

Abstract
The cytochrome P450 (CYP) enzymes are important in many areas, including pharmaceutical development. Subtle changes in the electronic structure of the active species, Compound I, have been postulated previously to account partly for the experimentally observed differences in reactivity between isoforms. Current predictive models of CYP metabolism typically assume an identical Compound I in all isoforms. Here we present a method to calculate the electronic structure and to estimate the Fe–O bond enthalpy of Compound I, and apply it to several human and bacterial CYP isoforms. Conformational flexibility is accounted for by sampling large numbers of structures from molecular dynamics simulations, which are subsequently optimized with density functional theory (B3LYP) based quantum mechanics/molecular mechanics. The observed differences in Compound I between human isoforms are small: They are generally smaller than the spread of values obtained for the same isoform starting from different initial structures. Hence, it is unlikely that the variation in activity between human isoforms is due to differences in the electronic structure of Compound I. A larger difference in electronic structure is observed between the human isoforms and P450cam and may be explained by the slightly different hydrogen-bonding environment surrounding the cysteinyl sulfur. The presence of substrate in the active site of all isoforms studied appears to cause a slight decrease in the Fe–O bond enthalpy, apparently due to displacement of water out of the active site, suggesting that Compound I is less stable in the presence of substrate.
Introduction
Prediction of substrate selectivity of human cytochrome P450 (CYP) enzymes is a challenging task. These enzymes are responsible for the oxidation of many different substrates (including drugs), often resulting in the formation of a variety of products, some of which are toxic.(1) Two major factors that influence selectivity are the steric and electrostatic interactions between the protein and substrate which control the substrate orientation, and the intrinsic reactivity of the substrate with respect to the active oxygenating species, Compound I (Cpd I).(2) The latter factor has been postulated to be dependent on the electronic structure of Cpd I.3,4 Better understanding of the factors that influence Cpd I reactivity could contribute significantly to drug development(5) and to other practical applications, such as use and engineering of CYPs for biocatalysis.(6)
The electronic structure of Cpd I has been shown previously to be sensitive to the electrostatic and hydrogen-bonding interactions within the protein as well as the identity of the axial ligand, which in the case of CYPs is a cysteinate.7−11 Such differences in the electronic structure of Cpd I include the amount of unpaired electron density located on the cysteinate sulfur, as is discussed below. Analogous differences have been observed between peroxidase enzymes, e.g., the different location of an unpaired electron observed in the electronic structures of Cpd I in cyctochrome c peroxidase and ascorbate peroxidase.(12) Observed differences in the electronic structure of CYP Cpd I have largely been based on quantum mechanical (QM) calculations, mostly using density functional theory (DFT), for a small model of the enzyme active site. It is also commonly believed that the electronic structure of Cpd I can be tuned by the enzyme, thus affecting the regioselectivity of oxidation for some substrates.4,13 The presence of substrate has been previously proposed to increase the stability of Cpd I in CYP3A4.(14) As different CYP isoforms vary in overall size, size of active site, and sequence of amino acid residues, it is conceivable that the electronic structure of Cpd I might vary between different isoforms. Previous work gives conflicting views on the significance of this effect,14,15 as is described below. Here the extent to which the electronic structure of Cpd I varies between isoforms is probed more rigorously.
Cpd I is a transient species and has only been isolated experimentally for the CYP119 isoform,(16) although it has been isolated in other enzymes, such as cytochrome c peroxidase(17) and horseradish peroxidase.(18) It is therefore presently not possible to compare the electronic structure of different CYP isoforms using experimental techniques. If the electronic structure of Cpd I does vary between isoforms, then it could be partially responsible for the different products formed in different isoforms. The proposed ability of the enzyme environment to tune the electronic structure of Cpd I has been termed “chameleon” behavior.3,10 The implications of this proposed behavior would be significant and may compromise models that aim to predict CYP reactivity based on Cpd I having identical behavior in all isoforms, e.g., MetaSite.(19) As multiple CYP isoforms in the human body are responsible for the metabolism of drugs, any possible difference in Cpd I reactivity between isoforms should be explored. It is also possible that the variations in the enzyme environment surrounding the cysteinate are not significant enough to cause a net change in the electronic structure of Cpd I and that observed changes in calculations on large models of P450 are due to thermal fluctuations in the enzyme environment. The main aim of this study is to test thoroughly which of these two interpretations is correct.
Electronic Structure of Cpd I
Cpd I is a triradicaloid species with three singly occupied molecular orbitals. These orbitals (shown in Figure 1) consist of two π* antibonding Fe–O orbitals and a third that is a combination of a porphyrin π orbital with idealized a2u symmetry and a p orbital on sulfur. Depending on the relative orientation of the spins of the two unpaired electrons on the Fe–O moiety and the third unpaired electron, one obtains a quartet or doublet spin state (denoted 4A2u and 2A2u), which are found to be near-degenerate in electronic structure calculations.15,20 In QM (DFT) calculations on small models, the distribution of the spin density in the a2u orbital varies, depending on the treatment of the cysteinate ligand and its environment. The inclusion of NH–S hydrogen bonds in these models increases the amount of spin density distributed over the porphyrin ring by effectively lowering the energy of the sulfur lone pair orbital.(3) These NH–S hydrogen bonds mimic the hydrogen bonds that are formed between the cysteinate sulfur and three neighboring amino acids (see Figure 2 and Table 1). The introduction of a polarizing electric field has a similar effect.(4) DFT calculations using a mercaptide (−SCH3) or cysteinate anion, without the inclusion of the above-mentioned hydrogen-bonding interactions, lead to the prediction of two further low-energy states, in which the third unpaired electron is located almost exclusively within a different p orbital on the sulfur atom, giving rise to 4,2ΠS states.8,21−23 These 4,2ΠS states are inconsistent with experimental studies of Cpd I in chloroperoxidase(24) and CYP119,(16) which are green in color and more consistent with 2,4A2u states. In the recent spectroscopic characterization of Cpd I in CYP119,(16) the UV–vis and Mössbauer spectra were discovered to be similar to those of chloroperoxidase, and the Cpd I species in CYPs is therefore expected to have 2,4A2u ground states.
Figure 1.

Frontier orbitals of Cpd I: (a) is the porphyrin a2u orbital with significant contribution from the sulfur p orbital; and (b) and (c) are the two π* antibonding Fe–O orbitals. Calculated in vacuo at the B3LYP/LACVP level.
Figure 2.
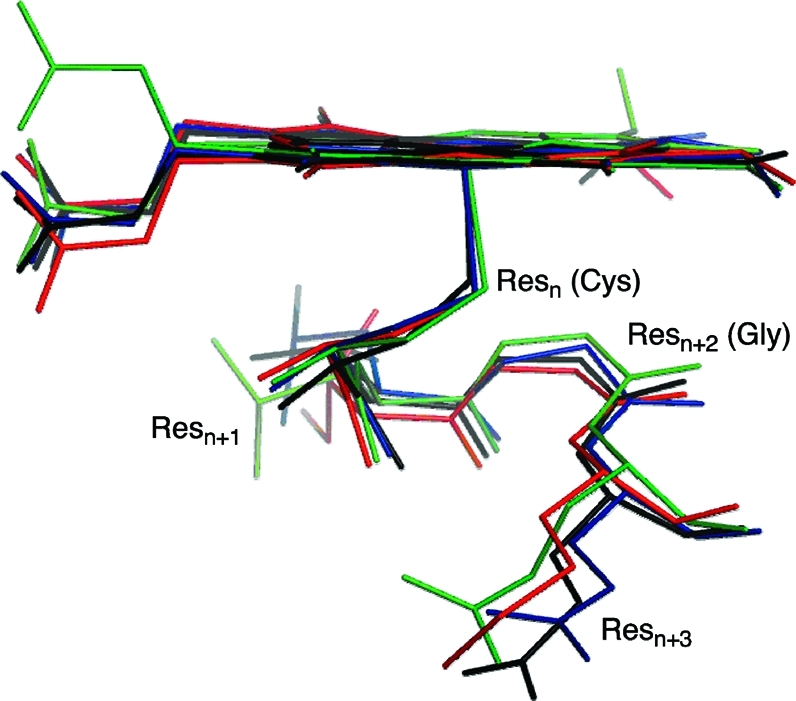
Residues surrounding the cysteinate sulfur in the crystal structures of P450cam (1DZ9, green), CYP2C9 (1OG5, black), CYP2D6 (2F9Q, blue), and CYP3A4 (1TQN, red).
Table 1. Residues in P450cam, CYP2C9, CYP2D6, and CYP3A4 (Resn+1, Resn+2, and Resn+3) Involved in the Hydrogen-Bond Network Surrounding the Fe-Bound Cysteinate (Resn)a.
| isoform | PDB code | Resn | Resn+1 | Resn+2 | Resn+3 |
|---|---|---|---|---|---|
| P450cam | 1DZ9 | Cys357 | Leu358 | Gly359 | Gln360 |
| CYP2C9 | 1OG5 | Cys435 | Val436 | Gly437 | Glu438 |
| CYP2D6 | 2F9Q | Cys443 | Leu444 | Gly445 | Glu446 |
| CYP3A4 | 1TQN | Cys442 | Ile443 | Gly444 | Met445 |
The Fe–S bond length also is sensitive to the environment and treatment of the cysteinate ligand. It shortens by 0.1 Å when hydrogen bonding and a polarizing electric field in the direction of the Fe–S bond are included in QM (DFT) calculations on small models.3,4,10,20 It was these observations that first led to the description of Cpd I as a chameleon species that changes its electronic structure according to its environment.(4)
The electronic structure of Cpd I in CYPs 2C9, 3A4, 2B4, and P450cam has been previously investigated using density functional quantum mechanics/molecular mechanics (QM/MM) methods by Bathelt et al.15,29 The sulfur spin density and Fe–S bond length were used in that work as a means of comparing the electronic structure of Cpd I in the isoforms studied. By enabling the treatment of much larger models, QM/MM calculations are able to describe the hydrogen bonding and electrostatic environment of the thiolate group better than typical small-model QM studies, and this is generally found to lead to shorter Fe–S optimized bond lengths and lower unpaired electron spin density on sulfur.9,15 The Fe–S bond lengths and sulfur spin densities in the QM/MM calculations were found to be insensitive to increasing the size of the QM region, to include either the porphyrin substituents or the full coordinating cysteinate.(15) The electronic structure of Cpd I was found to vary slightly between the isoforms. The largest difference was observed between the human CYPs and the bacterial P450cam. However, two calculations on the same isoform, starting from different initial structures (generated from molecular dynamics (MD) trajectories) exhibited similar differences in Fe–S bond length and sulfur spin density to those found between two calculations on different isoforms. A small change was also observed when comparing an isoform in the presence and absence of a substrate molecule in its active site cavity. It was suggested from those QM/MM calculations that the differences in electronic structure and Fe–S bond length were due to thermal motions of the protein, substrate, and solvent. These motions cause the hydrogen-bonding environment and electric field surrounding Cpd I to vary, but the preliminary conclusion of this early study was that there was no net difference between isoforms, given that the thermally induced fluctuations were as large or larger than the changes obtained from one isoform to another.
The effect of the presence of substrate in the active site of CYPs on the electronic structure of Cpd I has been investigated by others. The drug-metabolizing human CYP isoform, CYP3A4, has been associated with multiple substrate binding and possible allosteric mechanisms.30−33 Fishelovitch et al. performed QM/MM calculations on the electronic structure of Cpd I of CYP3A4 using various models in which the enzyme was either substrate-free or contained one or two substrate molecules.(14) They found that in the absence of substrate, the Fe–S bond tended to elongate thus localizing the unpaired electron on the sulfur. In the model where the substrate diazepam is located close to Cpd I, the NH–S interactions with the cysteinate ligand were found to be strengthened, with a resultant shortening of the Fe–S bond and delocalization of the unpaired electron over the porphyrin ring and cysteinate sulfur. However, the conclusion that the absence of substrate leads to an increase in Fe–S bond length was based on geometry optimization of relatively few snapshots obtained from a 500 ps MD simulation. Due to the many accessible energy minima in proteins, extensive conformational sampling must be performed, in order to reliably calculate structural, electronic, and energetic properties.34,35 In the work presented here, many more snapshots have been optimized with QM/MM, from longer MD simulations, to increase the amount of conformational sampling.
We also introduce here an approximate method for estimating the Fe–O bond enthalpy in Cpd I, which can be linked directly to the stability and oxidizing power of Cpd I. Previous work by de Visser showed that the calculated barriers to hydrogen abstraction by both heme and nonheme iron(IV)-oxo oxidants (including CYP) correlate with the energy of the Fe(III)O–H bond, which is formed after the abstraction step.(36) The energy of the Fe(III)O–H bond was shown to be sensitive to the metal ligands that are both cis and trans to the −OH ligand. It would not be straightforward to calculate the O–H bond energy associated with adding a hydrogen atom to Cpd I in a QM/MM framework. As hydrogen atom addition leads to an increase in the Fe–O bond length and a decrease in the formal Fe–O bond order, Fe–O and O–H bond strengths for Cpd I in different environments should be anticorrelated, and we focus on calculating the former. Further correlations have been observed, both experimentally(37) and computationally,36,38,39 between the barrier to substrate hydroxylation and the C–H bond energy involved in the hydrogen abstraction step. Recently, Kumar et al. reported a correlation between the rate constant of substrate epoxidation and the ionization potential of the substrate for Cpd I in CYPs and analogous Fe(IV)=O species.(39) Correlations between calculations and experimentally observed properties (such as rate constants) can provide a powerful tool for aiding the understanding of the reactivity in these and other enzymes(40) and could, for example, aid the design of biomimetic catalysts.
The electronic structure and Fe–O bond enthalpy is calculated for several different CYP isoforms and compared here. We focus on four important and well-studied isoforms: the human 2C9, 2D6, and 3A4 isoforms are the major isoforms responsible for the metabolism of 75% of drugs.41,42 The bacterial P450cam isoform from Pseudomonas putida was the first CYP to be crystallized (and hence is the most studied) and is also studied here.
Computational Details
X-ray Crystal Structures
The human isoforms CYP2C9, CYP2D6, and CYP3A4 were modeled in this work, along with the bacterial P450cam isoform. Two crystal structures, both obtained by Williams et al.,(43) were used for the calculations on CYP2C9; the 1OG2 structure was used for simulating CYP2C9 in the absence of substrate, while the 1OG5 structure was used for all simulations containing the substrate S-warfarin. The only currently available crystal structure (PDB code 2F9Q)(44) solved by Rowland et al. was used for studying CYP2D6. Several crystal structures of CYP3A4 are available, of which we used the structure (PDB code 1TQN) obtained by Yano et al.(45) The 1DZ9 structure, solved by Schlichting et al., was used as the starting structure for all calculations on P450cam.(25) In each case, where mutations had been introduced to facilitate crystallization, these mutations were reversed in silico with the SwissPDB program,(46) prior to MD and QM/MM calculations, to restore the wild-type enzyme.
MD Simulations
Stochastic boundary MD (and MM energy minimizations) were carried out using the CHARMM program, version 30b2(47) and the CHARMM27 MM force field.(48) Further details concerning program-specific options and parameters for nonstandard residues are contained in the Supporting Information.
Four simulations were carried out on CYP2C9. The simulation originating from the 1OG2 crystal structure in the absence of substrate will be referred to as 2C9_apo. Three separate simulations were carried out that originated from the 1OG5 crystal structure. The first of these, referred to as 2C9_dist, was derived from the original crystal structure, containing S-warfarin (Figure 2) in the distal cavity. The second simulation, referred to as 2C9_2warf, used the 1OG5 structure with a second molecule of S-warfarin docked into the active site, directly above Cpd I, in a position favoring oxidation at the C7 position. Hydroxylation at this carbon atom is the predominant oxidation pathway in metabolism by this isoform.(49) This second substrate molecule was docked into the active site cavity using the AUTODOCK program.(50) The third simulation originating from the 1OG5 crystal structure is referred to as 2C9_prox and refers to the 2C9_2warf structure with the distal molecule of S-warfarin removed, leaving the proximal molecule remaining.
Calculations on two further human CYP isoforms, 3A4 and 2D6, were also carried out. These were both studied in substrate-free states (3A4_apo and 2D6_apo) and also in complexes with a single molecule of dextromethorphan (Figure 2), bound in a proximal position relative to Cpd I (3A4_dex and 2D6_dex).(51) Metabolism of dextromethorphan by CYP2D6 occurs by demethylation of the oxygen atom, whereas in CYP3A4 metabolism occurs by demethylation at the nitrogen atom.(52)
Two simulations were performed on P450cam, using the 1DZ9 crystal structure. The natural substrate camphor was removed in both simulations. In one simulation, propene was docked (by hand) into the active site cavity (P450cam_prop). The other simulation was run in the absence of a substrate molecule docked into the active site (P450cam_apo). We have studied propene oxidation by P450cam previously,35,53 using MM MD and QM/MM methods, and observed that propene displays extensive mobility in the active site due to it is small size and absence of hydrogen-bonding groups.
Each simulation had a production phase of 5 ns and snapshots were taken at 200 ps intervals. Each snapshot was first minimized in CHARMM(47) using the same (CHARMM27) MM force field as in the MD simulations, with 500 steps of the steepest descent algorithm followed by 1500 steps of adapted basis Newton–Raphson minimization, prior to QM/MM minimization.
The crystal structure of CYP2D6 that was used(27) does not contain any active site water molecules directly above the heme. In the 2D6_apo model, it was found that only one water molecule had been added in the region above the Cpd I oxygen during the solvation protocol. This water molecule formed a hydrogen bond with the Cpd I ferryl oxygen at the start of the heating phase but left the active site prior to the equilibration phase. For the equilibration phase and first 3 ns of the production phase dynamics, the active site region above the Cpd I oxygen did not contain any water. After the 3 ns point, several water molecules flowed into the active site from the entrance channel, and the active site remained solvated for the rest of the simulation. For this reason, only the last 2 ns of these simulations were considered for the data presented here.
QM/MM Calculations
The structure of each MM-minimized snapshot described above was energy minimized using QM/MM. The QM region included the porphyrin ring without substituents and the cysteinate ligand, modeled as a methyl mercaptide, –SCH3. In calculations on models containing substrate, part or all of one substrate molecule, as described below, was included in the QM region, with the exception of the 3A4_dex calculations, where the dextromethorphan was treated purely at the MM level. As discussed below, the treatment of the substrate (i.e., whether it was included in the QM or MM region) was found to have no effect on the computed electronic structure of Cpd I. For the P450cam_prop system, the entire propene molecule was included in the QM region. For calculations including S-warfarin, the S-warfarin molecule was split into QM and MM regions, as shown in Figure 3. Similarly, in the case of the 2D6_dex calculations, the dextromethorphan molecule was split into QM and MM regions, as shown in Figure 3. The valences of the QM atoms at the QM/MM boundary were satisfied by the addition of link atoms, which were modeled as hydrogen atoms.(54) In the 2C9_2warf calculations, the proximal molecule of S-warfarin was split into two parts, one in the QM region and one in the MM region, while the distal substrate molecule was in the MM region.
Figure 3.
Division of S-warfarin (left) and dextromethorphan (right) into QM (black) and MM (red) regions, as applied in QM/MM calculations here. The N-protonated form of dextromethorphan, used in the 2D6 simulations, is shown here.(51)
In the 2D6_dex model, the N-protonated form of dextromethorphan was used (as shown in Figure 3),(51) because almost all substrates of CYP2D6 include a basic nitrogen which is thought to interact with one of the acidic residues (Asp301 or Glu216) of the active site.(44) In contrast, CYP3A4 is known to metabolize a wide range of substrates, many with large hydrophobic groups, and it is likely that dextromethorphan binds in a nonionized form to its active site.(55) Therefore in the 3A4_dex simulations, the unprotonated form of dextromethorphan was used.
The QM part of the QM/MM calculations was performed in Jaguar 5.5(56) using the B3LYP density functional.57−60 The LACVP basis set (and associated core potential) was used for iron and the 6-31G* basis set for all other atoms.(61) This combination of basis sets has been used in previous calculations and shown to agree well with calculations using the larger basis set LACV3P for iron and 6-311G* for all other atoms.(15) Since the quartet and doublet spin states lie within 1.0 kcal mol–1 of each other, only the quartet state was modeled in these calculations, using a restricted open-shell approach. The MM region was optimized at every QM step with the Tinker program(62) using the CHARMM27 force field.(48) The fixed charges from the MM region were included in the QM Hamiltonian, allowing for the QM region to be polarized by the rest of the protein. No nonbonded cutoff was employed in any QM/MM calculation here. The QM and MM calculations were linked using the QoMMMa interface.51,63 The same link atom approach described above for S-warfarin and dextromethorphan was used to satisfy the valence of the atoms at the QM/MM boundary on the heme and the cysteine. The MM charges on the link atoms, along with neighboring atoms, were set to zero to avoid any unphysical effects.(35) The charges on these atoms were shifted onto adjacent groups where necessary in order to maintain the same overall charge for each system.15,35,63−65 The QM/MM methodology used here has been shown previously to model reliably the hydrogen-bonding interactions between the QM and MM subsystems.(66)
Estimation of the Fe–O Bond Enthalpy in Cpd I
The QM/MM optimized snapshots mentioned above were used to estimate the bond enthalpy of the iron atom and ferryl oxygen of Cpd I. The main purpose of this was to assess whether a variation in this bond enthalpy exists between different CYP isoforms. This would indicate possible differences in the oxidizing power of Cpd I between different isoforms. Accurate calculation of bond enthalpies in systems with large numbers of atoms is not straightforward, due to the need to sample many different structures of the bonded system and the fragments.34,67 The approach used here is approximate but importantly includes significant averaging.
The importance of the Fe–O bond energy for reactivity is illustrated in the thermodynamic cycle in Figure 4. ΔE1 corresponds to the energy of oxidation of the substrate by Cpd I to form the product and resting state of the heme. The value of ΔE1 is dependent on the substrate, because it involves conversion of the substrate into product as well as breakage of the Fe–O bond of Cpd I. ΔE1 must be negative (or positive but small in magnitude) for oxidation to occur, and oxidation can be expected to be more facile the more negative ΔE1 is. This quantity can be expressed as ΔE1 = ΔE2 + ΔE3, where ΔE2 is the energy required to break the Fe–O bond in Cpd I, and ΔE3 is the energy released upon addition of the oxygen atom to the substrate to yield product. ΔE2 is to a first approximation independent of the substrate, and smaller values of this bond energy correspond to a more reactive and oxidizing state of Cpd I.
Figure 4.

Thermodynamic cycle showing the importance of the Fe–O bond energy ΔE2 in the reactivity of Cpd I with the substrate RH.
To estimate ΔE2 in a given enzyme environment, we started from each Cpd I structure optimized at the QM/MM level and its energy EQM/MM(FeO). Then, the QM/MM energy of this structure was recomputed after simply deleting the ferryl oxygen atom, yielding an unrelaxed Fe(III) species, which was treated as a low-spin (doublet) state, with energy EQM/MM(Fe*). The energy EQM(O) of the (triplet state) oxygen atom was also calculated, in vacuo. Next, the relaxation energy was calculated using a small model comprising only the porphyrin ring, iron, and –SH thiolate ligand, i.e., assuming that this contribution should be effectively equal for each isoform and each environment. We stress here again that the aim was to develop a quick and undemanding method to approximately calculate the relative reactivity of Cpd I. The difference in energy EQM(Fe) – EQM(Fe*), computed for the doublet state, was found to be −24.5 kcal mol–1 at the B3LYP/LACVP,6-31G* level of theory. Note that the true resting state of P450 enzymes usually involves either coordination of a water molecule to the heme iron(III) or a pentacoordinate iron(III) in the high-spin (sextet) state. Indeed, B3LYP calculations find that the ground state of the pentacoordinate states is the sextet.68,69 However, it can be assumed that the change in energy from one spin state to another, and upon binding water, is relatively small and approximately constant from one isoform to another, so can be neglected for the present purposes of examining the relative value of ΔE2 for different isoforms and for different heme group environments. In summary, ΔE2 is obtained, for each structure, as ΔE2 = EQM(O) + EQM(Fe) – EQM(Fe*) + EQM/MM(Fe*) – EQM/MM(FeO).
To explore the effect of the way in which the substrate is treated in these calculations (i.e., using QM or MM) on the calculated Fe–O bond enthalpies, test calculations of the type described above were carried out in both ways for the 2D6_dex system. In one set of calculations, the entire dextromethorphan molecule was placed in the MM region, whereas in the other, it was placed partly in the QM region and partly in the MM region, as shown in Figure 3. The calculated Fe–O bond energies using these two approaches were found to be the same, within 0.2 kcal mol–1, for each structure considered. Hence it is assumed that the treatment of the substrate does not have an effect on the calculated electronic structure of Cpd I.
Results and Discussion
Electronic Structure of Cpd I in Substrate-Free Systems
The electronic structure of Cpd I was compared between four different isoforms in the absence of substrate in the active site. These systems are the 2C9_apo, 2D6_apo, 3A4_apo, and P450cam_apo systems described above. Snapshots were taken from each MD simulation every 200 ps and subsequently optimized, first with MM and then with the QM/MM method, as described above. This yielded 26 QM/MM optimized geometries for each simulation (with the exception of the 2D6_apo simulation in which only 10 optimized geometries were calculated). The electronic structure of each optimized structure was inspected to ensure that the correct wave function for Cpd I had been found.(15)
Comparisons of the electronic structure of Cpd I between systems were made by observation of several quantities in the QM/MM optimized structures. These include the Fe–O and Fe–S bond lengths and Mulliken charges and spin densities of atoms in the Cpd I moiety. These are presented in detail in the Supporting Information. Large deviations in these quantities would reflect large differences in electronic structure between snapshots, including the amount of delocalization of the third unpaired electron between the sulfur p orbital and the porphine a2u orbital. Table 2 shows the average Fe–S bond lengths and Mulliken spin densities for the sulfur, oxygen, iron, and porphyrin, calculated for each set of snapshots in the substrate-free systems as well as the associated standard deviations. No indication of any overall change in the calculated quantities with respect to the time point in the MD run at which the initial snapshot was selected was noticed for any of the above observables (more detail is given in the Supporting Information). We note that if the MD simulation generates an equilibrium ensemble of structures, then no such time dependence would be expected, so the lack of a trend provides an indication that the MD simulations are well equilibrated for the present purposes. The Fe–S bond lengths (and Mulliken spin densities) are close to those observed in previous work.15,35 The average value of the Fe–S bond length falls within the range of the largest standard deviation obtained (0.08 Å) for all four isoforms, indicating that there is no appreciable difference in structure between these isoforms. The Fe–O bond length also shows no significant variation between different P450 systems; the average value of 1.62 Å is observed in all systems, with a standard deviation of 0. The average sulfur spin densities show a small degree of variation (less than 0.2), with the value found to increase from one isoform to another in proportion to the size of the active site cavity (P450cam < 2D6 < 2C9 < 3A4). However, this trend is probably not significant, because it is of the order of the standard deviations. An increase in sulfur spin density is accompanied by a decrease in porphyrin spin density, with the sum of the two quantities remaining close to 1. All average iron and oxygen spin densities lie within the standard deviations and hence appear not to vary between isoforms.
Table 2. Average Fe–S bond length (Å) and Mulliken Spin Densities (ρ) of Sulfur and Oxygen (with standard deviations, σ) for the QM/MM Optimized Snapshots of Cpd I.
| Fe–S | σ | Fe–O | σ | ρ(Fe) | σ | ρ(S) | σ | ρ(O) | σ | ρ(porph) | σ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2C9_apo | 2.67 | 0.08 | 1.62 | 0.0 | 1.17 | 0.03 | 0.43 | 0.07 | 0.77 | 0.03 | 0.63 | 0.06 |
| 2D6_apo | 2.69 | 0.05 | 1.62 | 0.0 | 1.15 | 0.03 | 0.41 | 0.04 | 0.76 | 0.02 | 0.62 | 0.03 |
| 3A4_apo | 2.60 | 0.04 | 1.62 | 0.0 | 1.16 | 0.02 | 0.47 | 0.05 | 0.78 | 0.03 | 0.56 | 0.05 |
| P450cam_apo | 2.63 | 0.06 | 1.62 | 0.0 | 1.15 | 0.04 | 0.34 | 0.05 | 0.80 | 0.04 | 0.70 | 0.05 |
| 2C9_prox | 2.61 | 0.03 | 1.62 | 0.0 | 1.18 | 0.03 | 0.46 | 0.06 | 0.76 | 0.03 | 0.56 | 0.06 |
| 2C9_2warf | 2.62 | 0.06 | 1.62 | 0.0 | 1.14 | 0.02 | 0.40 | 0.04 | 0.81 | 0.02 | 0.62 | 0.05 |
| 2C9_dist | 2.60 | 0.04 | 1.62 | 0.0 | 1.16 | 0.02 | 0.45 | 0.06 | 0.78 | 0.03 | 0.57 | 0.05 |
| 2D6_dex | 2.59 | 0.03 | 1.62 | 0.0 | 1.14 | 0.02 | 0.37 | 0.04 | 0.81 | 0.02 | 0.66 | 0.04 |
| 3A4_dex | 2.58 | 0.03 | 1.62 | 0.0 | 1.15 | 0.03 | 0.44 | 0.06 | 0.79 | 0.03 | 0.59 | 0.06 |
| P450cam_prop | 2.60 | 0.02 | 1.62 | 0.0 | 1.14 | 0.02 | 0.27 | 0.05 | 0.81 | 0.02 | 0.76 | 0.06 |
Electronic Structure of Cpd I in the Substrate-Bound Systems
In previous work,(14) it was observed that the absence of substrate in the active site of CYP3A4 leads to elongation of the Fe–S bond and localization of the radical on sulfur. In this work, the Fe–S bond lengths and Mulliken spin densities on the ferryl oxygen and cysteinate sulfur atoms were calculated for the substrate-bound systems, 2C9_dist, 2C9_prox, 2C9_2warf, 2d6_dex, 3A4_dex, and P450cam_prop. The average values and standard deviations for these quantities are presented in Table 2.
For all of the isoforms studied, there is a slight reduction in the average Fe–S bond length, compared with the corresponding substrate-free systems. However, this reduction is small compared to the standard deviation, σ. Similarly, the average spin density on sulfur is slightly reduced in the substrate-bound forms, although not in the case of 2C9. This is in agreement with previous work,(14) but it is difficult to draw any conclusions based on this observation alone. As with the substrate-free systems, there is no time dependence observed in any bond length or spin density (Supporting Information). This is in contrast to previous work,(14) where a time dependence was noted, but based on calculations performed on significantly fewer structures. This again stresses the need to sample many conformations of the protein in order to draw reliable conclusions from QM/MM calculations. The variation in the QM/MM optimized structures of Cpd I, and the residues surrounding the cysteinate sulfur, for the 2C9_dist simulation are illustrated in Figure 5. Figure 5 shows a comparable (if not larger) variation in protein structure surrounding the cysteinate ligand between structures of the same isoform, than between different isoforms (Figure 1). The root-mean-square deviation (rmsd) of the positions of the Cpd I, Cys435, Leu358, Gly359, and Gln360 atoms compared to the t = 0 ps snapshot are shown in Figure 6. From Figure 6 it can be noted that none of the snapshots vary by more than 0.7 Å compared to the initial QM/MM optimized structures and that there is no time dependence in the variation in these coordinates. Figures 5 and 6 provide evidence to suggest that the MD simulations are well-equilibrated and that an adequate ensemble of geometries has been studied.
Figure 5.
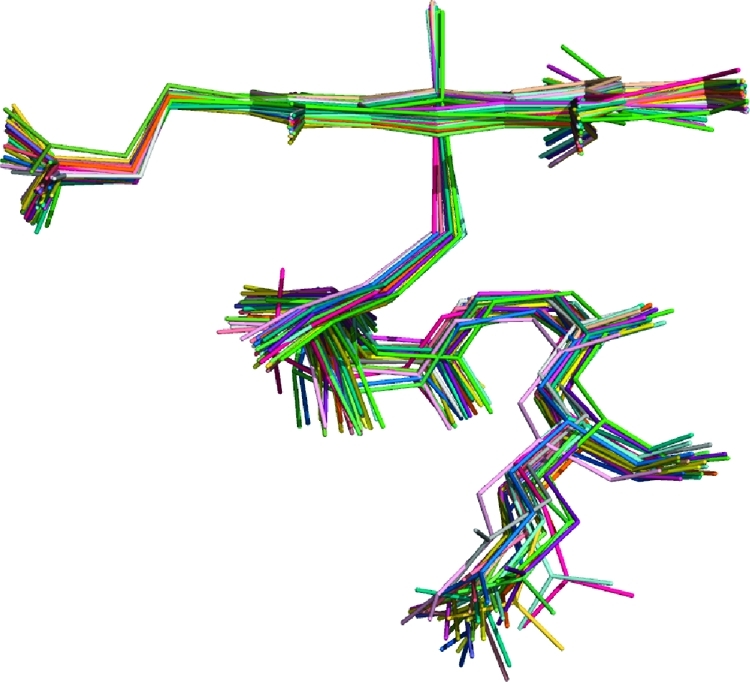
Overlay of QM/MM minimized structures from the 2C9_dist simulation. For clarity, only the Cpd I, Cys435, Leu358, Gly359, and Gln360 residues are shown.
Figure 6.

Root mean square deviation (rmsd) of the coordinates of the Cpd I, Cys435, Leu358, Gly359 and Gln360 residues, in the optimized QM/MM structures from the 2C9_dist simulation (relative to the t = 0 ps optimized geometry).
The calculations predict a difference between the electronic structure of Cpd I in the bacterial P450cam and human P450 isoforms, as indicated by the smaller spin density on sulfur in the bacterial isoform, both in substrate-bound and -free forms. Furthermore, this difference cannot be explained by the fluctuation observed in other properties, as reflected by the standard deviations. The spin density on sulfur is linked directly to the spin density on the porphyrin ring (see, e.g., the frontier orbital a2u in Figure 1) and could influence the reactivity of Cpd I.3,9,10 This difference between the bacterial and human isoforms is in agreement with our previous findings(15) and has been explained by the slightly different hydrogen-bonding environment surrounding the sulfur atom. As shown in Table 1, in all of the isoforms studied, the residue Resn+2 (where Resn corresponds to the cysteinate that is coordinated to the Fe) is a conserved glycine, which is common to all P450 isoforms.(70) The amide proton of this glycine forms a hydrogen bond with the cysteinate sulfur in all of the isoforms studied (see Figure 7). In the P450cam calculations, this hydrogen bond is found to be stronger, and shorter (by around 0.1 Å) than in the human isoforms. Hydrogen bonding is believed to alter the energy of the sulfur p orbital, relative to the porphyrin a2u, leading to a reduction in the amount of mixing between the two orbitals.(20)
Figure 7.
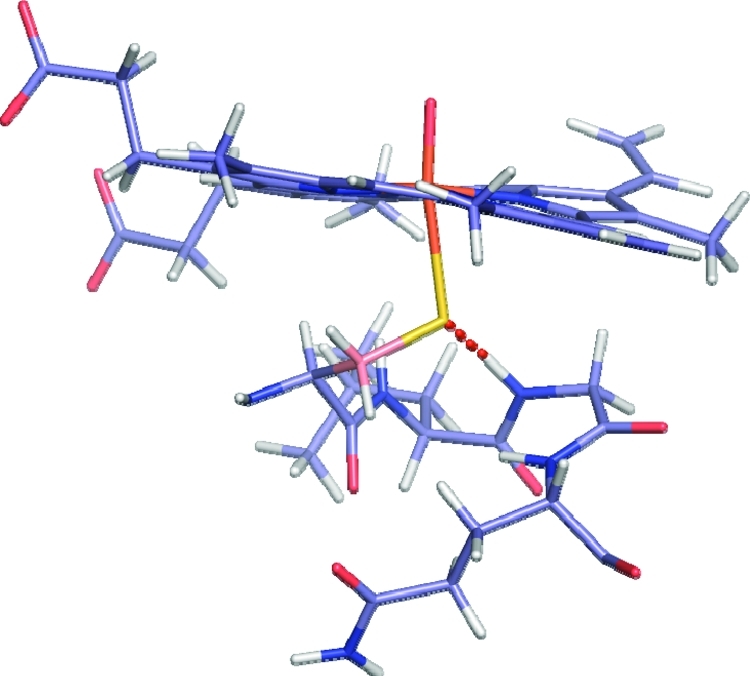
QM/MM optimized structural snapshot of Cpd I and residues surrounding the cysteinyl sulfur taken from the P450cam_prop simulation. The hydrogen bond between the amide proton of Gly359 and the sulfur of Cys357 is shown as a red dashed line. This hydrogen-bonding interaction is present in all of the isoforms studied in this work, however, the interaction is strongest in the P450cam isoform.
An interesting observation noted for the 2C9_prox and P450cam_prop MD simulations is that the conformation of the protein backbone for the conserved glycine located two amino acids along from the coordinating cysteine (Resn+2) flipped during the simulation. This resulted in the loss of the hydrogen-bonding interaction between the amide proton of the glycine and the cysteinate sulfur atom (shown for P450cam in Figure 7). For many snapshots, the absence of this hydrogen bond corresponds to an increase in spin density on the sulfur and to elongation (and, in some cases, breakage) of the Fe–S bond during the QM/MM optimization. While the change in conformation of the glycine is believed to be an artifact of the simulations, it highlights the importance of the hydrogen-bonding interaction between the amide proton of the glycine and the cysteine in the stabilization of Cpd I.
The spin density on the ferryl oxygen is linked directly to the spin density on iron. The observed standard deviation of these quantities (0.02–0.04) is smaller than that of the sulfur spin density (0.04–0.07), indicating a smaller sensitivity to the fluctuations of the surroundings in the former. It is possible that the presence of substrate may have a small effect on the ferryl oxygen spin density, as shown by the data for the 2D6, 3A4, and P450cam isoforms in Table 2. The amount of spin density is larger by approximately 0.01 in the models where substrate is present, compared to the apo enzymes. This difference is smaller than the standard deviation, so some caution is needed before interpreting it as meaningful. As discussed below, though, this quantity is found to correlate with the calculated bond energies, so it may be a genuine difference between the ligand-bound and -free forms.
Stability of Cpd I
We now consider the approximate Fe–O bond energies ΔE2. Table 3 displays the average values of ΔE2 calculated from QM/MM minimized structures taken from the 2C9_apo, 2C9_dist, 2C9_2warf, 2C9_prox, P450cam_prop, 2D6_apo, 2D6_dex, 3A4_apo, and 3A4_dex simulations (detailed information is in the Supporting Information). The majority of the Fe–O bond energies fall within the range 49–62 kcal mol–1. The energy ΔE3 for adding an oxygen atom into a C–H bond of methane or other simple substrates, such as propene, is of the order of −90 kcal mol–1.(72) Combined with the approximate value of ΔE2 of 50 kcal mol–1 in the presence of substrate, this suggests that the overall reaction energy for C–H bond oxidation by Cpd I should be of the order of −40 kcal mol–1, a value which agrees very well with typical calculated reaction energies.(20) This suggests that despite its approximate nature, the definition of the bond energy ΔE2 used here is physically reasonable.
Table 3. Average Fe–O Bond Enthalpies, ΔE2 [kcal mol–1], and Standard Deviation, σ, Calculated for QM/MM Optimized Snapshots.
| ΔE2 | σ | |
|---|---|---|
| 2C9_apo | 58.5 | 3.9 |
| 2C9_prox | 56.1 | 2.3 |
| 2C9_2warf | 49.9 | 2.3 |
| 2C9_dist | 55.1 | 3.8 |
| 2D6_apo | 61.5 | 3.5 |
| 2D6_dex | 49.3 | 2.5 |
| 3A4_apo | 55.0 | 3.3 |
| 3A4_dex | 54.9 | 2.7 |
| P450cam_apo | 54.9 | 4.1 |
| P450cam_prop | 52.1 | 1.6 |
There is considerable fluctuation in the values obtained over the course of all the simulations. The most extreme example of this case is the 2C9_prox case, where the Fe–O bond enthalpy varies between 46.2 and 76.8 kcal mol–1. The largest values are those where the Fe–S bond is broken (i.e., >3.0 Å), as discussed above and therefore are not included in the calculation of average values.
The values of the average ΔE2 for different systems all fall within the range of values obtained for any particular simulation, suggesting that there is no major difference in bond energy from one isoform to another, which is expected given the fairly constant chemical environment in each case. However, one possibly significant trend is that in the presence of a substrate molecule in the active site, the average value of ΔE2 is slightly lower than in its absence. For example, of the CYP2C9 models, the average Fe–O bond enthalpy follows the order 2C9_2warf < 2C9_dist ≈ 2C9_prox < 2C9_apo. This trend is mirrored in the P450cam and CYP2D6 simulations, where the presence of substrate significantly lowers the average Fe–O bond enthalpy.
The weakening effect of the substrate molecule on the Fe–O bond can be described in terms of several mostly complementary effects: (a) One is that the presence of the substrate could directly affect the polarization of the Fe–O bond, thus raising the energy of Cpd I relative to the ground state, so that less energy would be required for its breakage. The energy of formation of Cpd I from Cpd 0 has been calculated by Zheng et al.(73) to be around −4 kcal mol–1, hence the destabilization effect cannot be very large otherwise Cpd I formation would be unfavorable. A similar estimate of the energy of formation of Cpd I from Cpd 0 is obtained from an analysis of experimental data based on a thermodynamic cycle.(74) (b) Another possible explanation is that the presence of substrate stabilizes the resting state of the enzyme, lowering the energy required to break the Fe–O bond. (c) Linked to these two explanations, it is possible that the substrate has an indirect effect on the stability of either the resting state or Cpd I, by changing the preferred conformations of the surrounding protein and the water in the active site. This could affect the hydrogen-bonding interactions between the protein and Cpd I, which have been shown to have an effect on its electronic structure,3,4,10,20 as discussed in the Introduction. The possible effect (a) of substrate polarization of the Fe–O bond is explored in the next section. The most convincing explanation of the effect, in our view, is of type (c) and can be best expressed as saying that it is not the presence of the substrate that has a large effect but rather its absence. When no substrate molecule is bound, more water is able to enter the active site cavity. As a result, there are more hydrogen-bonding interactions between the Cpd I ferryl oxygen and the surrounding water molecules in the absence of substrate.
The average number of hydrogen bonds to the Cpd I ferryl oxygen, NHB(FeO), throughout all of the MD simulations was calculated in CHARMM, and these data are shown in Table 3. A hydrogen bond is defined as present between the atoms A–H–D if the distance between the donor (D) and acceptor (A) is less than 2.8 Å and if the A–H–D angle is greater than 90°.(75) N–H and O–H groups only were defined as potential hydrogen-bond donors. As expected, the presence of substrate in the active site displaces water and hence results in fewer hydrogen bonds, on average, to the ferryl oxygen. This effect is most apparent in the 2C9 systems: in the 2C9_apo and 2C9_dist simulations in which there is no substrate molecule close to Cpd I, NHB(FeO) is 1.05 and 1.06, respectively. In the 2C9_prox and 2C9_2warf simulations, where a substrate molecule is present directly above Cpd I, NHB(FeO) is significantly lower (0.50 and 0.14 respectively). Hydrogen bonding to neighboring water molecules will lower the amount of unpaired spin density on the Cpd I oxygen and hence stabilize Cpd I. In this interpretation, Cpd I is predicted to be slightly more oxidizing in the presence of substrate than in its absence, and this might help to account for its experimental elusiveness. The presence of substrate has a smaller effect on the hydrogen bonding between water and the Cpd I oxygen in the P450cam simulations. This is probably because propene is smaller than the other substrates modeled, and therefore, its presence does not prevent water from accessing Cpd I. We do not observe a hydrogen bond between the hydroxyl group of Thr309 and the Cpd I oxygen at any point in the CYP3A4 simulations, contrary to findings that were reported previously, based on MM MD simulations.(14)
In the catalytic cycle of CYPs, it is widely accepted that the substrate is required to be present in the active site in order to start the chain of steps leading to formation of Cpd I. Although it might be expected that Cpd I will not form in the absence of substrate, Cpd I in CYP119 has been recently isolated and analyzed spectroscopically in the absence of substrate.(16) It has been proposed that the presence of substrate in the active site of CYPs increases the stability of Cpd I,(14) which may at first seem to contradict our findings. However, this claim is made based on the presence of a substrate preventing Cpd I from performing hydrogen abstraction from active site side chains, such as Ala305 and Thr309 in CYP3A4.(14) Here we consider how the intrinsic stability of Cpd I may differ between the substrate-free and -bound systems.
It is to be noted that the protocol used here to estimate the Fe–O bond energy ΔE2 is only approximate. In particular, structural relaxation that occurs after breaking the Fe–O bond is treated here only for the heme group itself. The rest of the system would also undergo various structural changes, and this relaxation effect is not treated here. Given the larger number of hydrogen bonds to the ferryl oxygen in the systems without substrate in the active site, the water present in the active site would presumably undergo larger amplitude relaxation in the substrate-free cases. This would correspond to a decrease in the Fe–O bond energy, thereby bringing the substrate-bound and -free values closer together. Nevertheless, the results obtained here do suggest that the ferryl bond in P450 Cpd I is intrinsically somewhat more reactive when it is in a more hydrophobic environment, as when substrate is present. It has been suggested previously that hydrogen bonding to Cpd I by a water molecule can lower the barrier to hydrogen abstraction in some cases.76,77 This effect was rationalized by suggesting that the hydrogen bond increased in strength from the reactant complex to the transition state, as a greater negative charge is observed on the oxo atom in the transition state than the reactant complex. In this study, we do not consider transition states, so we cannot comment directly on whether this effect would be generally observed. We note, however, that the effect is quite small when using large basis sets,(77) and our calculations suggest that there is very little water present in the reactant complexes in the presence of bulky substrates. Two comments are needed here: First, previous studies36,37 have suggested that the key factor affecting reactivity in C–H bond abstraction by a metal oxo species such, as Cpd I, is the strength of the resulting O–H bond. We have argued above that for Cpd I in a range of different environments, a stronger Fe–O bond is likely to be correlated with a weaker O–H bond, thus the Fe–O bond energy provides some information on likely reactivity. As this anticorrelation will not be perfect, this will only provide a rough guideline. Second, our calculations include some cases where Cpd I has no close-lying water molecule. At first sight, this is inconsistent with the fact that Cpd I formation from the Cpd 0 precursor leads to formation of one molecule of water. However, between formation and reaction of Cpd I, the water molecule can diffuse away if it is favorable in free energy terms to do so. Various previous studies have shown that water diffusion is fairly rapid on the millisecond time scale for Cpd I turnover.(78) Our discussion here assumes that the ensemble of structures generated by the MD simulations is a realistic representation of Cpd I and that where the MD simulation suggests that no water is present, this represents the preferred environment.
Influence of the Ferryl Oxygen Spin Density on the Fe–O Bond Energy
The Fe–O bond energy is influenced by the spin polarization of the Fe–O bond, which is affected by the presence or absence of substrate. As mentioned above, the absence of a substrate molecule in the active site generally results in an increase in water molecules surrounding the ferryl oxygen. The increase in hydrogen-bonding interactions between the Cpd I ferryl oxygen and surrounding water molecules leads to a decrease in the spin polarization of the Fe–O bond, for the reasons discussed below. Therefore, in the substrate-free calculations, the ferryl oxygen spin density will be, in general, slightly lower than in the calculations where substrate is present. In Figure 8, the Fe–O bond enthalpies are plotted against the spin densities on the ferryl oxygen atom for the QM/MM optimized geometries of Cpd I. Figure 8 shows a direct correlation between the ferryl oxygen spin density and the Fe–O bond enthalpy calculated for a particular structural snapshot. The QM/MM optimized structures with the highest spin densities on oxygen (i.e., with the largest amount of Fe–O spin polarization) tend to have the lowest Fe–O bond enthalpies. The simulations where the substrate is in a position in which it can react with Cpd I, for example 2C9_2warf and P450cam_prop, have larger spin densities on the ferryl oxygen and also lower Fe–O bond enthalpies than those with a distal substrate or no substrate, e.g., 2C9_dist and 2C9_apo, for the reasons mentioned above (see Table 4).
Figure 8.

Fe–O bond enthalpy, E(Fe–O) [kcal mol–1] plotted versus Cpd I ferryl oxygen spin density (ρ(O)) for multiple structures for different CYP isoforms (indicated by different symbols and colors, as shown in the legend).
Table 4. Average Number of Hydrogen Bonds (NHB(FeO)) Formed between the Ferryl Oxygen and Active Site Water Molecules during Each of the 5 ns MD Simulations.
| NHB(FeO) | |
|---|---|
| 2C9_apo | 1.05 |
| 2C9_prox | 0.50 |
| 2C9_2warf | 0.14 |
| 2C9_dist | 1.06 |
| 2D6_apo | 0.92 |
| 2D6_dex | 0 |
| 3A4_apo | 1.04 |
| 3A4_dex | 1.15 |
| P450cam_apo | 1.16 |
| P450cam_prop | 0.97 |
The formation of Cpd I is usually preceded in the catalytic cycle by entrance of the substrate into the active site cavity, as the substrate is believed to displace the water molecule that is bound to the Fe(III) resting state. The displacement of this water molecule results in a spin-state change for the Fe(III), which facilitates the first electron transfer which precedes the subsequent steps to Cpd I formation. It therefore seems reasonable to imagine that the reactivity of Cpd I might somehow be affected by the presence of substrate. It would appear from Figure 4 that the difference in reactivity of Cpd I between the substrate-bound and -free forms is, at least in part, due to the substrate increasing the amount of unpaired electron density on the ferryl oxygen. This increase in spin density is most likely due to displacement of the active site water molecules by the substrate, resulting in fewer hydrogen-bonding interactions with the Cpd I oxygen. Iron–oxygen bonding involves two two-center three-electron π bonds, with the unpaired electrons residing in π* orbitals that are shared between the oxygen px (or py) and metal dxz (or dyz) orbitals. Hydrogen bonds to oxygen lower the energy of its p orbitals, thereby increasing the O character of the bonding π orbitals, decreasing the O character of the π* orbitals, and lowering the spin density on O. By displacing water from the active site, the substrate reverses this effect, so that there is more spin density on oxygen. This also has the effect of slightly increasing the Fe–O bond length, and presumably, decreasing its bond energy. Mn(V)=O complexes have been found to be more reactive when more spin is located on the oxo atom.79,80 Hence the oxidizing capability of Cpd I is enhanced, compared to the apo enzyme, in the presence of substrate. This effect may vary depending on the size of the substrate and the active site cavity, as these both influence the number of water molecules able to enter the active site. This is evident from Table 4 where in the case of model systems in which the active site and the substrate have similar sizes (e.g., 2D6_apo and 2C9_prox), fewer hydrogen-bonding interactions are observed between water and the Cpd I ferryl oxygen. Note also that we do not claim this effect to be catalytic; clearly it is not sensible to discuss the possibility of catalysis in the absence of substrate. Analyzing catalysis would require comparison of the reaction in different environments. Our results do indicate, however, that Cpd I has somewhat different properties depending on whether substrate is present or not, due to its interaction with water molecules (or lack thereof). This may have important implications for understanding its stability, formation, and/or reactivity in the absence of substrate.
Conclusions
The electronic structure of Cpd I displays little variation between the three human isoforms studied: 2C9, 3A4, and 2D6. There is more significant variation for any single isoform (i.e., between different conformations generated by MD) than between isoforms. This indicates that Cpd I is sensitive to changes in the polarization and hydrogen-bonding environment (this is what Shaik et al. have referred to as the “chameleon” effect).3,10 The bacterial CYP isoform P450cam shows a significantly sulfur spin density compared to the human isoforms. On average, the hydrogen-bonding environment is similar in all of the human isoforms studied here.
The presence of substrate in the active sites of the human and bacterial isoforms has an effect on the electronic structure of Cpd I. The presence of substrate results in a slight shortening of the Fe–S bond and a decrease in the amount of spin density on the cysteinate sulfur. This is likely to be due to the displacement of water molecules from the active site by the substrate.
An approximate method for estimating the Fe–O bond enthalpy of Cpd I is presented here and used to investigate the variation of the oxidizing ability of different CYP isoforms. Differences in the Fe–O bond enthalpy between isoforms are within the variation observed between different structures of the same isoform. This suggests that there is not a significant difference in oxidizing power between different isoforms. The presence of substrate in the active site cavity slightly lowers the energy required to break the Fe–O bond. This suggests that the substrate has a small effect on the electronic structure of Cpd I. This effect is due to the substrate restricting the number of water molecules that are able to hydrogen bond to the ferryl Cpd I oxygen. When substrate is bound, fewer hydrogen bonds are donated by water molecules to the ferryl oxygen. Without these solvent hydrogen bonds, the spin density on the oxygen increases, while the charge associated with it decreases. The Fe–O bond energy shows an approximately linear correlation with the spin density on the oxygen atom. QM/MM optimized structures with the largest amount of spin density on the ferryl oxygen of Cpd I tend to have the lowest Fe–O bond enthalpies.
From this work it appears that Cpd I has a very similar electronic structure in different isoforms, especially between the human isoforms studied here. It hence seems reasonable to assume Cpd I will show similar reactivity across different CYP isoforms.
The QM/MM calculations also show that the electronic structure of Cpd I varies due to thermal fluctuations, which lead to motion of protein residues, substrate, and water molecules, and hence to changes in the hydrogen-bonding environment surrounding the heme group and especially the cysteinate sulfur. The effect of the fluctuation of the surrounding environment may mean that conclusions about electronic structure based on single QM/MM optimized structures are not robust, and properties are better derived from averaging over a number of different conformers, as has also been found for calculating energy barriers for oxidation of alkenes in P450s.(35)
Acknowledgments
A.J.M. is an EPSRC Leadership Fellow (grant number EP/G007705/1) and (together with R.L. and J.N.H.) thanks the EPSRC for support. J.O. acknowledges receipt of an EU Marie Curie Fellowship (Project “Modelling CYPs”).
Supporting Information Available
Absolute energies of all QM/MM structures, Mulliken charges and spin densities, Fe–S and Fe–O bond lengths, hydrogen-bond distances to the cysteinate sulfur, and further details of setup for MD simulations (including (CHARMM27) MM parameters). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Materials Structure and Modeling Research Group of the Hungarian Academy of Sciences, Budapest University of Technology and Economics, P.O. Box 91, 1521 Budapest, Hungary
Supplementary Material
References
- Guengerich F. P. In The Ubiquitous Roles of Cytochrome P450 Proteins; John Wiley & Sons, Ltd: Chichester, U.K., 2007; Vol. 3, p 561–589. [Google Scholar]
- Guengerich F. P. Chem. Res. Toxicol. 2001, 14, 611–650. [DOI] [PubMed] [Google Scholar]
- Ogliaro F.; Cohen S.; de Visser S. P.; Shaik S. J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar]
- de Visser S. P.; Ogliaro F.; Sharma P. K.; Shaik S. Angew. Chem., Int. Ed. 2002, 41, 1947–+. [PubMed] [Google Scholar]
- Mulholland A. J. Drug Discovery Today 2005, 10, 1393–1402. [DOI] [PubMed] [Google Scholar]
- O’Reilly E.; Köhler V.; Flitsch S. L.; Turner N. J. Chem. Commun. 2011, 47, 2490–2490. [DOI] [PubMed] [Google Scholar]
- Loew G. H.; Harris D. L. Chem. Rev. 2000, 100, 407–420. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Sigfridsson E.; Ryde U. J. Biol. Inorg. Chem. 2004, 9, 203–223. [DOI] [PubMed] [Google Scholar]
- Schöneboom J. C.; Lin H.; Reuter N.; Thiel W.; Cohen S.; Ogliaro F.; Shaik S. J. Am. Chem. Soc. 2002, 124, 8142–8151. [DOI] [PubMed] [Google Scholar]
- Ogliaro F.; de Visser S. P.; Cohen S.; Kaneti J.; Shaik S. ChemBioChem 2001, 2, 848–851. [DOI] [PubMed] [Google Scholar]
- Ogliaro F.; Cohen S.; Filatov M.; Harris N.; Shaik S. Angew. Chem., Int. Ed. 2000, 39, 3851–3855. [DOI] [PubMed] [Google Scholar]
- Bathelt C. M.; Mulholland A. J.; Harvey J. N. Dalton Trans. 2005, 3470–3476. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Ogliaro F.; Sharma P. K.; Shaik S. J. Am. Chem. Soc. 2002, 124, 11809–11826. [DOI] [PubMed] [Google Scholar]
- Fishelovitch D.; Hazan C.; Hirao H.; Wolfson H. J.; Nussinov R.; Shaik S. J. Phys. Chem. B 2007, 111, 13822–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathelt C. M.; Zurek J.; Mulholland A. J.; Harvey J. N. J. Am. Chem. Soc. 2005, 127, 12900–12908. [DOI] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- Edwards S. L.; Nguyen Huu X.; Hamlin R. C.; Kraut J. Biochemistry 1987, 26, 1503–1511. [DOI] [PubMed] [Google Scholar]
- Dolphin D.; Forman A.; Borg D. C.; Fajer J.; Felton R. H. Proc. Natl. Acad. Sci. U.S.A. 1971, 68, 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciani G.; Carosati E.; De Boeck B.; Ethirajulu K.; Mackie C.; Howe T.; Vianello R. J. Med. Chem. 2005, 48, 6970–6979. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Kumar D.; de Visser S. P.; Altun A.; Thiel W. Chem. Rev. 2005, 105, 2279–2328. [DOI] [PubMed] [Google Scholar]
- Antony J.; Grodzicki M.; Trautwein A. X. J. Phys. Chem. A 1997, 101, 2692–2701. [Google Scholar]
- Green M. T. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar]
- Ohta T.; Matsuura K.; Yoshizawa K.; Morishima I. J. Inorg. Biochem. 2000, 82, 141–152. [DOI] [PubMed] [Google Scholar]
- Rutter R.; Hager L. P.; Dhonau H.; Hendrich M.; Valentine M.; Debrunner P. Biochemistry 1984, 23, 6809–6816. [DOI] [PubMed] [Google Scholar]
- Schlichting I.; Berendzen J.; Chu K.; Stock A. M.; Maves S. A.; Benson D. E.; Sweet R. M.; Ringe D.; Petsko G. A.; Sligar S. G. Science 2000, 287, 1615–1622. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Ward A.; Angove H. C.; Vinkovic D. M.; Jhoti H. Science 2004, 305, 683–686. [DOI] [PubMed] [Google Scholar]
- Rowland P.; Blaney F. E.; Smyth M. G.; Jones J. J.; Leydon V. R.; Oxbrow A. K.; Lewis C. J.; Tennant M. G.; Modi S.; Eggleston D. S.; Chenery R. J.; Bridges A. M. J. Biol. Chem. 2006, 281, 7614–7622. [DOI] [PubMed] [Google Scholar]
- Yano J. K.; Wester M. R.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. J. Biol. Chem. 2004, 279, 38091–38094. [DOI] [PubMed] [Google Scholar]
- Harvey J. N.; Bathelt C. M.; Mulholland A. J. J. Comput. Chem. 2006, 27, 1352–1362. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Vinkovic D. M.; Ward A.; Angove H. C.; Day P. J.; Vonrhein C.; Tickle I. J.; Jhoti H. Science 2004, 305, 683–686. [DOI] [PubMed] [Google Scholar]
- Fishelovitch D.; Hazan C.; Shaik S.; Wolfson H. J.; Nussinov R. J. Am. Chem. Soc. 2007, 129, 1602–1611. [DOI] [PubMed] [Google Scholar]
- Lampe J. N.; Atkins W. M. Biochemistry 2006, 45, 12204–12215. [DOI] [PubMed] [Google Scholar]
- Atkins W. M. Drug Discovery Today 2004, 9, 478. [DOI] [PubMed] [Google Scholar]
- Sharma P. K.; Chu Z. T.; Olsson M. H. M.; Warshel A. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 9661–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale R.; Harvey J. N.; Mulholland A. J. J. Phys. Chem. B 2010, 114, 1156–1162. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. J. Am. Chem. Soc. 2010, 132, 1087–1097. [DOI] [PubMed] [Google Scholar]
- Mayer J. M. Acc. Chem. Res. 1998, 31, 441–450. [Google Scholar]
- Shaik S.; Kumar; de Visser S. P. J. Am. Chem. Soc. 2008, 130, 14016–14016. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Karamzadeh B.; Sastry G. N.; de Visser S. P. J. Am. Chem. Soc. 2010, 132, 7656–7667. [DOI] [PubMed] [Google Scholar]
- Ridder L.; Mulholland A. J.; Rietjens I.; Vervoort J. J. Am. Chem. Soc. 2000, 122, 8728–8738. [Google Scholar]
- Evans W. E.; Relling M. V. Science 1999, 286, 487–491. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. In Cytochrome P450: Structure, Mechanism and Biochemistry; 3rd ed.; Ortiz de Montellano P. R., Ed.; Kluwer Academic/Plenum Publishers: New York, 2005, p 377. [Google Scholar]
- Williams P. A.; Cosme J.; Ward A.; Angova H. C.; Vinkovic D. M.; Jhoti H. Nature 2003, 424, 464–468. [DOI] [PubMed] [Google Scholar]
- Rowland P.; Blaney F. E.; Smyth M. G.; Jones J. J.; Leydon V. R.; Oxbrow A. K.; Lewis C. J.; Tennant M. G.; Modi S.; Eggleston D. S.; Chenery R. J.; Bridges A. M. J. Biol. Chem. 2006, 281, 7614–7622. [DOI] [PubMed] [Google Scholar]
- Yano J. K.; Wester M. R.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. J. Biol. Chem. 2004, 279, 38091–38094. [DOI] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C. Electrophoresis 1997, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar]
- MacKerell A. D. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Kaminsky L.; Zhang Z. Pharmacol. Ther. 1997, 73, 67–74. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- Oláh J.; Mulholland A. J.; Harvey J. N. Proc. Natl. Acad. Sci. U.S.A. 2011, 105, 6050–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Moltke L. L.; Greenblatt D. J.; Grassi J. M.; Granda B. W.; Venkatakrishnan K.; Schmider J.; Harmatz J. S.; Shade R. I. J. Pharm. Pharmacol. 1998, 50, 997–1004. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Harvey J. N.; Mulholland A. J. J. Phys. Chem. Lett. 2010, 1, 3232–3237. [Google Scholar]
- Singh U. C.; Kollman P. A. J. Comput. Chem. 1986, 7, 718–730. [Google Scholar]
- Paine M. J. I.; McLaughlin L. A.; Flanagan J. U.; Kemp C. A.; Sutcliffe M. J.; Roberts G. C. K.; Wolf C. R. J. Biol. Chem. 2003, 278, 4021–4027. [DOI] [PubMed] [Google Scholar]
- Jaguar, 5.5 ed.; Schrödinger, LLC: Portland, OR, 2003.
- Lee C.; Yang W.; Parr R. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Becke A., D. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar]
- Rassolov V. A.; Ratner M. A.; Pople J. A.; Redfern P. C.; Curtiss L. A. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar]
- Ponder J. W.Tinker - Software Tools for Molecular Design; Washington University School of Medicine: St. Louis, MO, 2004.
- Harvey J. N. Faraday Discuss. 2004, 127, 165–177. [DOI] [PubMed] [Google Scholar]
- Żurek J.; Foloppe N.; Harvey J. N.; Mulholland A. J. Org. Biomol. Chem. 2006, 4, 3931–3937. [DOI] [PubMed] [Google Scholar]
- Bathelt C. M.; Mulholland A. J.; Harvey J. N. J. Phys. Chem. A 2008, 112, 13149–13156. [DOI] [PubMed] [Google Scholar]
- Senthilkumar K.; Mujika J. I.; Ranaghan K. E.; Manby F.; Mulholland A. J.; Harvey J. N. J. R. Soc. Interface 2008, 5, S207–S216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland N.; Mulholland A. J.; Harvey J. N. Biophys. J. 2006, 90, L27–L29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun A.; Thiel W. J. Phys. Chem. B 2005, 109, 1268–1280. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Altun A.; Thiel W. J. Comput. Chem. 2007, 28, 2147–2158. [DOI] [PubMed] [Google Scholar]
- Nelson D. R.; Kamataki T.; Waxman D. J.; Guengerich F. P.; Estabrook R. W.; Feyereisen R.; Gonzalez F. J.; Coon M. J.; Gunsalus I. C.; Gotoh O.; Okuda K.; Nebert D. W. DNA Cell Biol. 1993, 12, 1–51. [DOI] [PubMed] [Google Scholar]
- Altun A.; Shaik S.; Thiel W. J. Comput. Chem. 2006, 27, 1324–1337. [DOI] [PubMed] [Google Scholar]
- Heats of formation for CH4, C3H6, O, CH3OH, and C3H5OH; NIST Chemistry WebBook. http://webbook.nist.gov/chemistry/ (accessed April 11, 2011).
- Zheng J.; Wang D.; Thiel W.; Shaik S. J. Am. Chem. Soc. 2006, 128, 13204–13215. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H. J. Am. Chem. Soc. 2007, 129, 9686–9690. [DOI] [PubMed] [Google Scholar]
- Ranaghan K. E.; Ridder L.; Szefczyk B.; Sokalski W. A.; Hermann J. C.; Mulholland A. J. Org. Biomol. Chem. 2004, 2, 968–980. [DOI] [PubMed] [Google Scholar]
- Altun A.; Guallar V.; Friesner R. A.; Shaik S.; Thiel W. J. Am. Chem. Soc. 2006, 128, 3924–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Altun A.; Shaik S.; Thiel W. Faraday Discuss. 2011, 148, 373–383. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Rod T. H.; Olsen L.; Ryde U. J. Phys. Chem. B 2007, 111, 5445–5457. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M.; Crabtree R. H. J. Am. Chem. Soc. 1998, 121, 117–127. [Google Scholar]
- Balcells D.; Raynaud C.; Crabtree R. H.; Eisenstein O. Chem. Commun. 2008, 744–746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.