

Abstract
We have previously demonstrated that glia maturation factor (GMF), a brain-specific protein isolated, sequenced and cloned in our laboratory, is a prominent mediator of inflammation in the central nervous system (CNS) leading to the death of neurons. In the present study we demonstrate, for the first time, a significant up regulation of the GMF protein in various regions of Alzheimer's disease (AD) brains compared to age matched non-demented (ND) control brains. We analyzed AD and ND brain samples by quantitative ELISA using a combination of highly specific monoclonal and polyclonal anti-GMF antibodies developed and characterized in our laboratory. For the comparison between ND controls and AD cases, we examined brain tissue from 12 AD cases (ages ranging from 78–92 years) and 8 age-matched ND controls (ages ranging from 76–88 years). We observed a significant increase in GMF concentration in entorhinal cortex, parietal cortex, frontal cortex, occipital cortex, perirhinal cortex, and temporal cortex of AD patients. Our results clearly demonstrate that the GMF protein levels are significantly higher in all AD affected brain regions than in ND controls. The immunohistochemistry analysis revealed co-localization of GMF with amyloid plaques (AP) and neurofibrillary tangles (NFTs) in AD brains. Our results imply that under conditions of neuro degeneration the expression of GMF is significantly upregulated.
Keywords: Alzheimer's disease (AD), Glia maturation factor (GMF), Amyloid plaques (APs), Neurofibrillary tangles (NFTs), Neuroinflammation, Neurodegeneration
INTRODUCTION
Alzheimer's disease (AD) is the most common form of dementia among older people, affecting over 5 million Americans and over 15 million individuals worldwide. The etiology of AD remains unknown and there is no definite treatment yet available. The pathology of AD is characterized by neuritic plaques containing aggregates of β-amyloid peptide, neurofibrillary tangles (NFTs), degenerating neurons, and abundance of reactive astrocytes and activated microglia. Neurofibrillary tangles are composed of excessively phosphorylated tau proteins. Sustained inflammation by glial activation through their production of pro-inflammatory cytokines, chemokines and free radicals (such as reactive oxygen species/reactive nitrogen species and the transcription factor NF-κB) play a significant role in AD pathophysiology (Strohmeyer and Rogers 2001; Cartier et al., 2005; Glass et al. 2010). However, the precise role of chronic inflammation in the central nervous system in contributing to neurodegeneration in AD is not clear and deserves careful investigation.
Glia maturation factor (GMF), a highly conserved brain-specific protein, was isolated, sequenced and cloned in our laboratory (Lim, R., et al., 1989; Lim, R., et al., 1990; Kaplan, R., et al., 1991). We have demonstrated earlier (Zaheer, A., et al., 1993; Zaheer, A., et al., 1995a; Zaheer, A., et al., 1995b; Lim, R., et al., 1998; Zaheer, A., et al., 2004; Zaheer, A., et al., 2007a; Zaheer, A., et al., 2007b) a pro-inflammatory and an immunomodulatory function for GMF. Previously, we have shown that overexpression of GMF in astrocytes leads to immune activation of microglia through secretion of granulocyte-macrophage-colony stimulating factor (GM-CSF) (Zaheer, A., et al., 2007b). Using DNA microarray analysis, we found that GMF activates a host of genes in the nervous system related to immuno/inflammatory activity, including tumor necrosis factor-α, GM-CSF, interleukin-1β, MIP-1β, complement protein C1q, class II major histocompatibility complex (MHC) proteins, 12-lipoxygenase and chemokine CX3C (A Zaheer & R. Lim, 2002). All of these factors are known to be associated with the pathophysiology of neurodegenerative disorders such as AD. We also reported the stimulation of p38 mitogen activated protein kinase pathway and nuclear factor kappa B (NF-κB) by GMF in primary astrocyte cultures (Lim, et al., 2000; Zaheer, A., et al., 2007b). Recently, we have demonstrated that GMF is a prominent mediator of inflammatory signal transduction in central nervous system leading to the death of neuronal cells (Zaheer, A., et al., 2007b). In the present study, we performed a quantitative estimation of GMF protein and also demonstrated a co-localization of GMF protein with NFT and APs in the AD affected brain regions.
EXPERIMENTAL PROCEDURES
Brain tissue and histology
Autopsy human brains included in the present study were obtained from the University of Iowa Deeded body program. All the cases were consistent with neuropathological criteria for definite AD. We used twelve AD donor brains with dementia and compared with eight age matched ND controls with no neurological or psychiatric disease. The post mortem delay (PMI) for all cases was less than 6 hrs. For all cases, the ventromedial temporal lobe was dissected, and the tissue samples were immediately frozen or immersed in cold solution of 4% paraformaldehyde and 15% picric acid and 0.1 M PB, for 24 hours at 4°C. After 24 hrs, the blocks were placed in 30% sucrose in PB solution at 4 °C until they sank. Thereafter the blocks were cut on a freezing microtome at a thickness of 50 μm and the sections were collected in PB.
Immunohistochemistry
Immunohistochemical procedure was performed according to our previously published methods (Thangavel et al, 2008a, 2008b, 2009a, and 2009b). In brief, free-floating sections from the entorhinal, perirhinal, inferior and superior cortical tissues were processed by using the standard avidin-biotin-peroxidase complex (ABC) technique. Tissue sections were first treated with 0.3% H2O2 in PBS for 20 min to eliminate endogenous peroxidase activity. After three washes in PBS, sections were placed in blocking buffer (5% normal goat serum (NGS) and 3 % bovine serum albumin (BSA) diluted in a phosphate buffered saline containing 0.1% Triton-X 100 (PBS-T) for 1 h. The sections were incubated overnight at 4°C with anti-GMF (G209 mouse monoclonal antibody; 1:100) or AT8 (mouse monoclonal, 1:2000; Innogenetics, Belgium) to detect GMF and the hyperphosphorylated tau protein containing neurofibrillary tangles (NFTs) and amyloid plaques (APs). Next, sections were incubated for 2 h with corresponding biotinylated secondary anti-mouse IgG (1:200) (Vector Laboratories, Burlingame, CA) and developed using an ABC standard staining kit (Vector Laboratories, Burlingame CA). After washes, sections were placed in peroxidase reaction solution with diaminobenzidine (DAB) or SG substrate (Vector labs, Burlingame, CA). For negative control, sections were processed without the primary antibody incubation. Finally, the sections were mounted on slides, dried, dehydrated in a graded series of ethanol, and cover slipped with Permount (Fisher Scientific). The sections were observed under a Nikon microscope equipped with Nikon DSFi1 camera.
Thioflavin-S histochemistry
Entorhinal, perirhinal, inferior and superior temporal cortical tissue sections from AD and ND control brains were stained in an aqueous 1% thioflavin-S (Sigma, St. Louis, MO) solution for 10 min. Sections were then differentiated with 70% ethanol for 5 min, then dehydrated in a graded series of alcohol, before being cleared with xylene and cover slipped with Permount (Fisher Scientific) Thioflavin-S stained positive NFTs and APs were evaluated under the microscope with fluorescent lighting using with FITC filter in a standard Nikon microscope equipped with Nikon DSFi1 camera
Triple labeling
After immunostaining with AT8 antibody using DAB substrate, sections were incubated with anti-GMF for overnight at 4°C followed by the corresponding Alexa 568-conjugated secondary antibody incubated at room temperature for 1 h. The sections were washed with PBS. Sections were incubated with 1% thioflavin-S (Sigma, St. Louis, MO) solution for 5 min. Sections were then differentiated with 70% ethanol for 5 min, rinsed with distilled water, and coverslipped with Fluorogel mounting medium (EM Sciences, Hatfield, PA). The sections were analyzed with Nikon microscope equipped with Nikon DSFi1 camera.
GMF-antibodies used
CT-11 was a monoclonal antibody (IgG1) against a synthetic peptide corresponding to the C-terminal 11 amino acid residues of GMF and was affinity-purified with protein-A. 91–01 was an affinity purified rabbit polyclonal antibody against human GMF. G2-09 was a monoclonal antibody (IgG1) against GMF and was affinity-purified with protein-G plus.
Enzyme-linked immunosorbent assay (ELISA)
The analysis of GMF protein concentration in the brain tissues was estimated by sandwich immuno-assay procedure as described earlier (Zaheer et al 2007b). The concentration of GMF was estimated from a standard curve generated using recombinant GMF with each run. The lower detection limits of these ELISA are in the range of 10–15 pg/ml. ELISA data are presented as mean values ± standard deviations and represent more than three independent experiments with similar results.
RESULTS
Human brain tissues were obtained from autopsy, with PMI less than 6 h, at University of Iowa Deeded Body Program (Body Donation Program). For the comparison, we examined brain tissue from 12 cases with clinically diagnosed and neuropathologically confirmed AD cases (five females, seven males) with a mean age (±SEM) of 83 ± 4.7 years (range 78–92 years) and 8 ND controls (4 females, 4 males) with a mean age (±SEM) of 83 ± 3.4 years (range 76–88 years). The AD cases examined had duration of clinical dementia ranging from 7 to 18 years. Consistent with the pathology our samples of AD brains showed numerous amyloid plaques and neurofibrillary tangles in all the cortical sections (entorhinal, superior temporal, inferior temporal and perirhinal) and rarely in non-AD normal brain sections. Representative images of Tau immunostaining with AT8 antibody showing the distribution of neurofibrillary tangles (NFTs) in the entorhinal cortex, superior temporal cortex and inferior temporal cortex (Fig. 1) of the autopsied brain from AD case (A, B, and C magnification at X4, X10, and X20, respectively) compared with age-matched ND control (D, E, and F magnification at X4, X10, and X20, respectively). Thioflavin-S histochemical staining showing the distribution of NFTs and APs in the perirhinal cortex, entorhinal cortex, superior temporal cortex and inferior temporal cortex of AD affected brain (A and B magnification at X10 and X20) and age-matched ND (C and D magnification at X10 and X20) controls (Fig. 2). These results revealed numerous NFTs and amyloid plaques (APs) in AD affected brains as compared to ND brains.
Figure 1.

Tau immunostaining with AT8 antibody showing the distribution of neurofibrillary tangles in the entorhinal cortex, superior temporal cortex, and inferior temporal cortex of a severe Alzheimer's disease case (A, B, and C) and an age-matched ND control case (D, E, and F). Magnification: A and D, X4; B and E, X10; C and F, X20.
Figure 2.

Thioflavin-S histochemical staining showing the distribution of neurofibrillary tangles and amyloid plaques in the perirhinal cortex, entorhinal cortex, superior temporal cortex, and inferior temporal cortex of a severe Alzheimer's disease case (A, B) and an age-matched ND control case (C, D). Magnification: A and C, X10; B and D, X20.
Next, we quantitatively estimated the protein levels of GMF in different regions of autopsied brains of AD cases in comparison to the control ND subjects, and we observed a significant increase in GMF concentration in entorhinal cortex (Ent Cx), parietal cortex (P Cx), frontal cortex (F Cx), occipital cortex (Occi Cx), perirhinal cortex (Pr Cx), and temporal cortex (T Cx) of AD cases (Table 1). In AD affected brains, GMF protein levels were significantly higher than in ND controls in all the brain regions examined. These results show an augmented expression of GMF in specific brain regions in AD affected cases. Statistical analysis of the mean GMF data from various cortices of individuals is presented in Table 2. In all the cortices, the GMF levels significantly increased to 6–7 fold in AD cases compared to the corresponding cortex of ND control group except in Occi Cx of AD showing a 9 fold increase, however the level of GMF in ND Occi Cx was about half of the other ND cortices.. The mean of GMF levels in the AD cases and ND control were 3625 ± 488 and 555 ± 488 pg/mg protein, respectively, showing an average of 6.5 fold increase in AD brains. In our AD population, the GMF levels in Ent Cx, Pr Cx, T Cx, F Cx, and Occi Cx were 3654 ± 422, 3746 ± 458, 3701 ± 473, 3704 ± 408, and 3136 ± 594 pg/mg protein, respectively. No significant difference in GMF levels among the different cortices from AD brains existed. In the ND brains, Ent Cx, Pr Cx, T Cx, F Cx, and Occi Cx had 655 ± 104, 628 ± 52, 589 ± 67, 538 ± 73, and 343 ± 196 pg GMF/mg protein, respectively. GMF levels did not vary significantly also among cortices of ND control brains.
Table 1.
GMF protein levels in various brain regions of AD patients in comparison to the age matched ND control
| Case | Age | Sex | GMF (pg/mg protein) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ent Cx | Pr Cx | T Cr | F Cx | Occi Cx | |||||||||||||
| AD-1 | 83 | F | 4487 | ± | 126 | 4581 | ± | 132 | 4620 | ± | 266 | 4069 | ± | 246 | NA | ||
| AD-2 | 89 | F | 3118 | ± | 340 | 3281 | ± | 315 | 3272 | ± | 390 | 3009 | ± | 184 | 2597 | ± | 140 |
| AD-3 | 88 | F | 3775 | ± | 242 | 3747 | ± | 94 | 3836 | ± | 161 | 3674 | ± | 253 | NA | ||
| AD-4 | 78 | M | 3860 | ± | 460 | 3876 | ± | 458 | 3882 | ± | 431 | NA | NA | ||||
| AD-5 | 79 | M | 3712 | ± | 200 | 3867 | ± | 87 | 3926 | ± | 244 | 3790 | ± | 213 | 2623 | ± | 136 |
| AD-6 | 82 | M | 3753 | ± | 146 | 3220 | ± | 581 | 3948 | ± | 70 | 3648 | ± | 238 | 3890 | ± | 72 |
| AD-7 | 82 | M | 3573 | ± | 245 | 4047 | ± | 400 | 3809 | ± | 586 | 4332 | ± | 195 | 3559 | ± | 232 |
| AD-8 | 82 | M | 4107 | ± | 380 | 4195 | ± | 193 | 3452 | ± | 159 | 3959 | ± | 397 | 3731 | ± | 356 |
| AD-9 | 78 | M | 3103 | ± | 227 | 3072 | ± | 130 | 2952 | ± | 293 | 3213 | ± | 372 | NA | ||
| AD-10 | 87 | M | 3060 | ± | 265 | 3242 | ± | 318 | 2952 | ± | 293 | NA | 2459 | ± | 189 | ||
| AD-11 | 92 | F | 3768 | ± | 238 | 3798 | ± | 377 | 3934 | ± | 186 | NA | 3090 | ± | 175 | ||
| AD-12 | 84 | F | 3508 | ± | 330 | 4026 | ± | 302 | 3830 | ± | 346 | 3629 | ± | 366 | NA | ||
| ND-1 | 76 | M | 596 | ± | 125 | 614 | ± | 117 | 552 | ± | 59 | 513 | ± | 103 | 658 | ± | 54 |
| ND-2 | 88 | F | NA | 551 | ± | 40 | 484 | ± | 76 | 446 | ± | 63 | 559 | ± | 45 | ||
| ND-3 | 79 | F | 689 | ± | 74 | 703 | ± | 76 | 656 | ± | 63 | NA | 233 | ± | 16 | ||
| ND-4 | 83 | M | 595 | ± | 105 | 573 | ± | 79 | 615 | ± | 104 | 606 | ± | 86 | 239 | ± | 33 |
| ND-5 | 80 | M | 528 | ± | 59 | 627 | ± | 78 | 567 | ± | 63 | NA | NA | ||||
| ND-6 | 81 | M | 703 | ± | 76 | 688 | ± | 72 | 523 | ± | 68 | NA | 354 | ± | 23 | ||
| ND-7 | 84 | F | 627 | ± | 90 | 647 | ± | 61 | 658 | ± | 104 | 585 | ± | 89 | 118 | ± | 17 |
| ND-8 | 87 | F | 850 | ± | 89 | 625 | ± | 64 | 653 | ± | 100 | NA | 243 | ± | 25 | ||
Entorhinal cortex (Ent Cx), perirhinal cortex (Pr Cx), temporal cortex (T Cx), frontal cortex (F Cx), and occipital cortex (Occi Cx). NA= sample not available
Table 2.
Statistical analysis of GMF in AD and ND brain regions
| Case | Total number (Male + female) | Mean Age Years ± SD | GMF protein levels (pg/mg protein ) Mean ± SD | |||||
|---|---|---|---|---|---|---|---|---|
| Ent Cx | Pr Cx | T Cx | F Cx | Occi Cx | Means (all cortex) | |||
| AD | 12(7 + 5) | 83.7 ±4.5 | 3654 ± 422 | 3746 ± 458 | 3701 ± 473 | 3704 ± 408 | 3136 ± 594 | 3625 ± 488 |
| ND | 8 (4 + 4) | 82.3 ±4.1 | 655 ± 104 | 628 ± 52 | 589 ± 67 | 538 ± 73 | 343 ± 196 | 555 ± 488 |
| Ratio (AD/ND) | 1.50 | 1.02 | 5.58 | 5.96 | 6.28 | 6.88 | 9.14 | 6.53 |
Entorhinal cortex (Ent Cx), perirhinal cortex (Pr Cx), temporal cortex (T Cx), frontal cortex (F Cx) and occipital cortex (Occi Cx)
To examine the association of GMF protein with APs and NFTs in AD-affected brain regions, double-immunofluorescence staining was performed. The results (Fig. 3) showed co-localization of GMF with thioflavin-S and AT-8 positive amyloid plaque and NFTs in the temporal cortex of an AD affected brain. Absorption of GMF-antibody with human recombinant GMF protein removed most of the GMF immunoreactivity, indicating the staining of GMF is specific and not due to cross-reactivity with other proteins as the absorption of GMF-antibody with an irrelevant human protein did not affect the immunoreactivity of GMF (data not shown).
Figure 3.
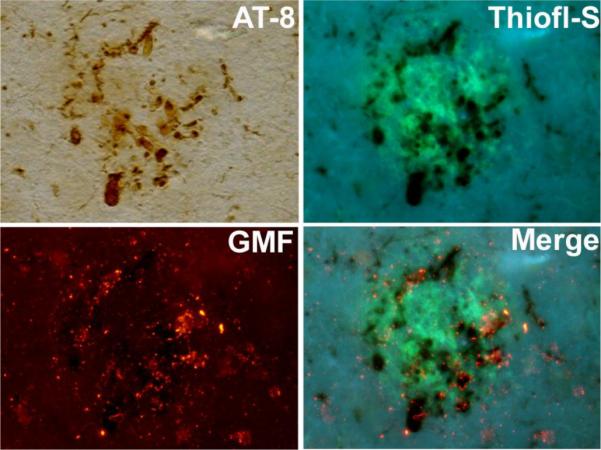
Immunohistochemical localization of GMF in the Alzheimer's disease brain. Temporal cortex of a severely AD affected brain immunostained with tau (AT8) and the same section stained with thioflavin-S for the presence of amyloid plaques and with anti-GMF antibody using immunofluorescence techniques. The merged photomicrographs illustrating colocalization of GMF with thioflavin-S and AT-8 stained neurofibrillary tangles and amyloid plaques.
DISCUSSION
In the present study we analyzed the expression of a novel CNS-specific inflammatory protein GMF in different cortical areas of AD and ND brains. Using quantitative enzyme-linked immunosorbent assay (ELISA) for measuring GMF protein in 12 AD cases (five females, seven males) with a mean age of 83 ± 4.7 years and 8 non-AD controls (4 females, 4 males) with a mean age of 83 ± 3.4 years, we have shown significantly higher levels of GMF protein in all effected regions of AD brain compared to age-matched ND controls. Using triple labeling immunohistochemical techniques, we demonstrated that a co-localization of GMF with thioflavin-S and AT-8 positive APs and NFTs in the temporal cortex of AD affected brain. Thus, our current results have clearly demonstrated that under conditions of neuro degeneration the expression of GMF is significantly upregulated. This is the first demonstration of up regulation of GMF protein in AD brains.
Glia Maturation Factor was isolated, sequenced and cloned in our laboratory (Lim et al. 1989; Lim et al. 1990; Kaplan et al. 1991). The GMF gene is localized to the long arm of human chromosome 14 by us and the Human Genome sequence Consortium (Lander et al. 2001). GMF is an intracellular inflammatory signal transduction regulator known to play a role in neuroinflammation and neurodegeneration. Our earlier studies have demonstrated that in cell-free assays, GMF is phosphorylated by protein kinase C (PKC), protein kinase A (PKA), casein kinase II (CKII), and ribosomal S6 kinase (RSK) (Lim and Zaheer 1995); and PKA-phosphorylated GMF is an inhibitor of the ERK isoform of MAP kinase (Zaheer and Lim 1996) while at the same time a stimulator of the p38 isoform (Lim and Zaheer 1996), which implicates GMF protein in stress-activated inflammatory responses. In C6 as well as in normal astrocytes, GMF overexpression stimulates p38 MAP kinase activity, and activates the redox enzyme CuZn superoxide dismutase (CuZnSOD) and the transcription factors NF-κB (Lim et al. 1998; Lim et al. 2000). Overexpression of GMF in PC12 pheochromocytoma cells activates p38 MAP kinase, its downstream MAPKAP-kinase 2, and the final effector tyrosine hydroxylase, accompanied by an increased phosphorylation of these proteins (Zaheer and Lim 1998). Kaimori et al. (Kaimori et al. 2003) demonstrated oxidative stress enhancing ability of GMF in the pathophysiology of a renal disease. Overexpression of GMF in proximal tubular cells leads to vulnerability to oxidative injury through p38 pathway and changes in antioxidant enzyme activities (Kaimori et al. 2003). A study by Baldwin et al. demonstrated that the transient overexpression of GMF in glioblastoma cells activated p38 MAPK signaling and also increased susceptibility of the tumor cells to cytotoxicity of cisplatin, a chemotherapeutic agent (Baldwin et al. 2006). In another study, we infected a mixed culture of primary glial cells with a replication-defective adenovirus carrying GMF cDNA (Zaheer et al. 2002). A comprehensive survey of gene expression by Affymetrix microarray analysis showed a big increase in several major histocompatibility complex (MHC) class II proteins and several classical proinflammatory cytokines. In more recent investigation, we studied pro inflammatory cytokine profiles of primary glial cells following GMF overexpression. The results demonstrated that overexpression of GMF is necessary for the induction of granulocyte-macrophage-colony stimulating factor (GM-CSF) in astrocytes and that GM-CSF is the key molecule for GMF-dependent production of inflammatory cytokines in microglial cells. The results also demonstrated that the increased release of GMF-dependent microglial proinflammatory cytokines is cytotoxic to oligodendrocytes, the myelin-forming cells, and neurons. GM-CSF is a hematopoietic cytokine that is not detected under normal conditions but can be induced by activated macrophages and glial cells (Malipiero et al. 1990). Its gene is under the transcriptional control of NF-κB (Shannon et al. 1997). It has been reported that GM-CSF enhances the expression of several pro inflammatory cytokine/chemokine, thus playing an important role in the amplification of immune and inflammatory processes. A report by Tarkowski et al. (2001) clearly demonstrated that levels of GM-CSF was significantly increased in patients with AD and with vascular dementia. Moreover, they also found a significant correlation between the enhanced levels of GM-CSF and that of Fas/APO-1 (proapoptotic factors) and Tau protein in dementia. Based on these results they suggested a crucial role for GM-CSF in neuronal cell damage in AD. According to Henze et al (2005), the proliferation of microglial cells in response to the dopaminergic neurotoxin 1-methyl-4-phenylpyridinium (MPP+) was mimicked by proinflammatory cytokine GM-CSF and was blocked by a neutralizing antibody to GM-CSF. They concluded that the microglial reaction observed following MPP+ exposure was dependent on GM-CSF and GM-CSF was likely to contribute to the observed microgliosis. Another report by Marusic et al. (2002) demonstrated that overexpression of GM-CSF in the CNS and not in the periphery affects the effector phase of experimental autoimmune encephalomyelitis (EAE), an animal model with clinical and pathological similarities to multiple sclerosis (MS), leading to chronic and severe disease. Recently, it has also been reported that GM-CSF knockout are resistant to EAE induction by immunization with MOG35–55 antigen (McQualter et al. 2001). These reports support our hypothesis that GMF-induced GM-CSF production in astrocytes leading to immune activation of microglia in CNS plays a crucial and deleterious role in pathogenesis of neurodegenerative diseases, including AD and MS.
Chronic production of inflammatory cytokines/chemokines by reactive astrocytes and activated microglia are suspected to launch the neuro inflammatory cascade in AD. We previously reported (Zaheer et al. 2008) that intraventricular infusion of amyloid beta peptide1–42 (Aβ1–42) in wild type mice brains caused activation of astrocytes and microglia, several fold increase in the production of proinflammatory cytokines/chemokines and memory deficit. Whereas, all these effects of Aβ1–42 infusion, including the memory deficit, were suppressed in GMF deficient mice. In conclusion, we provided the first time evidence for association of high level GMF with AD affected cortical regions of human brains where GMF was prominently localized in APs and NFTs, possibly to facilitate AD associated neuro inflammation and memory deficit.
In summary, our finding that NFTs and APs contained substantial amounts of GMF suggests that the co-localization of GMF with NFTs and APs is essential in AD pathology. It seems reasonable to contemplate that the chronic and significant levels of GMF association with NFTs/ APs may exacerbate neuroinflammation leading to neurodegeneration in AD.
Research highlights
-
❖
Significant up regulation of GMF protein in AD affected brain regions
-
❖
Co-localization of GMF with amyloid plaques (AP) and neurofibrillary tangles (NFTs) in AD brains
-
❖
GMF association with NFTs/ APs exacerbates neuroinflammation leading to neurodegeneration in AD
Acknowledgements
We thank Lavanya Ramamoorthy, Yanghong Wu, Marcus Ahrens, and Xi Yang for excellent technical help. We thank Paul Reimann for photography and Darrell Wilkins for tissue acquisition from the University of Iowa Deeded Body Program, Iowa City, Iowa. This work was supported by the Department of Veterans Affairs Merit Review award (to A.Z.) and by the National Institute of Neurological Disorders and Stroke grant NS-47145 (to A.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Baldwin RM, et al. Protection of glioblastoma cells from cisplatin cytotoxicity via protein kinase Ciota-mediated attenuation of p38 MAP kinase signaling. Oncogene. 2006;25:2909–2919. doi: 10.1038/sj.onc.1209312. [DOI] [PubMed] [Google Scholar]
- Henze C, et al. Proliferation of microglial cells induced by 1-methyl-4-phenylpyridinium in mesencephalic cultures results from an astrocyte-dependent mechanism: role of granulocyte macrophage colony-stimulating factor. J Neurochem. 2005;95:1069–1077. doi: 10.1111/j.1471-4159.2005.03416.x. [DOI] [PubMed] [Google Scholar]
- Kaplan R, et al. Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem. 1991;57:483–490. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- Kaimori JY, et al. Induction of glia maturation factor-beta in proximal tubular cells leads to vulnerability to oxidative injury through the p38 pathway and changes in antioxidant enzyme activities. The Journal of biological chemistry. 2003;278:33519–33527. doi: 10.1074/jbc.M301552200. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lim R, et al. Purification and characterization of glia maturation factor beta: a growth regulator for neurons and glia. Proc Natl Acad Sci U S A. 1989;86:3901–3905. doi: 10.1073/pnas.86.10.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, et al. Complete amino acid sequence of bovine glia maturation factor beta. Proc Natl Acad Sci U S A. 1990;87:5233–5237. doi: 10.1073/pnas.87.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R, Zaheer A. Phorbol ester stimulates rapid interacellular phosphorylation of glia maturation factor. Biochem. Biophys. Res. Comm. 1995;211:928–934. doi: 10.1006/bbrc.1995.1901. [DOI] [PubMed] [Google Scholar]
- Lim R, Zaheer A. In vitro enhancement of p38 mitogen-activated protein kinase activity by phosphorylated glia maturation factor. J. Biol. Chem. 1996;271:22953–22956. doi: 10.1074/jbc.271.38.22953. [DOI] [PubMed] [Google Scholar]
- Lim R, et al. Overexpression of glia maturation factor in C6 cells promotes differentiation and activates superoxide dismutase. Neurochem Res. 1998;23:1445–1451. doi: 10.1023/a:1020715126326. [DOI] [PubMed] [Google Scholar]
- Lim R, et al. Activation of nuclear factor-kB in C6 rat glioma cells after transfection with glia maturation factor. J. Neurochem. 2000;74:596–602. doi: 10.1046/j.1471-4159.2000.740596.x. [DOI] [PubMed] [Google Scholar]
- Malipiero UV, et al. Production of hemopoietic colony-stimulating factors by astrocytes. J Immunol. 1990;144:3816–3821. [PubMed] [Google Scholar]
- Marusic S, et al. Local delivery of granulocyte macrophage colony-stimulating factor by retrovirally transduced antigen-specific T cells leads to severe, chronic experimental autoimmune encephalomyelitis in mice. Neurosci. Lett. 2002;332:185–189. doi: 10.1016/s0304-3940(02)00947-3. [DOI] [PubMed] [Google Scholar]
- McQualter JL, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp. Med. 2001;194:873–882. doi: 10.1084/jem.194.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon MF, et al. Signals for activation of the GM-CSF promoter and enhancer in T cells. Critical reviews in immunology. 1997;17:301–323. doi: 10.1615/critrevimmunol.v17.i3-4.30. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, et al. Local and systemic GM-CSF increase in Alzheimer's disease and vascular dementia. Acta Neurol. Scand. 2001;103:166–174. doi: 10.1034/j.1600-0404.2001.103003166.x. [DOI] [PubMed] [Google Scholar]
- Thangavel R, et al. Modular and laminar pathology of Brodmann's area 37 in Alzheimer's disease. Neuroscience. 2008a;152:50–55. doi: 10.1016/j.neuroscience.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel R, et al. Posterior parahippocampal gyrus pathology in Alzheimer's disease. Neuroscience. 2008b;154:667–676. doi: 10.1016/j.neuroscience.2008.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel R, et al. Loss of nonphosphorylated neurofilament immunoreactivity in temporal cortical areas in Alzheimer's disease. Neuroscience. 2009a;160:427–433. doi: 10.1016/j.neuroscience.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel R, et al. The abnormally phosphorylated tau lesion of early Alzheimer's disease. Neurochem Res. 2009b;34:118–123. doi: 10.1007/s11064-008-9701-1. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Expression of glia maturation factor beta mRNA and protein in rat organs and cells. J Neurochem. 1993;60:914–920. doi: 10.1111/j.1471-4159.1993.tb03237.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Expression of mRNAs of multiple growth factors and receptors by neuronal cell lines: detection with RT-PCR. Neurochem Res. 1995a;20:1457–1463. doi: 10.1007/BF00970594. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Expression of mRNAs of multiple growth factors and receptors by astrocytes and glioma cells: detection with reverse transcription-polymerase. 1995b doi: 10.1007/BF02073330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer A, Lim R. In Vitro inhibition of MAP kinase (ERK1/ERK2) activity by phosphorylated glia maturation factor (GMF) Biochemistry. 1996;35:6282–6288. doi: 10.1021/bi960034c. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Lim R. Overexpression of GMF in PC12 cells activates p38 MAP kinase, MAPKAP kinase-2 and Tyrosine Hydroxylase. Biochem. Biophys. Res. Comm. 1998;250:278–282. doi: 10.1006/bbrc.1998.9301. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Overexpression of glia maturation factor in astrocytes leads to immune activation of microglia through secretion of granulocyte macrophage-colony stimulating factor. Biochem. Biophys. Res. Comm. 2002;294:238–244. doi: 10.1016/S0006-291X(02)00467-9. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Decreased copper-zinc superoxide dismutase activity and increased resistance to oxidative stress in glia maturation factor-null astrocytes. Neurochem Res. 2004;29:1473–1480. doi: 10.1023/b:nere.0000029558.82943.00. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Res. 2007a;1144:239–247. doi: 10.1016/j.brainres.2007.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer A, et al. A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem. 2007b;101:364–376. doi: 10.1111/j.1471-4159.2006.04385.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, et al. Glia maturation factor modulates β-amyloid-induced glial activation, inflammatory cytokine/chemokine production and neuronal damage. Brain Res. 2008;1208:192–203. doi: 10.1016/j.brainres.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]