

Abstract
Recent studies have clearly linked nuclear factor-kappaB (NF-κB), a transcription factor that plays a central role in regulating immune and inflammatory responses, to tumor development, progression, and metastasis as well as tumor therapy resistance. However, it still remains largely unknown on how the tightly regulated NF-κB becomes constitutively activated in tumorigenesis and how the original cancer immunosurveillance function of NF-κB is transformed to be tumorigenic. To address these important issues for cancer prevention and treatment, we discuss current understanding of the molecular mechanisms and molecules involved in the oncogenic activation of NF-κB. We also discuss current understanding of how NF-κB coordinates the inflammatory and malignant cells in tumorigenesis.
Keywords: A20, cancer, cancer immunology, cancer immunosurveillance, CYLD, deubiquitination, IκB, IKK, microRNA, NF-κB, oncogene, oncogenesis, PDLIM2, tumor, tumorigenesis, tumor suppressor, ubiquitination, WWOX
Introduction
The NF-κB family
Nuclear factor-κB (NF-κB) plays a central role in the regulation of diverse biological processes, including immune responses, development, cell proliferation and survival. Deregulated NF-κB has been linked to a variety of human diseases, particularly cancers [1-3]. The NF-κB family consists of five closely related DNA binding proteins: RelA (p65), RelB, c-Rel, NF-κB1/P50 and NF-κB2/P52, which function as various homodimers and heterodimers. All five NF-κB members share a highly conserved 300-amino-acid-long N-terminal Rel homology domain (RHD), which is responsible for their dimerization, nuclear translocation, DNA binding and also interaction with the inhibitors of NF-κB (IK Bs) (Figure 1). But they show huge difference in their C-terminal sequences and also synthesis modes. While RelA, RelB and c-Rel have trans-activating domain (TAD) at their C-termini and are synthesized directly as mature forms, p50 and p52 lack a TAD and are generated from large precursor proteins, p105 and p100, respectively. Interestingly, p105 and p100 contain a C-terminal ankyrin repeat domain (ARD), the characteristic domain of IκB. Indeed, both p105 and p100 function as kB-like inhibitors of NF-κ B[4, 5].
Figure 1.

Schematic representation of members of NF-κB and IKB families. The NF-κB family can be divided into two subfamilies. One subfamily consists of three members: RelA, RelB and c-Rel, which contain TADs at their C-termini and are synthesized directly as mature forms; the other one consists of two members: NF-κBl/p50 and NF-κB2/p52, which lack a TAD and are generated from large precursor proteins p105 and p100, respectively. Typical NF-κB dimers are usually composed of one member from each subfamily, such as RelA/p50 and RelB/p52, although all NF-κB members may form various homo- or hetero-dimers. Of note, the p50 or p52 homodimers repress NF-κB target gene expression due to lack of a TAD. The IκB family can be classified into three subfamilies: the typical IκB proteins (IκBα and IκBε), the precursor proteins (p100 and p105) and the atypical IKB proteins (BCL-3, IKB(3, IKBE and IKBNS). The typical subfamily just simply functions as NF-κB inhibitors. The precursor subfamily is also required for generation of the NF-κB members p50 and p52, besides being NF-κB inhibitors. While the processing of p105 to p50 is a constitutive event, the processing of p100 to p52 is tightly controlled. The atypical subfamily may function as co-activator or co-repressor of NF-κB depending on different situations [6, 7]. RHD: Rel homology domain; TAD: transactivating domain; ARD: ankyrin repeat domain; DD: death domain; LZ: leucine zipper; PEST: PEST containing sequence.
The IκB family
The IκB family comprises eight members and shares one common structural feature: presence of an ARD, which can bind to the RHD of NF-κB (Figure 1). While IκBCX and IκBG function as potent NF-κB inhibitors by sequestering NF-κ B dimers in the cytoplasm, other IκB proteins including IκBβ are not simple inhibitors of NF-κB but rather cofactors displaying both positive and negative effects on NF-κB-mediated gene transcription [6, 7]. For example, Bcl-3, which was originally identified as an oncogene from B-cell chronic lymphocytic leukemias (B-CLLs), has a typical TAD in its C-terminal. Bcl-3 is constitutively translocated into the nucleus where it interacts with p50 or p52 homodimers, which lack TAD, to facilitate transcription of NF-κB target genes. The nuclear translocation of Bcl-3 requires its K63-linked polyubiquitination (via the lysine 63 of ubiquitin) [8]. Bcl-3 may also repress transcription of NF-κB target genes. In this case, Bcl-3 promotes p50 homodimer occupancy of the κB site-containing promoters by inhibiting ubiquitination and degradation of p50, therefore preventing replacement by active NF-κB dimers [9].
NF-κB signaling pathways
In unstimulated cells, NF-κB dimers usually exist as latent complexes with the IκB proteins in the cytoplasm. There are two major mechanisms leading to NF-κB activation: the canonical and non-canonical NF-κB pathways, which are based on the inducible degradation of lκBα and processing of p100 to generate p52 (selective degradation of the C-terminal lκB-like sequence of p100), respectively (Figure 2).
Figure 2.

NF-κB signaling pathways. Although the canonical and non-canonical signaling pathways primarily activate the RelA/p50 and RelB/p52 dimers, respectively, all NF-κB members can be activated by either pathway or both. In fact, the RelA/p50 dimers may be sequestered in the cytoplasm by p100 and can be activated through p100 processing. On the other hand, NF-κB dimers containing p52 may be sequestered in the cytoplasm by IκBα and can be activated through IκBα degradation. Furthermore, activation of the canonical NF-κB signaling pathway can be induced through inducible degradation of IκBβ, IκBε and p105, a process similar to the inducible IκBα degradation, although their degradation dynamics can be different.
Pathways leading to NF-κB activation
Canonical NF-κB pathway:
The canonical pathway can be rapidly activated by a plethora of stimuli, such as inflammation cytokines and antigens. These NF-κB inducers induce assembly of a multimolecular complex that includes the RING-finger E3 ubiquitin ligase tumor necrosis factor receptor associated factor 6 (TRAF6), leading to TRAF6 auto-polyubiquitination [10, 11]. The K63 ubiquitinated TRAF6 then recruits and catalyses K63-linked ubiquitination of the transforming growth factor-β-activated kinase 1 (TAK1) and the IκB kinase (IKK) complex [which consists of two catalytic subunits, IKK1 (IKKα) and IKK2 (IKKβ), and a regulatory subunit, NEMO (NF-κB essential modulator, IKKγ)], so that TAK1 can phosphorylate and activate IKK. Once activated, IKK phosphorylates specific serines (S32 and S36) within lκBα, triggering its K48-linked ubiquitination by the E3 ubiquitin ligase β-transducin repeat-containing protein (β-TrCP) and degradation by the 26S proteasome. The released NF-κB translocates into the nucleus to regulate expression of a wide range of genes, particularly those involved in cell proliferation, survival, adhesion and migration. In addition to IκB degradation, many other regulatory mechanisms are also important for canonical NF-κB activation, such as phosphorylation, acetylation, prolyl isomerization and/or methylation of the prototypical NF-κB member RelA. These post-translational modifications prevent RelA from binding to IκBα, facilitate its DNA binding and transcriptional coactivator recruitment, and/or increase its stability [6].
Non-canonical NF-κB pathway:
In contrast to the canonical pathway, the noncanonical NF-κB pathway is induced only by a handful of stimuli including B-cell activating factor (BAFF), lymphotoxin β (LTβ), CD40 ligand, TNF-like weak inducer of apoptosis (TWEAK) and receptor activator of NF-κB ligand (RANKL) [6]. In addition, activation of the noncanonical NF-κB pathway is slow and depends on protein synthesis of NF-κB -inducing kinase (NIK) [12]. Although its mRNA expression is relatively abundant, the protein level of NIK is normally very low because of its constitutive degradation via a TRAF3-dependent mechanism [12, 13]. TRAF3 functions as a scaffold between NIK and TRAF2, which in turn recruits cellular inhibitors of apoptosis 1 and 2 (c-IAP1/2) into the NIK complex. Within the complex, c-IAP1 or c-IAP2 acts as the E3 ubiquitin ligase to mediate NIK polyubiquitination and proteolysis, thereby keeping its abundance below the threshold required for its function. In response to noncanonical NF-κB stimuli, either TRAF2 and TRAF3 or c-IAP1 and c-IAP2 are degraded by the proteasome, resulting in stabilization and accumulation of the newly synthesized NIK, thereby allowing NIK proteins to form oligomers and cross-phosphorylate each other for their activation [12-19]. Self-activated NIK in turn activates the IKK complex and specifically recruits IKK1 into the p100 complex to phosphorylate p100, leading to p100 ubiquitination by the β-TrCP E3 ubiquitin ligase and processing by the proteasome to generate p52 [20-23]. The processed product p52 together with its NF-κB binding partner translocates into the nucleus to induce or repress gene expression. Moreover, NIK-activated IKK may also induce IκBα degradation to activate the canonical NF-κB pathway [24].
Termination of NF-κB activation
Activation of the NF-κB pathways is tightly regulated and rapidly curtailed following the initial activating stimulus. Transient activation of NF-κ B is physiologically important because persistent activation can result in deleterious or even fatal conditions, such as acute inflammation, septic shock or at a cellular level, inappropriate cell growth and survival leading to cancer. It is therefore not surprising that feedback inhibition mechanisms to terminate NF-κB activation occur at almost all the levels or regulations that led to the initial activation.
Consistent with the central role of IKK in the activation of both canonical and non-canonical NF-κB pathways, several mechanisms are employed to inactivate IKK. Once activated, IKK also phosphorylates themselves and upstream activators, such as RIP in the canonical NF-κB pathway and NIK in the non-canonical NF-κB pathway, in addition to the IκB proteins. The autophosphorylation of the IKK catalytic sub-units at multiple C-terminal serines is supposed to cause IKK conformational alteration and phosphatase recruitment, resulting in dephos-phorylation of the IKK activation loops and IKK inactivation [25]. Phosphorylation of RIP and NIK, similar to lκB phosphorylation, leads to K48-linked ubiquitination and degradation of these IKK activators [26, 27]. The ubiquitination of RIP is mediated by A20 (TNFAIP3, TNFα-induced protein 3), a known target of NF-κB activation [28], providing a distinct feedback inhibition mechanism. In addition to as an E3 ubiquitin ligase for RIP K48-linked ubiquitination and degradation, A20 exerts at least two additional functions in the termination of NF-κB activation: on one hand functions as a deubiq-uitinase (DUB) to remove K63-linked ubiquitin chains from multiple NF-κB signaling molecules such as TRAF2/6, RIP, MALT1 and NEMO, and on the other hand blocks continuous K63-linked ubiquitination of these key NF-κB regulators by disrupting the interaction between the K63 ubiquitin ligases TRAF2/6 and their E2 ubiquitin conjugating enzymes Ubc13 and UbcH5c [26, 29-32]. As stated above and shown in Figure 2, K63-linked ubiquitination of NF-κB signaling molecules is critical for the assembly of signaling complex and subsequent activation of IKK/ NF-κB. The K48-ubiquitin ligase and the K63-ubiquitin deubiquitinase activities of A20 are mediated by its C-terminal zinc finger containing domain and N-terminal ovarian tumor (OTU) domain, respectively [26, 33, 34]. Interestingly, A20 is also a target of IKK activation for phoshorylation. In this case, IKK-mediated phos-phorylation increases the K63-specific DUB activity of A20, suggesting another level of feedback inhibition mechanism of IKK/NF-κB activation [35] (Figure 3). Besides A20, another deubiquitinase termed cylindromatosis (CYLD) also plays an important role in the termination of IKK/NF-κB activation. Like A20, CYLD is a target gene of NF-κB activation and can remove K63-linked ubiquitin chains from multiple activated IKK/NF-κB signaling molecules, including TRAF2/6, RIP, TAK, NEMO and Bcl-3 [36-38].
Figure 3.

Domain structure of A20. OUT: ovarian tumour domain; ZnF: zinc finger.
However, the best known and most critical feedback inhibition mechanism is to replenish the pool of IκB proteins via NF-κB activation itself. Same to many NF-κB repressors such as A20 and CYLD, all the IκB family members except IκBβ are direct targets of NF-κB. Newly synthesized IκB, particularly IκBα, enters the nucleus to bind to and transport NF-κB dimers back to the cytoplasm to reconstitute the status quo ante [39].
Recent studies, however, indicate that this feedback inhibition mechanism is neither sufficient nor necessary for the turnoff of NF-κB activation, at least in certain situations [40]. Instead, ubiquitination-mediated proteasomal degradation of activated NF-κB members directly in the nucleus provides a more rapid but also essential mechanism for NF-κB termination. Two different E3 ubiquitin ligases have been reported to be involved in the nuclear degradation of RelA: suppressor of cytokine signaling 1 (SOCS1) and PDZ-LIM domain-containing protein 2 (PDLIM2). The SOCS1 ligase complex, which consists of Elongins B and C, Cul2 and SOCS1, requires COMMD1 (MURR1) for its function in RelA ubiquitination and degradation [41-44]. COMMD1 binds to both SOCS1 and RelA and therefore enhances the interaction between SOCS1 and RelA. Accordingly, knockdown of COMMD1 stabilizes nuclear RelA and enhances NF-κB activity in response to TNF stimulation or certain stress stimuli [44, 45]. PDLIM2, a ubiquitously expressed nuclear protein with a strong shuttling activity between the nucleus and the cytoplasm, terminates NF-κB activation using two distinct but related mechanisms: it not only functions as an E3 ubiquitin ligase to promote nuclear RelA ubiquitination but also shuttles RelA to the nuclear matrix for the pro-teasome-mediated degradation [46, 47]. Importantly, PDLIM2 knockout mice are more sensitive to septic shock due to enhanced p65 activation and subsequently augmented production of inflammatory cytokines [46].
Crosstalk between NF-κB pathways
Although the canonical and non-canonical NF-κ B signaling pathways are fundamentally different, they do interact with each other at multiple levels. As described above, all known non-canonical NF-κB stimuli are also able to activate the canonical pathway, although most of canonical NF-κB stimuli are unable to activate the non-canonical pathway. However, activation of the canonical NF-κB pathway does facilitate activation of the non-canonical NF-κB pathway, e.g. transcriptional upregulation of p100 and non-canonical NF-κB stimuli such as CD40L and TWEAK. Interestingly, recent studies indicate that activation of the non-canonical NF-κB pathway may facilitate or repress activation of the canonical pathway depending on cell-context. For example, non-canonical NF-κB activation by the viral oncoprotein Tax leads to transcriptional repression of the tumor suppressor WWOX, which selectively inhibits Tax activation of the canonical NF-κB by blocking IKK1-mediated RelA phosphorylation [48]. On the other hand, NIK-dependent induction of CYLD by RANKL has been reported to be critical in repressing osteo-clastogenesis through downregulation of the TRAF6 signaling pathways including the canonical NF-κB activation [49]. Furthermore, both signaling pathways activate some common NF-κ B members and regulate some common target genes. In addition to the signaling interactions, the two pathways also cooperate functionally. Whereas one of the main functions of canonical NF-κB activation is to regulate innate immunity, the major function of non-canonical NF-κB activation is to control adaptive immunity. Another common property of the two NF-κB pathways is that they both have been linked to various human pathogenesis, particularly cancer, although it still remains largely unknown whether and how the two signaling pathways cooperate during tumorigenesis.
Yin-Yang imbalance of NF-κB activation in cancer
The involvement of NF-κB in oncogenesis has been long suggested since the discovery of c-Rel and its viral derivative v-Rel [50, 51]. The v-Rel oncoprotein induces acute lymphoid malignancies in young chickens as well as T-cell lymphomas in transgenic mice [52]. Subsequent work has indicated that persistent activation of NF-κB is associated with various human cancers. More recent studies involving genetically modified mice have clearly demonstrated the significance of the IKK/NF-κB signaling in tumorigenesis. For example, conditional deletion of IKK2 or RelA in intestinal or lung epithelial cells results in significant, although not complete, inhibition of tumor genesis and progression in mouse models of colitis-associated cancer and lung carcinomas, respectively [53, 54]. On the other hand, transgenic mice conditionally expressing c-Rel in mammary gland or a constitutive processing form of p100 in lymphocytes develop mammary tumors and lymphomas, respectively [55, 56]. However, the molecular mechanisms by which the NF-κB signaling pathways become constitutively activated during cancer pathogenesis still remain obscure. Both NF-κB pathways are tightly regulated by both negative (yin) and positive (yang) regulators. Logically, disruption of the delicate balance between those yin-yang factors due to excess of yang and/or shortage of yin should result in persistent activation of NF-κB. In some cancers, the constitutive activity of NF-κB is clearly caused by genetic alterations in genes encoding NF-κB members and their inhibitors IκBs. In most cases, however, the deregulated NF-κB activity is attributed to overactivation of the positive regulators and/or inactivation of the negative regulators of the IKK/NF-κB signaling.
NF-κB versus IκB
Activating mutations of NF-κB members and their co-factors:
As mentioned above, the link between NF-κB and cancer was initially suggested by the acute oncogenicity of the v-Rel oncoprotein, a close kinship to c-Rel. Soon thereafter, all five NF-κB members have been found to be overactivated in human cancers (Table 1). One mechanism leading to aberrant activation of NF-κB involves the genetic alterations of the NF-κB genes themselves, which include gene amplifications, mutations, deletions and chromosomal translocations, a major cause of oncogene activation.
Table 1.
Alteration of NF-κB and IκB in cancer
| Genes | Locus | Alteration | Tumor type | References |
|---|---|---|---|---|
| rela | 11q13 | amplification rearrangement splicing variant amino acid substitution overactivation | various types of lymphomas, including diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma and follicular large cell lymphoma; solid tumors such as squamous head and neck, breast and stomach adenocarcinoma B-cell non-Hodgkin's lymphoma and multiple myeloma non-small cell lung carcinoma multiple myeloma various types of cancers | [51,190-192] |
| relb | 19q13.32 | rearrangement | adult T-cell leukemia cell lines | [193] |
| c-rel | 2p13-p12 | amplification rearrangement overexpression | diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, and follicular large cell lymphoma follicular lymphoma and diffuse large B-cell lymphoma follicular lymphoma, diffuse large cell lymphoma and non-small cell lung carcinoma | [51,190,194,195]. |
| nf-kb1 | 4q24 | rearrangement overexpression | acute lymphoblastic leukemia various cancers including non-small cell lung carcinoma, colon, prostate, breast and brain cancer | [51,196] |
| nf-kb2 | 10q24 | rearrangement overexpression | cutaneous T-cell lymphoma, B-cell non-Hodgkin's lymphoma, B-cell chronic lymphocytic leukemia and multiple myeloma cutaneous T-cell lymphoma, breast and colon carcinoma | [51,196-199] |
| bcl3 | 19q13.1-q13.2 | rearrangement overexpression | B-cell chronic lymphocytic leukemia and B-cell non-Hodgkin's lymphoma B-cell chronic lymphocytic leukemia and B-cell non-Hodgkin's lymphoma | [51,200,201] |
| ikbα | 14q13 | mutation | Hodgkin's disease | [51,85] |
| ikbε | 6p11 | mutation | Hodgkin's disease | [88] |
Consistent with their global functions, genetic alterations of either the rela gene or the nf-κb1 gene are only infrequently found in human tumors, although overexpression and overactivation of both RelA and p50 (due to activation of the NF-κB signaling, see discussion below) are common not only in tumors but also in other human diseases. Currently, a definitive correlation between the genetically altered rela and nf-κb1 genes and human tumorigenesis remains to be established.
A much clearer link between nf-κb genetic alterations and tumorigenesis has been observed in the studies of c-rel and nf-κb2. Amplification of the c-rel gene has been often detected in various non-Hodgkin's B-cell lymphomas such as diffuse lymphomas with a large cell component (DLLC), follicular large cell lymphomas and mediastinal thymic B-cell lymphomas (Table 1). The c-rel gene also undergoes rearrangement in some DLLCs and in a few follicular lymphomas [51]. Interestingly, the resulting C-terminal deletion of c-Rel because of the gene rearrangement is reminiscent of that of the v-rel oncogene. Although the involved mechanism remains to be investigated, overexpression of c-Rel has often been reported in solid tumors as well, such as breast and lung cancers [57, 58]. Given the tumorigenesis in c-Rel transgenic mice and the oncogenic potentials of v-rel, there is no doubt that amplification and rearrangement of the c-rel gene contribute to, if not cause, the lymphomagenesis.
Unlike its closest relative nf-κb1, the human NF-κb2 gene is frequently involved in chromosomal translocations or small deletions associated with development of various lymphomas and leukemias, such as cutaneous T-cell lymphoma (CTL), B-cell non-Hodgkin lymphoma (B-NHL), B-cell chronic lymphocytic leukemia (B-CLL), multiple myeloma (MM) and adult T-cell leukemia/lymphoma (ATL) [51, 59]. In fact, the nf-κb2 gene is the first NF-κB member that was found to undergo genetic alterations in human tumors. In all cases studied, such gene rearrangements always lead to deletions of the C-terminal processing-regulatory sequences together with part of ankyrin repeats [6]. Since these C-terminal sequences are essential for repressing p100 nuclear translocation and constitutive processing, these truncated p100 mutants undergo constitutive processing in association with the κb site-containing promoters in the nucleus [20, 60, 61]. The genetic mutation of the nf-κb2 gene results in the loss of IκB-like function of p100 in two ways: genetic deletion and biochemical degradation (protein processing) of C-terminal ankyrin repeats. The genetic mutation also results in the gain of transcriptional function in two related mechanisms, because these p100 truncation proteins and their processed products p52 regulate transcription of common and distinct target genes [60]. Although these constitutive processing forms of p100 show strong oncogenicity both in vitro and in vivo [56, 60, 62], the involved mechanisms remains unclear. Homozygous knockout of the nf-κb2 gene leading to deficiency of both p100 and p52 proteins in mice is not tumorigenic [63, 64], suggesting that the simple loss of the IκB-like function is not sufficient to account for the oncogenicity of the truncated p100 proteins. Interestingly, overexpression of p52 in the absence of p100 in p100 knockin mice causes marked gastric and lymphocyte hyperplasia and early postnatal death [65]. On the other hand, p52 transgenic mice expressing wild type p100 only leads to development of thymoma at extremely low rate, although over 50% mice develop inflammatory autoimmune disease by 8-month age [66]. Furthermore, mice selectively expressing the human n1-κb2 gene in mammary epithelial cells by the β-lactoglobulin milk protein promoter exhibit ductal thickening and hyperplasia only when the transgene expression and p100 processing to p52 are repeatedly induced through multiple pregnancies [67]. Thus, it seems plausible that both loss of κB-like function of p100 and gain of transcriptional function of p52 contribute to the oncogenicity of C-terminally truncated p100 proteins. This idea is also consistent with the fact that aberrant processing to p52 of p100 has been found in many types of tumors [6]. The transcriptional function of truncated p100 proteins themselves also plays a role in the oncogenesis, given the strong transforming abilities of the mutants.
Another piece of evidence linking activating mutations of the nf-κb genes to tumorigenesis comes from chromosomal translocations of the Bcl-3 oncoprotein, a coactivator of p50 and p52 (Figure 1). Bcl-3 was originally identified through cloning of the t(14;19) breakpoint junction, which occurs in a subset of B-cell chronic lymphocytic leukemias (B-CLLs) [68]. Of note, the rearranged bcl-3 gene remains intact but is transcriptionally activated, resulting in overproduction of the Bcl-3 protein and presumably elevated transactivation activity of the p50 or p52 complexes. Overproduction of Bcl-3 independent of its gene translocation has also been observed in various tumors including breast cancer, colorectal cancer, hepatocellular carcinoma, melanoma, nasopharyngeal carcinoma, and different lymphomas [69-74]. In fact, overexpression of Bcl-3 is often associated with tumor progression and poor prognosis. The contribution of Bcl-3 overexpression to tumorigenesis is further suggested by the findings that expression of exogenous Bcl-3 leads to cell transformation in vitro and transgenic mice overexpressing Bcl-3 in B cells develop lymphadenopathy and splenomegaly with excess number of B cells [75, 76].
Inactivating mutations of IκB proteins:
The first hint to a tumor suppressive function of the prototypical NF-κB inhibitor IκBα came from the finding that inactivation of IκBα by iκbα antisense transcript is sufficient to transform mouse fibroblasts [77]. Although later studies showed that iκbα knockout fibroblasts are not transformed [78, 79], the tumor suppressive role of IκBα cannot be overlooked, as defective activity of IκBα is often associated with persistent NF-κB activation in many human tumors. Importantly, overexpression of a super-repressor form of IκBα induces tumor cell apoptosis and inhibits tumorigenesis in numerous animal models [48, 80-82]. In most cases, the I KBCX deficiency in tumors is because of the enhanced protein degradation mediated by consti-tutively activated IKK. However, a subset of Hodgkin's lymphomas (HLs) are associated with genetic mutations or deletions of the iκbα gene, which lead to generation of nonfunctional or unstable IκBα mutants [93-87]. Similar to iκbα, the iκbε gene also undergoes somatic mutations in some Hodgkin's lymphomas, generating nonfunctional lκBε mutants [88]. Interestingly, simultaneous inactivations of both the iκbα gene and the iκbε gene are also detected in certain Hodgkin/Reed-Sternberg (HRS) cells. Given the non-redundant functions of IκBα and IκBε in the control of a subset of NF-κB target genes [78, 89], these findings suggest the importance of coordination among NF-κB members and also their target genes in tumorigenesis.
NF-κB activators versus NF-κB terminators
Overactivation of NF-κB activators:
Although activating mutations of the NF-κB proteins and inactivating mutations of the IκB proteins have been defined in human tumors, they are mainly limited to lymphoid malignanices and only account for a small number of leukemias and lymphomas. On the other hand, constitutive degradation of IκBs due to the elevated activation of IKK, the primary NF-κB-activating kinase, has been found not only in lymphoid malignancies but also in most solid tumors, suggesting one common mechanism for the NF-κB oncogenic activation. As yet, however, no oncogenic mutations of IKK have been detected. Instead, persistent existence of NF-κB stimuli, particularly proinflammatory cytokines and growth factors in the tumor microenvironment, has been suggested to be involved in the oncogenic activation of IKK/NF-κB (Table 2). Interestingly, many of these NF-κB inducers are also target genes of NF-κB activation, thereby providing an autocrine or paracrine mechanism for persistent NF-κB activation.
Table 2.
NF-κB activating and adaptor molecules in cancer
| NF-κB pathway | Cancer linkage | References | |
|---|---|---|---|
| Growth Factors | |||
| EGF | canonical | stimulates tumor cells proliferation; modulates tumor-associated angiogenesis and bone metastasis; regulates resistance to chemotherapy | [202-204] |
| NGF | canonical | promotes survival and proliferation of breast cancer cells | [205] |
| TGFβ | canonical | causes adenoma and adenocarcinoma; induces epithelial to mesenchymal transition in cancer | [206-209] |
| Kinases | |||
| IRAK | canonical | its polymorphism correlates with prostate cancer risk; its expression correlates with lung cancer development | [210-212] |
| RIP | canonical | mediates proliferation of human head and neck squamous cell carcinoma | [213] |
| MEKKs | canonical | essential for cancer cell survival | [214, 215] |
| Tpl2 | canonical | promotes cell migration and transformation | [216-218] |
| TBK1 | canonical | highly expressed in cancer; essential for KRAS-dependent cancer cells survival | [219, 220] |
| MLK3 | canonical | critical for cancer cell migration and invasion; highly expressed in breast cancer cells | [221, 222] |
| Raf | canonical | oncoprotein involved in various cancers; essential for the progression of metastatic melanoma and breast epithelial cancer | [223, 224] |
| TAK1 | canonical | required for progression and metastasis of breast cancer cells; required for R-RAS mediated transformation of mammary epithelial cells; suppresses procarcinogenic pathway in liver cancer | [225-227] |
| PKCs | canonical | promotes tumor progression and invasion | [228-230] |
| AKT | canonical | activated in multiple types of cancer; promotes cancer progression | [231, 232] |
| PKR | canonical | promotes cancer progression and metastasis | [233, 234] |
| PAK1 | canonical | overexpression and/or hyperactivation in cancer; promotes tumor progression and invasion | [235, 236] |
| CK2 | canonical | promotes tumorigenesis | [237] |
| NIK | both canonical and noncanonical | shows oncogenicity in vitro; elevated expression in various types of cancer | [103, 104, 238, 239] |
| Adaptors | |||
| Ras | canonical | promotes cancer proliferation, metastasis and invasion; commonly mutated in various cancers | [240, 241] |
| FADD | canonical | elevated in and associated with aggressive lung cancer | [242] |
| MyD88 | canonical | crucial for tumour promotion in models of spontaneous and carcinogeninduced intestinal tumorigenesis; required for RAS-mediated carcinogenesis | [243, 244] |
| Bcl10 | canonical | aberrant expression found in primary cutaneous marginal zone B-cell lymphoma | [245] |
| MALT1 | canonical | contributes to tumorigenesis in diffuse large B-cell lymphoma of the activated B-cell subtype | [246] |
| CARMA1 | canonical | oncogenic mutation of CARMA1 is found in diffuse large B cell lymphoma; overexpressed in primary gastric B-cell lymphoma | [247, 248] |
| Viral oncoproteins | |||
| HTLV-1 Tax | both canonical and noncanonical | promotes cell transformation and tumor progress | [249] |
| EBV LMP-1 | both canonical and noncanonical | promotes cell transformation, tumor progress and migration in EBVassociated cancer | [250, 251] |
| Herpesvirus Tio | both canonical and noncanonical | essential for transformation of primary human T cells | [252-254] |
| KSHV K13 | both canonical and noncanonical | promotes cell transformation and tumor progress | [118, 255] |
EGF: epidermal growth factor; NGF: nerve growth factor; TGFβ: transforming growth factor beta; ROS: reactive oxygen species; HTLV-1: human T-cell lymphotropic virus type 1; EBV: Epstein-Bar virus; LMP-1: latent membrane protein 1; Tio: two in one; KSHV: Kaposi's sarcoma-associated herpesvirus; K13: FADD-like interleukin-1 beta-converting enzyme (FLICE) inhibitory protein (vFLIP)
Another important mechanism contributing to oncogenic NF-κB activation involves overexpression and activating mutations of IKK/NF-κB upstream activators. One example is the geneticmutations of the CARMA1/MALT1/Bcl10 complex, which is essential for antigen-induced IKK/ NF-κB activation. Chromosome translation of the bcl10 gene [t(1:14)(p22:q32)] and the malt1 gene [t(11:18)(q21:q21)] are frequently found in B-cell lymphomas of mucosa-associated lymphoid tissue (MALT), while missense mutations of the carma1 gene are detected in about 10% activated B cell-like (ABC) diffuse large B cell lymphomas (ABC-DLBCLs) and in about 4% germinal center B cell-like (GCB)-DLBCLs [90-98]. Interestingly, the genetic mutations of these signaling proteins always result in their overexpression and/or enhanced abilities in IKK/NF-κB activation, which is required for the survival of tumor cells [95-99].
As stated above, stabilization and accumulation of the NIK kinase will lead to activation of both canonical and non-canonical NF-κB pathways. It is thus not surprising that overexpression of the NIK protein has been linked to various tumors such as multiple myeloma, adult T-cell leukemia, melanoma, pancreatic carcinoma, breast cancer and lung cancer. Several mechanisms may contribute to the oncogenic expression of NIK: inactivating mutations of NIK negative regulators TRAF2/3 and c-IAP1/2, activating mutations of NIK and its positive activators CD40 and LtβR, as well as epigenetic activations of NIK mRNA transcription [100-102]. In support of the role of NIK overexpression in tumorigenesis, a recent study show that expression of exogenous NIK is sufficient to transform rat fibroblasts and knockdown of NIK reverses the tumor phenotype of those malignant cells with high expression of NIK [103, 104]. It is important to reiterate that the oncogenic action of NIK depends on NF-κB activation. Interestingly, NIK can also be stabilized and activated through K63-linked ubiquitination, which is mediated by an atypical E3 ubiquitin ligase termed zinc finger protein 91(ZFP91) [105]. Giving the findings that ZFP91 is overexpressed in 93% acute myelogenous leukemia (AML) and required for cancer survival [105, 106], ZFP91-mediated stabilization may stand for another mechanism of NIK oncogenic activation.
In addition to deregulation of those central NF-κ B signaling molecules, activation of many well-known oncoproteins also persistently activates IKK/NF-κB (Table 2). For instance, oncogenic mutations of the ras gene, which occur in over 30% of human tumors, induce IKK/NF-κB indirectly through the AKT and Raf kinases, another two famous oncogenes that can induce tumors when expressed in transgenic mice [107, 108]. It seems that activation of NF-κB is one crucial mechanism of Ras-mediated tumorigenesis, because inhibition of NF-κB activation by transgenically expressing the super-repressor form of IκBα or genetically silencing RelA significantly blocks Ras-mediated tumorigenesis and tumor progression [54, 80].
Similar to cellular oncogenes, many viral onco-proteins also target NF-κB for their oncogenicities [6, 59]. The first and best example is the Tax oncoprotein encoded by the human T-cell leukemia virus type I (HTLV-I), an etiological agent of an acute T-cell malignancy termed adult T-cell leukemia/lymphoma (ATL) [109]. By directly interacting with NEMO, Tax activates IKK to phosphorylate IκBα, resulting in IκBα degradation and canonical NF-κB activation [110-114]. In parallel, Tax specifically recruits IKK1 into the p100 complex to activate the non-canonical NF-κB pathway independent of NIK [115, 116]. The significance of NF-κB activation in Tax-mediated tumorigenesis has been well defined. Whereas HTLV-I or Tax mutants selectively defective in NF-κB activation lose the transforming ability, blockage of Tax activation by overexpression of p100 or the super-repressor form of IκBα prevents HTLV-I/Tax-mediated transformation [109]. However, the most impressive in vivo data supporting a role of NF-κB in Tax-mediated pathogenesis is from studies on Tax transgenic mice in the presence or absence of endogenous p100/p52 [48]. In this model, genetic knockout of the nf-κb2 gene, leading to no expression of both p100 and p52 proteins, significantly delays Tax-mediated tumorigenesis in mice. Later on, NF-κB has been found to be targeted for human tumorigenesis by many tumor viruses including Kaposi sarcoma-associated herpesvirus (KSHV), and Epstein-Barr virus (EBV) [6, 59]. KSHV can induce several different clinical variants of Kaposi's sarcoma, primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) through expression of a viral version of the cFLIP protein named vFLIP, which resembles Tax in the NF-κB activation [117, 118]. On the other hand, EBV encodes a potent oncoprotein named latent membrane protein 1 (LMP1), which acts like a constitutively activated member of the TNFR/CD40 superfamily for the activation of both canonical and non-canonical NF-κ B pathways [119].
Inactivation of NF-κB terminators:
NF-κB is controlled by a delicate counterbalance between its activators and terminators, guaranteeing an inducible but transient activation of NF-κB. Thus, disruption of this fine balance by inactivation of NF-κB terminators represents another important mechanism of oncogenic activation of NF-κB. In fact, rapidly increasing evidence indicates that many NF-κB terminators actually function as tumor suppressors (Table 3).
Table 3.
NF-κB signaling repressors in cancer
| Gene Name | Effects on NF-κB Module | Cancer linkage | References |
|---|---|---|---|
| CYLD | targets multiple NF-κB signaling molecules | tumor suppressor; mutated, deleted or downregulated in various cancers | [100, 130, 131, 134, 136-138] |
| A20 | targets multiple NF-κB signaling molecules | tumor suppressor; mutated, deleted or downregulated in various lymphomas | [124-128, 256] |
| PDLIM2 | promotes nuclear RelA degradation | potential tumor suppressor; epigenetically downregulated in various types of cancer | [144-147] |
| WWOX | inhibits HTLV-1 Tax induced RelA phosphorylation and NF-κB activation | inhibits tumor growth; deleted or downregulated in various types of cancer | [257-263] |
| CHFR | negatively regulates RelA transcriptional activity | potential tumor suppressor; silenced in various cancer | [264] |
| LZAP | impairs RelA phosphorylation and transcription activity | inhibits cellular proliferation and clonogenic growth; downregulated in human head and neck squamous cell carcinomas | [265] |
| NLBP | inhibits RelA transcription activity | inhibits cell invasion; downregulated in invasive cancer cells | [266] |
| PIAS1 | blocks the DNA binding activity of RelA | tumor suppression function; downregulated in multiple myeloma and colon cancer | [267-269] |
| LDOC1 | inhibits TNF-α and PMA induced NF-κB activation | sensitizes pancreatic cancer cells to apoptosis; downregulated in pancreatic cancer | [270] |
| OPTN | inhibits NF-κB activation; competes with NEMO for ubiquitinated RIP binding | its mutation is linked with some forms of glaucoma | [271, 272] |
| MENIN | interacts with RelA, p50 and p52; represses RelA transcriptional activity | tumor suppressor; is mutated or deleted in parathyroid tumors | [273] |
| ARF | represses RelA transcriptional activity by ATR and Chk1-dependent phosphorylation | central component of the cellular defense against oncogene activation | [274] |
| RKIP | interacts with NIK, TAK1 and IKKs; inhibits TNF-α induced IKK activation | inhibits prostate cancer metastasis | [275-277] |
| KEAP1 | induces IKK2 degradation and inhibits IKK2 phosphorylation | functions as tumor suppressor, and mutated in multiple types of cancer | [278-280] |
| PP2A | dephosphorylates MEKK3; inhibits LPS induced IKK2 and NF-κB activation | involved in growth suppression, enhances apoptosis, restores differentiation, impairs clonogenic potential | [281, 282] |
Abbreviation: CHFR: checkpoint with forkhead and ring finger domains; LZAP: LXXLL/leucine zipper-containing alternative reading frame (ARF)-binding protein; NLBP: novel LZAP-binding protein; PIAS1: protein inhibitor of activated STAT, 1; LDOC1: leucine zipper, down-regulated in cancer 1; OPTN: optineurin; MENIN: multiple endocrine neoplasia I; ARF: cyclin-dependent kinase inhibitor 2A (p16); ATR: ataxia telangiectasia mutated (ATM) and Rad3-related checkpoint kinases; Chk1: checkpoint kinase 1; RKIP: Raf-kinase inhibitor protein; KEAP1: kelch-like ECH-associated protein 1; PP2A: serine/threonine protein phosphotase 2A.
Consistent with its role in NF-κB termination, genetic knockout of the deubiquitinase A20 leads to severe spontaneous multiorgan inflammation and cachexia in mice and importantly inactivating mutations of A20 have been detected in several human autoimmune disorders including Crohn's disease, Coeliac disease, psoriasis, rheumatoid arthritis, systemic lupus erythematosus and diabetes [120, 121]. Although the premature death of A20 knockout mice prevented the detection of a potential tumorigenic effect, the tumor suppressor role of A20 has been well suggested recently. The first clue came from the identification of the a20 gene as a target gene of 6q23.3-q24.1 deletion in ocular adnexal marginal zone B cell lymphoma (MZBCL) [122]. Soon thereafter, A20 was found to be frequently inactivated by deletion, promoter methylation or somatic mutations in a variety of lymphomas, including Hodgkin's lymphoma (HL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), mucosa-associated lymphoid tissue (MALT) lymphoma, follicular lymphoma (FL), Burkitt's lymphoma, natural killer cell lymphoma and adult T cell leukaemia/lymphoma (ATL) [34, 123-126]. Interestingly, the inactivating mutations often involve both alleles of the gene, suggesting that a complete inactivation of A20 favors cell survival and tumorigenesis. Except the genetic and epigenetic inactivations, A20 is also negatively regulated at the protein level by the paracaspase MALT1, which cleaves and inactivates A20 to enhance NF-κB activation [127]. As we already discussed, MALT1 is a proto-oncoprotein that is constitutively activated in certain lymphomas. Notably, the oncogenicity of MALT1 is through targeting NF-κB. In direct support of the tumor suppressor role of A20, reconstitution of A20 decreases NF-κB activation and induces growth arrest and apoptosis of A20-deficient lymphoma cell lines [124, 128, 129].
CYLD, another deubiquitinase (DUB) that plays a key role in NF-κB termination, was originally identified as a tumor suppressor that is mutated in familial cylindromatosis, an autosomal-dominant predisposition to multiple tumors of skin appendages including multiple familial trichoepithelioma and Brooke-Spiegler syndromes [130]. The genetic mutations of the cyld gene involve loss of heterozygosity (LOH) and mutations (Figure 4). Interestingly, in all cases the allele lost is the wild-type allele inherited from the parent not carrying mutations and all mutations predict absence, truncation or mutation of the encoded protein, leading to loss of the DUB activity of CYLD [131-134]. These findings suggest the importance of the DUB activity in the tumor suppressor function of CYLD. However, the direct evidence came from a more recent study showing a strong tumorigenicity of a CYLD point mutant defective in the deubiquitinating function that mimics the identified mutations of cyld in human tumors [135]. In addition to skin cancers, inactivating mutation or down-regulation of CYLD has been detected in several human cancers including colon cancer, hepato-cellular carcinoma (HCC), T-cell acute lymphoblastic leukemia (T-ALL), multiple myeloma (MM) and melanoma, and is inversely correlated with NF-κB activation, tumor progression and patient's survival [100, 136-138]. Thus, inactivation of CYLD DUB catalytic activity by any mechanism contributes to tumorigenesis by promoting unchecked NF-κB activity and enhanced cell survival. Consistent with the tumor suppressor role of CYLD, cyld-deficient mice exhibit increased susceptibility to cilitis-associated tumorigenesis and chemically induced skin tumors [8, 139]. Mechanistic studies further indicate that CYLD deficiency leads to sustained NF-κB activity by increasing K63-linked ubiquitination and/or nuclear translocation of NF-κB activators/co-activator such as TRAF2, NEMO and Bcl-3. Interestingly, recent studies show that CYLD is negatively regulated by miR-181b, a microRNA (miRNA) that is upregulated during cellular transformation and in acute lymphocytic leukemia (ALL) [140, 141]. More importantly, expression of miR-181b inhibits CYLD, leading to increased NF-κB activity required to maintain cell transformation. These studies provide a different mechanism for the oncogenic deregulation of CYLD and NF-κB.
Figure 4.

Domain structure of CYLD. CAP: cytoskeletal-associated protein-glycine-conserved repeats; PR: proline-rich region; USP: ubiquitin carboxy-terminal hydrolases domain.
Like A20 and CYLD, PDLIM2 is also required for the termination of NF-κB, although the involved mechanisms are totally different [46] (Figure 5). Thus, it should be no surprise if PDLIM2 is linked to tumor suppression. Indeed, recent studies indicate that expression of PDLIM2 is significantly downregulated in several NF-κB-associated tumors, including adult T-cell leukemia/lymphoma (ATL), breast cancer and colorectal cancer [142-147]. More importantly, expression of exogenous PDLIM2 or reinduction of endogenous PDLIM2 inhibits constitutive NF-κB activation and suppresses the in vitro anchorage-independent growth and in vivo tumor formation of these malignant cells [144, 146, 147]. In contrast, PDLIM2 mutants defective in NF-κB repression lose the tumor suppressive function. It is noteworthy that in the case of ATL, which is mediated by the HTLV-I retrovirus, the tumor suppression role of PDLIM2 is much more complicated, since PDLIM2 also promotes ubiquitination and degradation of the viral onco-protein Tax, a potent NF-κB activator that is largely responsible for HTLV-I-mediated patho-genesis [144]. In fact, Tax inhibition plays a predominant role in PDLIM2-mediated ATL suppression [47, 109]. One mechanism contributing to the oncogenic PDLIM2 downregulation involves the methylation of the pdlim2 promoter [145-147]. Although not reported yet, the genetic alterations of the pdlim2 gene could also be involved in the PDLIM2 downregulation in tumors, as the pdlim2 gene is located at chromosome 8p21.1, a region that frequently undergoes allelic loss in a number of tumor types including breast, colon, live, lung, stomach, prostate and ovarian cancer [143, 148-157]. Furthermore, the activity of PDLIM2 is regulated by protein phosphorylation [158, 159], suggesting another potential mechanism for PDLIM2 inactivation in tumor.
Figure 5.
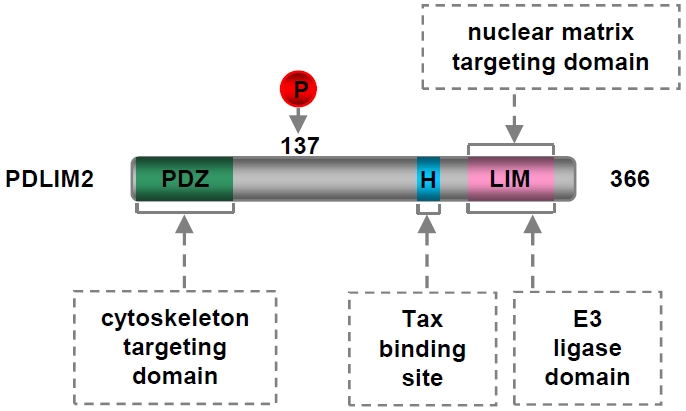
Domain structure of PDLIM2. PDZ: postsynaptic density 65-discs large-zonula occludens 1; H: putative α-helix motif; LIM: abnormal cell lineage 11-islet 1-mechanosensory abnormal 3.
In addition to those ‘traditional’ repressors, NF-κ B is negatively regulated by many miRNAs, which induce mRNA degradation or inhibit mRNA translation of NF-κB or its key signaling components (Table 4). Downregulation or deletion of these miRNAs has been detected in a broad range of human tumors including leukemias, lymphomas and solid tumors. For instance, miR-125b, which targets the bcl-3 onco-gene, is downregulated in multiple types of tumor, including hepatocellular carcinoma (HCC), breast cancer, oral cancer, bladder cancer, anaplastic thyroid carcinomas, metastatic cutaneous malignant melanoma, head and neck squamous cell carcinomas (HNSCC) and ovarian cancer [160-166]. In fact, the downregulation of miR-125b is often associated with tumor progression and patient's survival. In support of the tumor suppressor role of miR-125b, expression of exogenous miR-125b blocks the tumorigenicity of these malignant cells. Interestingly, the tumor suppression effect, at least the anti-growth activity of miR-125b can be antagonized by expression of Bcl-3 [166]. It should be pointed out that some NF-κB repressor miRNAs including miR-125b are already known target genes of NF-κB [167-171], further supporting the idea that disruption of feedback inhibition of NF-κB is a common and important mechanism of NF-κB pathogenic activation and human tumorigenesis.
Table 4.
NF-κB regulating microRNAs in cancer
| miRNA | Targets in NF-κB Signaling | Alteration in Cancer | Cancer linkage | Other Targets Linked to Cancer | References |
|---|---|---|---|---|---|
| NF-κB members | |||||
| miR-9 | NF-κB1 | epigenetically downregulated | gastric cancer and clear cell renal cell carcinoma | E-cadherin | [283-286] |
| miR-125b | Bcl-3 | downregulated | human liver cancer, melanoma, glioma, ovarian cancer, bladder cancer and breast cancer | LIN28B2; MUC1; E2F3; Endothelin-1; BAK1; BMF; CYP24; p53 | [165, 166, 170, 287-292] |
| NF-κB activators | |||||
| miR-15a, miR-16 | IKK1 | downregulated; deleted | chronic lymphocytic leukemia, multiple myeloma, lung squamous cell carcinoma and ovarian cancer | BMI-1 | [293-297] |
| miR-223 | IKK1 | downregulated | chronic lymphocytic leukemia and acute myeloid leukemia | E2F1 | [296, 298, 299] |
| miR-218 | IKK2 | downregulated | glioma, gastric cancer and lung squamous cell carcinoma | ROBO1; LASP1 | [300-304] |
| miR-199a | IKK2 | downregulated | breast cancer, melanoma and bladder cancer | MET; KRT7 | [163, 305-307] |
| miR-146a | IRAK1 TRAF6 | downregulated | breast cancer and pancreatic cancer | - | [308-310] |
| NF-κB inhibitors | |||||
| miR-181b-1 | CYLD | upregulated | acute lymphocytic leukemia | - | [140, 141] |
| miR-301a | NRF | upregulated | pancreatic tumor | - | [311] |
Oncogen ic interplay between NF-κB pathways
Both the canonical and non-canonical NF-κB pathways have been linked to tumorigenesis and in many cancers they are simultaneously deregulated. However, until recently there has been little progress on whether and how the two signaling pathways cooperate during tumorigenesis. Using the Tax viral oncoprotein as a model, a recent study provides the first example of how the deregulated canonical and non-canonical NF-κB pathways collaborate in tumorigenesis [48]. While Tax activation of the canonical NF-κB pathway induces p100 expression, Tax-induced p100 processing to generate p52 (activation of the non-canonical NF-κB pathway) leads to transcriptional down regulation of the WW domain-containing oxidoreductase (wwox), a tumor suppressor gene that has been linked to various tumors including breast cancer, ovarian tumor, lung cancer, gastric carcinoma, pancreatic adenocarci-noma, hematopoietic neoplasia and squamous cell carcinoma [172] (Figure 6). Notably, WWOX specifically inhibits Tax-induced activation of the canonical, but not the non-canonical NF-κB pathway. Mechanistic studies indicate that WWOX blocks Tax-induced IKK1 recruitment to RelA and subsequent RelA phosphorylation at ser-ine 536, which is required for RelA transcriptional activity. In contrast, WWOX Y33R, a mutant unable to block the IKK1 recruitment and RelA phosphorylation, loses the ability to inhibit Tax-mediated tumorigenesis. It can be speculated that many targets genes other than nf-κb2 and wwox may also contribute to the oncogenic coordination between the two pathways. Since both NF-κB activation and WWOX inactivation are associated with many different cancers in addition to Tax-associated tumorigenesis, it is of interest to investigate whether and how they cross-talk in general tumorigenesis.
Figure 6.
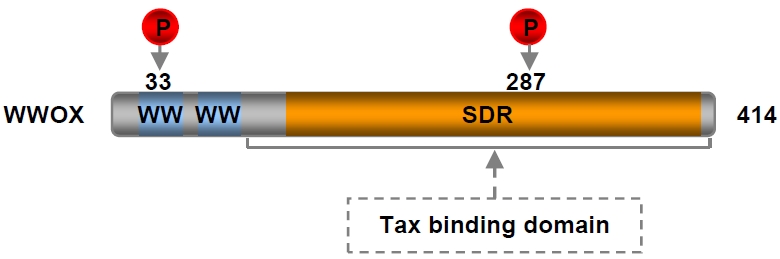
Domain structure of WWOX. WW: WW domain; SDR: short-chain dehydrogenase/reductase domain.
Role of NF-κB in tumorigenesis
As a transcription factor, NF-κB is involved in all stages of tumorigenesis from initiation all the way to metastasis by regulation of expression of various tumor-related genes (Figure 7). Like tumor itself, however, the role of NF-κB in tumorigenesis is complex and dynamic. During tumor initiation, NF-κB within pre-malignant cells and possibly also their neighbors is activated to induce expression of chemokines and cytokines, leading to the recruitment and activation of immune cells, particularly myeloid cells. Activated immune cells in turn produce a large amount of pro-inflammatory cytokines/ chemokines and growth factors, such as IL-1, IL-6, TNF, and EGF, which is also through NF-κB activation within the cells [1]. These secreted cytokines, growth factors and other bioactive molecules act on both malignant and inflammatory cells in an autocrine and/or paracrine manner, generating a complex inflammatory and protumorigenic microenvironment. The NF-κB-mediated inflammation contributes to DNA damage and induction of oncogenic mutations (activating mutations of oncogenes and/or inactivating mutations of tumor suppressor genes) in pre-malignant cells through both NF-κB dependent [induction of the ‘mutagenic’ enzyme activation-induced cytidine deaminase (AID) and suppression of DNA damage gatekeepers such as p53] and independent [production of reactive oxygen and nitrogen species (ROS and RNS)] mechanisms [173-176], facilitating tumor initiation and progression. Furthermore, NF-κB, which is activated by NF-κB-induced cytokines and growth factors as well as inflammation-induced ROS/RNS and DNA damage, regulate the transcription of genes involved in cell survival, proliferation, angiogenesis, invasion and metastasis, promoting tumor growth and progression. Thus, NF-κB participates in tumorigenesis in both extrinsic (inflammatory cells) and intrinsic (tumor cells) ways.
Figure 7.

Current model depicting the NF-KB-dependent interaction between inflammatory cells and malignant cells in tumorigenesis.
Conclusions and perspectives
There is no doubt that NF-κB plays a critical role in tumorigenesis. However, many key issues have not been addressed yet. First, the significance of the NF-κB members themselves in tumorigenesis has been rarely studied. Most evidences linking NF-κB to tumorigenesis are from studies on the knockout of NF-κB regulators. As we already know, almost all known NF-κB regulators, such as IKK, A20, CYLD, and PDLIM2, also regulate signaling pathways other than NF-κB, and many of them have already been linked to tumorigenesis [34, 177-180]. Thus, the functions of these regulators in tumorigenesis may not be attributed to NF-κB. Second, the mechanisms by which NF-κB interacts with other signaling pathways in tumorigenesis remain largely unknown. In fact, how the two NF-κB pathways cooperate in tumorigenesis still remains unclear. In this regard, overexpression of the super -repressor forms of IκBα or knockout of rela or nf-κb2 or even IKK components fails to completely block, although significantly reduces, tumor genesis and progression in different tumor models. On the other hand, NF-κB is known to crosstalk with many other tumor-related signaling pathways such as autophagy, STAT3 and p53 [1, 2, 177, 181-184]. Third, most studies are focused on the net effect of NF-κB activation on tumor tumorigenesis. As an old Chinese saying goes, everything has both yin and yang aspects, and so does NF-κB. Although NF-κB activation contributes to tumorigenesis in general, it may also play a negative role in certain stages of tumorigenesis and even exert a net negative effect on tumorigenesis in certain situations. One mechanism of NF-κB-mediated tumor suppression involves its original function in immunity and immunosurveillance. Currently, it is still unknown how the anti-tumor activity is suppressed and transformed to be protumorigenic. Paradoxically, the survival function of NF-κB may also contribute to tumor suppression by preventing tissue damage and oxidant accumulation as well as the activation of other signaling pathways such as c-Jun N-terminal kinase (JNK). Indeed, a tumor suppressive function of NF-κB has been suggested in several skin and liver cancer models [185-189]. Furthermore, very few of downstream targets of NF-κB that play a critical role in tumorigenesis have been clearly and comprehensively identified. Future genetic studies, particularly those involved in the induc-ible and conditional transgenic mice, and computational modeling analysis will help understand the complex and dynamic role of NF-κB in tumorigenesis and help design personalized treatments for cancer patients.
Acknowledgments
This study was supported in part by National Institutes of Health/National Cancer Institute (NIH/NCI) grant R01 CA116616, American Cancer Society (ACS) Research Scholar grant RSG-06-066-01-MGO and Hillman Innovative Cancer Research Award to G. Xiao. The authors apologize to those investigators whose work was not cited because of space limitations.
References
- 1.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 3.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice NR, Ernst MK. In vivo control of NF-kappa B activation by I kappa B alpha. Embo J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercurio F, DiDonato JA, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-kappa B-mediated signal transduction. Genes Dev. 1993;7:705–718. doi: 10.1101/gad.7.4.705. [DOI] [PubMed] [Google Scholar]
- 6.Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17:281–293. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Kamata H, Tsuchiya Y, Asano T. IkappaB-beta is a positive and negative regulator of NF-kappaB activity during inflammation. Cell Res. 2010;20:1178–1180. doi: 10.1038/cr.2010.147. [DOI] [PubMed] [Google Scholar]
- 8.Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 9.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317:675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 10.Wuerzberger-Davis SM, Miyamoto S. TAK-ling IKK activation: “Ub” the judge. Sci Signal. 2010;3:pe3. doi: 10.1126/scisignal.3105pe3. [DOI] [PubMed] [Google Scholar]
- 11.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qing G, Qu Z, Xiao G. Stabilization of basally translated NF-kappaB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-kappaB2 p100. J Biol Chem. 2005;280:40578–40582. doi: 10.1074/jbc.M508776200. [DOI] [PubMed] [Google Scholar]
- 13.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 14.He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, Dempsey PW, Cheng G. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med. 2006;203:2413–2418. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 16.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 17.Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity. 2004;21:629–642. doi: 10.1016/j.immuni.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Moore CR, Bishop GA. Differential regulation of CD40-mediated TNF receptor-associated factor degradation in B lymphocytes. J Immunol. 2005;175:3780–3789. doi: 10.4049/jimmunol.175.6.3780. [DOI] [PubMed] [Google Scholar]
- 19.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391–401. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 21.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 22.Fong A, Sun SC. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of NF-kappa B2/p100. J Biol Chem. 2002;277:22111–22114. doi: 10.1074/jbc.C200151200. [DOI] [PubMed] [Google Scholar]
- 23.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 24.Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105:3503–3508. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 26.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 27.Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, Loo JA, Cheng G. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKal-pha-mediated phosphorylation. Sci Signal. 2010;3:ra41. doi: 10.1126/scisignal.2000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267:17971–17976. [PubMed] [Google Scholar]
- 29.Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 31.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 32.Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, Leonardi A. ABIN-1 binds to NEMO/ IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J Biol Chem. 2006;281:18482–18488. doi: 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 33.Malynn BA, Ma A. A20 takes on tumors: tumor suppression by an ubiquitin-editing enzyme. J Exp Med. 2009;206:977–980. doi: 10.1084/jem.20090765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nat Rev Cancer. 2010;10:332–341. doi: 10.1038/nrc2775. [DOI] [PubMed] [Google Scholar]
- 35.Hutti JE, Turk BE, Asara JM, Ma A, Cantley LC, Abbott DW. IkappaB kinase beta phos-phorylates the K63 deubiquitinase A20 to cause feedback inhibition of the NF-kappaB pathway. Mol Cell Biol. 2007;27:7451–7461. doi: 10.1128/MCB.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 37.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 38.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 39.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappaB controls expression of inhibitor I kappa B alpha: evidence for an inducible auto-regulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 40.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor kappaB response. J Exp Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 42.Ganesh L, Burstein E, Guha-Niyogi A, Louder MK, Mascola JR, Klomp LW, Wijmenga C, Duckett CS, Nabel GJ. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature. 2003;426:853–857. doi: 10.1038/nature02171. [DOI] [PubMed] [Google Scholar]
- 43.Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, Maine GN, Wilkinson JC, Mayo MW, Duckett CS. COMMD proteins, a novel family of structural and functional homologs of MURR1. J Biol Chem. 2005;280:22222–22232. doi: 10.1074/jbc.M501928200. [DOI] [PubMed] [Google Scholar]
- 44.Maine GN, Mao X, Komarck CM, Burstein E. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. Embo J. 2007;26:436–447. doi: 10.1038/sj.emboj.7601489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thoms HC, Loveridge CJ, Simpson J, Clipson A, Reinhardt K, Dunlop MG, Stark LA. Nucleo-lar targeting of RelA(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res. 2010;70:139–149. doi: 10.1158/0008-5472.CAN-09-1397. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF -kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–591. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 47.Fu J, Yan P, Li S, Qu Z, Xiao G. Molecular determinants of PDLIM2 in suppressing HTLV-I Tax-mediated tumorigenesis. Oncogene. 2010;29:6499–6507. doi: 10.1038/onc.2010.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu J, Qu Z, Yan P, Ishikawa C, Aqeilan R, Rabson AB, Xiao G. The tumor suppressor gene wwox links the canonical and non-canonical NF-kappaB pathways in HTLV-I Tax-mediated tumorigenesis. Blood. 2011 doi: 10.1182/blood-2010-08-303073. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin W, Chang M, Paul EM, Babu G, Lee AJ, Reiley W, Wright A, Zhang M, You J, Sun SC. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–1866. doi: 10.1172/JCI34257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilmore TD. Multiple mutations contribute to the oncogenicity of the retroviral oncoprotein v-Rel. Oncogene. 1999;18:6925–6937. doi: 10.1038/sj.onc.1203222. [DOI] [PubMed] [Google Scholar]
- 51.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–6947. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 52.Carrasco D, Rizzo CA, Dorfman K, Bravo R. The v-rel oncogene promotes malignant T-cell leukemia/lymphoma in transgenic mice. Embo J. 1996;15:3640–3650. [PMC free article] [PubMed] [Google Scholar]
- 53.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 54.Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romieu-Mourez R, Kim DW, Shin SM, Demicco EG, Landesman-Bollag E, Seldin DC, Cardiff RD, Sonenshein GE. Mouse mammary tumor virus c-rel transgenic mice develop mammary tumors. Mol Cell Biol. 2003;23:5738–5754. doi: 10.1128/MCB.23.16.5738-5754.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang B, Wang Z, Li T, Tsitsikov EN, Ding HF. NF-kappaB2 mutation targets TRAF1 to induce lymphomagenesis. Blood. 2007;110:743–751. doi: 10.1182/blood-2006-11-058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mukhopadhyay T, Roth JA, Maxwell SA. Altered expression of the p50 subunit of the NF -kappa B transcription factor complex in non-small cell lung carcinoma. Oncogene. 1995;11:999–1003. [PubMed] [Google Scholar]
- 58.Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun SC, Xiao G. Deregulation of NF-kappaB and its upstream kinases in cancer. Cancer Metastasis Rev. 2003;22:405–422. doi: 10.1023/a:1023733231406. [DOI] [PubMed] [Google Scholar]
- 60.Qing G, Qu Z, Xiao G. Endoproteolytic processing of C-terminally truncated NF-kappaB2 precursors at kappaB-containing promoters. Proc Natl Acad Sci U S A. 2007;104:5324–5329. doi: 10.1073/pnas.0609914104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qing G, Qu Z, Xiao G. Regulation of NF-kappa B2 p100 processing by its cis-acting domain. J Biol Chem. 2005;280:18–27. doi: 10.1074/jbc.M406619200. [DOI] [PubMed] [Google Scholar]
- 62.Ciana P, Neri A, Cappellini C, Cavallo F, Pomati M, Chang CC, Maiolo AT, Lombardi L. Constitutive expression of lymphoma-associated NFKB-2/Lyt-10 proteins is tumorigenic in mur-ine fibroblasts. Oncogene. 1997;14:1805–1810. doi: 10.1038/sj.onc.1201015. [DOI] [PubMed] [Google Scholar]
- 63.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, Bravo R. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, Love P, Brown K, Siebenlist U. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-kappaB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Z, Zhang B, Yang L, Ding J, Ding HF. Constitutive production of NF-kappaB2 p52 is not tumorigenic but predisposes mice to inflammatory autoimmune disease by repressing Bim expression. J Biol Chem. 2008;283:10698–10706. doi: 10.1074/jbc.M800806200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Connelly L, Robinson-Benion C, Chont M, Saint-Jean L, Li H, Polosukhin VV, Blackwell TS, Yull FE. A transgenic model reveals important roles for the NF-kappa B alternative pathway (p100/p52) in mammary development and links to tumorigenesis. J Biol Chem. 2007;282:10028–10035. doi: 10.1074/jbc.M611300200. [DOI] [PubMed] [Google Scholar]
- 68.McKeithan TW, Takimoto GS, Ohno H, Bjorling VS, Morgan R, Hecht BK, Dube I, Sandberg AA, Rowley JD. BCL3 rearrangements and t (14;19) in chronic lymphocytic leukemia and other B-cell malignancies: a molecular and cytogenetic study. Genes Chromosomes Cancer. 1997;20:64–72. [PubMed] [Google Scholar]
- 69.Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS., Jr Selective activation of NF-kappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. 2000;19:1123–1131. doi: 10.1038/sj.onc.1203412. [DOI] [PubMed] [Google Scholar]
- 70.Thornburg NJ, Pathmanathan R, Raab-Traub N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in naso-pharyngeal carcinoma. Cancer Res. 2003;63:8293–8301. [PubMed] [Google Scholar]
- 71.Canoz O, Rassidakis GZ, Admirand JH, Medeiros LJ. Immunohistochemical detection of BCL-3 in lymphoid neoplasms: a survey of 353 cases. Mod Pathol. 2004;17:911–917. doi: 10.1038/modpathol.3800140. [DOI] [PubMed] [Google Scholar]
- 72.O'Neil BH, Buzkova P, Farrah H, Kashatus D, Sanoff H, Goldberg RM, Baldwin AS, Funkhouser WK. Expression of nuclear factor-kappaB family proteins in hepatocellular carcinomas. Oncology. 2007;72:97–104. doi: 10.1159/000111116. [DOI] [PubMed] [Google Scholar]
- 73.Puvvada SD, Funkhouser WK, Greene K, Deal A, Chu H, Baldwin AS, Tepper JE, O'Neil BH. NF-κB and Bcl-3 activation are prognostic in metastatic colorectal cancer. Oncology. 2010;78:181–188. doi: 10.1159/000313697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brenne AT, Fagerli UM, Shaughnessy JD, Jr, Vatsveen TK, Ro TB, Hella H, Zhan F, Barlogie B, Sundan A, Borset M, Waage A. High expression of BCL3 in human myeloma cells is associated with increased proliferation and inferior prognosis. Eur J Haematol. 2009;82:354–363. doi: 10.1111/j.1600-0609.2009.01225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, Piette J, Siebenlist U, Bours V, Chariot A. GSK3-mediated BCL-3 phosphorylation modulates its degradation and its onco-genicity. Mol Cell. 2004;16:35–45. doi: 10.1016/j.molcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 76.Ong ST, Hackbarth ML, Degenstein LC, Baunoch DA, Anastasi J, McKeithan TW. Lymphadenopathy, splenomegaly, and altered immunoglobulin production in BCL3 transgenic mice. Oncogene. 1998;16:2333–2343. doi: 10.1038/sj.onc.1201771. [DOI] [PubMed] [Google Scholar]
- 77.Beauparlant P, Kwan I, Bitar R, Chou P, Koromilas AE, Sonenberg N, Hiscott J. Disruption of I kappa B alpha regulation by antisense RNA expression leads to malignant transformation. Oncogene. 1994;9:3189–3197. [PubMed] [Google Scholar]
- 78.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 79.Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA, Stewart CL. IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 82.Lind MH, Rozell B, Wallin RP, van Hogerlinden M, Ljunggren HG, Toftgard R, Sur I. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proc Natl Acad Sci U S A. 2004;101:4972–4977. doi: 10.1073/pnas.0307106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wood KM, Roff M, Hay RT. Defective IkappaBalpha in Hodgkin cell lines with constitu-tively active NF-kappaB. Oncogene. 1998;16:2131–2139. doi: 10.1038/sj.onc.1201735. [DOI] [PubMed] [Google Scholar]
- 84.Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, Mathas S, Krappmann D, Scheidereit C, Stein H, Dorken B. Overex-pression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood. 1999;94:3129–3134. [PubMed] [Google Scholar]
- 85.Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IκBa gene in Hodg-kin's disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. 1999;18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- 86.Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dorken B, Scheidereit C. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene. 1999;18:943–953. doi: 10.1038/sj.onc.1202351. [DOI] [PubMed] [Google Scholar]
- 87.Jungnickel B, Staratschek-Jox A, Brauninger A, Spieker T, Wolf J, Diehl V, Hansmann ML, Rajewsky K, Kuppers R. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin's lymphoma. J Exp Med. 2000;191:395–402. doi: 10.1084/jem.191.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emmerich F, Theurich S, Hummel M, Haeffker A, Vry MS, Dohner K, Bommert K, Stein H, Dorken B. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol. 2003;201:413–420. doi: 10.1002/path.1454. [DOI] [PubMed] [Google Scholar]
- 89.Memet S, Laouini D, Epinat JC, Whiteside ST, Goudeau B, Philpott D, Kayal S, Sansonetti PJ, Berche P, Kanellopoulos J, Israel A. Ikappa-Bepsilon-deficient mice: reduction of one T cell precursor subspecies and enhanced Ig isotype switching and cytokine synthesis. J Immunol. 1999;163:5994–6005. [PubMed] [Google Scholar]
- 90.Willis TG, Jadayel DM, Du MQ, Peng H, Perry AR, Abdul-Rauf M, Price H, Karran L, Majekodunmi O, Wlodarska I, Pan L, Crook T, Hamoudi R, Isaacson PG, Dyer MJ. Bcl10 is involved in t (1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell. 1999;96:35–45. doi: 10.1016/s0092-8674(00)80957-5. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Q, Siebert R, Yan M, Hinzmann B, Cui X, Xue L, Rakestraw KM, Naeve CW, Beckmann G, Weisenburger DD, Sanger WG, Nowotny H, Vesely M, Callet-Bauchu E, Salles G, Dixit VM, Rosenthal A, Schlegelberger B, Morris SW. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t (1;14)(p22;q32) Nat Genet. 1999;22:63–68. doi: 10.1038/8767. [DOI] [PubMed] [Google Scholar]
- 92.Akagi T, Motegi M, Tamura A, Suzuki R, Hosokawa Y, Suzuki H, Ota H, Nakamura S, Morishima Y, Taniwaki M, Seto M. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene. 1999;18:5785–5794. doi: 10.1038/sj.onc.1203018. [DOI] [PubMed] [Google Scholar]
- 93.Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, De Wolf-Peeters C, Hagemeijer A, Van den Berghe H, Marynen P. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood. 1999;93:3601–3609. [PubMed] [Google Scholar]
- 94.Morgan JA, Yin Y, Borowsky AD, Kuo F, Nourmand N, Koontz JI, Reynolds C, Soreng L, Griffin CA, Graeme-Cook F, Harris NL, Weisenburger D, Pinkus GS, Fletcher JA, Sklar J. Breakpoints of the t(11;18)(q21;q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res. 1999;59:6205–6213. [PubMed] [Google Scholar]
- 95.Uren AG, O'Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell. 2000;6:961–967. doi: 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 96.Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, Yamaoka S, Seto M, Nunez G. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. 2001;276:19012–19019. doi: 10.1074/jbc.M009984200. [DOI] [PubMed] [Google Scholar]
- 97.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, Xu W, Shaffer AL, Wright G, Xiao W, Powell J, Jiang JK, Thomas CJ, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Johnson NA, Rimsza LM, Campo E, Jaffe ES, Wilson WH, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pierce SK, Staudt LM. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 99.Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 100.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, Zhao H, Stephens O, Santra M, Williams DR, Dang L, Barlogie B, Shaughnessy JD, Jr, Kuehl WM, Staudt LM. Frequent engagement of the classical and alternative NF-kappaB path ways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yamamoto M, Ito T, Shimizu T, Ishida T, Semba K, Watanabe S, Yamaguchi N, Inoue J. Epi-genetic alteration of the NF-kappaB-inducing kinase (NIK) gene is involved in enhanced NIK expression in basal-like breast cancer. Cancer Sci. 2010;101:2391–2397. doi: 10.1111/j.1349-7006.2010.01685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saitoh Y, Yamamoto N, Dewan MZ, Sugimoto H, Martinez Bruyn VJ, Iwasaki Y, Matsubara K, Qi X, Saitoh T, Imoto I, Inazawa J, Utsunomiya A, Watanabe T, Masuda T, Yamamoto N, Yamaoka S. Overexpressed NF-kappaB-inducing kinase contributes to the tumorigenesis of adult T-cell leukemia and Hodgkin Reed-Sternberg cells. Blood. 2008;111:5118–5129. doi: 10.1182/blood-2007-09-110635. [DOI] [PubMed] [Google Scholar]
- 104.Saitoh Y, Martinez Bruyn VJ, Uota S, Hasegawa A, Yamamoto N, Imoto I, Inazawa J, Yamaoka S. Overexpression of NF-kappaB inducing kinase underlies constitutive NF-kappaB activation in lung cancer cells. Lung Cancer. 2010;70:263–270. doi: 10.1016/j.lungcan.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 105.Jin X, Jin HR, Jung HS, Lee SJ, Lee JH, Lee JJ. An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J Biol Chem. 2010;285:30539–30547. doi: 10.1074/jbc.M110.129551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Unoki M, Okutsu J, Nakamura Y. Identification of a novel human gene, ZFP91, involved in acute myelogenous leukemia. IntJ Oncol. 2003;22:1217–1223. [PubMed] [Google Scholar]
- 107.Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- 108.Karreth FA, Tuveson DA. Modelling oncogenic Ras/Raf signalling in the mouse. Curr Opin Genet Dev. 2009;19:4–11. doi: 10.1016/j.gde.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Qu Z, Xiao G. Human T-cell lymphotropic virus: a model of NF-kappaB associated tumorigenesis. Viruses. 2011 doi: 10.3390/v3060714. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chu ZL, DiDonato JA, Hawiger J, Ballard DW. The tax oncoprotein of human T-cell leukemia virus type 1 associates with and persistently activates IkappaB kinases containing IKKalpha and IKKbeta. J Biol Chem. 1998;273:15891–15894. doi: 10.1074/jbc.273.26.15891. [DOI] [PubMed] [Google Scholar]
- 111.Jin DY, Giordano V, Kibler KV, Nakano H, Jeang KT. Role of adapter function in oncopro-tein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J Biol Chem. 1999;274:17402–17405. doi: 10.1074/jbc.274.25.17402. [DOI] [PubMed] [Google Scholar]
- 112.Harhaj EW, Sun SC. IKKgamma serves as a docking subunit of the IkappaB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J Biol Chem. 1999;274:22911–22914. doi: 10.1074/jbc.274.33.22911. [DOI] [PubMed] [Google Scholar]
- 113.Xiao G, Sun SC. Activation of IKKalpha and IKKbeta through their fusion with HTLV-I tax protein. Oncogene. 2000;19:5198–5203. doi: 10.1038/sj.onc.1203894. [DOI] [PubMed] [Google Scholar]
- 114.Xiao G, Harhaj EW, Sun SC. Domain-specific interaction with the I kappa B kinase (IKK) regulatory subunit IKK gamma is an essential step in tax-mediated activation of IKK. J Biol Chem. 2000;275:34060–34067. doi: 10.1074/jbc.M002970200. [DOI] [PubMed] [Google Scholar]
- 115.Xiao G, Cvijic ME, Fong A, Harhaj EW, Uhlik MT, Waterfield M, Sun SC. Retroviral oncoprotein Tax induces processing of NF-kappaB2/ p100 in T cells: evidence for the involvement of IKKalpha. EmboJ. 2001;20:6805–6815. doi: 10.1093/emboj/20.23.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Qu Z, Qing G, Rabson A, Xiao G. Tax deregulation of NF-kappaB2 p100 processing involves both beta-TrCP-dependent and -independent mechanisms. J Biol Chem. 2004;279:44563–44572. doi: 10.1074/jbc.M403689200. [DOI] [PubMed] [Google Scholar]
- 117.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J Biol Chem. 2002;277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- 118.Matta H, Chaudhary PM. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP) Proc Natl Acad Sci U S A. 2004;101:9399–9404. doi: 10.1073/pnas.0308016101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Soni V, Cahir-McFarland E, Kieff E. LMP1 TRAFficking activates growth and survival pathways. Adv Exp Med Biol. 2007;597:173–187. doi: 10.1007/978-0-387-70630-6_14. [DOI] [PubMed] [Google Scholar]
- 120.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vereecke L, Beyaert R, van Loo G. The ubiq-uitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30:383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 122.Honma K, Tsuzuki S, Nakagawa M, Karnan S, Aizawa Y, Kim WS, Kim YD, Ko YH, Seto M. TNFAIP3 is the target gene of chromosome band 6q23.3-q24.1 loss in ocular adnexal marginal zone B cell lymphoma. Genes Chromosomes Cancer. 2008;47:1–7. doi: 10.1002/gcc.20499. [DOI] [PubMed] [Google Scholar]
- 123.Chanudet E, Ye H, Ferry J, Bacon CM, Adam P, Muller-Hermelink HK, Radford J, Pileri SA, Ichimura K, Collins VP, Hamoudi RA, Nicholson AG, Wotherspoon AC, Isaacson PG, Du MQ. A20 deletion is associated with copy number gain at the TNFA/B/C locus and occurs preferentially in translocation-negative MALT lymphoma of the ocular adnexa and salivary glands. J Pathol. 2009;217:420–430. doi: 10.1002/path.2466. [DOI] [PubMed] [Google Scholar]
- 124.Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, Seto M. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009;114:2467–2475. doi: 10.1182/blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 125.Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J, Asakura Y, Muto S, Tamura A, Iio M, Akatsuka Y, Hayashi Y, Mori H, Igarashi T, Kurokawa M, Chiba S, Mori S, Ishikawa Y, Okamoto K, Tobinai K, Nakagama H, Nakahata T, Yoshino T, Kobayashi Y, Ogawa S. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 126.Novak U, Rinaldi A, Kwee I, Nandula SV, Rancoita PM, Compagno M, Cerri M, Rossi D, Murty VV, Zucca E, Gaidano G, Dalla-Favera R, Pasqualucci L, Bhagat G, Bertoni F. The NF-{kappa}B negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood. 2009;113:4918–4921. doi: 10.1182/blood-2008-08-174110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, Sun L, Chen ZJ, Marynen P, Beyaert R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. 2008;9:263–271. doi: 10.1038/ni1561. [DOI] [PubMed] [Google Scholar]
- 128.Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, Klapper W, Vater I, Giefing M, Gesk S, Stanelle J, Siebert R, Kuppers R. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–989. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, Bhagat G, Chadburn A, Dalla-Favera R, Pasqualucci L. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, Jones C, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Den Ouweland A, Halley D, Delpech B, Cleveland MG, Leigh I, Leisti J, Rasmussen S. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160–165. doi: 10.1038/76006. [DOI] [PubMed] [Google Scholar]
- 131.Lee DA, Grossman ME, Schneiderman P, Celebi JT. Genetics of skin appendage neoplasms and related syndromes. J Med Genet. 2005;42:811–819. doi: 10.1136/jmg.2004.025577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Saggar S, Chernoff KA, Lodha S, Horev L, Kohl S, Honjo RS, Brandt HR, Hartmann K, Celebi JT. CYLD mutations in familial skin appendage tumours. J Med Genet. 2008;45:298–302. doi: 10.1136/jmg.2007.056127. [DOI] [PubMed] [Google Scholar]
- 133.Young AL, Kellermayer R, Szigeti R, Teszas A, Azmi S, Celebi JT. CYLD mutations underlie Brooke-Spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet. 2006;70:246–249. doi: 10.1111/j.1399-0004.2006.00667.x. [DOI] [PubMed] [Google Scholar]
- 134.Zhang G, Huang Y, Yan K, Li W, Fan X, Liang Y, Sun L, Li H, Zhang S, Gao M, Du W, Yang S, Liu J, Zhang X. Diverse phenotype of Brooke-Spiegler syndrome associated with a nonsense mutation in the CYLD tumor suppressor gene. Exp Dermatol. 2006;15:966–970. doi: 10.1111/j.1600-0625.2006.00501.x. [DOI] [PubMed] [Google Scholar]
- 135.Alameda JP, Moreno-Maldonado R, Navarro M, Bravo A, Ramirez A, Page A, Jorcano JL, Fernan-dez-Acenero MJ, Casanova ML. An inactivating CYLD mutation promotes skin tumor progression by conferring enhanced proliferative, survival and angiogenic properties to epidermal cancer cells. Oncogene. 2010 doi: 10.1038/onc.2010.378. [DOI] [PubMed] [Google Scholar]
- 136.Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK. Reduced expression of CYLD in human colon and hepa-tocellular carcinomas. Carcinogenesis. 2007;28:21–27. doi: 10.1093/carcin/bgl081. [DOI] [PubMed] [Google Scholar]
- 137.Espinosa L, Cathelin S, D'Altri T, Trimarchi T, Statnikov A, Guiu J, Rodilla V, Ingles-Esteve J, Nomdedeu J, Bellosillo B, Besses C, Abdel-Wahab O, Kucine N, Sun SC, Song G, Mullighan CC, Levine RL, Rajewsky K, Aifantis I, Bigas A. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268–281. doi: 10.1016/j.ccr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T, Pfeifer A, Fassler R, Bosserhoff AK. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med. 2009;206:221–232. doi: 10.1084/jem.20082044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang J, Stirling B, Temmerman ST, Ma CA, Fuss IJ, Derry JM, Jain A. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006;116:3042–3049. doi: 10.1172/JCI28746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39:493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zanette DL, Rivadavia F, Molfetta GA, Barbuzano FG, Proto-Siqueira R, Silva-Jr WA, Falcao RP, Zago MA. miRNA expression profiles in chronic lymphocytic and acute lymphocytic leukemia. Braz J Med Biol Res. 2007;40:1435–1440. doi: 10.1590/s0100-879x2007001100003. [DOI] [PubMed] [Google Scholar]
- 142.Loughran G, Healy NC, Kiely PA, Huigsloot M, Kedersha NL, O'Connor R. Mystique is a new insulin-like growth factor-I-regulated PDZ-LIM domain protein that promotes cell attachment and migration and suppresses Anchorage -independent growth. Mol Biol Cell. 2005;16:1811–1822. doi: 10.1091/mbc.E04-12-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Macartney-Coxson DP, Hood KA, Shi HJ, Ward T, Wiles A, O'Connor R, Hall DA, Lea RA, Royds JA, Stubbs RS, Rooker S. Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metasta-ses: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer. 2008;8:187. doi: 10.1186/1471-2407-8-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yan P, Fu J, Qu Z, Li S, Tanaka T, Grusby MJ, Xiao G. PDLIM2 suppresses human T-cell leukemia virus type I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for protea-somal degradation. Blood. 2009;113:4370–4380. doi: 10.1182/blood-2008-10-185660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yan P, Qu Z, Ishikawa C, Mori N, Xiao G. Human T-cell leukemia virus type I-mediated repression of PDZ-LIM domain-containing protein 2 involves DNA methylation but independent of the viral oncoprotein tax. Neoplasia. 2009;11:1036–1041. doi: 10.1593/neo.09752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Qu Z, Fu J, Yan P, Hu J, Cheng SY, Xiao G. Epigenetic repression of PDZ-LIM domain-containing protein 2: implications for the biology and treatment of breast cancer. J Biol Chem. 2010;285:11786–11792. doi: 10.1074/jbc.M109.086561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Qu Z, Yan P, Fu J, Jiang J, Grusby MJ, Smithgall TE, Xiao G. DNA methylation-dependent repression of PDZ-LIM domain-containing protein 2 in colon cancer and its role as a potential therapeutic target. Cancer Res. 2010;70:1766–1772. doi: 10.1158/0008-5472.CAN-09-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Emi Y, Kohnoe S, Yoshida M, Takahashi I, Maehara Y, Sugimachi K. Hyperthermia enhances the inhibition of tumor growth by 1-(2-tetrahydrofuryl)-5-fluorouracil/uracil (1:4) in tumors in mice and humans. Cancer. 1992;70:1177–1182. doi: 10.1002/1097-0142(19920901)70:5<1177::aid-cncr2820700525>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 149.Ohata H, Emi M, Fujiwara Y, Higashino K, Nakagawa K, Futagami R, Tsuchiya E, Nakamura Y. Deletion mapping of the short arm of chromosome 8 in non-small cell lung carcinoma. Genes Chromosomes Cancer. 1993;7:85–88. doi: 10.1002/gcc.2870070204. [DOI] [PubMed] [Google Scholar]
- 150.Pykett MJ, Murphy ME, Harnish PR, Muenke M, Marks J, George DL. Loss of chromosome 8p sequences in human breast carcinoma cell lines. Cancer Genet Cytogenet. 1994;76:23–28. doi: 10.1016/0165-4608(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 151.Arai T, Akiyama Y, Yamamura A, Hosoi T, Shibata T, Saitoh K, Okabe S, Yuasa Y. Allelo-type analysis of early colorectal cancers with lymph node metastasis. Int J Cancer. 1998;79:418–423. doi: 10.1002/(sici)1097-0215(19980821)79:4<418::aid-ijc18>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 152.Brown MR, Chuaqui R, Vocke CD, Berchuck A, Middleton LP, Emmert-Buck MR, Kohn EC. Allelic loss on chromosome arm 8p: analysis of sporadic epithelial ovarian tumors. Gynecol Oncol. 1999;74:98–102. doi: 10.1006/gyno.1999.5439. [DOI] [PubMed] [Google Scholar]
- 153.Lutchman M, Pack S, Kim AC, Azim A, Emmert-Buck M, van Huffel C, Zhuang Z, Chishti AH. Loss of heterozygosity on 8p in prostate cancer implicates a role for dematin in tumor progression. Cancer Genet Cytogenet. 1999;115:65–69. doi: 10.1016/s0165-4608(99)00081-3. [DOI] [PubMed] [Google Scholar]
- 154.Wistuba, Behrens C, Virmani AK, Milchgrub S, Syed S, Lam S, Mackay B, Minna JD, Gazdar AF. Allelic losses at chromosome 8p21-23 are early and frequent events in the patho-genesis of lung cancer. Cancer Res. 1999;59:1973–1979. [PubMed] [Google Scholar]
- 155.Kurimoto F, Gemma A, Hosoya Y, Seike M, Takenaka K, Uematsu K, Yoshimura A, Shibuya M, Kudoh S. Unchanged frequency of loss of heterozygosity and size of the deleted region at 8p21-23 during metastasis of lung cancer. Int J Mol Med. 2001;8:89–93. doi: 10.3892/ijmm.8.1.89. [DOI] [PubMed] [Google Scholar]
- 156.Swalwell JI, Vocke CD, Yang Y, Walker JR, Grouse L, Myers SH, Gillespie JW, Bostwick DG, Duray PH, Linehan WM, Emmert-Buck MR. Determination of a minimal deletion interval on chromosome band 8p21 in sporadic prostate cancer. Genes Chromosomes Cancer. 2002;33:201–205. doi: 10.1002/gcc.10015. [DOI] [PubMed] [Google Scholar]
- 157.French AJ, Petroni G, Thibideau SN, Smolkin M, Bissonette E, Roviello F, Harper JC, Koch BR, Anderson SA, Hebbring SJ, Powell SM. Allelic imbalance of 8p indicates poor survival in gastric cancer. J Mol Diagn. 2004;6:243–252. doi: 10.1016/S1525-1578(10)60517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Healy NC, O'Connor R. Sequestration of PDLIM2 in the cytoplasm of monocytic/ macrophage cells is associated with adhesion and increased nuclear activity of NF-kappaB. J Leukoc Biol. 2009;85:481–490. doi: 10.1189/jlb.0408238. [DOI] [PubMed] [Google Scholar]
- 159.Guo H, Mi Z, Bowles DE, Bhattacharya SD, Kuo PC. Osteopontin and protein kinase C regulate PDLIM2 activation and STAT1 ubiquitination in LPS-treated murine macrophages. J Biol Chem. 2010 doi: 10.1074/jbc.M110.161869. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 160.Li W, Xie L, He X, Li J, Tu K, Wei L, Wu J, Guo Y, Ma X, Zhang P, Pan Z, Hu X, Zhao Y, Xie H, Jiang G, Chen T, Wang J, Zheng S, Cheng J, Wan D, Yang S, Li Y, Gu J. Diagnostic and prognostic implications of microRNAs in human hepato-cellular carcinoma. Int J Cancer. 2008;123:1616–1622. doi: 10.1002/ijc.23693. [DOI] [PubMed] [Google Scholar]
- 161.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 162.Henson BJ, Bhattacharjee S, O'Dee DM, Feingold E, Gollin SM. Decreased expression of miR-125b and miR-100 in oral cancer cells contributes to malignancy. Genes Chromosomes Cancer. 2009;48:569–582. doi: 10.1002/gcc.20666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ichimi T, Enokida H, Okuno Y, Kunimoto R, Chiyomaru T, Kawamoto K, Kawahara K, Toki K, Kawakami K, Nishiyama K, Tsujimoto G, Nakagawa M, Seki N. Identification of novel mi-croRNA targets based on microRNA signatures in bladder cancer. Int J Cancer. 2009;125:345–352. doi: 10.1002/ijc.24390. [DOI] [PubMed] [Google Scholar]
- 164.Visone R, Pallante P, Vecchione A, Cirombella R, Ferracin M, Ferraro A, Volinia S, Coluzzi S, Leone V, Borbone E, Liu CG, Petrocca F, Troncone G, Calin GA, Scarpa A, Colato C, Tallini G, Santoro M, Croce CM, Fusco A. Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene. 2007;26:7590–7595. doi: 10.1038/sj.onc.1210564. [DOI] [PubMed] [Google Scholar]
- 165.Glud M, Rossing M, Hother C, Holst L, Hastrup N, Nielsen FC, Gniadecki R, Drzewiecki KT. Downregulation of miR-125b in metastatic cutaneous malignant melanoma. Melanoma Res. 2010;20:479–484. doi: 10.1097/CMR.0b013e32833e32a1. [DOI] [PubMed] [Google Scholar]
- 166.Guan Y, Yao H, Zheng Z, Qiu G, Sun K. MiR-125b targets BCL3 and suppresses ovarian cancer proliferation. Int J Cancer. 2010 doi: 10.1002/ijc.25575. [DOI] [PubMed] [Google Scholar]
- 167.Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM. Modulation of miR-155 and miR-125b levels following lipopolysaccha-ride/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 168.Pichler K, Schneider G, Grassmann R. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology. 2008;5:100. doi: 10.1186/1742-4690-5-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-kappaB p65-dependent transacti-vation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog. 2009;5:e1000681. doi: 10.1371/journal.ppat.1000681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, Xiong W, Li G, Lu J, Fodstad O, Riker AI, Tan M. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 2010;285:21496–21507. doi: 10.1074/jbc.M109.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR-9 in human mono-cytes and neutrophils exposed to proinflamma-tory signals. Proc Natl Acad Sci U S A. 2009;106:5282–5287. doi: 10.1073/pnas.0810909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Aqeilan RI, Croce CM. WWOX in biological control and tumorigenesis. J Cell Physiol. 2007;212:307–310. doi: 10.1002/jcp.21099. [DOI] [PubMed] [Google Scholar]
- 173.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 174.Takai A, Toyoshima T, Uemura M, Kitawaki Y, Marusawa H, Hiai H, Yamada S, Okazaki IM, Honjo T, Chiba T, Kinoshita K. A novel mouse model of hepatocarcinogenesis triggered by AID causing deleterious p53 mutations. Oncogene. 2009;28:469–478. doi: 10.1038/onc.2008.415. [DOI] [PubMed] [Google Scholar]
- 175.Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 176.Ak P, Levine AJ. p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. Faseb J. 2010;24:3643–3652. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 177.Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 178.Sun SC. CYLD: a tumor suppressor deubiquiti-nase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17:25–34. doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Torrado M, Senatorov VV, Trivedi R, Fariss RN, Tomarev SI. Pdlim2, a novel PDZ-LIM domain protein, interacts with alpha-actinins and filamin A. Invest Ophthalmol Vis Sci. 2004;45:3955–3963. doi: 10.1167/iovs.04-0721. [DOI] [PubMed] [Google Scholar]
- 180.Tanaka T, Soriano MA, Grusby MJ. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immunity. 2005;22:729–736. doi: 10.1016/j.immuni.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 181.Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK) Cell Res. 2006;16:895–901. doi: 10.1038/sj.cr.7310109. [DOI] [PubMed] [Google Scholar]
- 182.Qing G, Yan P, Qu Z, Liu H, Xiao G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007;17:520–530. doi: 10.1038/cr.2007.47. [DOI] [PubMed] [Google Scholar]
- 183.Xiao G. Autophagy and NF-kappaB: fight for fate. Cytokine Growth Factor Rev. 2007;18:233–243. doi: 10.1016/j.cytogfr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yan P, Qing G, Qu Z, Wu CC, Rabson A, Xiao G. Targeting autophagic regulation of NFkap-paB in HTLV-I transformed cells by geldanamycimplications for therapeutic interventions. Autophagy. 2007;3:600–603. doi: 10.4161/auto.4761. [DOI] [PubMed] [Google Scholar]
- 185.Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, Khavari PA. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 186.Seitz CS, Lin Q, Deng H, Khavari PA. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc Natl Acad Sci U S A. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.van Hogerlinden M, Rozell BL, Ahrlund-Richter L, Toftgard R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999;59:3299–3303. [PubMed] [Google Scholar]
- 188.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 189.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 190.Houldsworth J, Mathew S, Rao PH, Dyomina K, Louie DC, Parsa N, Offit K, Chaganti RS. REL proto-oncogene is frequently amplified in extranodal diffuse large cell lymphoma. Blood. 1996;87:25–29. [PubMed] [Google Scholar]
- 191.Lai JL, Zandecki M, Mary JY, Bernardi F, Izydorczyk V, Flactif M, Morel P, Jouet JP, Bauters F, Facon T. Improved cytogenetics in multiple myeloma: a study of 151 patients including 117 patients at diagnosis. Blood. 1995;85:2490–2497. [PubMed] [Google Scholar]
- 192.Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, Royer HD, Scheidereit C, Dorken B. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood. 1996;87:4340–4347. [PubMed] [Google Scholar]
- 193.Isogawa M, Higuchi M, Takahashi M, Oie M, Mori N, Tanaka Y, Aoyagi Y, Fujii M. Rearranged NF-kappa B2 gene in an adult T-cell leukemia cell line. Cancer Sci. 2008;99:792–798. doi: 10.1111/j.1349-7006.2008.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lu D, Thompson JD, Gorski GK, Rice NR, Mayer MG, Yunis JJ. Alterations at the rel locus in human lymphoma. Oncogene. 1991;6:1235–1241. [PubMed] [Google Scholar]
- 195.Joos S, Otano-Joos MI, Ziegler S, Bruderlein S, du Manoir S, Bentz M, Moller P, Lichter P. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood. 1996;87:1571–1578. [PubMed] [Google Scholar]
- 196.Liptay S, Schmid RM, Perkins ND, Meltzer P, Altherr MR, McPherson JD, Wasmuth JJ, Nabel GJ. Related subunits of NF-kappa B map to two distinct loci associated with translocations in leukemia, NFKB1 and NFKB2. Genomics. 1992;13:287–292. doi: 10.1016/0888-7543(92)90244-m. [DOI] [PubMed] [Google Scholar]
- 197.Thakur S, Lin HC, Tseng WT, Kumar S, Bravo R, Foss F, Gelinas C, Rabson AB. Rearrangement and altered expression of the NFKB-2 gene in human cutaneous T-lymphoma cells. Oncogene. 1994;9:2335–2344. [PubMed] [Google Scholar]
- 198.Migliazza A, Lombardi L, Rocchi M, Trecca D, Chang CC, Antonacci R, Fracchiolla NS, Ciana P, Maiolo AT, Neri A. Heterogeneous chromosomal aberrations generate 3' truncations of the NFKB2/lyt-10 gene in lymphoid malignancies. Blood. 1994;84:3850–3860. [PubMed] [Google Scholar]
- 199.Chang CC, Zhang J, Lombardi L, Neri A, Dalla-Favera R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol. 1995;15:5180–5187. doi: 10.1128/mcb.15.9.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Michaux L, Mecucci C, Stul M, Wlodarska I, Hernandez JM, Meeus P, Michaux JL, Scheiff JM, Noel H, Louwagie A, Criel A, Boogaerts M, Van Orshoven A, Cassiman JJ, Van Den Berghe H. BCL3 rearrangement and t(14;19) (q32;q13) in lymphoproliferative disorders. Genes Chromosomes Cancer. 1996;15:38–47. doi: 10.1002/(SICI)1098-2264(199601)15:1<38::AID-GCC6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 201.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 202.Sethi G, Ahn KS, Chaturvedi MM, Aggarwal BB. Epidermal growth factor (EGF) activates nuclear factor-kappaB through IkappaBalpha kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IkappaBalpha. Oncogene. 2007;26:7324–7332. doi: 10.1038/sj.onc.1210544. [DOI] [PubMed] [Google Scholar]
- 203.De Luca A, Carotenuto A, Rachiglio A, Gallo M, Maiello MR, Aldinucci D, Pinto A, Normanno N. The role of the EGFR signaling in tumor mi-croenvironment. J Cell Physiol. 2008;214:559–567. doi: 10.1002/jcp.21260. [DOI] [PubMed] [Google Scholar]
- 204.Lu X, Kang Y. Epidermal growth factor signalling and bone metastasis. BrJ Cancer. 2010;102:457–461. doi: 10.1038/sj.bjc.6605490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Descamps S, Toillon RA, Adriaenssens E, Pawlowski V, Cool SM, Nurcombe V, Le Bourhis X, Boilly B, Peyrat JP, Hondermarck H. Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J Biol Chem. 2001;276:17864–17870. doi: 10.1074/jbc.M010499200. [DOI] [PubMed] [Google Scholar]
- 206.Konig HG, Kogel D, Rami A, Prehn JH. TGF-{beta}1 activates two distinct type I receptors in neurons: implications for neuronal NF-{kappa}B signaling. J Cell Biol. 2005;168:1077–1086. doi: 10.1083/jcb.200407027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lu T, Burdelya LG, Swiatkowski SM, Boiko AD, Howe PH, Stark GR, Gudkov AV. Secreted transforming growth factor beta2 activates NF-kappaB, blocks apoptosis, and is essential for the survival of some tumor cells. Proc Natl Acad Sci U S A. 2004;101:7112–7117. doi: 10.1073/pnas.0402048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lu T, Tian L, Han Y, Vogelbaum M, Stark GR. Dose-dependent cross-talk between the transforming growth factor-beta and interleukin -1 signaling pathways. Proc Natl Acad Sci U S A. 2007;104:4365–4370. doi: 10.1073/pnas.0700118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Matsui Y, Halter SA, Holt JT, Hogan BL, Coffey RJ. Development of mammary hyperplasia and neoplasia in MMTV-TGF alpha transgenic mice. Cell. 1990;61:1147–1155. doi: 10.1016/0092-8674(90)90077-r. [DOI] [PubMed] [Google Scholar]
- 210.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 211.Behrens C, Feng L, Kadara H, Kim HJ, Lee JJ, Mehran R, Hong WK, Lotan R, Wistuba II. Expression of interleukin-1 receptor-associated kinase-1 in non-small cell lung carcinoma and preneoplastic lesions. Clin Cancer Res. 2010;16:34–44. doi: 10.1158/1078-0432.CCR-09-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sun J, Wiklund F, Hsu FC, Balter K, Zheng SL, Johansson JE, Chang B, Liu W, Li T, Turner AR, Li L, Li G, Adami HO, Isaacs WB, Xu J, Gronberg H. Interactions of sequence variants in interleukin-1 receptor-associated kinase4 and the toll-like receptor 6-1-10 gene cluster increase prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2006;15:480–485. doi: 10.1158/1055-9965.EPI-05-0645. [DOI] [PubMed] [Google Scholar]
- 213.Jackson-Bernitsas DG, Ichikawa H, Takada Y, Myers JN, Lin XL, Darnay BG, Chaturvedi MM, Aggarwal BB. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene. 2007;26:1385–1397. doi: 10.1038/sj.onc.1209945. [DOI] [PubMed] [Google Scholar]
- 214.Samanta AK, Huang HJ, Le XF, Mao W, Lu KH, Bast RC, Jr, Liao WS. MEKK3 expression correlates with nuclear factor kappa B activity and with expression of antiapoptotic genes in serous ovarian carcinoma. Cancer. 2009;115:3897–3908. doi: 10.1002/cncr.24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hirano T, Shino Y, Saito T, Komoda F, Okutomi Y, Takeda A, Ishihara T, Yamaguchi T, Saisho H, Shirasawa H. Dominant negative MEKK1 inhibits survival of pancreatic cancer cells. Oncogene. 2002;21:5923–5928. doi: 10.1038/sj.onc.1205643. [DOI] [PubMed] [Google Scholar]
- 216.Babu G, Waterfield M, Chang M, Wu X, Sun SC. Deregulated activation of oncoprotein kinase Tpl2/Cot in HTLV-I-transformed T cells. J Biol Chem. 2006;281:14041–14047. doi: 10.1074/jbc.M512375200. [DOI] [PubMed] [Google Scholar]
- 217.Choi HS, Kang BS, Shim JH, Cho YY, Choi BY, Bode AM, Dong Z. Cot, a novel kinase of histone H3, induces cellular transformation through up-regulation of c-fos transcriptional activity. Faseb J. 2008;22:113–126. doi: 10.1096/fj.07-9078com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Rodriguez C, Lopez P, Pozo M, Duce AM, Lopez-Pelaez M, Fernandez M, Alemany S. COX2 expression and Erk1/Erk2 activity mediate Cot-induced cell migration. Cell Signal. 2008;20:1625–1631. doi: 10.1016/j.cellsig.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 219.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T, Hahn WC. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Korherr C, Gille H, Schafer R, Koenig-Hoffmann K, Dixelius J, Egland KA, Pastan I, Brinkmann U. Identification of proangiogenic genes and pathways by high-throughput functional genomics: TBK1 and the IRF3 pathway. Proc Natl Acad Sci U S A. 2006;103:4240–4245. doi: 10.1073/pnas.0511319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chen J, Miller EM, Gallo KA. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene. 2010;29:4399–4411. doi: 10.1038/onc.2010.198. [DOI] [PubMed] [Google Scholar]
- 222.Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol. 2010;24:598–607. doi: 10.1210/me.2009-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Baumann B, Weber CK, Troppmair J, Whiteside S, Israel A, Rapp UR, Wirth T. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc Natl Acad Sci U S A. 2000;97:4615–4620. doi: 10.1073/pnas.080583397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A. 2010;107:844–849. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Safina A, Ren MQ, Vandette E, Bakin AV. TAK1 is required forTGF-beta 1-mediated regulation of matrix metalloproteinase-9 and metastasis. Oncogene. 2008;27:1198–1207. doi: 10.1038/sj.onc.1210768. [DOI] [PubMed] [Google Scholar]
- 227.Erdogan M, Pozzi A, Bhowmick N, Moses HL, Zent R. Transforming growth factor-beta (TGF-beta) and TGF-beta-associated kinase 1 are required for R-Ras-mediated transformation of mammary epithelial cells. Cancer Res. 2008;68:6224–6231. doi: 10.1158/0008-5472.CAN-08-0513. [DOI] [PubMed] [Google Scholar]
- 228.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, Kato RM, Kang S, Patrone L, Wall R, Teitell M, Leitges M, Kawakami T, Rawlings DJ. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3:780–786. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 229.Mauro LV, Grossoni VC, Urtreger AJ, Yang C, Colombo LL, Morandi A, Pallotta MG, Kazanietz MG, Bal de Kier Joffe ED, Puricelli LL. PKC Delta (PKCdelta) promotes tumoral progression of human ductal pancreatic cancer. Pancreas. 2010;39:e31–41. doi: 10.1097/MPA.0b013e3181bce796. [DOI] [PubMed] [Google Scholar]
- 230.Aziz MH, Hafeez BB, Sand JM, Pierce DB, Aziz SW, Dreckschmidt NE, Verma AK. Protein kinase Cvarepsilon mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2) Oncogene. 2010;29:3100–3109. doi: 10.1038/onc.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 232.Shant J, Cheng K, Marasa BS, Wang JY, Raufman JP. Akt-dependent NF-kappaB activation is required for bile acids to rescue colon cancer cells from stress-induced apoptosis. Exp Cell Res. 2009;315:432–450. doi: 10.1016/j.yexcr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Delgado Andre N, De Lucca FL. Knockdown of PKR expression by RNAi reduces pulmonary metastatic potential of B16-F10 melanoma cells in mice: possible role of NF-kappaB. Cancer Lett. 2007;258:118–125. doi: 10.1016/j.canlet.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 234.Kim SH, Gunnery S, Choe JK, Mathews MB. Neoplastic progression in melanoma and colon cancer is associated with increased expression and activity of the interferon-inducible protein kinase, PKR. Oncogene. 2002;21:8741–8748. doi: 10.1038/sj.onc.1205987. [DOI] [PubMed] [Google Scholar]
- 235.Coniglio SJ, Zavarella S, Symons MH. Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol Cell Biol. 2008;28:4162–4172. doi: 10.1128/MCB.01532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci. 2009;66:1850–1857. doi: 10.1007/s00018-009-9153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Demchenko YN, Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010;1:59–68. doi: 10.18632/oncotarget.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Thu YM, Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010;21:213–226. doi: 10.1016/j.cytogfr.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kim EY, Seo JM, Cho KJ, Kim JH. Ras-induced invasion and metastasis are regulated by a leukotriene B4 receptor BLT2-linked pathway. Oncogene. 2010;29:1167–1178. doi: 10.1038/onc.2009.412. [DOI] [PubMed] [Google Scholar]
- 241.Downward J. Cancer: A tumour gene's fatal flaws. Nature. 2009;462:44–45. doi: 10.1038/462044a. [DOI] [PubMed] [Google Scholar]
- 242.Bhojani MS, Chen G, Ross BD, Beer DG, Rehemtulla A. Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer. Cell Cycle. 2005;4:1478–1481. doi: 10.4161/cc.4.11.2188. [DOI] [PubMed] [Google Scholar]
- 243.Coste I, Le Corf K, Kfoury A, Hmitou I, Druillennec S, Hainaut P, Eychene A, Lebecque S, Renno T. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest. 2010;120:3663–3667. doi: 10.1172/JCI42771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 245.Gallardo F, Bellosillo B, Espinet B, Pujol RM, Estrach T, Servitje O, Romagosa V, Barranco C, Boluda S, Garcia M, Sole F, Ariza A, Serrano S. Aberrant nuclear BCL10 expression and lack of t(11;18)(q21;q21) in primary cutaneous marginal zone B-cell lymphoma. Hum Pathol. 2006;37:867–873. doi: 10.1016/j.humpath.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 246.Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, Penas EM, Dierlamm J, Chan WC, Staudt LM, Thome M. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:19946–19951. doi: 10.1073/pnas.0907511106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lamason RL, McCully RR, Lew SM, Pomerantz JL. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry. 2010;49:8240–8250. doi: 10.1021/bi101052d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Nakamura S, Nakamura S, Matsumoto T, Yada S, Hirahashi M, Suekane H, Yao T, Goda K, Iida M. Overexpression of caspase recruitment domain (CARD) membrane-associated guanylate kinase 1 (CARMA1) and CARD9 in primary gastric B-cell lymphoma. Cancer. 2005;104:1885–1893. doi: 10.1002/cncr.21421. [DOI] [PubMed] [Google Scholar]
- 249.Giam CZ, Jeang KT. HTLV-1 Tax and adult T-cell leukemia. Front Biosci. 2007;12:1496–1507. doi: 10.2741/2163. [DOI] [PubMed] [Google Scholar]
- 250.Keller SA, Hernandez-Hopkins D, Vider J, Ponomarev V, Hyjek E, Schattner EJ, Cesarman E. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 2006;107:3295–3302. doi: 10.1182/blood-2005-07-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Shair KH, Schnegg CI, Raab-Traub N. EBV latent membrane protein 1 effects on plakoglo-bin, cell growth, and migration. Cancer Res. 2008;68:6997–7005. doi: 10.1158/0008-5472.CAN-08-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.de Jong SJ, Albrecht JC, Schmidt M, Muller-Fleckenstein I, Biesinger B. Activation of noncanonical NF-kappaB signaling by the onco-protein Tio. J Biol Chem. 2010;285:16495–16503. doi: 10.1074/jbc.M110.102848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Heinemann S, Biesinger B, Fleckenstein B, Albrecht JC. NFkappaB signaling is induced by the oncoprotein Tio through direct interaction with TRAF6. J Biol Chem. 2006;281:8565–8572. doi: 10.1074/jbc.M510891200. [DOI] [PubMed] [Google Scholar]
- 254.Albrecht JC, Muller-Fleckenstein I, Schmidt M, Fleckenstein B, Biesinger B. Tyrosine phos-phorylation of the Tio oncoprotein is essential for transformation of primary human T cells. J Virol. 2005;79:10507–10513. doi: 10.1128/JVI.79.16.10507-10513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Sun Q, Zachariah S, Chaudhary PM. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J Biol Chem. 2003;278:52437–52445. doi: 10.1074/jbc.M304199200. [DOI] [PubMed] [Google Scholar]
- 256.Chanudet E, Huang Y, Ichimura K, Dong G, Hamoudi RA, Radford J, Wotherspoon AC, Isaacson PG, Ferry J, Du MQ. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 2010;24:483–487. doi: 10.1038/leu.2009.234. [DOI] [PubMed] [Google Scholar]
- 257.Aqeilan RI, Pekarsky Y, Herrero JJ, Palamarchuk A, Letofsky J, Druck T, Trapasso F, Han SY, Melino G, Huebner K, Croce CM. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci U S A. 2004;101:4401–4406. doi: 10.1073/pnas.0400805101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, Hagan JP, Zanesi N, Kaou M, Stein GS, Lian JB, Croce CM. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci U S A. 2007;104:3949–3954. doi: 10.1073/pnas.0609783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Chang NS, Doherty J, Ensign A, Schultz L, Hsu U, Hong Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes ser-ine 46-phosphorylated p53. J Biol Chem. 2005;280:43100–43108. doi: 10.1074/jbc.M505590200. [DOI] [PubMed] [Google Scholar]
- 260.Chang NS, Hsu LJ, Lin YS, Lai FJ, Sheu HM. WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol Med. 2007;13:12–22. doi: 10.1016/j.molmed.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 261.Fabbri M, Iliopoulos D, Trapasso F, Aqeilan RI, Cimmino A, Zanesi N, Yendamuri S, Han SY, Amadori D, Huebner K, Croce CM. WWOX gene restoration prevents lung cancer growth in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:15611–15616. doi: 10.1073/pnas.0505485102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Fu J, Qu Z, Yan P, Ishikawa C, Aqeilan R, Rabson AB, Xiao G. The tumor suppressor gene wwox links the canonical and non-canonical NFkappaB pathways in HTLV-I Tax-mediated tu-morigenesis. Blood. 2010 doi: 10.1182/blood-2010-08-303073. in press: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kurek KC, Del Mare S, Salah Z, Abdeen S, Sadiq H, Lee SH, Gaudio E, Zanesi N, Jones KB, DeYoung B, Amir G, Gebhardt M, Warman M, Stein GS, Stein JL, Lian JB, Aqeilan RI. Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res. 2010;70:5577–5586. doi: 10.1158/0008-5472.CAN-09-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kashima L, Toyota M, Mita H, Suzuki H, Idogawa M, Ogi K, Sasaki Y, Tokino T. CHFR, a potential tumor suppressor, downregulates interleukin-8 through the inhibition of NFkappaB. Oncogene. 2009;28:2643–2653. doi: 10.1038/onc.2009.123. [DOI] [PubMed] [Google Scholar]
- 265.Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG. LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell. 2007;12:239–251. doi: 10.1016/j.ccr.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 266.Kwon J, Cho HJ, Han SH, No JG, Kwon JY, Kim H. A novel LZAP-binding protein, NLBP, inhibits cell invasion. J Biol Chem. 2010;285:12232–12240. doi: 10.1074/jbc.M109.065920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Liu B, Yang R, Wong KA, Getman C, Stein N, Teitell MA, Cheng G, Wu H, Shuai K. Negative regulation of NF-kappaB signaling by PI-AS1. Mol Cell Biol. 2005;25:1113–1123. doi: 10.1128/MCB.25.3.1113-1123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Coppola D, Parikh V, Boulware D, Blanck G. Substantially reduced expression of PIAS1 is associated with colon cancer development. J Cancer Res Clin Oncol. 2009;135:1287–1291. doi: 10.1007/s00432-009-0570-z. [DOI] [PubMed] [Google Scholar]
- 269.Driscoll JJ, Pelluru D, Lefkimmiatis K, Fulciniti M, Prabhala RH, Greipp PR, Barlogie B, Tai YT, Anderson KC, Shaughnessy JD, Jr, Annunziata CM, Munshi NC. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood. 2010;115:2827–2834. doi: 10.1182/blood-2009-03-211045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Nagasaki K, Schem C, von Kaisenberg C, Biallek M, Rosel F, Jonat W, Maass N. Leu-cine-zipper protein, LDOC1, inhibits NF-kappaB activation and sensitizes pancreatic cancer cells to apoptosis. Int J Cancer. 2003;105:454–458. doi: 10.1002/ijc.11122. [DOI] [PubMed] [Google Scholar]
- 271.Chalasani ML, Swarup G, Balasubramanian D. Optineurin and its mutants: molecules associated with some forms of glaucoma. Ophthalmic Res. 2009;42:176–184. doi: 10.1159/000232400. [DOI] [PubMed] [Google Scholar]
- 272.Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17:1438–1443. doi: 10.1016/j.cub.2007.07.041. [DOI] [PubMed] [Google Scholar]
- 273.Heppner C, Bilimoria KY, Agarwal SK, Kester M, Whitty LJ, Guru SC, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene. 2001;20:4917–4925. doi: 10.1038/sj.onc.1204529. [DOI] [PubMed] [Google Scholar]
- 274.Rocha S, Garrett MD, Campbell KJ, Schumm K, Perkins ND. Regulation of NF-kappaB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. Embo J. 2005;24:1157–1169. doi: 10.1038/sj.emboj.7600608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21:7207–7217. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Keller ET, Fu Z, Yeung K, Brennan M. Raf kinase inhibitor protea prostate cancer metastasis suppressor gene. Cancer Lett. 2004;207:131–137. doi: 10.1016/j.canlet.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 277.Tang H, Park S, Sun SC, Trumbly R, Ren G, Tsung E, Yeung KC. RKIP inhibits NF-kappaB in cancer cells by regulating upstream signaling components of the IkappaB kinase complex. FEBS Lett. 2010;584:662–668. doi: 10.1016/j.febslet.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kim JE, You DJ, Lee C, Ahn C, Seong JY, Hwang JI. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phos-phorylation. Cell Signal. 2010;22:1645–1654. doi: 10.1016/j.cellsig.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 279.Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, Shen J, Chen CT, Huo L, Hsu MC, Li CW, Ding Q, Liao TL, Lai CC, Lin AC, Chang YH, Tsai SF, Li LY, Hung MC. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell. 2009;36:131–140. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG, Biswal S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 2010;9:336–346. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Sun W, Wang H, Zhao X, Yu Y, Fan Y, Wang H, Wang X, Lu X, Zhang G, Fu S, Yang J. Protein phosphatase 2A acts as a mitogen-activated protein kinase kinase kinase 3 (MEKK3) phosphatase to inhibit lysophos-phatidic acid-induced IkappaB kinase beta/ nuclear factor-kappaB activation. J Biol Chem. 2010;285:21341–21348. doi: 10.1074/jbc.M110.104224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, Caligiuri MA, Bloomfield CD, Marcucci G, Perrotti D. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 283.Guo LM, Pu Y, Han Z, Liu T, Li YX, Liu M, Li X, Tang H. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-kappaB1. Febs J. 2009;276:5537–5546. doi: 10.1111/j.1742-4658.2009.07237.x. [DOI] [PubMed] [Google Scholar]
- 284.Hildebrandt MA, Gu J, Lin J, Ye Y, Tan W, Tamboli P, Wood CG, Wu X. Hsa-miR-9 methylation status is associated with cancer development and metastatic recurrence in patients with clear cell renal cell carcinoma. Oncogene. 2010;29:5724–5728. doi: 10.1038/onc.2010.305. [DOI] [PubMed] [Google Scholar]
- 285.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wan HY, Guo LM, Liu T, Liu M, Li X, Tang H. Regulation of the transcription factor NF-kappaB1 by microRNA-9 in human gastric ade-nocarcinoma. Mol Cancer. 2010;9:16. doi: 10.1186/1476-4598-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Huang L, Luo J, Cai Q, Pan Q, Zeng H, Guo Z, Dong W, Huang J, Lin T. MicroRNA-125b suppresses the development of bladder cancer by targeting E2F3. IntJ Cancer. 2010 doi: 10.1002/ijc.25509. [DOI] [PubMed] [Google Scholar]
- 288.Li D, Yang P, Xiong Q, Song X, Yang X, Liu L, Yuan W, Rui YC. MicroRNA-125a/b-5p inhibits endothelin-1 expression in vascular endo-thelial cells. J Hypertens. 2010;28:1646–1654. doi: 10.1097/HJH.0b013e32833a4922. [DOI] [PubMed] [Google Scholar]
- 289.Liang L, Wong CM, Ying Q, Fan DN, Huang S, Ding J, Yao J, Yan M, Li J, Yao M, Ng IO, He X. MicroRNA-125b suppressesed human liver cancer cell proliferation and metastasis by directly targeting oncogene LIN28B2. Hepatology. 2010;52:1731–1740. doi: 10.1002/hep.23904. [DOI] [PubMed] [Google Scholar]
- 290.Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, Lodish HF, Lim B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Xia HF, He TZ, Liu CM, Cui Y, Song PP, Jin XH, Ma X. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell Physiol Biochem. 2009;23:347–358. doi: 10.1159/000218181. [DOI] [PubMed] [Google Scholar]
- 292.Komagata S, Nakajima M, Takagi S, Mohri T, Taniya T, Yokoi T. Human CYP24 catalyzing the inactivation of calcitriol is post-transcriptionally regulated by miR-125b. Mol Pharmacol. 2009;76:702–709. doi: 10.1124/mol.109.056986. [DOI] [PubMed] [Google Scholar]
- 293.Bhattacharya R, Nicoloso M, Arvizo R, Wang E, Cortez A, Rossi S, Calin GA, Mukherjee P. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009;69:9090–9095. doi: 10.1158/0008-5472.CAN-09-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Corthals SL, Jongen-Lavrencic M, de Knegt Y, Peeters JK, Beverloo HB, Lokhorst HM, Sonneveld P. Micro-RNA-15a and micro-RNA-16 expression and chromosome 13 deletions in multiple myeloma. Leuk Res. 2010;34:677–681. doi: 10.1016/j.leukres.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 295.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi-Impiombato A, Califano A, Migliazza A, Bhagat G, Dalla-Favera R. The DLEU2/ miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 296.Li T, Morgan MJ, Choksi S, Zhang Y, Kim YS, Liu ZG. MicroRNAs modulate the noncanonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation. Nat Immunol. 2010;11:799–805. doi: 10.1038/ni.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Yang Y, Li X, Yang Q, Wang X, Zhou Y, Jiang T, Ma Q, Wang YJ. The role of microRNA in human lung squamous cell carcinoma. Cancer Genet Cytogenet. 2010;200:127–133. doi: 10.1016/j.cancergencyto.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 298.Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Muller-Tidow C, Bohlander SK, Tenen DG, Behre G. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood. 2010;115:1768–1778. doi: 10.1182/blood-2009-08-240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Stamatopoulos B, Meuleman N, Haibe-Kains B, Saussoy P, Van Den Neste E, Michaux L, Heimann P, Martiat P, Bron D, Lagneaux L. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood. 2009;113:5237–5245. doi: 10.1182/blood-2008-11-189407. [DOI] [PubMed] [Google Scholar]
- 300.Davidson MR, Larsen JE, Yang IA, Hayward NK, Clarke BE, Duhig EE, Passmore LH, Bowman RV, Fong KM. MicroRNA-218 is deleted and downregulated in lung squamous cell carcinoma. PLoS One. 2010;5:e12560. doi: 10.1371/journal.pone.0012560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Gao C, Zhang Z, Liu W, Xiao S, Gu W, Lu H. Reduced microRNA-218 expression is associated with high nuclear factor kappa B activation in gastric cancer. Cancer. 2010;116:41–49. doi: 10.1002/cncr.24743. [DOI] [PubMed] [Google Scholar]
- 302.Song L, Huang Q, Chen K, Liu L, Lin C, Dai T, Yu C, Wu Z, Li J. miR-218 inhibits the invasive ability of glioma cells by direct downregulation of IKK-beta. Biochem Biophys Res Commun. 2010;402:135–140. doi: 10.1016/j.bbrc.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 303.Chiyomaru T, Enokida H, Kawakami K, Tatarano S, Uchida Y, Kawahara K, Nishiyama K, Seki N, Nakagawa M. Functional role of LASP1 in cell viability and its regulation by microRNAs in bladder cancer. Urol Oncol. 2010 doi: 10.1016/j.urolonc.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 304.Tie J, Pan Y, Zhao L, Wu K, Liu J, Sun S, Guo X, Wang B, Gang Y, Zhang Y, Li Q, Qiao T, Zhao Q, Nie Y, Fan D. MiR-218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet. 2010;6:e1000879. doi: 10.1371/journal.pgen.1000879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, Leiser A, Schwartz PE, Rutherford T, Mor G. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 2008;27:4712–4723. doi: 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Wang F, Zheng Z, Guo J, Ding X. Correlation and quantitation of microRNA aberrant expression in tissues and sera from patients with breast tumor. Gynecol Oncol. 2010;119:586–593. doi: 10.1016/j.ygyno.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 307.Worley LA, Long MD, Onken MD, Harbour JW. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed mi-croarray profiling. Melanoma Res. 2008;18:184–190. doi: 10.1097/CMR.0b013e3282feeac6. [DOI] [PubMed] [Google Scholar]
- 308.Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC. Expression of mi-croRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008;27:5643–5647. doi: 10.1038/onc.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69:1279–1283. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Li Y, Vandenboom TG, 2nd, Wang Z, Kong D, Ali S, Philip PA, Sarkar FH. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, Young KH, Martin R, Li Y. miR-301a as an NF-kappaB activator in pancreatic cancer cells. Embo J. 2010 doi: 10.1038/emboj.2010.296. [DOI] [PMC free article] [PubMed] [Google Scholar]