

Abstract
Motivation: In eukaryotic cells, alternative splicing expands the diversity of RNA transcripts and plays an important role in tissue-specific differentiation, and can be misregulated in disease. To understand these processes, there is a great need for methods to detect differential transcription between samples. Our focus is on samples observed using short-read RNA sequencing (RNA-seq).
Methods: We characterize differential transcription between two samples as the difference in the relative abundance of the transcript isoforms present in the samples. The magnitude of differential transcription of a gene between two samples can be measured by the square root of the Jensen Shannon Divergence (JSD*) between the gene's transcript abundance vectors in each sample. We define a weighted splice-graph representation of RNA-seq data, summarizing in compact form the alignment of RNA-seq reads to a reference genome. The flow difference metric (FDM) identifies regions of differential RNA transcript expression between pairs of splice graphs, without need for an underlying gene model or catalog of transcripts. We present a novel non-parametric statistical test between splice graphs to assess the significance of differential transcription, and extend it to group-wise comparison incorporating sample replicates.
Results: Using simulated RNA-seq data consisting of four technical replicates of two samples with varying transcription between genes, we show that (i) the FDM is highly correlated with JSD* (r=0.82) when average RNA-seq coverage of the transcripts is sufficiently deep; and (ii) the FDM is able to identify 90% of genes with differential transcription when JSD* >0.28 and coverage >7. This represents higher sensitivity than Cufflinks (without annotations) and rDiff (MMD), which respectively identified 69 and 49% of the genes in this region as differential transcribed. Using annotations identifying the transcripts, Cufflinks was able to identify 86% of the genes in this region as differentially transcribed. Using experimental data consisting of four replicates each for two cancer cell lines (MCF7 and SUM102), FDM identified 1425 genes as significantly different in transcription. Subsequent study of the samples using quantitative real time polymerase chain reaction (qRT-PCR) of several differential transcription sites identified by FDM, confirmed significant differences at these sites.
Availability: http://csbio-linux001.cs.unc.edu/nextgen/software/FDM
Contact: darshan@email.unc.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The transcriptome is a key vantage point for a molecular biologist's study of phenotypic differences between cells that result from environmental factors, cell specialization or disease. Classically, this study has been conducted largely by observing differential gene expression levels using microarrays or high-throughput RNA sequencing technologies. However, detailed analysis of the transcriptome has shown that significant variation is also encoded in the diversity and relative abundance of a gene's constituent transcripts (Kwan et al., 2008; Sultan et al., 2008; Wang et al., 2008). Consequently, beyond measuring differences in overall expression of genes between samples, there is a need to measure differences in expression at the transcript level.
We define differential transcription of a gene between samples as a difference in the relative abundance of the gene's transcript isoforms in the samples. In this manner, differential transcription is independent of the overall gene expression in the samples.
Short-read RNA sequencing technologies (RNA-seq) have evolved rapidly to sample the transcriptome at increasing depth and accuracy (Wang et al., 2009). Using RNA-seq datasets obtained from samples, the locus and depth of coverage by reads aligned to a reference genome provide the starting point for the detection of differential transcription (Pan et al., 2008).
Recently, two approaches have emerged to detect differential transcription between samples. The first approach is based on transcript inference and abundance estimation of the transcripts, as performed by tools like Cufflinks (Trapnell et al., 2010), rQuant (Bohnert and Rätsch, 2010), Trans-Abyss (Robertson et al., 2010) and Scripture (Guttman et al., 2010). Applying these methods to each of two samples, differential transcription can be determined directly for each gene using the estimated relative abundances of the gene's transcripts in the two samples. However, transcript inference algorithms rely on heuristics to resolve the transcript structure, because the inference problem is, in general, underdetermined. As a result, some transcripts may be missed or inferred incorrectly. Abundance estimation, in turn, is not able to correctly explicate the observed distribution of read alignments when starting from an incomplete or incorrect transcript model. Thus, differential transcription measured in this fashion may be inaccurate.
The second approach to detect differential transcription is based on observing loci in the reference genome at which reads from the two datasets align with different depth of coverage (after appropriate normalization for differing gene expression). The idea is that differential transcription should be revealed by different utilization of some exons. Stegle et al. (2010) describe two methods along these lines. The first is based on a priori analysis of annotated transcripts to identify regions that could reveal differential transcription. In each region, a Poisson statistical test is applied. The second method is without dependence on known transcript structure, and uses a non-parametric kernel-based statistical test called maximum mean discrepancy. Using synthetic data, both methods are shown by Stegle et al. (2010) to give accurate detection of differential transcription.
In this article, we introduce an approach that does not depend on annotations and instead leverages the splicing structure of a gene uncovered by spliced read alignments using tools like TopHat (Trapnell et al., 2009), MapSplice (Wang et al., 2010) or PALMapper (Jean et al., 2010). Using the read alignments from these tools, a splice graph is constructed with edges corresponding to transcribed intervals or splices, weighted by read coverage. We introduce the flow difference metric (FDM) to measure the difference between two graphs in the relative utilization of edges at splicing points. Using synthetic samples, for which we know the transcripts and their relative abundances, we show the FDM between two samples is highly correlated with the JSD*, provided coverage of the edges is sufficient. Hence, the FDM can serve as a metric of differential transcription, without need to infer the underlying transcripts or need for any annotation.
To interpret the significance of the FDM, we define a permutation test that can be efficiently implemented on the splice graph representation of the RNA-seq data. Since pairwise comparison of two samples is often insufficient to draw robust conclusions about differential transcription between two biological conditions, we extend the statistical test to incorporate replicates in each condition, when they are available. The test identifies differential transcription that is significant between conditions more often than it is significant within replicates.
2 METHODS
2.1 Jensen–Shannon divergence as a measure of differential transcription
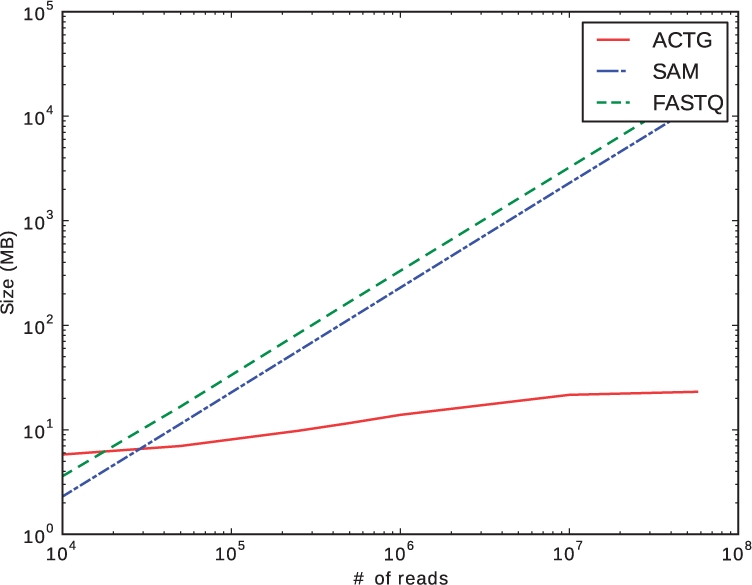
Let G be a gene with n different transcripts. In a given sample, the transcript abundance vector for G gives the relative abundance of each transcript isoform, i.e. the fraction of each isoform among all isoforms of G. One measure of differential transcription between two samples A and B, with transcript abundance vectors VA and VB, is the Jensen–Shannon Divergence
where H(V) is the Shannon entropy. The JSD itself is not a metric, but JSD* 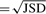 does satisfy the properties of a metric.
does satisfy the properties of a metric.
We adopt JSD* to measure differential transcription in this article, because it defines an objective measure of difference in transcript populations that is independent of the computational methods we examine. It has also been used to report differential transcription in other methods, e.g. CuffDiff (Trapnell et al., 2010).
2.2 Aligned cumulative transcript graph
The alignment of RNA-seq reads to a reference genome provides (i) the genomic coordinates of transcribed bases and (ii) the start and end coordinates of splices. As a consequence of alternative splicing, transcribed bases and splices may be part of multiple RNA transcripts and hence their coverage by aligned reads reflects their total utilization by all transcripts.
In the literature, transcripts have been mostly represented as paths in an acyclic directed graph with exons as nodes and splices as edges, e.g. Heber et al. (2002) and Sammeth's Flux Capacitor (http://flux.sammeth.net/capacitor.html). Analyzing the read coverage information with this data structure has limitations. First, this representation can only be used if all exons are known beforehand, which is usually not the case. Second, if two or more exons overlap in a region (e.g. in the case of alternative 5′ donor sites or 3′ acceptor sites), the read coverage needs to be determined separately for each of those exons. Our graph representation addresses these limitations.
The Aligned Cumulative Transcript Graph (ACT-Graph) is a weighted directed acyclic multigraph in which nodes correspond to genomic coordinates of splice start or end sites or to transcription start or end sites. Edges correspond to transcribed intervals (exonic edge) or to spliced-out intervals (splice edge). The weight of an exonic edge is its average coverage over the genomic interval it spans, and the weight of a splice edge is the number of reads that include the splice. The direction of the edges is the direction of transcription. Each exonic edge is transcribed as whole, i.e. it is included in its entirety in a transcript or not at all.
In principle, an ACT-Graph is the sum of weighted paths (flows), each of which is a transcript with some specific abundance. Therefore, we named the graph the ACT-Graph. Figure 1 shows an example of ACT-Graph. In practice, since reads are sampled non-uniformly from transcripts due to various biases, we use average coverage as an approximation of the total abundance.
Fig. 1.
ACT-Graph: the nodes are genome coordinates. A solid (blue) edge represents an exon or part of an exon labeled with the average depth of read coverage along the interval. A dashed (green) edge is a splice edge and is labeled by the number of reads that include the splice. Alternative splicing features such as mutually exclusive exons, a retained intron and a skipped exon are illustrated. Nodes drawn as boxes, circles and hexagons, respectively, represent annotated-only positions, novel-only splice positions and both annotated and novel positions.
2.2.1 ACT-Graph construction
The following describes the step-by-step construction of an ACT-Graph from RNA-seq data:
Spliced Alignment: RNA-seq reads are aligned to the reference genome using a gapped aligner such as MapSplice (Wang et al., 2010).
ACT-Graph nodes: the ACT-Graph nodes are created using one of the following—(i) Splices: genomic coordinates of splice start and end locations are obtained from spliced alignments; (ii) interpreting start and end sites of transcripts: we can use inference or annotations to identify these sites. We can infer the start of a transcript based on the observation that the first (ℓ − 1) bases following the start coordinate, where ℓ is the RNA-seq read length, show a characteristic ramp of increasing coverage as there are increasingly many ways for a read to sample bases further away from the start of a transcript. A transcription end site is inferred similarly. Alternatively transcript start and end coordinates can be taken from gene annotations, if available. Nodes introduced in this fashion are not harmful if the transcripts happen not to be expressed.
ACT-Graph edges and weights: (i) a splice edge is inferred from a spliced alignment. The weight of the splice edge is the number of reads that support the splice. The direction of the edge is inferred from the orientation of the flanking bases in the intron for canonical splices or it can be inferred from the direction of other splices in the gene. (ii) An exonic edge connects two adjacent nodes (from the sorted list of nodes) if the genomic interval is fully covered or nearly fully covered and has an average coverage above threshold. We use a threshold of 1. The weight of an exonic edge is the average coverage of that genomic region. Averaging over the genomic region gives a better estimate of the number of transcripts that use that genomic region.
ACT-Graph genes and transcribed regions: a transcribed region is a connected component in the ACT-Graph when edges are considered as undirected, and typically would correspond to genes. If gene annotations are available, the regions can be restricted to known genes. The coverage of a gene is defined as average base coverage over all the bases of the exonic regions in the gene.
2.2.2 ACT-Graph compressed representation
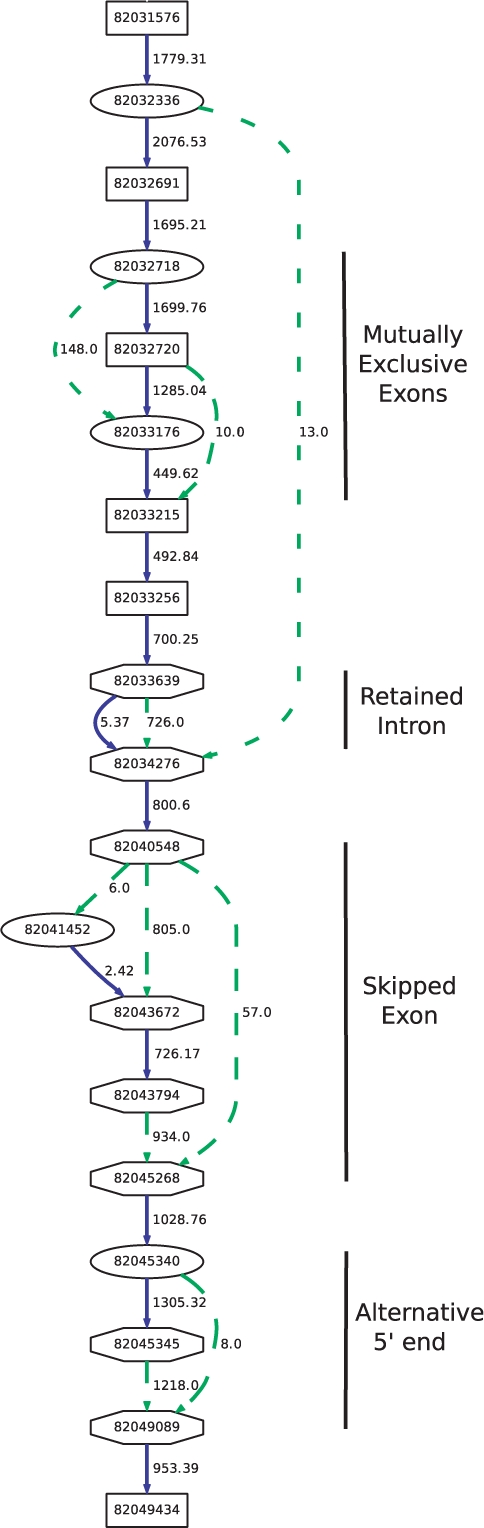
The ACT-Graph is stored in the standard GFF format. The field TYPE tells if the line describes a node, a splice edge or an exonic edge. The field SCORE is used for weight of the edges. The ACT-Graph format is a concise summary of alignments, and is powerful representation for quantitative analysis of alternative splicing. Figure 2 shows the compression achieved by the ACT-Graph representation as a function of the number of reads. The ACT-Graph is typically two to three orders of magnitude smaller than the SAM file or the raw reads, depending on the number of reads in the dataset and can be used for a number of downstream analyses, such as differential transcription.
Fig. 2.
ACT-Graph Compression (Section 2.2.2): plot of file sizes of ACT-Graph (ACTG), FastQ file (FASTQ) and the alignment file (SAM). As the number of reads increases, the storage used by ACT-Graph increases orders of magnitude more slowly than other representations.
2.2.3 Alternative splicing features in ACT-Graph
The ACT-Graph can be used to identify various alternative splicing features in a gene. Each alternative splicing feature can be represented by a subgraph which can be searched in the ACT-Graph. Figure 1 shows examples of various such features in a gene.
2.3 FDM
In this section, we describe the FDM, which uses the ACT-Graph to find genes with differential transcription. As stated earlier, the ACT-Graph can be viewed as the sum of weighted paths or flows, each of which corresponds to a transcript with some abundance. ACT-Graph nodes that have m>1 incoming or outgoing edges indicate that at least m transcripts use that node. These nodes are called divergence nodes. Divergence nodes imply alternative splicing. The m incoming/outgoing edges are called the divergence edges. The weights of divergence edges signify the relative abundances of alternative transcripts passing through the divergence node. The normalized weights of all the divergence edges of a node are grouped together in a vector called the flow vector for the node. The difference between flow vectors in ACT-Graphs constructed from different samples indicates the magnitude of differential transcription between the two samples.
We measure the difference in flow vectors using a metric called the FDM which is defined as follows. Assume an ACT-Graph has n divergence nodes. The flow vector for divergence node i of sample A is defined as VAi=[e(a,i)1,…, e(a,i)m] where m is the number of edges at node i and e(a,i)j is the normalized coverage at edge j, such that ∑j=1m e(a,i)j=1. The flow difference between samples A and B at divergence node i is
The FDM is computed as
as illustrated in Figure 3.
Fig. 3.

FDM and JSD illustration: an example for a gene in two samples A and B is shown. The gene has two transcripts with expression ratio of 1:4 and 4:1 in the two samples, respectively. The FDM is computed using the two ACT-Graphs. The ACT-Graphs have two divergence nodes: node n2 has outdegree 2 and node n5 has indegree 2. FDM(A,B) = 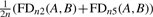 =
= 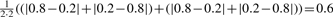 . The JSD is computed using the ground truth knowledge of the transcript abundance vectors. VA = [0.2,0.8] and VB = [0.8,0.2].
. The JSD is computed using the ground truth knowledge of the transcript abundance vectors. VA = [0.2,0.8] and VB = [0.8,0.2]. 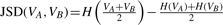 = 0.28. Thus, JSD* = 0.53 is the magnitude of differential transcription representing ground truth.
= 0.28. Thus, JSD* = 0.53 is the magnitude of differential transcription representing ground truth.
It is important that ACT-Graphs of both samples have identical nodes and edges. If a node or edge is present in only one ACT-Graph, it is added to the other one with weight zero. The weights of exonic edges split by added nodes are recomputed using the alignments.
2.3.1 FDM properties
Lemma: the FDM is between 0 and 1
Lemma: FDM is a metric
FDM(A,B) ≥ 0
FDM(A,B) = 0 if and only if A = B
FDM(A,B) = FDM(B,A)
FDM(A,B) ≤ FDM(A,C) + FDM(B,C)
The proofs of both lemmas are provided in the Supplementary Materials.
2.3.2 FDM usage
FDM may be applied between ACT-Graphs without need for normalization by the number of reads or read length, because the FDM is based on ratios of coverage, and these factors scale coverages linearly. Using synthetic data, we show that FDM has a high correlation with JSD*. The details of this are in Section 3.1. Since we do not know the transcripts or their relative abundance, we use the FDM as a metric for differential transcription.
2.4 Statistical tests for differential transcription
2.4.1 Statistical test to find genes with significant differential transcription.
We use the FDM as a test statistic to find genes with significant differential transcription between two samples. The ACT-Graph of each gene is different, so the range of FDM values differs from one gene to another. Thus, the FDM value for a gene is in itself not sufficient to tell if the differential transcription is significant. Instead, we devised a non-parametric test to determine whether differential transcription is significant. We create the null distribution of FDM for a gene, and test if the FDM value for the two samples has a significant P-value. The null hypothesis is that the gene has no differential transcription in two samples. The process of creating the FDM null distribution is illustrated in Figure 6 in the Supplementary Materials. Assume that there are N aligned reads in both the sample datasets. Create ACT-Graphs for the two samples such that nodes and edges are identical. The reads are partitioned into p equal-sized groups in both samples, and an ACT-Graph is created from the alignments of each group of N / p reads. Thus, for each sample we have p ACT-Graphs. The 2 p ACT-Graphs are randomly shuffled into two groups of p partitions each and a composite ACT-Graph for each group is created by simply adding the edge weights of the p ACT-Graphs in the group. Now the FDM is computed between ACT-Graphs of these two groups. This gives a value of the random variable which follows the null FDM distribution. By shuffling partitions a sufficient number of times, we get a null distribution of the FDM. In this fashion, the FDM null distribution is created for each gene, and the P-value for the specific partition that corresponds to the reads of the two samples can be computed. Section 1.5 in the Supplementary Materials provides details on sensitivity to the choice of p and the number of permutations.
Fig. 6.

UCSC browser: Gene CD46 in MCF7 and SUM102 (Section 3.2). The first four samples are from MCF7 and next four samples are from SUM102. This gene was identified as a gene with differential expression using FDM methodology. Note that the middle exon is skipped in different ratios in MCF7 and SUM102. This result was verifed by qRT-PCR. Additional figures are provided in Supplementary Material.
2.4.2 Statistical test for multiple replicates
A single pairwise comparison is often insufficient to draw robust conclusions about differential transcription. Due to several uncontrolled factors, RNA-seq replicates may vary considerably more than predicted from sampling error alone. Thus, pairwise comparison between replicates may yield false positives. If we have multiple replicates of the two samples, we can apply one more level of permutation test to further filter the false positives. Let us assume that there are r replicates for each of the two samples. Replicates from first sample are called Group 1, and replicates from other sample are called Group 2. The FDM pairwise statistical test can be applied to all 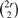 pairs. Out of those, r2 pairs are between replicates in different groups, and the rest are between replicates in the same group. Now, if a gene has significant differential transcription between groups more often than within groups, it is likely to be true positive. The difference between groups and within groups is used as the test statistic. By permuting the group label of the replicates, we get the null distribution of the test statistic. The P-value of the statistic is computed for the original labeling and tested for significance.
pairs. Out of those, r2 pairs are between replicates in different groups, and the rest are between replicates in the same group. Now, if a gene has significant differential transcription between groups more often than within groups, it is likely to be true positive. The difference between groups and within groups is used as the test statistic. By permuting the group label of the replicates, we get the null distribution of the test statistic. The P-value of the statistic is computed for the original labeling and tested for significance.
3 RESULTS
3.1 Experiments with simulated data
In biological data, we typically do not know the exact set of transcripts and their relative abundance in a sample, using which we could calculate the JSD*. Hence ,we use synthetic data, for which we know the exact transcript expression vectors for each gene, to determine (i) the correlation of the FDM and the JSD* metrics; (ii) the power of the FDM method when used as a classifier for a particular value of JSD* under various levels of read coverage; and (iii) the advantage of the groupwise significance test.
The RNA-seq dataset is simulated as follows. We use the annotated transcripts for human genome as a reference. Genes which have at least two transcripts are selected. Each of the genes is assigned an expression level sampled from an empirical distribution of observed expression levels in human genes. The individual transcripts of the genes are each assigned a relative abundance so that their sum is 1. The vector of relative abundances is called the transcript expression vector. For example, a gene with two transcripts T1 and T2 and a transcript expression vector of [p1, p2] indicates that p1% of transcripts are T1 and p2% are T2. A read of size ℓ from a transcript is a random segment of size ℓ taken from the transcript sequence generated using the reference DNA. The number of reads generated from a transcript is proportional to the product of gene coverage, transcript expression and the length of the transcript. The alignment for every read is known, and hence the input SAM datasets consist of reads that are uniquely and perfectly aligned. Additional details on the datasets created can be found in the Supplementary Materials.
3.1.1 FDM correlation with JSD*
We create three pairs of simulated RNA-seq datasets each with different gene coverages. The three pairs of datasets have 1500 genes each. They are generated by varying gene coverages over three ranges [0,5], [10–15] and 20 or higher. The JSD* for the genes is varied over the range 0.0–1.0.
The ACT-Graph is created for all the genes for both the samples in the pair. The FDM is computed for each gene in the pair. From the transcript expression vectors of the genes, the JSD*, which represents the ground truth of differential transcription, is computed.
In Figure 4, we see that the correlation of FDM and JSD* increases as read coverage of the gene increases. This is as expected; when gene coverage is lower, the ACT-Graph edges will have lower weights. Since ratios are used, a small change in edge weight caused by random effects would affect the FDM considerably.
Fig. 4.

Sensitivity and specificity of the FDM as a function of read coverage (Section 3.1.1 & 3.1.2) : Synthetic data of three sample pairs of 1500 genes each is analyzed. The first sample pairs have low gene coverage (coverage = [0,5]), the second sample pairs have medium gene coverage (coverage = [10,15]), and the third sample pairs have high gene coverage (coverage of 20 or higher). (A) JSD* - FDM Correlation: The points in the scatter plots correspond to (JSD*, FDM) values for a gene, where JSD* is ground truth and FDM is computed from ACT-Graphs. When the average gene coverage is high, the correlation between JSD* and FDM is high. For average coverage higher than 20, the correlation is 0.819. (B) FDM as a classifier for JSD*: a gene is marked positive for differential transcription if JSD* is more than 0.22 and negative otherwise. FDM is used to classify genes as positive or negative. Thus for each value of FDM, we get some true positives and some false positives. By varying FDM, the complete curve is plotted. The FDM values of (0.01,0.02,0.04,0.08,0.16,0.32.0.64) are marked on the curve. With coverage of 20 or higher, 90 % of true positives can be identified with ~10% false positives.
3.1.2 FDM as a classifier for JSD*
We tested if FDM can classify genes as high JSD* genes and low JSD* genes. We call a gene positive for high JSD* if the JSD* is >0.22 and negative otherwise. This threshold is arbitrary; we obtained similar results for other values. For each gene, we create ACT-Graphs for two samples and compute the FDM. For a constant c, if FDM >c, we classify the gene as positive. Some of the positives are true positives (using JSD definition) and some false positives. For each c, we get true positives and false positives. By varying c from 0.01 to 0.99 over a step of 0.01, we get the complete receiver operating characteristic curve (ROC). Figure 4 shows that with high coverage, 90% of true positives can be identified with ~10% of false positives.
3.1.3 FDM method over synthetic replicates
We created two synthetic tissues over 2100 genes with at least two transcripts. The JSD* between genes in the two tissues varies randomly over the range 0.01–1.00. The distribution of JSD* and log(Coverage) are given in Figure 1e and f, respectively, in Supplementary Materials. Four replicates were created for each of the tissues resulting in eight samples. FDM method was applied over all the 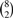 pairs of which 16 pairs were between group and 12 were within group comparisons. We used P ≥0.05 as significant. For creating FDM null distribution, the number of partitions we used was 30 and the number of permutations was 1000. Section 1.5 in the Supplementary Materials shows that increasing the number of partitions and permutations has little effect on the results. The method finds 90% of the genes which have JSD* >0.28 and coverage >7 as significant.
pairs of which 16 pairs were between group and 12 were within group comparisons. We used P ≥0.05 as significant. For creating FDM null distribution, the number of partitions we used was 30 and the number of permutations was 1000. Section 1.5 in the Supplementary Materials shows that increasing the number of partitions and permutations has little effect on the results. The method finds 90% of the genes which have JSD* >0.28 and coverage >7 as significant.
3.1.4 Comparison with other methods
In Figure 5, the results of FDM are compared against other methods not using annotations, namely Cuffdiff (without annotations) and rDiff (MMD), using synthetic RNA-seq datasets defined in the previous section. We ran Cuffdiff as included in release 1.0.3 of the Cufflinks software. Since the data are synthetic and without sampling bias, we deactivated the bias correction module. We used the upper quartile normalization option in order to improve the accuracy of the abundance estimation. All genes with P ≤0.05 were marked as significant. We ran rDiff.web as provided in http://galaxy.tuebingen.mpg.de/. The only option available for the software is which method to use: we used the ‘MMD-based’ method. All the genes with P ≤0.05 were marked as significant. The scatter plots in 5 show the results. For genes with JSD* >0.28 and coverage >7, FDM was able to identify 90% of the genes as differentially transcribed. This represents higher sensitivity than Cuffdiff (without annotation) and rDiff (MMD), which identified differential transcription between 68% and 49% of the genes in this region, respectively. For comparison, we also ran Cuffdiff with gene annotations, which identified differential transcription in 86% of the genes in this region.
Fig. 5.

Detection of differential transcription by different methods. The circles in scatterplots (a–d) represent 2100 genes in two samples with varying differential transcription (measured by JSD*) and varying depth of RNA-seq sampling (measured by the average coverage per transcribed nucleotide). Filled circles correspond to genes with significant differential transcription according to each of the methods. (a) FDM consistently identifies differential transcription when coverage is high or JSD* is high. For example, for genes with JSD* >0.28 and log(coverage) >0.85 (coverage >7), FDM was able to identify 90% of the genes as differentially transcribed. Two other methods not using annotations, (c) Cuffdiff (Trapnell et al. (2010) without annotations) and (d) rDiff (MMD) Stegle et al. (2010), had lower sensitivity, identifying differential transcription in 68 and 49% of the genes in this region, respectively. (b) For comparison, we also ran Cuffdiff with gene annotations, which identified differential transcription in 86% of the genes in this region.
3.2 Experiments with biological data
We used RNA-seq data for four replicates each of the cancer cell lines MCF7 and SUM102. Each dataset has ~80 million single-ended reads of length 100 nt.
We used the FDM method to find genes with differential transcription between SUM102 and MCF7. We used MapSplice to align the RNA-seq datasets. Using these alignments, we created ACT-Graphs for all the known genes. We applied the FDM statistical test to all the 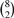 pairs of replicates. Out of these 28 pairs, 6 pairs were of MCF7-MCF7, another 6 for SUM102-SUM102 and 16 were MCF7-SUM102. The number of significantly different genes in single pair comparison are as follows:
pairs of replicates. Out of these 28 pairs, 6 pairs were of MCF7-MCF7, another 6 for SUM102-SUM102 and 16 were MCF7-SUM102. The number of significantly different genes in single pair comparison are as follows:
MCF7-MCF7 : 1949 (average over six pairs).
SUM102-SUM102: 1966 (average over six pairs).
MCF7-SUM102: 2727 (average over 16 pairs).
Next we applied the statistical test for replicates to get the most significant genes. After applying the replicates statistical test, 1425 genes were judged to have significant differential transcription between MCF7 and SUM102. CD46 is one of the genes found to be significantly different. The UCSC browser bedgraph tracks for gene CD46 (Fig. 6) shows that the middle exon has a different skipping ratio in MCF7 and SUM102. Additional examples can be found in the Supplementary Materials.
We performed quantitative real time polymerase chain reaction (qRT-PCR) on three genes to validate the FDM results. Details for the method can be found in Section 1.4 of the Supplementary Materials. For CD46, the skipped exon (chr1:207963598-207963690) was found to be expressed >2-fold higher in SUM102 than in MCF7 as measured by qRT-PCR. Working from the ACT-Graphs, average skipping ratios in the MCF7 samples were 0.16 and in the SUM102 samples were 0.5 predicting an average 3.1-fold change. For NPC2 (shown in the Supplementary Materials), the retained intron (chr14:74946991-74947405) was expressed at least 10-fold more in MCF7 than in SUM102 as measured by qRT-PCR. Working from the ACT-Graphs, an average fold change of 25 was predicted. Both experimental results were in congruence with the FDM results. Using Cuffdiff with annotations on our dataset, NPC2 was judged to have significant differential transcription, but the test for CD46 failed and thus was inconclusive.
A third gene ZNF408 (shown in the Supplementary Materials) gave a different result in the biological experiment than predicted by the FDM method. We directly resequenced cDNA derived from the mRNA from both cell lines and genomic DNA from both cell lines. The region of interest (chr11:46724721-46724734) has a high number of mutations in MCF7 compared with the reference genome, a common observation for cancer cell lines and cell lines that have been propagated extensively. This caused reads from a region of MCF7 to not align to the reference genome, and present a difference in the ACT-Graphs. Thus, the incorrect result is due to alignment limitations, rather than to FDM.
4 DISCUSSION
4.1 FDM - JSD* correlation
Although Figure 4 shows a high correlation between FDM and JSD*, there still are genes with high FDM and low JSD*. These genes are artifacts of low coverage at some divergence nodes and could be filtered out. Since FDM uses ratios, a variance in small edge weights can cause high variance in the flow difference.
There are also some genes with high JSD* but low FDM. These can be due to complex gene models with many transcripts giving rise to many divergence nodes. When most transcripts have low abundance and are unchanged between samples and just a few similar transcripts have larger abundance changes, then JSD* can be large, yet only a few divergence nodes observe large flow changes, and these are attenuated by the remaining unchanged nodes to create an FDM value that is not exceptional under permutation testing. Focusing on divergence nodes with flow differences could improve detection of these cases.
4.2 FDM and sequencing bias
Sample preparation protocols can introduce significant deviations from the assumption of uniform sampling of reads along transcript isoforms, in ways which are not fully understood. It is useful to consider how such sampling bias would affect FDM. (Roberts et al., 2011) cite two types of sampling bias.
Sequence-specific bias (Hansen et al., 2010) is related to the underlying sequence of nucleotides in a transcript, resulting in preferential locations for read starts. Sequence-specific bias affects the count of reads whose alignment starts within an exonic edge in the ACT-Graph the same way for all transcripts utilizing the exonic edge. Associating average coverage with such an edge both smooths local variation due to sequence-specific bias, and is independent of the underlying transcripts involved. In effect, sequence-specific bias is minimized in this fashion.
Position-specific bias (Bohnert and Rätsch, 2010) is related to position in the transcript, and results in increased sampling at transcript starts and ends. Position-specific bias affects both exonic and spliced edge coverage according to the specific transcript utilizing the edge, and this will change as the relative abundance of transcripts changes, which will alter the magnitude of the flow difference in a divergence node. However, we have indicated that the magnitude of a gene's FDM signal varies by gene, and for this reason a non-parametric test is used to determine significance. Thus, we believe the effect of position-specific bias will not substantially affect the determination of significance. In summary, while further investigation and validation is needed, we expect FDM to be largely insensitive to sequence-specific and position-specific sampling bias.
4.3 FDM and read length
The FDM method is specifically designed to detect differential transcription with short reads (35–100 nt), for which transcript reconstruction can be unreliable and, we would argue, is not needed. As we increase read length, read alignments become more accurate and the coverage on ACT-Graph edges increases, both of which improve the accuracy of the method. At the same time, if increased read length comes at the expense of deep sampling (under a fixed throughput assumption), then sensitivity would be expected to decrease.
Paired-end reads can improve FDM accuracy depending on the operation of the underlying RNA-seq aligner. At the least, paired-end reads yield higher quality alignments, because of the extra constraints on mate pair distance and alignment orientation. MapSplice aligns paired-end reads using these constraints and also incorporates a maximum likelihood method operating on the splice graph to infer the alignment of the complete insert, including the unsequenced fragment, given the distribution of insert lengths (Hu et al., 2010). This results in an effective increase in read length and coverage and hence can improve the accuracy of FDM.
5 CONCLUSION
While splice graphs were introduced nearly a decade ago (Heber et al., 2002), our definition is intended to record RNA-seq read coverage in such a graph (this is also the approach taken in the Flux Capacitor). To make such graphs efficient to analyze, we choose a specific representation that differs from classic splice graphs. Nodes are labeled with genomic coordinates which are unique and help address the ambiguities caused by overlapping exons and unannotated genomic regions. The node labels are also well suited for computing the union of graphs from which the edge set for comparison of coverages is easy to determine. The ACT-Graph representation can dramatically decrease the data storage requirement for RNA-seq data. It is not a lossless compression as the underlying reads cannot be recovered from the ACT-Graph, but it does suffice for the analysis of differential expression and transcription.
The FSM captures the signal of differential transcription directly from a pair of ACT-Graphs, without knowledge or inference of the underlying transcripts or need for normalization. The FDM has high correlation with JSD*, which is an independent measure of differential transcription. We showed that FDM can be used as classifier for differential transcription. We presented a statistical method using a permutation test on ACT-Graphs to find genes with significant differential transcription between pairs of samples or between groups of replicates.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the referees for their insightful questions and comments, and thank Charles Perou for RNA samples from the MCF-7 and SUM-102 cell lines and Anais Monroy for qRT-PCR validation.
Funding: National Science Foundation (ABI/EF grant number 0850237 to J.L. and J.F.P.); National Institutes of Health: NCI TCGA (grant number CA143848 to Charles Perou); NCRR Idea (INBRE Grant P20RR016481 to N. Cooper); NCI GI SPORE Developmental Project Award (P50CA106991 to D.Y.C.); Alfred P. Sloan Foundation fellowship (to D.Y.C.).
Conflict of Interest: none declared.
REFERENCES
- Bohnert R., Rätsch G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 2010;38(Suppl. 2):W348–W351. doi: 10.1093/nar/gkq448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M., et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.D., et al. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber S., et al. Splicing graphs and EST assembly problem. Bioinformatics. 2002;18(Suppl. 1):S181–S188. doi: 10.1093/bioinformatics/18.suppl_1.s181. [DOI] [PubMed] [Google Scholar]
- Hu Y., et al. A probabilistic framework for aligning paired-end RNA-seq data. Bioinformatics. 2010;26:1950–1957. doi: 10.1093/bioinformatics/btq336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean G., et al. RNA-Seq Read Alignments with PALMapper. Current Protocols in Bioinformatics. 2010:32:11.6.1–32:11.6.37. doi: 10.1002/0471250953.bi1106s32. Available at http://www.hpa.org.uk./infections/topics_az/antimicrobial_resistance/amr.pdf. [DOI] [PubMed] [Google Scholar]
- Kwan T., et al. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- Pan Q., et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Robertson G., et al. De novo assembly and analysis of RNA-seq data. Nat. Methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- Roberts A., et al. Improving rna-seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O., et al. Statistical tests for detecting differential rna-transcript expression from read counts. Nat. Preced. 2010 [Epub ahead of print, doi:10.1038/npre.2010.4437.1, May 11, 2010] [Google Scholar]
- Sultan M., et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- Trapnell C., et al. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.