021/bi00487a001. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279:16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet. 2001;10:2605–2617. doi: 10.1093/hmg/10.23.2605. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Inagaki H, Ohye T, Kogo H, Kato T, Emanuel BS. Chromosomal translocations mediated by palindromic DNA. Cell Cycle. 2006;5:1297–1303. doi: 10.4161/cc.5.12.2809. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, Tong M, Emanuel BS. The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin Genet. 2010;78:299–309. doi: 10.1111/j.1399-0004.2010.01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvikstad EM, Chiaromonte F, Makova KD. Ride the wavelet: a multiscale analysis of genomic contexts flanking small insertions and deletions. Genome Res. 2009;19:1153–1164. doi: 10.1101/gr.088922.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laken SJ, Petersen GM, Gruber SB, Oddoux C, Ostrer H, Giardiello FM, Hamilton SR, Hampel H, Markowitz A, Klimstra D, Jhanwar S, Winawer S, Offit K, Luce MC, Kinzler KW, Vogelstein B. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet. 1997;17:79–83. doi: 10.1038/ng0997-79. [DOI] [PubMed] [Google Scholar]
- Lam HY, Mu XJ, Stutz AM, Tanzer A, Cayting PD, Snyder M, Kim PM, Korbel JO, Gerstein MB. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol. 2010;28:47–55. doi: 10.1038/nbt.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, Wei CL. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gac G, Gourlaouen I, Ronsin C, Geromel V, Bourgarit A, Parquet N, Quemener S, Le Maréchal C, Chen JM, Férec C. Homozygous deletion of HFE produces a phenotype similar to the HFE p.C282Y/p.C282Y genotype. Blood. 2008;112:5238–5240. doi: 10.1182/blood-2008-07-167296. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Lee J, Jang SJ, Benoit N, Hoque MO, Califano JA, Trink B, Sidransky D, Mao L, Moon C. Presence of 5-methylcytosine in CpNpG trinucleotides in the human genome. Genomics. 2010;96:67–72. doi: 10.1016/j.ygeno.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol. 2011;18:75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Gao Y, Aach J, Zhang K, Kryukov GV, Xie B, Ahlford A, Yoon JK, Rosenbaum AM, Zaranek AW, LeProust E, Sunyaev SR, Church GM. Multiplex padlock targeted sequencing reveals human hypermutable CpG variations. Genome Res. 2009;19:1606–1615. doi: 10.1101/gr.092213.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Dent SY, Wilson JH, Wells RD, Napierala M. R loops stimulate genetic instability of CTG.CAG repeats. Proc Natl Acad Sci USA. 2010;107:692–697. doi: 10.1073/pnas.0909740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liskay RM, Letsou A, Stachelek JL. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics. 1987;115:161–167. doi: 10.1093/genetics/115.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Demple B. DNA repair in mammalian mitochondria: much more than we thought? Environ Mol Mutagen. 2010;51:417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- Liu G, Chen X, Bissler JJ, Sinden RR, Leffak M. Replication-dependent instability at (CTG) × (CAG) repeat hairpins in human cells. Nat Chem Biol. 2010;6:652–659. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Monnat RJ. DNA polymerases and human disease. Nat Rev Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]
- Longman-Jacobsen N, Williamson JF, Dawkins RL, Gaudieri S. In polymorphic genomic regions indels cluster with nucleotide polymorphism: Quantum Genomics. Gene. 2003;312:257–261. doi: 10.1016/s0378-1119(03)00621-8. [DOI] [PubMed] [Google Scholar]
- Lopez Castel A, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- Lopez-Correa C, Dorschner M, Brems H, Lazaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, Fryns JP, Marynen P, Stephens K, Legius E. Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet. 2001;10:1387–1392. doi: 10.1093/hmg/10.13.1387. [DOI] [PubMed] [Google Scholar]
- Lyons DM, O’Brien PJ. Human base excision repair creates a bias toward −1 frameshift mutations. J Biol Chem. 2010;285:25203–25212. doi: 10.1074/jbc.M110.118596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIver LJ, Fondon JW, 3rd, Skinner MA, Garner HR. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics. 2011;97:193–199. doi: 10.1016/j.ygeno.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhang T, Huang Z, Jiang X, Tao S. Patterns of nucleotides that flank substitutions in human orthologous genes. BMC Genomics. 2010;11:416. doi: 10.1186/1471-2164-11-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini D, Singh S, Ainsworth P, Rodenhiser D. Constitutively methylated CpG dinucleotides as mutation hot spots in the retinoblastoma gene (RB1) Am J Hum Genet. 1997;61:80–87. doi: 10.1086/513898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Sànchez-Ruiz J, Armengol L, Khaja R, Bertranpetit J, Lopez-Bigas N, Rocchi M, Gazave E, Navarro A. On the association between chromosomal rearrangements and genic evolution in humans and chimpanzees. Genome Biol. 2007;8:R230. doi: 10.1186/gb-2007-8-10-r230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, Eichler EE. The evolution of human segmental duplications and the core duplicon hypothesis. Cold Spring Harb Symp Quant Biol. 2009;74:355–362. doi: 10.1101/sqb.2009.74.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson E, Le Maréchal C, Levy P, Chuzhanova N, Ruszniewski P, Cooper DN, Chen JM, Férec C. Co-inheritance of a novel deletion of the entire SPINK1 gene with a CFTR missense mutation (L997F) in a family with chronic pancreatitis. Mol Genet Metab. 2007;92:168–175. doi: 10.1016/j.ymgme.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Mathews CK. DNA precursor metabolism and genomic stability. FASEB J. 2006;20:1300–1314. doi: 10.1096/fj.06-5730rev. [DOI] [PubMed] [Google Scholar]
- Mazurek A, Johnson CN, Germann MW, Fishel R. Sequence context effect for hMSH2-hMSH6 mismatch-dependent activation. Proc Natl Acad Sci USA. 2009;106:4177–4182. doi: 10.1073/pnas.0808572106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaburn KJ, Misteli T, Soutoglou E. Spatial genome organization in the formation of chromosomal translocations. Semin Cancer Biol. 2007;17:80–90. doi: 10.1016/j.semcancer.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, Tommerup N, Eichler EE, Bellanne-Chantelot C. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet. 2007;81:1057–1069. doi: 10.1086/522591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV, Parkin G, Fichera M, Reitano S, Lo Giudice M, Li KE, Casuga I, Broomer A, Conrad B, Schwerzmann M, Räber L, Gallati S, Striano P, Coppola A, Tolmie JL, Tobias ES, Lilley C, Armengol L, Spysschaert Y, Verloo P, De Coene A, Goossens L, Mortier G, Speleman F, van Binsbergen E, Nelen MR, Hochstenbach R, Poot M, Gallagher L, Gill M, McClellan J, King MC, Regan R, Skinner C, Stevenson RE, Antonarakis SE, Chen C, Estivill X, Menten B, Gimelli G, Gribble S, Schwartz S, Sutcliffe JS, Walsh T, Knight SJ, Sebat J, Romano C, Schwartz CE, Veltman JA, de Vries BB, Vermeesch JR, Barber JC, Willatt L, Tassabehji M, Eichler EE. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaed C, Rouleau GA. Molecular mechanisms underlying polyalanine diseases. Neurobiol Dis. 2009;34:397–405. doi: 10.1016/j.nbd.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C, Kehrer-Sawatzki H. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32:213–219. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- Messer PW, Arndt PF. The majority of recent short DNA insertions in the human genome are tandem duplications. Mol Biol Evol. 2007;24:1190–1197. doi: 10.1093/molbev/msm035. [DOI] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52:643–645. [PubMed] [Google Scholar]
- Millar DS, Tysoe C, Lazarou LP, Pilz DT, Mohammed S, Anderson K, Chuzhanova N, Cooper DN, Butler R. An isolated case of lissencephaly caused by the insertion of a mitochondrial genome-derived DNA sequence into the 5′ untranslated region of the PAFAH1B1 (LIS1) gene. Hum Genomics. 2010;4:384–393. doi: 10.1186/1479-7364-4-6-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stütz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Ye K, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO, 1000 Genomes Project Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miné M, Chen JM, Brivet M, Desguerre I, Marchant D, de Lonlay P, Bernard A, Férec C, Abitbol M, Ricquier D, Marsac C. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Hum Mutat. 2007;28:137–142. doi: 10.1002/humu.20449. [DOI] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Misawa K, Kikuno RF. Evaluation of the effect of CpG hypermutability on human codon substitution. Gene. 2009;431:18–22. doi: 10.1016/j.gene.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Brandon M, Wallace DC. Mitochondrial DNA-like sequences in the nucleus (NUMTs): insights into our African origins and the mechanism of foreign DNA integration. Hum Mutat. 2004;23:125–133. doi: 10.1002/humu.10304. [DOI] [PubMed] [Google Scholar]
- Molla M, Delcher A, Sunyaev S, Cantor C, Kasif S. Triplet repeat length bias and variation in the human transcriptome. Proc Natl Acad Sci USA. 2009;106:17095–17100. doi: 10.1073/pnas.0907112106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisada N, Rendtorff ND, Nozu K, Morishita T, Miyakawa T, Matsumoto T, Hisano S, Iijima K, Tranebjaerg L, Shirahata A, Matsuo M, Kusuhara K. Branchio-oto-renal syndrome caused by partial EYA1 deletion due to LINE-1 insertion. Pediatr Nephrol. 2010;25:1343–1348. doi: 10.1007/s00467-010-1445-x. [DOI] [PubMed] [Google Scholar]
- Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugal CF, von Grünberg HH, Peifer M. Transcription-induced mutational strand bias and its effect on substitution rates in human genes. Mol Biol Evol. 2009;26:131–142. doi: 10.1093/molbev/msn245. [DOI] [PubMed] [Google Scholar]
- Muniappan BP, Thilly WG. The DNA polymerase beta replication error spectrum in the adenomatous polyposis coli gene contains human colon tumor mutational hotspots. Cancer Res. 2002;62:3271–3275. [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- Musova Z, Hedvicakova P, Mohrmann M, Tesarova M, Krepelova A, Zeman J, Sedlacek Z. A novel insertion of a rearranged L1 element in exon 44 of the dystrophin gene: further evidence for possible bias in retroposon integration. Biochem Biophys Res Commun. 2006;347:145–149. doi: 10.1016/j.bbrc.2006.06.071. [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakken S, Rødland EA, Hovig E. Impact of DNA physical properties on local sequence bias of human mutation. Hum Mutat. 2010;31:1316–1325. doi: 10.1002/humu.21371. [DOI] [PubMed] [Google Scholar]
- Napierala M, Bacolla A, Wells RD. Increased negative superhelical density in vivo enhances the genetic instability of triplet repeat sequences. J Biol Chem. 2005;280:37366–37376. doi: 10.1074/jbc.M508065200. [DOI] [PubMed] [Google Scholar]
- Narita N, Nishio H, Kitoh Y, Ishikawa Y, Minami R, Nakamura H, Matsuo M. Insertion of a 5′ truncated L1 element into the 3′ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J Clin Invest. 1993;91:1862–1867. doi: 10.1172/JCI116402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necşulea A, Popa A, Cooper DN, Stenson PD, Mouchiroud D, Gautier C, Duret L. Meiotic recombination favors the spreading of deleterious mutations in human populations. Hum Mutat. 2011;32:198–206. doi: 10.1002/humu.21407. [DOI] [PubMed] [Google Scholar]
- Neiman M, Taylor DR. The causes of mutation accumulation in mitochondrial genomes. Proc Biol Sci. 2009;276:1201–1209. doi: 10.1098/rspb.2008.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevarez PA, DeBoever CM, Freeland BJ, Quitt MA, Bush EC. Context dependent substitution biases vary within the human genome. BMC Bioinformatics. 2010;11:462. doi: 10.1186/1471-2105-11-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol Edamura K, Leonard MR, Pearson CE. Role of replication and CpG methylation in fragile X syndrome CGG deletions in primate cells. Am J Hum Genet. 2005;76:302–311. doi: 10.1086/427928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- Nobile C, Toffolatti L, Rizzi F, Simionati B, Nigro V, Cardazzo B, Patarnello T, Valle G, Danieli GA. Analysis of 22 deletion breakpoints in dystrophin intron 49. Hum Genet. 2002;110:418–421. doi: 10.1007/s00439-002-0721-7. [DOI] [PubMed] [Google Scholar]
- O’Neill JP, Finette BA. Transition mutations at CpG dinucleotides are the most frequent in vivo spontaneous single-based substitution mutation in the human HPRT gene. Environ Mol Mutagen. 1998;32:188–191. doi: 10.1002/(sici)1098-2280(1998)32:2<188::aid-em16>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Okubo M, Horinishi A, Saito M, Ebara T, Endo Y, Kaku K, Murase T, Eto M. A novel complex deletion-insertion mutation mediated by Alu repetitive elements leads to lipoprotein lipase deficiency. Mol Genet Metab. 2007;92:229–233. doi: 10.1016/j.ymgme.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Oldridge M, Zackai EH, McDonald-McGinn DM, Iseki S, Morriss-Kay GM, Twigg SR, Johnson D, Wall SA, Jiang W, Theda C, Jabs EW, Wilkie AO. De novo alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am J Hum Genet. 1999;64:446–461. doi: 10.1086/302245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, Mandel A, Costa T, Grebe T, Cox S, Tsui LC, Scherer SW. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–325. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima J, Magner DB, Lee JA, Breman AM, Schmitt ES, White LD, Crowe CA, Merrill M, Jayakar P, Rajadhyaksha A, Eng CM, del Gaudio D. Regional genomic instability predisposes to complex dystrophin gene rearrangements. Hum Genet. 2009;126:411–423. doi: 10.1007/s00439-009-0679-9. [DOI] [PubMed] [Google Scholar]
- Ostertag EM, DeBerardinis RJ, Goodier JL, Zhang Y, Yang N, Gerton GL, Kazazian HH., Jr. A mouse model of human L1 retrotransposition. Nat Genet. 2002;32:655–660. doi: 10.1038/ng1022. [DOI] [PubMed] [Google Scholar]
- Ou Z, Stankiewicz P, Xia Z, Breman AM, Dawson B, Wiszniewska J, Szafranski P, Cooper ML, Rao M, Shao L, South ST, Coleman K, Fernhoff PM, Deray MJ, Rosengren S, Roeder ER, Enciso VB, Chinault AC, Patel A, Kang SH, Shaw CA, Lupski JR, Cheung SW. Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomes. Genome Res. 2011;21:33–46. doi: 10.1101/gr.111609.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi GB, Slean MM, Simard JP, Gileadi O, Pearson CE. Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSbeta, but clustered slip-outs are poorly repaired. Proc Natl Acad Sci USA. 2010;107:12593–12598. doi: 10.1073/pnas.0909087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier R, Krasilnikova MM, Samadashwily GM, Lahue R, Mirkin SM. Replication and expansion of trinucleotide repeats in yeast. Mol Cell Biol. 2003;23:1349–1357. doi: 10.1128/MCB.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DJ, Carrell RW. CpG dinucleotides are “hotspots” for mutation in the antithrombin III gene. Twelve variants identified using the polymerase chain reaction. Mol Biol Med. 1989;6:239–243. [PubMed] [Google Scholar]
- Peters JP, 3rd, Maher LJ. DNA curvature and flexibility in vitro and in vivo. Q Rev Biophys. 2010;43:23–63. doi: 10.1017/S0033583510000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP. Mutagenesis at methylated CpG sequences. Curr Top Microbiol Immunol. 2006;301:259–281. doi: 10.1007/3-540-31390-7_10. [DOI] [PubMed] [Google Scholar]
- Pfeifer GP, Besaratinia A. Mutational spectra of human cancer. Hum Genet. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard V, Chen JM, Tardy B, Aillaud MF, Boiteux-Vergnes C, Dreyfus M, Emmerich J, Lavenu-Bombled C, Nowak-Gottl U, Trillot N, Aiach M, Alhenc-Gelas M. Detection and characterisation of large SERPINC1 deletions in type I inherited antithrombin deficiency. Hum Genet. 2010;127:45–53. doi: 10.1007/s00439-009-0742-6. [DOI] [PubMed] [Google Scholar]
- Polak P, Arndt PF. Transcription induces strand-specific mutations at the 5′ end of human genes. Genome Res. 2008;18:1216–1223. doi: 10.1101/gr.076570.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potaman VN, Bissler JJ, Hashem VI, Oussatcheva EA, Lu L, Shlyakhtenko LS, Lyubchenko YL, Matsuura T, Ashizawa T, Leffak M, Benham CJ, Sinden RR. Unpaired structures in SCA10 (ATTCT)n.(AGAAT)n repeats. J Mol Biol. 2003;326:1095–1111. doi: 10.1016/s0022-2836(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- Punga T, Buhler M. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med. 2010;2:120–129. doi: 10.1002/emmm.201000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quemener S, Chen JM, Chuzhanova N, Benech C, Casals T, Macek M, Jr., Bienvenu T, McDevitt T, Farrell PM, Loumi O, Messaoud T, Cuppens H, Cutting GR, Stenson PD, Giteau K, Audrézet MP, Cooper DN, Férec C. Complete ascertainment of intragenic copy number mutations (CNMs) in the CFTR gene and its implications for CNM formation at other autosomal loci. Hum Mutat. 2010;31:421–428. doi: 10.1002/humu.21196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quental R, Azevedo L, Rubio V, Diogo L, Amorim A. Molecular mechanisms underlying large genomic deletions in ornithine transcarbamylase (OTC) gene. Clin Genet. 2009;75:457–464. doi: 10.1111/j.1399-0004.2009.01172.x. [DOI] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PN, Li W, Vissers LE, Veltman JA, Ophoff RA. Recurrent inversion events at 17q21.31 microdeletion locus are linked to the MAPT H2 haplotype. Cytogenet Genome Res. 2010;129:275–279. doi: 10.1159/000315901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renciuk D, Kypr J, Vorlickova M. CGG repeats associated with fragile X chromosome form left-handed Z-DNA structure. Biopolymers. 2011;95:174–181. doi: 10.1002/bip.21555. [DOI] [PubMed] [Google Scholar]
- Resta N, Giorda R, Bagnulo R, Beri S, Della Mina E, Stella A, Piglionica M, Susca FC, Guanti G, Zuffardi O, Ciccone R. Breakpoint determination of 15 large deletions in Peutz-Jeghers subjects. Hum Genet. 2010;128:373–382. doi: 10.1007/s00439-010-0859-7. [DOI] [PubMed] [Google Scholar]
- Rhodes CH, Call KM, Budarf ML, Barnoski BL, Bell CJ, Emanuel BS, Bigner SH, Park JP, Mohandas TK. Molecular studies of an ependymoma-associated constitutional t(1;22)(p22;q11.2) Cytogenet Cell Genet. 1997;78:247–252. doi: 10.1159/000134667. [DOI] [PubMed] [Google Scholar]
- Ricchetti M, Tekaia F, Dujon B. Continued colonization of the human genome by mitochondrial DNA. PLoS Biol. 2004;2:E273. doi: 10.1371/journal.pbio.0020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout WM, 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990;249:1288–1290. doi: 10.1126/science.1697983. [DOI] [PubMed] [Google Scholar]
- Rodenhiser DI, Andrews JD, Mancini DN, Jung JH, Singh SM. Homonucleotide tracts, short repeats and CpG/CpNpG motifs are frequent sites for heterogeneous mutations in the neurofibromatosis type 1 (NF1) tumour-suppressor gene. Mutat Res. 1997;373:185–195. doi: 10.1016/s0027-5107(96)00171-6. [DOI] [PubMed] [Google Scholar]
- Roehl AC, Vogt J, Mussotter T, Zickler AN, Spoti H, Hogel J, Chuzhanova NA, Wimmer K, Kluwe L, Mautner VF, Cooper DN, Kehrer-Sawatzki H. Intrachromosomal mitotic nonallelic homologous recombination is the major molecular mechanism underlying type-2 NF1 deletions. Hum Mutat. 2010;31:1163–1173. doi: 10.1002/humu.21340. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res. 2003;544:65–85. doi: 10.1016/s1383-5742(03)00032-2. [DOI] [PubMed] [Google Scholar]
- Rooms L, Reyniers E, Kooy RF. Diverse chromosome breakage mechanisms underlie subtelomeric rearrangements, a common cause of mental retardation. Hum Mutat. 2007;28:177–182. doi: 10.1002/humu.20421. [DOI] [PubMed] [Google Scholar]
- Rudiger NS, Gregersen N, Kielland-Brandt MC. One short well conserved region of Alu-sequences is involved in human gene rearrangements and has homology with prokaryotic chi. Nucleic Acids Res. 1995;23:256–260. doi: 10.1093/nar/23.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikovic B, Wang J, El-Hattab A, Landsverk M, Douglas G, Brundage EK, Craigen WJ, Schmitt ES, Wong L-JC. Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes. PLoS ONE. 2010;5:e15687. doi: 10.1371/journal.pone.0015687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, Wells RD. Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- Samuels DC, Schon EA, Chinnery PF. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004;20:393–398. doi: 10.1016/j.tig.2004.07.003. [DOI] [PubMed] [Google Scholar]
- SantaLucia J, Jr, Hicks D. The thermodynamics of DNA structural motifs. Annu Rev Biophys Biomol Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- Schollen E, Keldermans L, Foulquier F, Briones P, Chabas A, Sanchez-Valverde F, Adamowicz M, Pronicka E, Wevers R, Matthijs G. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Mol Genet Metab. 2007;90:408–413. doi: 10.1016/j.ymgme.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Scott SA, Cohen N, Brandt T, Warburton PE, Edelmann L. Large inverted repeats within Xp11.2 are present at the breakpoints of isodicentric X chromosomes in Turner syndrome. Hum Mol Genet. 2010;19:3383–3393. doi: 10.1093/hmg/ddq250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res. 2010;704:152–159. doi: 10.1016/j.mrrev.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert E, Ross JB, Osman R. Role of DNA flexibility in sequence-dependent activity of uracil DNA glycosylase. Biochemistry. 2002;41:10976–10984. doi: 10.1021/bi026121o. [DOI] [PubMed] [Google Scholar]
- Seibert E, Ross JB, Osman R. Contribution of opening and bending dynamics to specific recognition of DNA damage. J Mol Biol. 2003;330:687–703. doi: 10.1016/s0022-2836(03)00598-9. [DOI] [PubMed] [Google Scholar]
- Shannon M, Richardson L, Christian A, Handel MA, Thelen MP. Differential gene expression of mammalian SPO11/TOP6A homologs during meiosis. FEBS Lett. 1999;462:329–334. doi: 10.1016/s0014-5793(99)01546-x. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJ, Eichler EE. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, Murday V, Mansour S, Fichera M, Castiglia L, Failla P, Ventura M, Jiang Z, Cooper GM, Knight SJ, Romano C, Zuffardi O, Chen C, Schwartz CE, Eichler EE. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, Porter K, Prigmore E, Krepischi-Santos AC, Varela MC, Koiffmann CP, Lees AJ, Rosenberg C, Firth HV, de Silva R, Carter NP. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- Sheen CR, Jewell UR, Morris CM, Brennan SO, Férec C, George PM, Smith MP, Chen JM. Double complex mutations involving F8 and FUNDC2 caused by distinct break-induced replication. Hum Mutat. 2007;28:1198–1206. doi: 10.1002/humu.20591. [DOI] [PubMed] [Google Scholar]
- Shen JC, Rideout WM, 3rd, Jones PA. High frequency mutagenesis by a DNA methyltransferase. Cell. 1992;71:1073–1080. doi: 10.1016/s0092-8674(05)80057-1. [DOI] [PubMed] [Google Scholar]
- Shen JC, Rideout WM, 3rd, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994;22:972–976. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan MB, Kato T, Haldeman-Englert C, Jalali GR, Milunsky JM, Zou Y, Klaes R, Gimelli G, Gimelli S, Gemmill RM, Drabkin HA, Hacker AM, Brown J, Tomkins D, Shaikh TH, Kurahashi H, Zackai EH, Emanuel BS. A palindrome-mediated recurrent translocation with 3:1 meiotic nondisjunction: the t(8;22)(q24.13;q11.21) Am J Hum Genet. 2010;87:209–218. doi: 10.1016/j.ajhg.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimajiri S, Arima N, Tanimoto A, Murata Y, Hamada T, Wang KY, Sasaguri Y. Shortened microsatellite d(CA)21 sequence down-regulates promoter activity of matrix metalloproteinase 9 gene. FEBS Lett. 1999;455:70–74. doi: 10.1016/s0014-5793(99)00863-7. [DOI] [PubMed] [Google Scholar]
- Shiroeda H, Tahara T, Shibata T, Nakamura M, Yamada H, Nomura T, Hayashi R, Saito T, Fukuyama T, Otsuka T, Yano H, Ozaki K, Tsuchishima M, Tsutsumi M, Arisawa T. Functional promoter polymorphisms of macrophage migration inhibitory factor in peptic ulcer diseases. Int J Mol Med. 2010;26:707–711. doi: 10.3892/ijmm_00000517. [DOI] [PubMed] [Google Scholar]
- Shlien A, Baskin B, Achatz MI, Stavropoulos DJ, Nichols KE, Hudgins L, Morel CF, Adam MP, Zhukova N, Rotin L, Novokmet A, Druker H, Shago M, Ray PN, Hainaut P, Malkin D. A common molecular mechanism underlies two phenotypically distinct 17p13.1 microdeletion syndromes. Am J Hum Genet. 2010;87:631–642. doi: 10.1016/j.ajhg.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddle KJ, Goodship JA, Keavney B, Santibanez-Koref MF. Bases adjacent to mononucleotide repeats show an increased single nucleotide polymorphism frequency in the human genome. Bioinformatics. 2011;27:895–898. doi: 10.1093/bioinformatics/btr067. [DOI] [PubMed] [Google Scholar]
- Siepel A, Haussler D. Phylogenetic likelihood estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjödin P, Bataillon T, Schierup MH. Insertion and deletion processes in recent human history. PLoS ONE. 2010;5:e8650. doi: 10.1371/journal.pone.0008650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GR. Chi hotspots of generalized recombination. Cell. 1983;34:709–710. doi: 10.1016/0092-8674(83)90525-1. [DOI] [PubMed] [Google Scholar]
- Soejima M, Fujihara J, Takeshita H, Koda Y. Sec1-FUT2-Sec1 hybrid allele generated by interlocus gene conversion. Transfusion. 2008;48:488–492. doi: 10.1111/j.1537-2995.2007.01553.x. [DOI] [PubMed] [Google Scholar]
- Son LS, Bacolla A, Wells RD. Sticky DNA: in vivo formation in E. coli and in vitro association of long GAA*TTC tracts to generate two independent supercoiled domains. J Mol Biol. 2006;360:267–284. doi: 10.1016/j.jmb.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Shaw CJ, Dapper JD, Wakui K, Shaffer LG, Withers M, Elizondo L, Park SS, Lupski JR. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet. 2003;72:1101–1116. doi: 10.1086/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltzfus A. Evidence for a predominant role of oxidative damage in germline mutation in mammals. Mutat Res. 2008;644:71–73. doi: 10.1016/j.mrfmmm.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Sutton MD. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim Biophys Acta. 2010;1804:1167–1179. doi: 10.1016/j.bbapap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata A, Sheng JS, Ushikai M, Song YZ, Gao HZ, Lu YB, Okumura F, Iijima M, Mutoh K, Kishida S, Saheki T, Kobayashi K. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. 2008;53:534–545. doi: 10.1007/s10038-008-0282-2. [DOI] [PubMed] [Google Scholar]
- Takasu M, Hayashi R, Maruya E, Ota M, Imura K, Kougo K, Kobayashi C, Saji H, Ishikawa Y, Asai T, Tokunaga K. Deletion of entire HLA-A gene accompanied by an insertion of a retrotransposon. Tissue Antigens. 2007;70:144–150. doi: 10.1111/j.1399-0039.2007.00870.x. [DOI] [PubMed] [Google Scholar]
- Tan EC, Li H. Characterization of frequencies and distribution of single nucleotide insertions/deletions in the human genome. Gene. 2006;376:268–280. doi: 10.1016/j.gene.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Tanay A, Siggia ED. Sequence context affects the rate of short insertions and deletions in flies and primates. Genome Biol. 2008;9:R37. doi: 10.1186/gb-2008-9-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Ponting CP, Copley RR. Occurrence and consequences of coding sequence insertions and deletions in mammalian genomes. Genome Res. 2004;14:555–566. doi: 10.1101/gr.1977804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Wang Q, Zhang P, Araki H, Yang S, Kreitman M, Nagylaki T, Hudson R, Bergelson J, Chen JQ. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature. 2008;455:105–108. doi: 10.1038/nature07175. [DOI] [PubMed] [Google Scholar]
- Tippin B, Kobayashi S, Bertram JG, Goodman MF. To slip or skip, visualizing frameshift mutation dynamics for error-prone DNA polymerases. J Biol Chem. 2004;279:45360–45368. doi: 10.1074/jbc.M408600200. [DOI] [PubMed] [Google Scholar]
- Toffolatti L, Cardazzo B, Nobile C, Danieli GA, Gualandi F, Muntoni F, Abbs S, Zanetti P, Angelini C, Ferlini A, Fanin M, Patarnello T. Investigating the mechanism of chromosomal deletion: characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics. 2002;80:523–530. [PubMed] [Google Scholar]
- Tolstorukov MY, Volfovsky N, Stephens RM, Park PJ. Impact of chromatin structure on sequence variability in the human genome. Nat Struct Mol Biol. 2011;18:510–515. doi: 10.1038/nsmb.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomé S, Panigrahi GB, López Castel A, Foiry L, Melton DW, Gourdon G, Pearson CE. Maternal germline-specific effect of DNA ligase I on CTG/CAG instability. Hum Mol Genet. 2011;20:2131–2143. doi: 10.1093/hmg/ddr099. [DOI] [PubMed] [Google Scholar]
- Tomso DJ, Bell DA. Sequence context at human single nucleotide polymorphisms: overrepresentation of CpG dinucleotide at polymorphic sites and suppression of variation in CpG islands. J Mol Biol. 2003;327:303–308. doi: 10.1016/s0022-2836(03)00120-7. [DOI] [PubMed] [Google Scholar]
- Tornaletti S. Transcriptional processing of G4 DNA. Mol Carcinog. 2009;48:326–335. doi: 10.1002/mc.20513. [DOI] [PubMed] [Google Scholar]
- Truong HT, Dudding T, Blanchard CL, Elsea SH. Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature. BMC Med Genet. 2010;11:142. doi: 10.1186/1471-2350-11-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuohy TM, Done MW, Lewandowski MS, Shires PM, Saraiya DS, Huang SC, Neklason DW, Burt RW. Large intron 14 rearrangement in APC results in splice defect and attenuated FAP. Hum Genet. 2010;127:359–369. doi: 10.1007/s00439-009-0776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner C, Killoran C, Thomas NS, Rosenberg M, Chuzhanova NA, Johnston J, Kemel Y, Cooper DN, Biesecker LG. Human genetic disease caused by de novo mitochondrial-nuclear DNA transfer. Hum Genet. 2003;112:303–309. doi: 10.1007/s00439-002-0892-2. [DOI] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin K, Woodford KJ. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995;23:4202–4209. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk JA, Meij IC, Seleme MC, Kano H, Nikopoulos K, Hoefsloot LH, Sistermans EA, de Wijs IJ, Mukhopadhyay A, Plomp AS, de Jong PT, Kazazian HH, Cremers FP. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet. 2007;16:1587–1592. doi: 10.1093/hmg/ddm108. [DOI] [PubMed] [Google Scholar]
- Védrine SM, Vourc’h P, Tabagh R, Mignon L, Höfflin S, Cherpi-Antar C, Mbarek O, Paubel A, Moraine C, Raynaud M, Andres CR. A functional tetranucleotide (AAAT) polymorphism in an Alu element in the NF1 gene is associated with mental retardation. Neurosci Lett. 2011;491:118–121. doi: 10.1016/j.neulet.2011.01.019. [DOI] [PubMed] [Google Scholar]
- Vidaud D, Vidaud M, Bahnak BR, Siguret V, Gispert Sanchez S, Laurian Y, Meyer D, Goossens M, Lavergne JM. Haemophilia B due to a de novo insertion of a human-specific Alu subfamily member within the coding region of the factor IX gene. Eur J Hum Genet. 1993;1:30–36. doi: 10.1159/000472385. [DOI] [PubMed] [Google Scholar]
- Vikman S, Brena RM, Armstrong P, Hartiala J, Stephensen CB, Allayee H. Functional analysis of 5-lipoxygenase promoter repeat variants. Hum Mol Genet. 2009;18:4521–4529. doi: 10.1093/hmg/ddp414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser R, Shimokawa O, Harada N, Kinoshita A, Ohta T, Niikawa N, Matsumoto N. Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. Am J Hum Genet. 2005;76:52–67. doi: 10.1086/426950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, Bhatt SS, Janssen IM, Xia Z, Lalani SR, Pfundt R, Derwinska K, de Vries BB, Gilissen C, Hoischen A, Nesteruk M, Wisniowiecka-Kowalnik B, Smyk M, Brunner HG, Cheung SW, van Kessel AG, Veltman JA, Stankiewicz P. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum Mol Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- Völker J, Plum GE, Klump HH, Breslauer KJ. Energy crosstalk between DNA lesions: implications for allosteric coupling of DNA repair and triplet repeat expansion pathways. J Am Chem Soc. 2010;132:4095–4097. doi: 10.1021/ja1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- Walser JC, Ponger L, Furano AV. CpG dinucleotides and the mutation rate of non-CpG DNA. Genome Res. 2008;18:1403–1414. doi: 10.1101/gr.076455.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walser JC, Furano AV. The mutational spectrum of non-CpG DNA varies with CpG content. Genome Res. 2010;20:875–882. doi: 10.1101/gr.103283.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CP, Xu GL. Cytosine methylation and DNA repair. Curr Top Microbiol Immunol. 2006;301:283–315. doi: 10.1007/3-540-31390-7_11. [DOI] [PubMed] [Google Scholar]
- Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci USA. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst. 2008;100:1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Vasquez KM. Models for chromosomal replication-independent non-B DNA structure-induced genetic instability. Mol Carcinog. 2009;48:286–298. doi: 10.1002/mc.20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang Y, Schofield MJ, Du C, Fridman Y, Lee SD, Larson ED, Drummond JT, Alani E, Hsieh P, Erie DA. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc Natl Acad Sci USA. 2003;100:14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006a;27:323–329. doi: 10.1002/humu.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Christensen LA, Vasquez KM. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci USA. 2006b;103:2677–2682. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst. 2008;100:1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanrooij S, Falkenberg M. The human mitochondrial replication fork in health and disease. Biochim Biophys Acta. 2010;1797:1378–1388. doi: 10.1016/j.bbabio.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Warren ST. Polyalanine expansion in synpolydactyly might result from unequal crossing-over of HOXD13. Science. 1997;275:408–409. doi: 10.1126/science.275.5298.408. [DOI] [PubMed] [Google Scholar]
- Waters TR, Swann PF. Thymine-DNA glycosylase and G to A transition mutations at CpG sites. Mutat Res. 2000;462:137–147. doi: 10.1016/s1383-5742(00)00031-4. [DOI] [PubMed] [Google Scholar]
- Wells RD. Non-B DNA conformations, mutagenesis and disease. Trends Biochem Sci. 2007;32:271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Wells RD, Ashizawa T. Genetic Instabilities and Neurological Diseases. Elsevier/Academic Press; 2006. [Google Scholar]
- Wells RD, Collier DA, Hanvey JC, Shimizu M, Wohlrab F. The chemistry and biology of unusual DNA structures adopted by oligopurine.oligopyrimidine sequences. FASEB J. 1988;2:2939–2949. [PubMed] [Google Scholar]
- Wessels MW, Kuchinka B, Heydanus R, Smit BJ, Dooijes D, de Krijger RR, Lequin MH, de Jong EM, Husen M, Willems PJ, Casey B. Polyalanine expansion in the ZIC3 gene leading to X-linked heterotaxy with VACTERL association: a new polyalanine disorder? J Med Genet. 2010;47:351–355. doi: 10.1136/jmg.2008.060913. [DOI] [PubMed] [Google Scholar]
- Wienholz BL, Kareta MS, Moarefi AH, Gordon CA, Ginno PA, Chédin F. DNMT3L modulates significant and distinct flanking sequence preference for DNA methylation by DNMT3A and DNMT3B in vivo. PLoS Genet. 2010;6:e1001106. doi: 10.1371/journal.pgen.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijchers PJ, de Laat W. Genome organization influences partner selection for chromosomal rearrangements. Trends Genet. 2011;27:63–71. doi: 10.1016/j.tig.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Witherspoon DJ, Watkins WS, Zhang Y, Xing J, Tolpinrud WL, Hedges DJ, Batzer MA, Jorde LB. Alu repeats increase local recombination rates. BMC Genomics. 2009;10:530. doi: 10.1186/1471-2164-10-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf A, Millar DS, Caliebe A, Horan M, Newsway V, Kumpf D, Steinmann K, Chee IS, Lee YH, Mutirangura A, Pepe G, Rickards O, Schmidtke J, Schempp W, Chuzhanova N, Kehrer-Sawatzki H, Krawczak M, Cooper DN. A gene conversion hotspot in the human growth hormone (GH1) gene promoter. Hum Mutat. 2009;30:239–247. doi: 10.1002/humu.20850. [DOI] [PubMed] [Google Scholar]
- Wolfle WT, Washington MT, Prakash L, Prakash S. Human DNA polymerase kappa uses template-primer misalignment as a novel means for extending mispaired termini and for generating single-base deletions. Genes Dev. 2003;17:2191–2199. doi: 10.1101/gad.1108603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock DM, Crowther PJ, Diver WP. The majority of methylated deoxycytidines in human DNA are not in the CpG dinucleotide. Biochem Biophys Res Commun. 1987;145:888–894. doi: 10.1016/0006-291x(87)91048-5. [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Arand J, Sebastiano V, Lepikhov K, Boiani M, Reinhardt R, Scholer H, Walter J. Dynamic link of DNA demethylation, DNA strand breaks and repair in mouse zygotes. EMBO J. 2010;29:1877–1888. doi: 10.1038/emboj.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Mohideen K, Vasudevan D, Davey CA. Structural insight into the sequence dependence of nucleosome positioning. Structure. 2010;18:528–536. doi: 10.1016/j.str.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Wulff K, Gazda H, Schroder W, Robicka-Milewska R, Herrmann FH. Identification of a novel large F9 gene mutation-an insertion of an Alu repeated DNA element in exon e of the factor 9 gene. Hum Mutat. 2000;15:299. doi: 10.1002/(SICI)1098-1004(200003)15:3<299::AID-HUMU23>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, Jorde LB. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19:1516–1526. doi: 10.1101/gr.091827.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, Rajewsky K, Alt FW. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Yang Z, Lau R, Marcadier JL, Chitayat D, Pearson CE. Replication inhibitors modulate instability of an expanded trinucleotide repeat at the myotonic dystrophy type 1 disease locus in human cells. Am J Hum Genet. 2003;73:1092–1105. doi: 10.1086/379523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Smit AF, Schwartz S, Chiaromonte F, Roskin KM, Haussler D, Miller W, Hardison RC. Patterns of insertions and their covariation with substitutions in the rat, mouse, and human genomes. Genome Res. 2004;14:517–527. doi: 10.1101/gr.1984404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. Structure and mechanism for DNA lesion recognition. Cell Res. 2008;18:184–197. doi: 10.1038/cr.2007.116. [DOI] [PubMed] [Google Scholar]
- Yang Z, Funke BH, Cripe LH, Vick GW, 3rd, Mancini-Dinardo D, Pena LS, Kanter RJ, Wong B, Westerfield BH, Varela JJ, Fan Y, Towbin JA, Vatta M. LAMP2 microdeletions in patients with Danon disease. Circ Cardiovasc Genet. 2010;3:129–137. doi: 10.1161/CIRCGENETICS.109.901785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsenko SA, Brundage EK, Roney EK, Cheung SW, Chinault AC, Lupski JR. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum Mol Genet. 2009;18:1924–1936. doi: 10.1093/hmg/ddp114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Epps J, Williams R, Huttley G. Evidence that localized variation in primate sequence divergence arises from an influence of nucleosome placement on DNA repair. Mol Biol Evol. 2010;27:637–649. doi: 10.1093/molbev/msp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You YH, Pfeifer GP. Similarities in sunlight-induced mutational spectra of CpG-methylated transgenes and the p53 gene in skin cancer point to an important role of 5-methylcytosine residues in solar UV mutagenesis. J Mol Biol. 305:389–399. doi: 10.1006/jmbi.2000.4322. [DOI] [PubMed] [Google Scholar]
- Youssoufian H, Kazazian HH, Jr, Phillips DG, Aronis S, Tsiftis G, Brown VA, Antonarakis SE. Recurrent mutations in haemophilia A give evidence for CpG mutation hotspots. Nature. 1986;324:380–382. doi: 10.1038/324380a0. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gerstein M. Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 2003;31:5338–5348. doi: 10.1093/nar/gkg745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Mathews CK. Effect of DNA cytosine methylation upon deamination-induced mutagenesis in a natural target sequence in duplex DNA. J Biol Chem. 1994;269:7066–7069. [PubMed] [Google Scholar]
- Zhang F, Zhao Z. The influence of neighboring-nucleotide composition on single nucleotide polymorphisms (SNPs) in the mouse genome and its comparison with human SNPs. Genomics. 2004;84:785–795. doi: 10.1016/j.ygeno.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Zhang QM, Dianov GL. DNA repair fidelity of base excision repair pathways in human cell extracts. DNA Repair. 2005;4:263–270. doi: 10.1016/j.dnarep.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bouffard GG, Wallace SS, Bond JP, NISC Comparative Sequencing Program Estimation of DNA sequence context-dependent mutation rates using primate genomic sequences. J Mol Evol. 2007;65:207–214. doi: 10.1007/s00239-007-9000-5. [DOI] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Seeman P, Liu P, Weterman MA, Gonzaga-Jauregui C, Towne CF, Batish SD, De Vriendt E, De Jonghe P, Rautenstrauss B, Krause KH, Khajavi M, Posadka J, Vandenberghe A, Palau F, Van Maldergem L, Baas F, Timmerman V, Lupski JR. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet. 2010;86:892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Boerwinkle E. Neighboring-nucleotide effects on single nucleotide polymorphisms: a study of 2.6 million polymorphisms across the human genome. Genome Res. 2002;12:1679–1686. doi: 10.1101/gr.287302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zhang F. Sequence context analysis of 8.2 million single nucleotide polymorphisms in the human genome. Gene. 2006;366:316–324. doi: 10.1016/j.gene.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Zuffardi O, Bonaglia M, Ciccone R, Giorda R. Inverted duplications deletions: underdiagnosed rearrangements? Clin Genet. 2009;75:505–513. doi: 10.1111/j.1399-0004.2009.01187.x. [DOI] [PubMed] [Google Scholar]