

Abstract
Macrophages and dendritic cells play key roles in viral infections, providing virus reservoirs that frequently resist anti-viral therapies and linking innate virus detection to anti-viral adaptive immune responses1,2. HIV-1 fails to transduce dendritic cells and has a reduced ability to transduce macrophages, due to an as yet uncharacterized mechanism that inhibits infection by interfering with efficient synthesis of viral cDNA3,4. In contrast, HIV-2 and related simian immunodeficiency viruses (SIVsm/mac) transduce myeloid cells efficiently owing to their virion-associated Vpx accessory proteins, which counteract the restrictive mechanism5,6. Here we show that the inhibition of HIV-1 infection in macrophages involves the cellular SAM domain HD domain-containing protein 1 (SAMHD1). Vpx relieves the inhibition of lentivirus infection in macrophages by loading SAMHD1 onto the CRL4DCAF1 E3 ubiquitin ligase, leading to highly efficient proteasome-dependent degradation of the protein. Mutations in SAMHD1 cause Aicardi-Goutieres syndrome (AGS), a disease that produces a phenotype that mimics the effects of a congenital viral infection7,8. Failure to dispose of endogenous nucleic acid debris in AGS results in inappropriate triggering of innate immune responses via cytosolic nucleic acids sensors9,10. Thus, our findings reveal that macrophages are defended from HIV-1 infection by a mechanism that prevents an unwanted interferon response triggered by self nucleic acids, and uncover an intricate relationship between innate immune mechanisms that control response to self and to retroviral pathogens.
Vpx associates with the DCAF1/VprBP substrate receptor subunit of the Cullin4-based E3 ubiquitin ligase CRL4DCAF1,(11). Failure to infect macrophages and dendritic cells by HIV-2 and SIVmac viruses encoding mutant Vpx proteins deficient in association with this E3 suggested that Vpx usurps CRL4DCAF1 to overcome the inhibition of infection in myeloid cells12,13. The inhibition of HIV-1 infection in monocyte-derived macrophages (MDM) can be overcome by co-infection with Vpx-containing SIVsm virus-like particles (SIV VLP)4,14. These SIV VLP do not contain any viral genetic material and can be used to deliver Vpx, which is naturally packaged into virion cores through its interaction with Gag15,16, to target cells. To assess whether Vpx-mediated relief of the inhibition of HIV-1 infection in macrophages is also linked to CRL4DCAF1, we co-infected human MDM with VSV-G-pseudotyped HIV-1 NL4-3-derived single cycle reporter virus encoding the GFP reporter (HIV-1–GFP) in the presence of VSV-G-pseudotyped SIV VLP loaded with HIV-2 Rod Vpx (SIV VLP(Vpx)) or with a Vpx variant (VpxQ76A), that does not bind to DCAF112. Wild type Vpx greatly enhanced MDM transduction by HIV-1–GFP, whereas the VpxQ76A did not have such a stimulatory effect (Figure 1a), and viral cDNA synthesis was inefficient both in the absence of Vpx and upon co-infection with VpxQ76A (Figure 1b), as also observed with vpx-defective single cycle HIV-2–GFP and SIVmac–GFP reporter viruses12 (Supplementary Figure 1). This evidence supports the model in which Vpx relieves the inhibition of HIV-1 infection in MDM by targeting an unknown anti-viral inhibitory protein for proteasome-dependent degradation, via the CRL4DCAF1 E3 ubiquitin ligase.
Figure 1. Vpx-mediated relief of the inhibition of HIV-1 infection in MDM requires Vpx glutamine Q76.

a. Alleviation of the inhibition of HIV-1 infection in MDM requires Vpx glutamine Q76. MDM were infected with single cycle HIV-1–GFP reporter virus alone, or co infected with HIV-1–GFP and SIV VLP loaded with wild type or Q76A substituted HIV-2 Rod Vpx. Percent fractions of GFP-positive cells are indicated.
b. VpxQ76A does not rescue HIV-1 cDNA synthesis. Quantification of HIV-1 “early” (light bars) and “late” (black bars) cDNA products in MDM co-infected with HIV-1–GFP and SIV VLP loaded with HIV-2 wild type Vpx or VpxQ76A. (Boil): boiled HIV-1/SIV VLP control.
c. SIV VLP contain similar amounts of wild type and Q76A substituted Vpx. Two-fold dilutions of SIV VLP(Vpx), SIV VLP(VpxQ76A) or SIV VLP with no Vpx (none) were immunoblotted for Vpx and p27 capsid.
To identify Vpx-recruited substrates for the CRL4DCAF1 ubiquitin ligase, a proteomic screen was used to search for the cellular protein(s) that associate with the CRL4DCAF1 complex only in the presence of Vpx. Specifically, we directed the assembly of three sets of protein complexes in HEK 293T cells for their subsequent proteomic characterization (Figure 2a). First, to capture the cellular proteins bound by Vpx, we transiently expressed a functional SIVmac 239 Vpx protein tagged with FLAG- and HA- epitopes in tandem. Second, to trap proteins recruited by Vpx to CRL4DCAF1, we assembled quaternary complexes comprising HA-Cullin4, DDB1, DCAF1 and FLAG-Vpx, in cells transiently expressing epitope-tagged Cullin4 and Vpx subunits. The placement of epitope tags on Cullin4 and Vpx, subunits that are the most distally located in the quaternary complex17, ensured that partial complexes lacking one or more subunits, or comprising CRL4 substrate receptors other than DCAF1, would not co-purify with the dually-tagged complexes. Third, as a negative control, we assembled and purified a ternary HA-Cullin4, DDB1, FLAG-DCAF1 complex, lacking Vpx. All protein complexes were purified by two sequential immunoprecipitations of epitope tags, under native conditions, and their subunit composition was analyzed by Multidimensional Protein Identification Technology (MudPIT)18.
Figure 2. Vpx recruits SAMHD1 to DDB1-DCAF1 module of CRL4DCAF1 E3 complex.
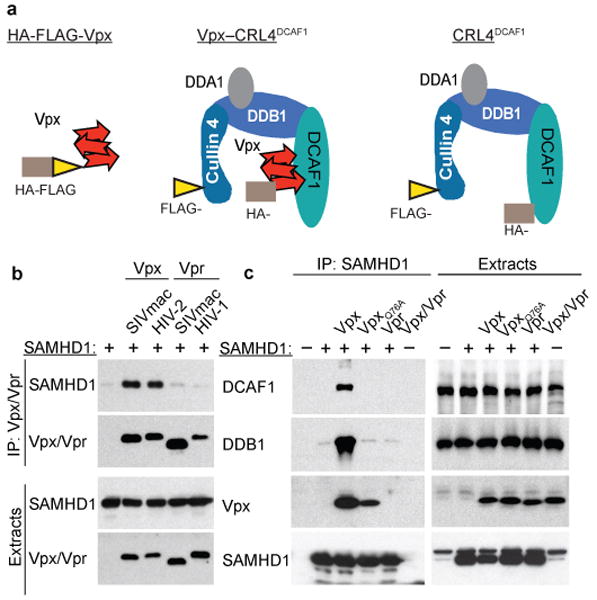
a. Proteins and complexes purified for MudPIT analyses. Architecture of the quaternary Cullin4-DDB1(DDA1)-DCAF1 complex alone, and with bound Vpx, and the placements of HA- (brown rectangle) and FLAG- (yellow triangle) epitope tags is shown. DDA1 is a DDB1-associated protein that co-purifies with DCAF1-DDB1 sub-complex19,20.
b. HIV-2 and SIVmac Vpx bind SAMHD1. The indicated FLAG-tagged Vpx and Vpr proteins were transiently co-expressed with myc-tagged SAMHD1 in HEK 293T cells. Cell extracts and α-FLAG immune complexes were immunoblotted for SAMHD1 and Vpx/Vpr.
c. Vpx recruits SAMHD1 to DCAF1-DDB1 complex. FLAG-SAMHD1 was transiently co-expressed with myc-tagged SIVmac Vpx, VpxQ76A, or HIV-1 Vpr, in HEK293T cells. α-FLAG-SAMHD1 immune complexes were immunoblotted for endogenous DCAF1, DDB1, and ectopically expressed SAMHD1 and Vpr/Vpx.
The most abundant of the candidate proteins uncovered in this screen was the innate immune SAM-domain HD-domain containing protein 1 (SAMHD1), which co-purified with both Vpx (6 spectra) and Vpx-CRL4DCAF1 complex (37 spectra), but not with CRL4DCAF1 complex alone (Supplementary Table I). Interestingly, SAMHD1 mutations are associated with Aicardi-Goutieres syndrome (AGS), an autoimmune disease caused by an abnormal innate immune response to endogenous nucleic acids7,8. Besides SAMHD1, approximately 150 cellular proteins were found associated with Cullin4 in the presence of Vpx. Because these proteins were low quality hits represented by three or fewer spectra, and/or were found previously to be frequent contaminants, they were not considered to be viable candidates.
To assess whether SAMHD1 binding and recruitment to the DCAF-linked complex are conserved functions of Vpx, we investigated HIV-2 Rod Vpx, which, as SIVmac 239 Vpx, is also functional6,12,13. As a negative control we selected the Vpr accessory proteins of HIV-1 and SIVmac, because they are closely related to Vpx, sharing roughly 25% and 50% amino acid identity, and can load cellular substrate proteins onto CRL4DCAF1,(21) but, unlike Vpx, Vpr does not alleviate the inhibition of lentivirus infection in MDM. Human epitope-tagged SAMHD1 was co-expressed with Vpx, or Vpr, in HEK 293T cells and Vpx/Vpr immune complexes were analyzed by Western blotting. SAMHD1 co-precipitated efficiently with both Vpx variants, but not with Vpr (Figure 2b). Next, we confirmed that Vpx specifically links SAMHD1 to the CRL4DCAF1 E3 complex. SAMHD1 was co-expressed with wild type SIVmac Vpx, a VpxQ76A variant that does not bind DCAF1, or HIV-1 Vpr that does not bind SAMHD1, in HEK 293T cells. Analysis of SAMHD1 immune complexes revealed that SAMHD1 directed assembly of a protein complex containing Vpx, and the DCAF1- and DDB1- subunits of CRL4DCAF1 complex (Figure 2c). In contrast, neither VpxQ76A, which exhibited reduced binding to SAMHD1, nor Vpr, formed such complexes. We conclude that Vpx specifically binds to, and recruits SAMHD1 to the DCAF1-DDB1 module of CRL4DCAF1 E3 ubiquitin ligase.
Because SAMHD1 was originally identified in HEK 293T cells, it was important to confirm that it is both expressed and targeted by Vpx for degradation via CRL4DCAF1 in MDM. We therefore performed Western blot analysis of MDM lysates and measured steady state SAMHD1 levels following MDM infection with SIV VLP loaded with either Vpx or the VpxQ76A variant control (Figure 3a). We observed that wild type Vpx readily depleted endogenous SAMHD1 levels. In contrast, VpxQ76A, which is defective for DCAF1 binding12, did not decrease SAMHD1 levels. Importantly, the effect of Vpx was blocked when macrophages were exposed to the proteasome inhibitor MG132 prior to and during infection with Vpx-loaded SIV VLP (Figure 3b). We conclude that Vpx programs SAMHD1 for destruction in MDM.
Figure 3. Vpx programs SAMHD1 for proteasomal degradation in MDM, via CRL4DCAF1 E3.

a. SAMHD1 levels in MDM infected with 3-fold serial dilutions of SIV VLP loaded with wild type or Q76A substituted HIV-2 Rod Vpx, or in mock infected MDM (M). α-tubulin served as loading control.
b. SAMHD1 levels in MDM infected with 3-fold dilutions of SIVmac Vpx-loaded SIV VLP and cultured in the presence or absence of MG132 proteasome inhibitor (1 μg/ml).
c. Vpx depletes SAMHD1 levels in MDM via DCAF1. MDM were subjected to RNAi targeting DCAF1, SAMHD1 or non-targeting RNAi (NT1, NT2), and infected (+) or not (-) with SIV VLP loaded with SIVmac Vpx, two days later. DCAF1 and SAMHD1 were revealed by Western blotting after additional two days.
Our above biochemical and functional studies with VpxQ76A variants suggested that Vpx uses the CRL4DCAF1 E3 ubiquitin ligase to program SAMHD1 degradation. To test this directly, we first depleted the DCAF1 subunit of CRL4DCAF1 by RNA interference (RNAi), and then characterized the effects of this knockdown on Vpx-mediated SAMHD1 depletion in MDM. MDM were transfected with siRNA targeting DCAF1 or with non-targeting control siRNAs (NT1, NT2). DCAF1-specific siRNA caused a large reduction in DCAF1 levels (Figure 3c). Two days following the initiation of RNAi, the cells were infected with Vpx-loaded SIV VLP and the SAMHD1 levels were assessed 2 days later. In the absence of Vpx (left panels) there was no significant change in SAMHD1 levels following treatment with either the DCAF1, or control siRNAs. Significantly, Vpx induced an almost complete degradation of SAMHD1 in cells treated with the control siRNAs. By contrast, in the DCAF1 depleted cells degradation of SAMHD1 induced by Vpx was significantly inhibited providing further evidence for our hypothesis that Vpx utilizes CRL4DCAF1 E3 to program SAMHD1 degradation.
Then we set out to investigate the role of SAMHD1 in HIV-1 infection in MDM. Unfortunately, RNAi-mediated knockdown of SAMHD1 was only weakly effective in MDM (Figure 3c, left panel). We believe that SAMHD1 levels in MDM are resistant to depletion by RNAi because the half-life of SAMHD1 is long, and RNAi can only prevent new protein synthesis; thus, due to slow turnover rate, it cannot achieve depletion of existing SAMHD1 protein. Hence, we developed a two-step protocol to manipulate SAMHD1 levels over a wide concentration range by using a combination of RNAi and SIV VLP to deliver to cells low doses of Vpx. In the first step, macrophages were infected with low doses of Vpx-loaded SIV VLP to deplete the pre-existing SAMHD1 pool. In the second step we blocked SAMHD1 re-synthesis by RNAi.
As shown in Figure 4, freshly isolated CD14+ monocytes cultured in the presence of M-CSF were infected with increasing, but suboptimal, doses of Vpx-loaded SIV VLP. RNAi targeted to SAMHD1 or a non-targeting RNAi was initiated 4 days later. Three days following the initiation of RNAi the macrophages were challenged with HIV-1–GFP reporter virus. We observed that RNAi to SAMHD1 stimulated MDM transduction by several-fold, compared to non-targeting RNAi, throughout the entire titration range for Vpx, and the permissiveness to HIV-1–GFP transduction showed good, but not simple, inverse correlation with SAMHD1 levels in MDM (Figure 4a,b, see also Supplementary Figures 2, 3 and 4). The most dramatic increases in MDM permissiveness were revealed at low doses of Vpx that on their own barely stimulated HIV-1 transduction. Quantification of viral reverse transcription intermediates showed that depleting SAMHD1 levels stimulated synthesis of viral full length cDNA by approximately 10-fold (Figure 4c), and again this effect was well pronounced even at the lowest doses of Vpx. Specifically, it can be clearly seen that the “early” (“E”) vs. “late” (“L”) cDNA PCR readouts showed selective repression of “late” reverse transcription products at high but not low SAMHD1 levels. Furthermore, MDM transduction by Vpx-defective HIV-2vpx-–GFP and SIVmacvpx-–GFP reporter constructs was also enhanced up to 12-fold, by siRNA to SAMHD1 (Figure 4d). This evidence clearly demonstrates that SAMHD1 inhibits MDM infection by HIV-1 and other primate lentiviruses by disrupting synthesis of viral cDNA.
Figure 4. SAMHD1 inhibits HIV-1 infection in macrophages.
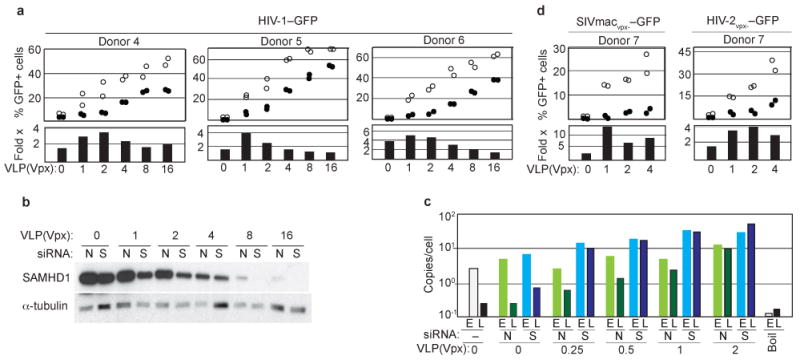
a. RNAi mediated SAMHD1 depletion relieves inhibition of HIV-1 infection in MDM. CD14+ monocytes isolated from three donors were infected with decreasing doses of Vpx-loaded SIV VLP, differentiated into MDM, subjected to RNAi targeting SAMHD1 (open circles), or non-targeting RNAi (filled circles), and infected with HIV-1–GFP reporter virus. GFP+ MDM were quantified 3 days later (upper panels). Fold-stimulation (Fold x) of HIV-1–GFP transduction of MDM, following RNAi targeting SAMHD1, compared to non-targeting RNAi, is also shown (bottom panels).
b. SAMHD1 and α-tubulin control levels in MDM from a typical experiment shown in panel (a). N – non-targeting RNAi, S – RNAi to SAMHD1.
c. SAMHD1 depletion relieves HIV-1 cDNA synthesis. “early” (E) and “late” (L) viral cDNA products in MDM exposed to low doses of Vpx-loaded SIV VLP and transfected with siRNA to SAMHD1 (S) or non-targeting siRNA (N), and then challenged with HIV-1–GFP. (Boil): boiled HIV-1/SIV VLP control.
d. SAMHD1 inhibits MDM transduction by vpx-defective HIV-2vpx-–GFP and SIVmacvpx-–GFP single cycle reporter viruses.
In summary, our results show that SAMHD1 interferes with HIV infection of macrophages by preventing efficient viral cDNA synthesis. The Vpx accessory protein of HIV-2/SIVsm lineage viruses removes this inhibition by targeting SAMHD1 for proteasome-dependent degradation via CRL4DCAF1 E3 ubiquitin ligase (Supplementary Figure 5). Since HIV-2 and related simian viruses have evolved the Vpx function to counteract SAMHD1, they are able to infect macrophages much more readily than HIV-1. Indeed, vpx-deficient SIVsm/mac viruses are attenuated in simian models of AIDS, and show evidence for defective macrophage-dependent dissemination at early stages of infection and the apparent absence of macrophage-driven encephalitis at later stages22,23. Our discovery that the SAMHD1 protein inhibits HIV infection in macrophages unveils a novel and unexpected link between unconventional cell-intrinsic innate immune mechanisms and the anti-viral defence. Notably, in addition to SAMHD1, two nucleases TREX1 and RNAse H2 have been linked to Aicardi-Goutieres syndrome8. Recent studies of TREX1 suggest that it plays a role in the control of retrotransposon and endogenous retroviral elements, and in inhibiting the innate immune response to HIV-1 DNA in T cells and macrophages by clearing excess of viral cDNA9,24. However, the reported effects of TREX1 on HIV-1 were not associated with an overt restriction of HIV-1 infection in either cell type, in contrast to our findings with SAMHD1.
One unanswered question is how myeloid cell specificity of SAMHD1-mediated inhibition of HIV-1 infection is achieved? SAMHD1 is expressed in HEK 293T cells, undifferentiated THP-1 cells and other non-myeloid cell types that do not possess a Vpx-sensitive mechanism restricting primate lentivirus infection (Supplementary Figures 6 and 7). One possible explanation is that SAMHD1-mediated restriction may require a myeloid cell-specific molecule. Alternatively, cell type specific differences in early post-entry events involved in viral core uncoating and/or reverse transcription complex function may make HIV/SIV prone to SAMHD1 specifically in myeloid cells25,26. Of note, we observed that SAMHD1 levels are depleted by Vpx in both MDM and monocyte-derived dendritic cells in a Vpx glutamine Q76 dependent manner (data not shown) implicating SAMHD1 as the key AGS protein inhibiting lentivirus infection in myeloid cells. Whether SAMHD1 functions autonomously, what are the other key components of a putative SAMHD1 pathway, and what are the salient features of SAMHD1 that endow this protein with anti-HIV activity, requires further investigation.
Methods Summary
Blood from adult healthy volunteer donors was collected at Case CFAR Clinical Core according to Institutional Review Board (IRB) approved protocol. Monocytes isolated from PBMCs by positive selection for CD14 using CD14 microbeads (Miltenyi Biotec Inc.) were differentiated into macrophages by culturing in DMEM supplemented with 10% heat inactivated foetal bovine serum (FBS), 10 ng/ml Macrophage-Colony Stimulating Factor (M-CSF, R&D Systems) for the first 2 days and 20 ng/ml thereafter, in 24 well plates at 3.0-3.5×105 cells/well. Cells were fed every alternate day by replacing one half of the cell culture medium with fresh medium. Purity of CD14+ cells obtained by positive selection usually ranged between 95% and 99% while the final purity of the adherent macrophage population was typically greater than 99%. Infections were carried out between day 6 and day 9. Flow cytometry analysis of GFP expression was performed 3 to 4 days post infection. MDM were detached from wells by trypsin treatment, resuspended in PBS containing 1% FBS, fixed by addition of paraformaldehyde to 1% and GFP expression analyzed by flow cytometry.
Methods
Expression plasmids
pCG plasmids expressing epitope tagged DCAF1, DDB1, wild type and mutant HIV-2 Rod and SIVmac 239 Vpx proteins were previously described20. Human SAMHD1 protein coding sequence was amplified by PCR from an EST clone (MHS1010-7295666, Open Biosystems) using 5′-ggaagaatctagaatgcagcgagccgattccg-3′ and 5′-ggaagaaacgcgtcacattgggtcatctttaaaaagctgg-3′ primers and subcloned into pCG vectors encoding N-terminal epitope tags.
Single cycle proviral reporter constructs
HIV-1-derived single cycle pHR′-EGFP-IRES-nef proviral clone and SIVmac 239(GFP) reporter viruses, kindly provided by J. Karn and F. Kirchhoff, respectively, were previously described12,27. HIV-2 Rod GFP proviral clone was constructed by M. Emerman and kindly provided by N. Manel6,28. SIVmac 251 virus-like particles (SIV VLP) for Vpx delivery were produced using SIV3+ plasmid, provided by A. Cimarelli3. The vpx gene in the SIV3+ plasmid was inactivated by converting its initiation codon to a stop codon to give SIV3+vpx-, using Quikchange XLII kit (Stratagene).
Viruses and virus like particles (SIV VLP)
VSV-G pseudotyped, single cycle pHR′-EGFP-IRES-Nef (referred to as HIV-1–GFP) was produced from HEK 293T cells co-transfected with the proviral clone, BH10 φ–env– packaging and VSV-G expression plasmids, by calcium phosphate co-precipitation method. SIVmac–GFP and HIV-2–GFP reporter viruses were produced from HEK 293T cells co-transfected with the respective proviral clone and VSV-G expression plasmid. SIV VLP were similarly produced from HEK 293T cells co-transfected with SIV3+ and VSV-G expression plasmid. SIV VLP loaded with wild type or mutant SIVmac 239, or HIV-2 Rod Vpx proteins were produced from HEK 293T cells co-transfected with pSIV3+vpx-, which contains an inactive vpx open reading frame, and an appropriate pCG vector expressing wild type or Q76A mutated SIVmac 239, or HIV-2 Rod Vpx12. Virus- and SIV VLP- containing cell culture media were harvested one day after transfection, treated with DNAse I (Roche) at 20 u/ml, for 20 minutes at 30°C and virions were concentrated and partially purified by pelleting through 20% sucrose in TNE buffer (10 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA) cushion, for 3 hours at 30,000 rpm, 4°C in SW32TI rotor16. Virion pellets were suspended in 1% of the original volume of TNE, aliquoted, and stored at -80°C. Virus preparations were normalized based on their infectivities to HEK 293T and/or Jurkat T cells. SIV VLP were normalized by Western blotting for p27 capsid and/or Vpx.
Monocyte-derived macrophage culture
Blood from adult healthy volunteer donors was collected at Case CFAR Clinical Core. Monocytes isolated from PBMCs by positive selection for CD14 using CD14 microbeads (Miltenyi Biotec Inc.) were differentiated into macrophages by culturing in DMEM supplemented with 10% heat inactivated foetal bovine serum (FBS), 2 mM glutamine, 100 u/ml Penicillin-G, 100 μg/ml Streptomycin, 10 ng/ml Macrophage-Colony Stimulating Factor (M-CSF, R&D Systems) for the first 2 days and 20 ng/ml thereafter, in 24 well plates at 3.0-3.5×105 cells/well. Cells were fed every alternate day by replacing one half of the cell culture medium with fresh medium. Purity of CD14+ cells obtained by positive selection usually ranged between 95% and 99% whereas the final purity of the adherent macrophage population was typically greater than 99%. Infections were carried out between day 6 and day 9. Flow cytometry analysis of GFP expression was performed 3 to 4 days post infection. Macrophages were detached from wells by trypsin treatment, resuspended in PBS containing 1% FBS, fixed by addition of paraformaldehyde to 1% and GFP fluorescence analyzed by flow cytometry.
Transient expression assays and immunoprecipitations
HEK 293T cells were transfected with pCG plasmids expressing proteins of interest, by calcium phosphate co-precipitation method. Detergent extracts and anti-FLAG immune complexes were prepared and analyzed by immunoblotting as previously described20.
Antibodies and immunoblot analyses
Antibodies to human SAMHD1 and α-tubulin were purchased from Sigma-Aldrich. Antibodies to DCAF1, DDB1, Vpx (6D2.6), SIVmac 251 capsid (13-112-100), and those to FLAG- (M2), HA- (12CA5) and myc- (9E10) epitopes, were previously described12,20. For western blot analyses, proteins were separated by SDS-PAGE, transferred to PVDF membrane (Millipore) in a semi-dry blotter (BioRad), incubated with appropriate primary and secondary antibodies, and immune complexes visualized by enhanced chemiluminescence (GE Healthcare), as previously described12,20.
Immunoaffinity purifications of protein complexes and MudPIT analyses
Protein complexes were purified from HEK 293T cells transiently expressing epitope tagged subunits. The FLAG-Cullin4–DDB1(DDA1)–HA-DCAF1 complex was purified from HEK 293T cells transiently expressing FLAG-Cullin4 and HA-DCAF1. The FLAG-Cullin4–DDB1(DDA1)-DCAF1-HA-Vpx complex was purified from HEK 293T cells transiently expressing FLAG-Cullin4 and HA-Vpx. Protein complexes were purified by two sequential precipitations via HA- and then FLAG-epitope tags each followed by competitive elution with the respective epitope peptide under native conditions, from approximately 3 grams of wet cell mass. MudPIT analyses of purified protein complexes were performed as we previously described20. Distributed normalized spectral abundance factors (dNSAF) were calculated for each detected protein as described29.
RNA interference
MDM cultured in 24 well plates (Becton & Dickinson) were transferred into antibiotic free DMEM supplemented with 10% FBS 24 hrs before transfection. Cells were transfected with 40 pmol aliquots of siRNA per well, using Lipofectamine 2000 (Invitrogen). siRNA pools targeting SAMHD1 (L-013950-01), DCAF1 (L-021119-01) and control non-targeting siRNA pools “NT1” (D-001206-14-05) and “NT2” (D-001810-10), were purchased from Dharmacon. The SAMHD1-targeting pool comprised the siRNAs to the following target sequences: J-013950-09: GACAAUGAGUUGCGUAUUU; J-013950-10: CAUGUUUGAUGGACGAAUUU; J-013950-11: AAGUAUUGCUAGACGUGAA and J-013950-12: UUAGUUAUAUCCAGCGAUU. The DCAF1-targeting pool targeted the following DCAF1 sequences: J-021119-09: GGAGGGAAUUGUCGAGAAU; J-021119-10: CCACAGAAUUUGUUGCGCA; J-021119-11: GGAAUGACACUGUGCGCUU; J-021119-12: CGGAGUUGGAGGAGGACGA. Liposomes were formed using 2 μl Lipofectamine 2000 per well, according to manufacturer's instructions, as previously described12.
Depletion of SAMHD1 by Vpx-loaded SIV VLP and siRNA
CD14+ monocytes isolated and cultured as described above, were infected with various doses of Vpx-loaded SIV VLP two days after plating. on day 6, adherent macrophages were transfected with siRNA, using Lipofectamine 2000. Two to 3 days following the initiation of RNAi cells were harvested for immunoblot analyses, or infected with reporter viruses. Cells were harvested 1 day post-infection for real time analysis of viral cDNA synthesis, or three days post-infection for flow cytometry analysis of GFP fluorescence in the infected cells. For studies with proteasome inhibitor MG132, MDM were cultured in the presence or absence of MG132 (1 μg/ml) for one hour and then infected with 3-fold dilutions of SIVmac Vpx-loaded SIV VLP for additional 3 hours, in the continuous presence or absence of MG132, and harvested for western blot analysis of SAMHD1 levels.
Real time PCR
Reverse transcription products were quantified using BioRad MyiQ Single Color Real-Time PCR Detection System. A typical reaction contained 50 ng of DNA isolated with DNAeasy Kit (Qiagen) from infected or control cells and SYBR Green PCR master mix in a total volume of 25 μl (Applied Biosystems). HIV-1 HR reverse transcription products were amplified with ert2f and ert2r (early), and MH532 and MH531 (late), primers as previously described30. The levels of reverse transcription products were calculated by comparison to standard curves generated with serially diluted proviral clones. The variances between triplicate data points were less than 5%.
Supplementary Material
Acknowledgments
We thank the Karn, McDonald and Bernstein labs, Cathy Carlin, Robert Asaad, Morgan Reuter, and Alice Valentin-Torres, for reagents and help at various stages of this project. We acknowledge the excellent assistance of Chuanping Wang with fluorescence microscopy. We also thank Michael Emerman and Nicolas Manel for HIV-2 Rod proviral constructs, Frank Kirchhoff for the SIVmac 239-GFP proviral clone, Michael Lederman and David McDonald for discussions, Jonathan Karn and Michael Greenberg for discussions and comments on the manuscript. We are grateful to Linda Van Aelst and Dan Littman for advice, discussions and critical reading of the manuscript. This work in JS laboratory was supported by NIH grants R01 AI077459 and R21 AI084694 and by Case CFAR developmental funds. SKS, LF and MPW are supported by The Stowers Institute for Medical Research.
Footnotes
Full Methods and any associated references are available in the online version of the paper on www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author Contributions: KH, CH, MG, and J.S. designed the study. KH, CH, MG, MKB, SKS, and JS performed the experiments and analyzed the data. SS, LF, MPW provided expertise and analyzed the data. All authors discussed results and edited the manuscript.
The RAW mass spectrometry and SEQUEST results files for the MudPIT analyses reported in Supplementary Table I may be downloaded from ProteomeCommons.org Tranche using the following hash:+qNEgtnB3fXlq2awdE8X67zEY8D2mQbK/tYXKaA1lWve32mRnGBMOZ3Lkoy+y9HeooRuLwE8aIvPMYEx9xqBDzciIscAAAAAAAAK0w==This data can be accessed using the passphrase Hrecka&SAMHD1.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–593. doi: 10.1146/annurev.med.53.082901.104024. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 3.Nègre D, et al. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 2000;7:1613–1623. doi: 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- 4.Kaushik R, Zhu X, Stranska R, Wu Y, Stevenson M. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus gammaretrovirus infection. Cell Host Microbe. 2009;6:68–80. doi: 10.1016/j.chom.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu XF, Yu QC, Essex M, Lee TH. The vpx gene of simian immunodeficiency virus facilitates efficient viral replication in fresh lymphocytes and macrophage. J Virol. 1991;65:5088–5091. doi: 10.1128/jvi.65.9.5088-5091.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guyader M, Emerman M, Montagnier L, Peden K. VPX mutants of HIV-2 are infectious in established cell lines but display a severe defect in peripheral blood lymphocytes. EMBO J. 1989;8:1169–75. doi: 10.1002/j.1460-2075.1989.tb03488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rice GI, et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–32. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18(R2):R130–136. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 11.Le Rouzic E, et al. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle. 2007;6:182–188. doi: 10.4161/cc.6.2.3732. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava S, et al. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008;4:e1000059. doi: 10.1371/journal.ppat.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergamaschi A, et al. The human immunodeficiency virus type 2 Vpx protein usurps the CUL4A-DDB1 DCAF1 ubiquitin ligase to overcome a postentry block in macrophage infection. J Virol. 2009;83:4854–4860. doi: 10.1128/JVI.00187-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goujon C, et al. SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology. 2007;4:2. doi: 10.1186/1742-4690-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horton R, Spearman P, Ratner L. HIV-2 viral protein X association with the GAG p27 capsid protein. Virology. 1994;199:453–457. doi: 10.1006/viro.1994.1144. [DOI] [PubMed] [Google Scholar]
- 16.Kewalramani VN, Emerman M. Vpx Association with Mature Core Structures of HIV-2. Virology. 1996;218:159–168. doi: 10.1006/viro.1996.0176. [DOI] [PubMed] [Google Scholar]
- 17.Angers S, et al. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 18.Florens L, Washburn MP. Proteomic analysis by multidimensional protein identification technology. Methods Mol Biol. 2006;328:159–175. doi: 10.1385/1-59745-026-X:159. [DOI] [PubMed] [Google Scholar]
- 19.Pick E, et al. Mammalian DET1 regulates Cul4A activity and forms stable complexes with E2 ubiquitin-conjugating enzymes. Mol Cell Biol. 27:4708–4719. doi: 10.1128/MCB.02432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hrecka K, et al. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci USA. 2007;104:11778–11783. doi: 10.1073/pnas.0702102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn J. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J Biol Chem. 2010;285:37333–37341. doi: 10.1074/jbc.M110.133181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirsch VM, et al. Vpx is required for dissemination and pathogenesis of SIV(SM) PBj: evidence of macrophage-dependent viral amplification. Nat Med. 1998;4:1401–1408. doi: 10.1038/3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibbs JS, et al. Progression to AIDS in the absence of a gene for vpr or vpx. J Virol. 1995;69:2378–2383. doi: 10.1128/jvi.69.4.2378-2383.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita M, Perez O, Hope TJ, Emerman M. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog. 2007;3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita M, Emerman M. Cellular restriction targeting viral capsids perturbs human immunodeficiency virus type 1 infection of nondividing cells. J Virol. 2009;83:9835–9843. doi: 10.1128/JVI.01084-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 28.Manel N, et al. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Wen Z, Washburn MP, Florens L. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem. 2010;82:2272–2281. doi: 10.1021/ac9023999. [DOI] [PubMed] [Google Scholar]
- 30.Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.