

Abstract
Wip1, a human protein Ser/Thr phosphatase also called PPM1D, stands for wild-type p53 induced phosphatase 1. Emerging evidences indicate that Wip1 can act as an oncogene largely by turning off DNA damage checkpoint responses. Here we report an unrecognized role of Wipl in normally growing cells. Wip1 can be induced by wild-type p53 under not only stressed but also non-stressed conditions. It can trigger G2/M arrest in wild-type p53 containing cells, which was attributed to the decreased Cdc2 kinase activity resulting at least partly from a high level of inhibitory tyrosine phosphorylation on Cdc2 protein at Tyr-15. Furthermore, we also found that Wip1 not only causes G2/M arrest but also decreases cell death triggered by microtubule assembly inhibitor in mouse fibroblasts when wild-type p53 function was restored. These results indicate that Wip1 can provide ample time for wild-type p53-containing cells to prepare entry into mitosis and avoid encountering mitotic catastrophe. Therefore, Wipl may play important roles in cell/tissue homeostasis maintained by wild-type p53 under normal conditions, enhancing our understanding of how p53 makes cell-fate decisions.
Key words: p53, Wip1, cell homeostasis, cell arrest, cell death
Introduction
Nearly all aspects of cell life are regulated by reversible protein phosphorylation. Some examples include metabolic processes, gene regulation, cell cycle control, DNA damage responses, the organization of the cytoskeleton and cell adhesion.1–8 There are no replacements for protein serine/threonine phosphatases, which play a crucial role in cell life. Protein phosphatase 2C is one of four major types of protein serine/threonine phophatases [PP1, 2A, 2B and 2C] in eukaryotic cells. They in general have 11 motifs conserved within the subfamily,9 and these motifs could be identified in sequences of numerous other proteins, which are called PP2C-related proteins. It has been known that the PP2C family and its related proteins are essential for regulating cellular stress responses in eukaryotes.10,11
The p53 is known to play crucial roles in cellular stress responses.12,13 p53 guardian function has been implicated in various aspects of cellular life besides stress conditions.14–16 The function of p53 is largely mediated through its transactivation activity to activate its target genes and fulfill its function.17–23 Such as waf1/p21 is a cyclin-dependent kinase inhibitor that is capable of inhibiting cdks' activities and plays a major role in p53-dependent G1 cell cycle checkpoint.24–26 Puma, Noxa, and others play important roles in mediating p53-dependent apoptotic responses.27 G1 and G2 checkpoints are major genomic surveillance mechanisms, which exist at G1/S and G2/M transitions respectively.28,29 Unlike p53's role in G1 checkpoint responses, largely relying on transactivating p21/waf1,24 a role for p53 in G2 cell cycle arrest is less well defined. Cells that do not contain wtp53 are deficient for G1 checkpoint response, but can undergo a G2 arrest.30–32 However, elevated steady-state level of p53 by ectopic expression is sufficient to induce cell cycle arrest at both G1/S and G2/M transitions by inducing p21/waf1, 14-3-3δ, and/or Gadd45.33–39 Taken together, these results suggest that the role for p53 surveillance in G2 phase of cell cycle is much more complicated and much less well defined compared with its role in controlling the progression of cells in G1 phase of cell cycle.
Wip1 was identified from the screen of p53 targeted genes in response to ionizing radiation (IR) and has close similarity to a number of known type2C phosphatases.40 The initial information that Wip1 expression in response to ionizing radiation (IR) is in a wtp53 dependent manner and ectopic expression of Wip1 can suppress colony formation, suggested that Wip1 might be involved in growth inhibitory pathways activated in response to DNA damage to contribute to p53 tumor suppressor activity.40 However, the studies reported in the following decade indicate that Wipl most likely functions as an oncogene. Analysis of primary human breast tumors provided the first hint for the role of Wip1 as an oncoprotein; Wip1 gene, also called PPM1D gene, is amplified and Wip1/PPM1D mRNA is overexpressed in 15% of breast cancers.41,42 Substantial experimental evidence indicates that Wip1 can act as a negative regulator of several tumor suppressors through removing phosphate from these proteins, including ATM, CHK2, p38MAPK, p53, INK4A and ARF.43–48 Modulation of these key checkpoint proteins by Wipl ultimately can attenuate p53 tumor suppressor responses.43,45,46,49 Here we report that Wip1, working in concert with the steady-state level of functional p53, provides ample time for cells to be ready for mitosis, which is at least partly mediated through increased inhibitory phosphotyrosine level on Cdc2. This study advances our understanding of how wtp53 maintains cells at G2/M boundary and how Wip1 functions as a target to contribute to p53 surveillance function as a cell-fate decision maker.
Results
Upregulation of Wip1 by a higher level of p53 expression in both stress and non-stressed cells.
Wip1 was identified in cells exposed to ironing radiation (IR) in a wtp53-dependent manner.40 To define how specifically Wipl induction relies on p53 status upon other stresses, DNA damage agent 5-FU (Fluorouracil) was used, which is often used clinically as an anti-cancer drug.50 Human fibroblast WI-38 cell line and several tumor cell lines were examined for Wip1 expression following exposure to 5-FU. Four of them (Lovo, HCT116, RKO, U2OS) carry a wt p53 gene as does WI-38. The rest of them (HT29, Calu-6, Saos-2, T98G and RKOE6) harbor mutations or functional defects of p53. Wip1 mRNA expression was determined by RNA gel blot analysis of RNAs from all cell lines with or without exposure to 5-FU. As shown in Figure 1, Wip1 expression was clearly detectable in 5-FU treated cells carrying wtp53, but not in untreated corresponding cells or in cells with deficient p53. This indicates that the induction of Wip1 in response to 5-FU treatment is dependent on the existence of wtp53. Taken together, wtp53 status appears to be essential for induction of Wip1 in stressed cells, consistent with findings that wip1 is a target of p53,40,48 which may partly mediate p53 function.
Figure 1.

The expression of Wip1 depends on wtp53 status. Total RNA isolated from WI-38, RKO, U2OS, HCT-116 and Lovo, which all carry wtp53, and from RKO-E6, Saos-2, Calu-6, T98G and HT29, which all harbor mtp53 without (−) or with (+) the treatment of 5-FU respectively was hybridized to the 32P-labeled Wip1 cDNA or GAPDH cDNA fragment. Wip1 mRNA level is substantially elevated only in cell lines having wtp53 following 5-FU treatment (+). (GAPDH used as RNA loading control). P21/waf1 mRNA expression was detected in the same batch of RNA samples used. The pattern of P21/waf1 mRNA expression is similar to the one of wip1 mRNA expression (data not shown), confirming the activity of wtp53.
The induction of Wip1 in response to DNA damage triggered by IR or 5-FU demonstrates the coexistence between the increased expression of Wip1 and wtp53 in stressed cells, suggesting their functions may partly depend on each other. To further reveal Wipl expression relies on wtp53 status, we asked whether an increased expression of Wip1 depends on wtp53 is also true in non-stressed cells. GM47.23 cell line,51 a derivative of T98G that carries a hormone-inducible wild-type p53 transgene was used. RNA gel blot analysis of RNA from induced (wild-type p53+) and non-induced (endogenous mutant p53−) GM47.23 cells were performed. We found that Wip1 mRNA was detectable only in cells induced to express wtp53 (Fig. 2A). Wtp53 function was confirmed by the expression of p21, which was clearly detected only in Dex-induced GM cells, but not in non-Dex added GM cells or in GM parental cell line—T98G cells with or without Dex addition (Fig. 2B). These results demonstrate that wtp53 alone can upregulate the Wip1 expression, suggesting that a steady-state level of p53 also can regulate Wip1 expression, which is not solely depending on stress-activated wtp53.
Figure 2.
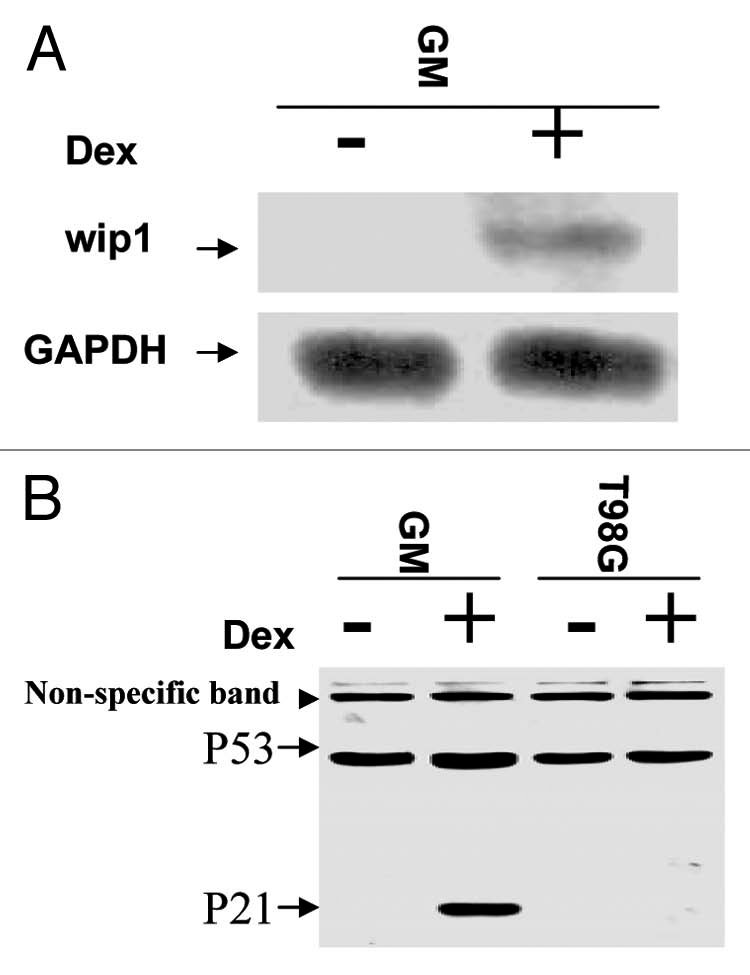
The Wip1 is induced by an elevated steady-state level of wtp53. (A) Autoradiogram of RNA gel blots using either Wip1 cDNA or GAPDH as probe. Lanes: marked (−), GM47.23 cells without Dex (control, no induction of wtp53); Marked (+), GM47.23 cells with Dex (for the induction of wtp53). (GAPDH used as a RNA loading control). The Wip1 mRNA can be detected in GM cells with the wtp53 induction. (B) Western blots analysis of p53 and p21 proteins in both GM and its parental cell line T98G cells. The p21 is only detected in GM cells with the addition of Dex (+), indicating the activity of wtp53.
Wip1 induction of G2/M arrest.
Past 30 y studies demonstrate that p53 is a critical checkpoint protein upon a variety of genotoxic stresses to guard genomic stability and suppress tumor development.13,52–54 However, emerging studies indicate that p53 also plays essential roles in cells that do not receive clear genotoxic stresses. But this aspect of p53 function remains much under-investigated compared with its function triggered by genotoxic stresses. Above study indicated that a higher steady-state level of wtp53 is also responsible for a higher level of Wip1 expression. We therefore anticipated that Wipl may play important roles in mediating p53 function in cells growing under normal conditions. To this end, we decided to investigate how a changed magnitude of Wip1 expression affects non-stress cell behavior, in hope of providing new insights into roles of p53 in cells growing under normal conditions. U2OS-15 and TWIF stable cells, derived from parental cells carrying wtp53 or non-functional p53 respectively, were established, in which Wipl transgene can be conditionally expressed upon the addition of a conditional inducer, Dex (Fig. 3A and B). The cell cycle analysis was subsequently performed, and Wip1 was found to be capable of inducing G2/M arrest in U2–15 cells (Fig. 3C) but not in TWIF cells (Fig. 3D), suggesting Wip1 can provide enough time for wtp53-containg cells to prepare entry into mitosis, and thus mediate the function of p53 in G2/M check under normal growth conditions.
Figure 3.

The Wip1 induces G2/M arrest in cells carrying wtp53 but not in cells harbouring mtp53. (A and B) Establishment of conditional Wip1 expression cells. RNA gel blots were prepared using total RNA isolated from un-induced (−Dex) and induced (+Dex) clone U2–15 (A) and TW1F cells (B) derived from U2OS (wtp53) and T98G (mtp53) cell lines respectively, detected by 32P-labeled Wip1 or GAPDH cDNA fragment. The Wip1 mRNA is increased substantially in cells with the addition of Dex. (GAPDH is loading control). The corresponding levels of Wip1 protein expression were also detected. (C) Wip1 induction of G2/M arrest in U2–15 cells. Synchronized U2–15 cells (left part) were released and incubated with (+) or without (−) Dex. Cells were collected at the time of 8 h in 2 h interval respectively after starvation synchronization (left part). The fitting curves from Flow Cytometric Analysis showed (right part) are cell cycle profiles for cells tested at 12 and 14 h points. At 14 h Wip1 induction, U2–15 cells could not move from G2/M into G1 as they did in the situation of non-induction of Wip1. (D) Induction of Wip1 has no effect on TW1F cells. Synchronized TW1F cells (left part) were released and incubated with (+) or without (−) Dex. Cells were collected at the time of 14 h in 2 h interval respectively. The fitting curves are cell cycle profiles of 18 h and 20 h points (right part). The transition from G2/M peak to G1 peak is similarly shown in both Wip1 induced (+) and non-induced (−) TW1F cells. (pMSG-PP2C stable cells were established, and used to conduct a similar cell cycle analysis; but induction of PP2C has no clear effect on cell cycle progression (data not shown), indicating G2/M arrest triggered by Wip1 is not a general effect of PP2C phosphatase family).
Wip1 induction of G2/M arrest was also found in mouse cells with “normal” genetic background.
Tumor cell lines may differ in ways other than only a p53 status. Therefore, it needs to be further validated that Wip1 induction of G2/M arrest found in tumor cells shows a wtp53 dependent manner. We next tried to use two known genetic background cell lines—mouse fibroblasts with p53−/− or p53+/+ derived from p53 knockout mice and normal mice respectively, as parental cell lines to establish two stable cell lines that can conditionally express Wip1 transgene. As shown in Figure 4A, the derivative cells of mouse fibroblasts with null p53 (named MF-41) were established and capable of conditionally expressing Wip1 transgene, but not those carrying wtp53, because the mouse fibroblasts with p53+/+ only survived a few passages. To directly study the relationship between wtp53 and the function of Wip1 seemed to be a problem when using mouse fibroblasts with wtp53, consistent with the concept that wtp53 can lead to cell arrest or/and apoptosis. Tsp53Val135 is murine p53 class II mutant, which holds wild-type (wt) conformation at 32°C and turns into mutant (mt) conformation at 37°C. It has been suggested as a tool to analyze whether, and how cells were growth-arrested or driven into apoptosis by wtp53.34,55,56 We thereby put tsp53 into MF41 stable cells, in which both Wip1 and wtp53 can be conditionally expressed (Fig. 4B). The clone 23 cells, derived from MF41, were established to conditionally express p53 protein at a level of about two-fold of endogenous p53 (Fig. 4B). Also, clone 23 cells at 37°C are basically like its parental cells MF41 wherein p53 is non-functional and Wip1 can be conditionally expressed; but at 32°C, the function of Wip1 can be studied on the basis of functional p53. Cell cycle studies were subsequently performed to define the relationship between wtp53 and Wip1 induction of G2/M arrest in these cells carrying known genetic background. As usual, cells should be synchronized first for cell cycle analysis. Mouse fibroblasts, however, are too rigid to be synchronized when using conventional starvation method. The exponentially growing cells were thus tentatively used to prepare for flow cytometry. The cell cycle analysis (Fig. 4C) on asynchronized clone 23 cells (wip1+, tsp53+) at 32°C shows G2 arrest under the condition of the expression of wip1, but, not at 37°C, indicating Wip1 induction of G2/M arrest in normal cells also depends on wtp53.
Figure 4.

Wip1 induction induces G2/M arrest in un-synchronized mouse cells carrying functional p53. (A) The Wip1 mRNA expression in mouse fibroblast (p53−/−) derivative cells (MF-41). RNA gel blot was prepared using total RNA isolated from the induced (+) and un-induced (−) No.41 clone, which is named MF41, and detected by 32P-Wip1 cDNA fragment. The Wip1 signal was increased in lane marked (+). (B) Western blot was prepared using 50 µg/lane of cell soluble protein from mouse fibroblast (MF) (p53+/+). MF 41 (wip1+, p53−/−) and clone 23 derived from MF41 (tsp53+; wip1+), detected with primary antibodies against p53 and actin respectively. The p53 protein signal is shown in MEF and clone 23, but not in MF41 cells. (The actin signal used as loading control). (C and D) Asynchronized MF-41 and clone 23 cells were collected after O/N induction (+) or non-induction (−) of Wip1 at both 32°C (wtp53) and 37°C (mtp53). The cell cycle profiles presented in DNA content histogram show that the Wip1 induction can clearly increase cell population in G2 phase when functional p53 is present (at 32°C). (Red arrow head marks a higher G2% of cells compared with other cells tested).
Wip1 not only induces G2/M arrest but also decreases cellular apoptotic response.
Although we found consistent results between mouse fibroblasts carrying normal genetic background and human tumor cells with genomic instability, we did not stop to find a way of synchronization for these mouse cells used. Nishii et al. reported that microtubule formation inhibitor Nocodazole can be used to synchronize cells at early M phase reversibly with variable levels of apoptosis among cell types. Indeed, these mouse cells showed a relatively better G2-rich population with a considerable percentage of cells undergoing apoptosis when treated with Nocodazole (Fig. 5A). Next, Both MF41 and Clone 23 cells were synchronized with Nocodazole and prepared for cell cycle study. We found that, at the 9-h point, the induction of Wip1 did not show any effect on cell population distribution in comparison with non-induction of wip1 at both 32°C and 37°C; however, at the 12-h point, Wip1 induction induced a significant increase in G2 cell percentage and a decreased percentage of sub-G1 at 32°C, but, not at 37°C (Fig. 5B). These results indicate that Wip1 can induce G2/M arrest and decrease cell death response when functional p53 is present (32°C). The corresponding controls were provided to make certain that Wipl effect did not result from the temperature switch that may misinterpret the dependence on wtp53. As seen in Figure 5B, induction of Wip1 in MF41 cells has no effect on cell cycle progression either at 32°C or 37°C. Furthermore, western blotting analysis indicates that Wip1 protein expression responds to inducer Dex to indicate that the conditional expression system remains to be intact; Figure 5C, once again, suggests that the effect of Wip1 observed in the systems used is wtp53-dependent, and not a non-specific effect.
Figure 5.

Wip1 induces G2/M arrest but also decreases cellular apoptosis in Mouse fibroblasts. (A) Mouse fibroblasts were treated with Nocadazole (0.1 µg/ml) for 40 h. The cell cycle profile of these treated cells shows a relatively nice synchronization at G2 population. (B) Cell cycle analysis on Nocodazole-synchronized clone 23 cells shows a better G2 arrest triggered by Wip1 in cells carrying functional p53. The cell cycle profiles presented here in DNA content histogram are those of 9 h and 12 h at 32°C (left part) and 37°C (right part). Wip1 induction (+) can increase cell population in G2 phase and decrease sub-G1 peak (arrow pointed) at time of 12 h at 32°C, but not at 37°C. (C) Wip1 induction has no effect on synchronized MF-41 cells. (D) Wip1 protein expression in both MF41 cells and clone 23 cells upon the Dex addition. Left part is for cells at 32°C and right part is for cells at 37°C, and both show the correct levels of Wip1 protein expression in cells used for cell cycle analysis, indicating the conditional system remains intact.
Wip1 induced G2/M arrest is at least attributed to the decreased Cdc2 kinase activity.
Induction of G2/M arrest by a higher level of Wip1 expression was found in both tumor and normal cells carrying wtp53, indicating Wipl must play substantial roles in regulating G2/M cell phase transition of wtp53-containing cells. The transition from G2 to M phase is controlled by Cdc2 kinase activity, and G2 arrest is associated tightly with an inhibition of Cdc2 activity. Therefore, Cdc2 kinase activity was examined in U2–15 and MF-tstp53 cells (Clone 23 cells) with functional p53 (Fig. 6A and B) and non-functional p53 (Fig. 6C). Cdc2/cyclinB1 complex immunoprecipitated from the soluble fraction of cells by using monoclonal antibody anti-Cdc2, was mixed with γ-32P-ATP and bovine histone H1 to perform a standard H1 kinase assay.58 We found that the induction of Wip1 can decrease the activity of Cdc2/cyclinB1 in functional p53-containing cells (Fig. 6A and B), not in cells carrying non-functional p53 (Fig. 6C) in comparison with the non-Wip1 induced cells, which is not a result of a different amount of Cdc2 protein used in the assay. Therefore, consistent with cell cycle analysis on tumor and mouse cells, the reduction in Cdc2 kinase activity was also found in tumor cells and mouse fibroblasts carrying normal genetic background, which appears to be a plausible cause of G2/M arrest induced by Wip1 (Fig. 6).
Figure 6.
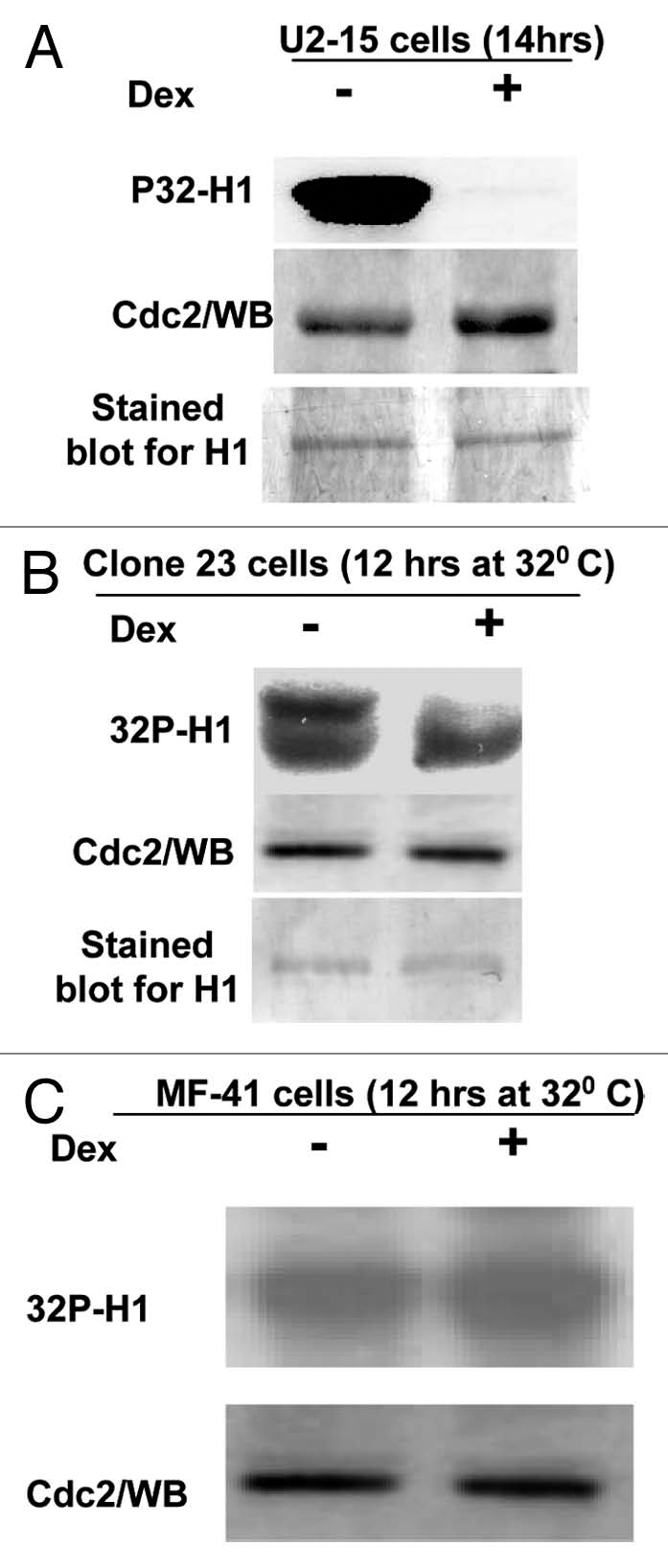
Induction of Wip1 inhibits Cdc2 kinase activity. (A) Wip1 modulates the kinase activity of Cdc2/cyclinB. Immune complexes from the total soluble protein of 12 h un-induced (Lane−) and induced (lane+) U2–15 cells using anti-cdc2 monoclonal antibodies were mixed with histone H1 and γ-32P-ATP at 30°C for 30 min. Autoradiogram (top part) shows much less phosphorylated histone H1 in Lane+ than in Lane−. [Wb (middle part) for the control of cdc2 protein used in the assay and Coomassie blue-stained blot (bottom part) for H1 loading control]. (B and C) Cdc2 kinase activity is decreased upon Wip1 induction in mouse cells when functional p53 is restored. Similar to the experiment conducted in (A), the corresponding mouse cells used for cell cycle analysis showing a nice G2/M arrest were used for preparing cell lysates, and subsequently Cdc2 IP and kinase assay were performed. The decreased Cdc2 kinase activity was found when Wip1 was induced in clone 23 cells (B) but not in MF-41 cells (C) at 32°C.
Increased level of tyrosine phosphorylation on Cdc2 may partially contribute to the decreased Cdc2 kinase activity.
Cdc2 kinase activity basically can be affected by expression levels of Cdc2 itself or/and its partner cyclin B. To this point, we decided to examine expression levels of both proteins in these interested cells with or without overexpressed Wip1. Protein lysates, prepared from the same batch of U2–15 cells used for FACS and kinase assay,58 were used for protein gel blot analysis on the levels of cdc2 and cyclin B protein expression. As shown in Figure 7A, no significant difference in the levels of these proteins is detected when Wip1 is induced compared with un-induced control cells. Therefore, both levels of cdc2 protein and cyclin B1 protein did not appear to be a cause of decreased Cdc2 kinase activity. At G2/M transition, Cdc2 kinase activity in not only determined by the levels of Cdc2 and cyclin B proteins but also by many post-translational modifications on Cdc2 protein, including an inhibitory tyrosine-15 phosphorylation level on Cdc2 protein.59 The relative level of tyrosine-15 phosphorylation on Cdc2 protein was thereby examined by immunoprecipitation using an anti Cdc2 monoclonal antibody and western blotting of the immunoprecipitated Cdc2 by using a monoclonal antibody specifically for phosphorylated tyrosine-15 on Cdc2. The result showed that the level of tyrosine-15 phosphorylation was significantly increased on Cdc2 in Wip1 induced cells (Fig. 7B). Again, mouse clone 23 cells were correspondently treated and prepared for cyclin B and Cdc2 western blotting and detection of tyrosine phosphorylation level on Cdc2 protein, and the results were consistent with what shown in Figure 7B (data not shown). Collectively, Wip1 can promote an inhibitory level of tyrosine phosphorylation on Cdc2, which is able to interfere with Cdc2 kinase activity conferring a G2/M arrest.
Figure 7.
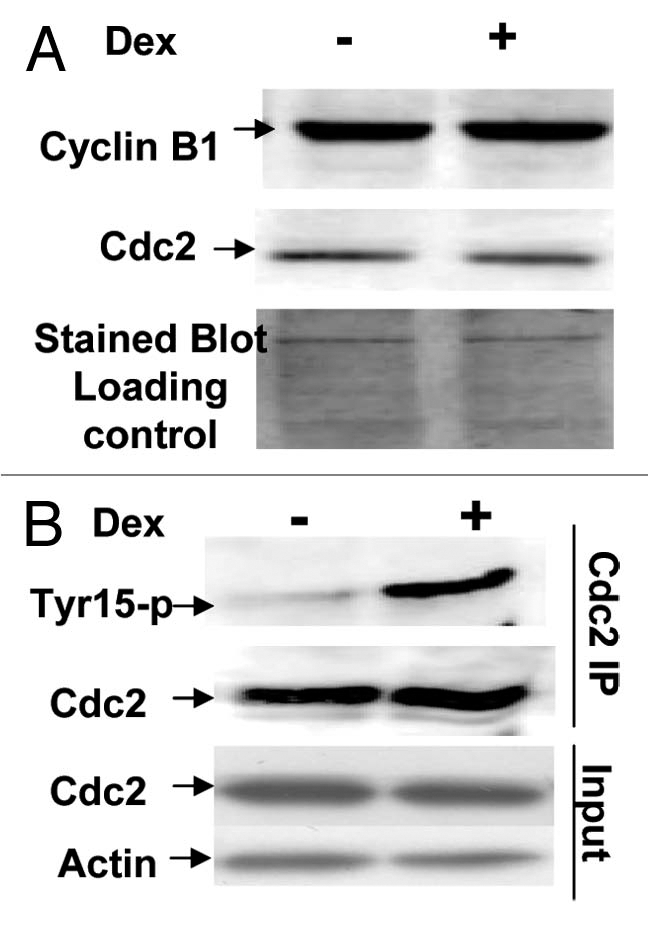
Induction of Wip1 increases the level of inhibitory tyrosine phosphorylation on Cdc2 protein. (A) The levels of nuclear cyclin B and cdc2 proteins are the same between the Wip1 induced and uninduced cells at time of 12 h. The western blot prepared from the nuclear fraction of Wip1 induced (+) and uninduced (−) U2–15 cells at time of 12 h, was blotted by cyclinB and cdc2 antibodies, respectively. There is no detectable difference in both protein levels of Cdc2 and cyclin B between lane (−) and lane (+). Cytoplasmic levels of both proteins were also the same between Wip1 induced and uninduced cells (data not shown) (the stained blot for the control of protein loading). (B) The level of inhibitory tyrosine phosphorylation on Cdc2 is increased when wip1 is induced. The immunoprecipitate from the total soluble protein of uninduced (lane−) and induced (lane+) U2–15 cells at time of 12 h using anti Cdc2 antibody was detected by anti-phosphotyrosine-15 monoclonal antibody. There is an increased level of inhibitory phosphotyrosine on Cdc2 protein in Lane (+). The same blot shown in the top part of (B) was stripped and detected using anti-Cdc2 antibody. There is no detectable difference in Cdc2 protein level between lane (−) and lane (+) (the middle part of B). The IP input control is provided (the bottom part of B).
Discussion
Since the discovery of p53 over 30 y ago, it is now recognized that all cell fate pathways of live cells are virtually under the control of p53. Such extensive involvement indicates that p53 protein is acting as a major traffic controller in the cell signaling network.12,27,60 But still how p53 acts at a cross road of live or death cell signaling pathway is difficult to answer. Our finding demonstrates Wip1 is involved in such a pivotal, complicated task of p53. As shown in Figures 1 and 2, both stressed-activated p53 and elevated steady-state level of p53 can induce Wip1, indicating p53 function is attributed at least partly to Wip1 under both stressed and non-stressed conditions. A large body of evidences indicates p53 exerts pivotal roles in guarding genomic stability upon many stress conditions.12,54,61 However, it is less well understood for its role in protecting cells under non-stress conditions. We revealed that, in non-stressed cells, Wip1 can induce G2/M arrest and decrease basal level cell death in wtp53-containing cells, suggesting Wip1 plays important roles in p53 maintaining cell/tissue homeostasis, consistent with the recent findings that p53 can maintain oocyte genomic stability via its target LIF (Leukemia inhibitory factor).14,62 Furthermore, our study revealed the mechanisms by which Wip1 induces G2/M arrest. We found a decreased Cdc2 kinase activity, leading to a G2/M arrest, is at least attributed to an elevated level of inhibitory tyrosine phosphorylation on Cdc2. At the moment, we remain unknown of how Wip1 caused elevated levels of tyrosine phosphorylation on Cdc2 protein. Further studies are needed on revealing more in-depth molecular controls by which Wipl induces G2/M arrest. Nonetheless, our study suggests that Wip1 can provide necessary time needed for wtp53-containing cells to have a regular cell cycle progression from G2 to M; otherwise, cells will be rushed into M phase of cell cycle and commit suicide owing to a limited preparation time at G2. Therefore, Wip1 plays an important role in maintaining cells carrying wtp53 particularly at G2/M cell cycle transition; and cells harbor mutated p53, or a low, null level of Wip1, would not have ample of time at G2 phase of cell cycle, and thereby encounter a mitotic catastrophe.
Accumulated studies indicate that Wip1 can inactivate ATM, CHK2, p38MAPK, MDM2 and p53 itself upon a variety of genotoxic stresses.43,45,46 Therefore Wip1 appears to act as an oncogene, and promotes tumor formation. We believe that Wipl plays an important role in p53 guarding the steady-state levels of cellular responses. This feature of Wipl can be turned into either good or bad in a cell context dependent manner. As we discussed above, when Wipl is not around under normal condition, wtp53 containing cells may lose a proper time needed before entry into mitosis, which was in agreement with our antisense study in U2OS cells showing a little faster G2/M transition (data not shown). Understandably, these normal cells with proper cellular checkpoint responses would die when rushed into M phases. On the other hand, if cells are undergoing neoplastic transformation, the loss of Wipl may favor the tumor suppression, as a result of cell rushing into M phase of cell cycle and encountering the death check suggested from our study (Fig. 5A), thereby Wip1 appears to act as an oncogene. This is also in agreement with an earlier reported study that silencing Wip1 suppressed tumor formation.45 We believe tumor suppression activity of Wip1-loss is at least partly attributed to a shorter-G2 preparation, which renders cells to commit a suicide. When Wipl is overexpressed again in cells with a neoplastic context, the decreased apoptotic potential caused by a high level of Wip1 (Fig. 5B) would promote tumor formation. Amplification of wip1 gene functioning in breast tumorigenesis might be a scenario we discussed here, under which Wip1 helps tumors cells escaped from the elimination presumably enforced by a mitotic checkpoint. Therefore our study provides cellular behaviors influenced by Wip1, a part of which is consistent with the initial finding regarding the function of Wip1 at the cellular level, capable of inhibiting colony formation in tumor cells tested. We revealed that, in tumor cells, Wipl can induce G2/M arrest (Figs. 3C and 4C), which apparently contributes to colony formation; however, a decreased apoptotic response resulting from Wip1 (Fig. 4C), probably unable to show clearly owing to a low level of basal apoptotic response in some tumor cells, contributes little to promoting the colony growth. Cancer cells in general harbor a compromised apoptotic response that may lead to a disguised phenotype when study gene functions in tumor cells. Indeed, when we reexamined Wip1 function in cells with normal genetic background, it turned that Wipl not only causes G2/M arrest but also decreases substantial apoptotic activity, which is consistent with the concept that Wip1 acts as an oncogene, thinking that apoptotic potential is a major determinant for tumor suppression. Here, the function of Wip1 we found is important for p53 guiding tissue homeostasis, because normal cells or tissues need to have ample of time to prepare entry into mitosis rather than rushing into M phase to be eliminated by surveillance mechanism. Collectively, Wip1 may perform a further check function for normal cells before entry into mitosis; otherwise cells would die in mitosis. This study reveals unrecognized roles of Wip1 in p53 guarding the steady-state levels of cellular responses, which can be easily extended and aligned with its known effects on cells carrying genetic instability, thereby providing further understanding of how p53 orchestrates cellular fate decisions to die or to live, and how these functions of p53 can be swung around by its targets in a cell context dependent manner.
Materials and Methods
Cell lines and cell culture.
The following eight cell lines were obtained from the American Type Culture Collection (ATCC): Saos-2, U-20S, T98G, HT-29, Lovo, HCT116, Calo-6 and WI-38. The RKO and RKO-E6 were generously provided by Dr. Vogelstein at the Johns Hopkins University. All the above cell lines were maintained routinely in MEM medium supplemented with 10% fetal calf serum (FCS), penicillin (100 U/ml) and streptomycin (100 µg/ml). Mouse fibroblasts with p53+/+ and p53−/− were obtained from the laboratory of Dr. Lawrence A. Donehower. These cell lines were maintained routinely in DMEM/F-12 medium supplemented with 10% fetal calf serum (FCS), penicillin (100 U/ml) and streptomycin (100 µg/ml). All cell lines were incubated in 37°C incubator with 5% CO2 ambient as described in reference 51. For a specific culture condition, the incubation temperature is 32°C, which will be specified in the experiment. For trypsin treatment, cells were washed in PBS-CMF (calcium and magnesium free) twice, followed by appropriate incubation time with 0.25% (m/v) trypsin at RT. Trypsin activity was quenched by addition of complete medium. GM47.23 cells are derivative cells of T98G that can conditionally express wtp53 upon the addition of Dexamethasone (Dex).51
Plasmids and sub-cloning.
pMSG (purchased from Pharmacia) is an eukaryotic expression plasmid containing the dexmethasone (Dex)-inducible mouse mammary tumor virus (MMTV) promoter and the gene encoding Eco-gpt selectable marker driven by the SV40 early promoter.63,64 Wip1 cDNA was inserted at EcoRI site of MCS of pMSG, and under the control of MMTV promoter. The plasmid pMSG-PP2C was a kind gift from Dr. Appella (NCI). pBabe-tsp53V135 was a kind gift from Dr. Skorski (Temple University).
RNA gel blotting analysis and the probe preparation.
Total RNA isolation and RNA gel blotting were performed as described in references 23 and 51. Briefly, 15 µg of total RNA from each cell line was electrophoresed through a 1.0% agarose gel. The RNA was transferred to a nylon membrane, and UV-crosslinked. The membrane was pre-hybridized and hybridized with 106 cpm/ml of radiolabeled cDNA fragments, and subsequently washed. The RNA-cDNA hybrids were exposed to X-ray films. To normalize the loading of RNA, the blot was stripped in the boiling water and rehybridized with radiolabeled cDNA fragment of GAPDH. 2.4 kb wip1 cDNA fragment and GAPDH fragment were labeled with a random hexamer primed synthesis kit (Promega).
Transfection and the stable clone(s) selection.
The whole procedures were performed as described in reference 51. Briefly, 10 µg of pMSG-wip1 plasmid was transfected into a 100 mm dish of U2OS cells, T98G cells, Mouse fibroblasts (p53−/−) or (p53+/+), or 10 µg of pBabe-tsp53V135 was transfected into a 100 mm dish of MF41 cells. 48–72 h later, cells were reseeded in gpt+ medium51 for the selection of cells containing pMSG-wip1 or 2 µg/ml puromycin was added for selection of pBabe-tsp53V135. After 3–5 weeks of selection, each individual clone was isolated and expanded until further verified by RNA gel blot analysis and western blot analysis.
Western blot analysis and immunoprecipitation.
As performed essentially in,64,65 primary antibodies targeting Cdc2, cyclinB1, phosphotyrosine-15 of CDC2, Wip1, actin, p53 or p21 (Santa Cruz) all were used at a ratio of 1:1,000 dilution, and the 2nd antibodies—horseradish peroxidase conjugated anti-rabbit or mouse IgG (Amersham) were used at a ratio of 1:10,000 dilution. The membrane was stained with Coomassie blue for the verification of the sample loading if actin signal was not detected. For IP, 1 µg of anti Cdc2 monoclonal Ab agarose conjugate (Santa Cruz) was added to 500 µg of cell lysates, and the mixture was rotated for binding at 4°C for 3 h or overnight. The pellet was washed two times with lysis buffer, another two times with kinase reaction buffer (if the precipitates needs for kinase assay). If not, it was washed four times with lysis buffer, collecting agarose beads each time by centrifuging (CFG) at 2.5 k rpm for 5 min at 4°C.
Cell sycronization, flow cytometry and cell cycle analysis.
To synchronize the cells in G0, exponentially proliferating cells at 80% confluence were starved in 0.1% serum medium for 72 h. To synchronize the cells in G2, exponentially proliferating cells at 80% confluence were grown in the medium with 0.1 µg/ml Nocodazole medium for 40 h. 106 cells per time point were trypsinized, and further prepared for flow cytometry.
Cdc2 kinase assay.
The detail procedures were performed as described in reference 66. Briefly, the immunoprecipitates equilibrated with kinase buffer were mixed with 10 µl of kinase buffer containing 50 µm ATP, 1.25 µCi [γ-32P] ATP, and 1 ug of his-tone H1 (Roche). The mixture then was incubated at 30°C for 30 min, and reactions were terminated by the addition of SDS-sample buffer. The samples were analyzed by SDS-PAGE, followed by drying gel and autoradiography.
Acknowledgments
We thank Dr. Ettore Appela for providing Wip1 cDNA and pMSG-PP2C, Dr. Tomasz Skorski for pBabe-tsp53V135, Dr. Lawrence A. Donehower for MEFs and Dr. Bert Vogelstein for RKO and RKO-E6 cells. P.F. would like to thank Dr. W. Edward Mercer (passed away in 2008) for his guidance in graduate school to initiate a part of the study. This study was partly supported by Department of Laboratory Medicine and Pathology, Mayo Clinic, as well as by the Hormel Institute, University of Minnesota.
References
- 1.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature reviews. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 2.Motoyama N, Naka K. DNA damage tumor suppressor genes and genomic instability. Curr Opin Genet Dev. 2004;14:11–16. doi: 10.1016/j. gde.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol. 2007;8:234–244. doi: 10.1038/nrm2126. [DOI] [PubMed] [Google Scholar]
- 4.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j. molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Nolt JK, Rice LM, Gallo-Ebert C, Bisher ME, Nickels JT. PP2A (Cdc55) is required for multiple events during meiosis I. Cell Cycle. 2011;10:1420–1434. doi: 10.4161/cc.10.9.15485. [DOI] [PubMed] [Google Scholar]
- 6.Zhong J, Liao J, Liu X, Wang P, Liu J, Hou W, et al. Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell cycle. 2010;10:1411–1419. doi: 10.4161/cc.10.9.15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuntziger T, Landsverk HB, Collas P, Syljuasen RG. Protein phosphatase 1 regulators in DNA damage signaling. Cell cycle. 2010;10:1356–1362. doi: 10.4161/cc.10.9.15442. [DOI] [PubMed] [Google Scholar]
- 8.Smolka M. Fine-tuning the DNA damage response: protein phosphatase 2A checks on CHK2. Cell cycle. 2010;9:862–863. [PubMed] [Google Scholar]
- 9.Bork P, Brown NP, Hegyi H, Schultz J. The protein phosphatase 2C (PP2C) superfamily: detection of bacterial homologues. Protein Sci. 1996;5:1421–1425. doi: 10.1002/pro.5560050720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu G, Wang Y. Functional diversity of mammalian type 2C protein phosphatase isoforms: new tales from an old family. Clin Exp Pharmacol Physiol. 2008;35:107–112. doi: 10.1111/j.1440681.2007.04843.x. [DOI] [PubMed] [Google Scholar]
- 11.Heideker J, Lis ET, Romesberg FE. Phosphatases, DNA damage checkpoints and checkpoint deactivation. Cell cycle (Georgetown, Tex) 2007;6:3058–3064. doi: 10.4161/cc.6.24.5100. [DOI] [PubMed] [Google Scholar]
- 12.Fei P, El-Deiry WS. p53 and radiation responses. Oncogene. 2003;22:5774–5783. doi: 10.1038/sj.onc.1206677. [DOI] [PubMed] [Google Scholar]
- 13.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 14.Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450:721–724. doi: 10.1038/ nature05993. [DOI] [PubMed] [Google Scholar]
- 15.Hu W, Feng Z, Atwal GS, Levine AJ. p53: a new player in reproduction. Cell Cycle. 2008;7:848–852. doi: 10.4161/cc.7.7.5658. [DOI] [PubMed] [Google Scholar]
- 16.Kang HJ, Feng Z, Sun Y, Atwal G, Murphy ME, Rebbeck TR, et al. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci USA. 2009;106:9761–9766. doi: 10.1073/pnas.0904280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science (New York, NY) 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 18.Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/S0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 19.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/S10972765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 20.Fei P, Bernhard EJ, El-Deiry WS. Tissue-specific induction of p53 targets in vivo. Cancer Res. 2002;62:7316–7327. [PubMed] [Google Scholar]
- 21.Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–849. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 22.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science (New York, NY) 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 23.Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell. 2004;6:597–609. doi: 10.1016/j. ccr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 24.el-Deiry WS, Harper JW, O'Connor PM, Velculescu VE, Canman CE, Jackman J, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 25.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/00928674(93)90499-G. [DOI] [PubMed] [Google Scholar]
- 26.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 27.Sebastian S, Azzariti A, Silvestris N, Porcelli L, Russo A, Paradiso A. p53 as the main traffic controller of the cell signaling network. Front Biosci. 2010;15:1172–1190. doi: 10.2741/3669. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, et al. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–3124. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang B, Xiao Z, Ko HL, Ren EC. The p53 response element and transcriptional repression. Cell cycle. 2010;9:870–879. doi: 10.4161/cc.9.5.10825. [DOI] [PubMed] [Google Scholar]
- 30.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 31.Li CY, Nagasawa H, Dahlberg WK, Little JB. Diminished capacity for p53 in mediating a radiation-induced G1 arrest in established human tumor cell lines. Oncogene. 1995;11:1885–1892. [PubMed] [Google Scholar]
- 32.Chung JH, Bunz F. Cdk2 is required for p53-independent G2/M checkpoint control. PLoS Genet. 2010;6:1000863. doi: 10.1371/journal. pgen.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA. 1995;92:8493–8497. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart N, Hicks GG, Paraskevas F, Mowat M. Evidence for a second cell cycle block at G2/M by p53. Oncogene. 1995;10:109–115. [PubMed] [Google Scholar]
- 35.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 36.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 37.Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA. 1999;96:3706–3711. doi: 10.1073/ pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nature Rev. 2007;7:861–869. doi: 10.1038/nrc2248. [DOI] [PubMed] [Google Scholar]
- 39.Meng S, Arbit T, Veeriah S, Mellinghoff IK, Fang F, Vivanco I, et al. 14-3-3sigma and p21 synergize to determine DNA damage response following Chk2 inhibition. Cell cycle. 2009;8:2238–2246. doi: 10.4161/cc.8.14.8998. [DOI] [PubMed] [Google Scholar]
- 40.Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci USA. 1997;94:6048–6053. doi: 10.1073/ pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bulavin DV, Demidov ON, Saito S, Kauraniemi P, Phillips C, Amundson SA, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–215. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Yang Y, Peng Y, Austin RJ, van Eyndhoven WG, Nguyen KC, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002;31:133–134. doi: 10.1038/ng888. [DOI] [PubMed] [Google Scholar]
- 43.Lu X, Nguyen TA, Donehower LA. Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle. 2005;4:1060–1064. [PubMed] [Google Scholar]
- 44.Le Guezennec X, Bulavin DV. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem Sci. 35:109–114. doi: 10.1016/j. tibs.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 46.Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–764. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 47.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–135. doi: 10.1007/ s10555-008-9127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu X, Nguyen TA, Appella E, Donehower LA. Homeostatic regulation of base excision repair by a p53-induced phosphatase: linking stress response pathways with DNA repair proteins. Cell Cycle. 2004;3:1363–1366. doi: 10.4161/cc.3.11.1241. [DOI] [PubMed] [Google Scholar]
- 50.Kinsella TJ. Radiosensitization and cell kinetics: clinical implications for S-phase-specific radiosensitizers. Semin Oncol. 1992;19:41–47. [PubMed] [Google Scholar]
- 51.Mercer WE, Shields MT, Amin M, Sauve GJ, Appella E, Romano JW, et al. Negative growth regulation in a glioblastoma tumor cell line that conditionally expresses human wild-type p53. Proc Natl Acad Sci USA. 1990;87:6166–6170. doi: 10.1073/ pnas.87.16.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 53.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 54.Talos F, Moll UM. Role of the p53 family in stabilizing the genome and preventing polyploidization. Adv Exp Med Biol. 2010;676:73–91. doi: 10.1007/978-1-4419-6199-0_5. [DOI] [PubMed] [Google Scholar]
- 55.Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62:671–680. doi: 10.1016/00928674(90)90113-S. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, Guo XY, Hu GY, Liu WB, Shay JW, Deisseroth AB. A temperature-sensitive mutant of human p53. EMBO J. 1994;13:2535–2544. doi: 10.1002/j.1460-2075.1994.tb06543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishii K, Kabarowski JH, Gibbons DL, Griffiths SD, Titley I, Wiedemann LM, et al. ts BCR-ABL kinase activation confers increased resistance to genotoxic damage via cell cycle block. Oncogene. 1996;13:2225–2234. [PubMed] [Google Scholar]
- 58.Dash BC, El-Deiry WS. Phosphorylation of p21 in G2/M promotes cyclin B-Cdc2 kinase activity. Mol Cell Biol. 2005;25:3364–3387. doi: 10.1128/MCB.25.8.3364-3387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan M, Jing T, Lan KH, Neal CL, Li P, Lee S, et al. Phosphorylation on tyrosine-15 of p34(Cdc2) by ErbB2 inhibits p34(Cdc2) activation and is involved in resistance to taxol-induced apoptosis. Mol Cell. 2002;9:993–1004. doi: 10.1016/ S1097-2765(02)00510-5. [DOI] [PubMed] [Google Scholar]
- 60.Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med. 2011;3:64. doi: 10.1126/scitranslmed.3001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chernova OB, Chernov MV, Agarwal ML, Taylor WR, Stark GR. The role of p53 in regulating genomic stability when DNA and RNA synthesis are inhibited. Trends Biochem Sci. 1995;20:431–434. doi: 10.1016/S0968-0004(00)89094-5. [DOI] [PubMed] [Google Scholar]
- 62.Hu W, Feng Z, Levine AJ. The regulation of human reproduction by p53 and its pathway. Cell Cycle. 2009;8:3621–3622. doi: 10.4161/cc.8.22.9938. [DOI] [PubMed] [Google Scholar]
- 63.Mulligan RC, Berg P. Selection for animal cells that express the Escherichia coli gene coding for xanthineguanine phosphoribosyltransferase. Proc Natl Acad Sci USA. 1981;78:2072–2076. doi: 10.1073/pnas.78.4.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang J, Zhao D, Park HK, Wang H, Dyer RB, Liu W, et al. FAVL elevation in human tumors disrupts Fanconi anemia pathway signaling and promotes genomic instability and tumor growth. J Clin Invest. 2010;120:1524–1534. doi: 10.1172/JCI40908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Zhao D, Wang H, Lin CJ, Fei P. FANCD2 monoubiquitination provides a link between the HHR6 and FA-BRCA pathways. Cell Cycle. 2008;7:407–413. doi: 10.4161/cc.7.3.5156. [DOI] [PubMed] [Google Scholar]
- 66.Poon RY, Chau MS, Yamashita K, Hunter T. The role of Cdc2 feedback loop control in the DNA damage checkpoint in mammalian cells. Cancer Res. 1997;57:5168–5178. [PubMed] [Google Scholar]