

Abstract
Platelet-activating factor acetylhydrolase type-II (PAFAH-II) is an intracellular phospholipase A2 enzyme that hydrolyzes platelet-activating factor and oxidatively fragmented phospholipids. This N-terminally myristoylated protein becomes associated with cytoplasmic facing cell membranes under oxidative stress. Structural requirements for PAFAH-II binding to membranes in response to oxidative stress are unknown. To begin elucidating the mechanism of trafficking and stress response, a homology model of PAFAH-II was constructed. From the predicted membrane orientation of PAFAH-II, the N-terminal myristoyl group and a hydrophobic patch are hypothesized to be involved in membrane binding. Localization studies of human PAFAH-II in HEK293 cells indicated that an unmyristoylated mutant remained cytoplasmic under stressed and unstressed conditions. The myristoylated wild-type enzyme was partially localized to the cytoplasmic membranes prior to stress, and became more localized to these membranes upon stress. A triple mutation of three hydrophobic patch residues of the membrane binding region likewise did not localize to membranes following stress. These results indicate that both the myristoyl group and the hydrophobic patch are essential for proper enzyme trafficking to the membranes following oxidative stress. Additionally, colocalization studies using organelle-specific proteins demonstrate that PAFAH-II traffics to the membranes of both the endoplasmic reticulum and Golgi apparatus.
Platelet-activating factor acetylhydrolase type-II (PAFAH-II) inactivates the potent phospholipid mediator platelet-activating factor (PAF) and other structurally similar bioactive lipids produced in response to oxidative damage. It is a 44 kDa, intracellular, calcium-independent protein, conserved in a wide number of organisms and found primarily in platelets, lymphocytes, neutrophils (1), skin (2), liver, and kidney cells (3, 4). PAFAH-II inactivates PAF by catalyzing a phospholipase A2 esterolysis at the sn-2 position, producing lyso-PAF and acetate (4). Relatively low levels of PAF (10−10 to 10−12 M) are able to stimulate a response via the PAF receptor (5, 6). The regulation of PAF levels is involved in a number of pathways, including: inflammatory response, anaphylaxis, hypotension, smooth muscle contraction, reproduction, and fetal development (4–6).
PAFAH-II has been shown to recognize and cleave oxidatively-fragmented phospholipids (1) produced by oxidative damage (4, 7). Reactive oxygen species (ROS) induce free radical reactions that oxidize polyunsaturated fatty acids and can produce a shortened acyl group of glycerophospholipids (6). Often, these fragmented phospholipids closely resemble PAF, allowing them to bind to the PAF receptor as agonists (8). Fragmented phospholipids also affect the cell by disrupting the morphology of the membranes that they constitute (9). As in the case of PAF, PAFAH-II hydrolyzes these fragmented phospholipids at the sn-2 position (4), preferring substrates with chains fewer than ten carbons (6).
In non-stressed cells, PAFAH-II has been observed in both the cytosol and in association with the cytoplasmic membranes (2, 10). However, upon oxidative stress, PAFAH-II shifts the balance of its localization to the cytoplasmic membranes, seemingly sensing the redox state of the cell (2, 10). It has been suggested that PAFAH-II maintains the integrity of the cell by hydrolyzing fragmented phospholipids in the membranes (2–6, 9, 10). Experiments with mammalian cell culture, yeast and knockout mice have also confirmed the importance of this enzyme in oxidative stress survival and prevention of apoptosis (2, 7, 10, 11). Nevertheless, the structural requirements for PAFAH-II binding to cytoplasmic facing membranes in response to oxidative stress are still unknown. Also, the specifics of which organelles PAFAH-II localizes with has not yet been revealed.
PAFAH-II contains the N-terminal myristoylation motif MGXXXS (3, 10) that signals the eukaryotic machinery to cleave the first methionine and to conjugate a 14-carbon myristic acid chain onto the glycine residue during protein translation (12). Even though the myristoylated form of PAFAH-II has been identified in MDBK cells (10), the exact role of the myristoyl group has not yet been elucidated.
The mechanism of PAFAH-II membrane binding has been difficult to predict without a structure. PAFAH-II shares 43% sequence identity with plasma PAFAH (pPAFAH), whose crystal structure has been recently solved (13). Furthermore, the substrate specificity and predicted enzyme function of these two enzymes are similar (3). However, pPAFAH is associated with LDL and HDL in blood plasma and is never myristoylated at the N-terminus (3).
Our PAFAH-II homology model, which was generated using pPAFAH as a template, allowed us to predict possible sites of enzyme interaction with the membrane: the myristoyl group and a series of hydrophobic residues, located near the entrance into the active site. With the human form of the enzyme, we show that both the myristoyl group and the hydrophobic patch residues are essential for PAFAH-II membrane trafficking through mutagenesis and localization experiments in HEK293 cells. In addition, we provide direct evidence that membrane-bound PAFAH-II is localized to the membranes of both the endoplasmic reticulum (ER) and the Golgi apparatus.
MATERIALS AND METHODS
Homology Modeling of PAFAH-II
We generated a structurally-influenced alignment of PAFAH-II to pPAFAH (Figure S1 of the Supporting Information) using the SALIGN function of the program MODELLER (14). This approach applies a penalty for the placement of alignment gaps in regions where the template has elements of secondary structure. Based on this alignment, 20 models were built using the structure of pPAFAH as the template (13) and scored using the discrete optimized potential energy (DOPE) algorithm included as part of the program MODELLER (15). The best-scoring model was subjected to energy minimization with the program CNS (16), and the final model rapidly converged to an energy minimum. The PAFAH-II model was then analyzed by Dr. Andrei Lomize (University of Michigan), who applied his previously reported method (17) to predict the membrane binding orientation and affinity of unmyristoylated PAFAH-II.
Cloning of PAFAH-II Yellow and Cyan Fluorescent Protein Fusions
cDNAs encoding cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) were generous gifts of Dr. Piston (Vanderbilt University, TN) and Dr. Tsien (UCSD, CA), respectively. To create CFP-pCEP4 and YFP-pCEP4 constructs, CFP and YFP genes were amplified using a forward primer that introduced an XhoI restriction site, and a reverse primer that introduced a BamHI site and two stop codons. The pCEP4 vector (Invitrogen) along with CFP and YFP PCR products were digested with BamHI and XhoI (Invitrogen) and ligated using T4 ligase (Invitrogen). The human PAFAH-II gene was amplified from a purchased clone (Invitrogen), where the forward primer introduced a 5′ Kozak sequence (GCCACCATG) and a HindIII restriction site, and the reverse primer contained an XhoI restriction site. The PCR product was cloned into CFP/YFP-pCEP4 as described above, constructing WT-PAFAH-II-YFP (Figure 1A). In order to express CFP and YFP without PAFAH-II, CFP-pCEP4 (+Kozak) and YFP-pCEP4 (+ Kozak) were created using a forward primer that contained a 5′ Kozak sequence upstream of the XhoI restriction site. To produce an unmyristoylated PAFAH-II, Gly-2 was mutated to Ala (G2A-PAFAH-II-YFP, Figure 1A) using the Quick Change site-directed mutagenesis kit (Stratagene) and the following primers:
Figure 1.
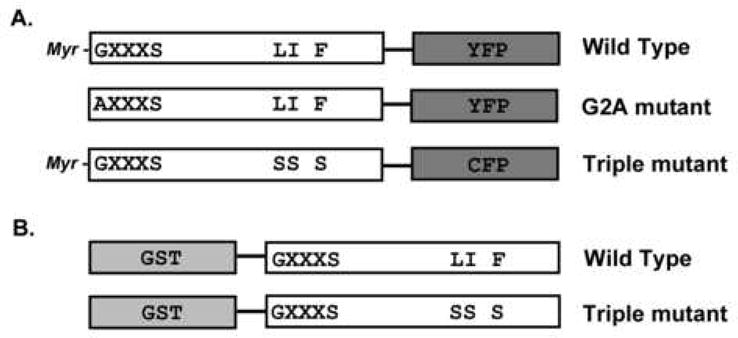
PAFAH-II DNA constructs. The PAFAH-II portion of the construct is in white, while the fusion tag is in grey. The myristoylation motif (GXXXS) and hydrophobic patch residues (L327, I328, F331) are shown by single letter amino acid code, along with the mutations of these areas (LI F versus SS S). (A) PAFAH-II constructs expressed in HEK293 cells with CFP/YFP C-terminal fusions. PAFAH-II constructs that would become myristoylated in vivo on the glycine residue are identified. (B) PAFAH-II constructs expressed in E. coli with GST N-terminal fusion.
5′GCTTGCCACCATGGCGGTCAACCAGTC3′
5′GACTGGTTGACCGCCATGGTGGCAAGC3′.
The triple mutation L327S/I328S/F331S of the membrane binding surface (Triple-mutant) was made by site-directed mutagenesis. The construct Triple-mutant-PAFAH-II-CFP (Figure 1A) as well as single mutants were generated one mutation at a time using the following primers:
L327S:
5′GCTTTTGTGACTGGCAACAGTATTGGTAAATTCTTCTCCAC3′
5′GTGGAGAAGAATTTACCAATACTGTTGCCAGTCACAAAAGC3′
I328S:
5′GTGACTGGCAACAGTAGTGGTAAATTCTTCTCC3′
5′GGAGAAGAATTTACCACTACTGTTGCCAGTCAC3′
F331S:
5′GGCAACAGTAGTGGTAAAAGCTTCTCCACTGAAACCC3′
5′GGGTTTCAGTGGAGAAGCTTTTACCACTACTGTTGCC3′
DNA sequences for all constructs were confirmed at the University of Delaware core sequencing facility.
GST-PAFAH-II Cloning, E. coli Expression and Purification
The PAFAH-II gene was amplified from a purchased PAFAH-II clone (Invitrogen), where the forward primer introduced an EcoRI restriction site, and the reverse primer contained a SalI restriction site. PAFAH-II was cloned into pGEX-4T-3 (GE Healthcare) by digesting the PAFAH-II PCR product and vector with EcoRI and SalI and ligated using T4 DNA ligase. The N-terminus of this construct is missing the start methionine and has five non-native residues (GSPNS) followed by the PAFAH-II sequence (GVNQ…). Since E. coli are incapable of myristoylating proteins, this WT-PAFAH-II-pGEX-4T-3 construct is predicted to function in a similar manner to the G2A-mutant (Figure 1B). The Triple-mutant-PAFAH-II-pGEX-4T-3 construct was made by site-directed mutagenesis as described above (Figure 1B).
GST-PAFAH-II constructs were transformed into BL21 (DE3) E. coli cells (Invitrogen) for protein expression. Cells were grown while shaking in LB media at 37 °C with 50 mg/L carbenicillin (Sigma-Aldrich) until the OD600nm reached 0.6. Cultures were then induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (Fisher) and allowed to grow at room temperature for 20 h while shaking. Cells were harvested by centrifugation and lysed by sonication in the following lysis buffer: 50 mM Tris-Cl, pH 8.0, containing 150 mM NaCl, 1 mM EDTA and 0.5 mM DTT. Lysed cells were then incubated in 20 mM CHAPS with gentle stirring for 30 min at 4 °C. Lysates were then cleared by centrifugation at 8,000 rpm and the supernatant was incubated with 10 mM ATP and 20 mM MgCl2 for 20 min at room temperature to eliminate co-purification of a contaminate, bacterial chaperonin GroEL (18). PAFAH-II was purified by GST affinity chromatography using a batch column method (GE Healthcare). Glutathione resin (GE Healthcare) was washed extensively with 50 mM Tris-Cl, pH 8.0 containing 150 mM NaCl, 1 mM EDTA, 0.5 mM DTT and 10mM CHAPS at 4 °C and GST fusions were eluted with wash buffer containing 10 mM reduced glutathione (Sigma-Aldrich) at room temperature. Protein fractions containing PAFAH-II were concentrated, dialyzed into storage buffer (50 mM Tris-Cl, pH 8.0, containing 1 mM EDTA and150mM NaCl), and stored at −20 °C.
Mammalian Cell Culture, Transfections and Oxidative Stress
HEK293 cells were maintained in Dubelcco’s modified Eagle medium (Invitrogen) supplemented with 10% cosmic calf serum (HyClone), 100 U/ml penicillin and 0.1 mg/ml streptomycin (Invitrogen) at 37 °C, 5% CO2. Transient transfections were carried out using Lipofectamine2000 (Invitrogen) according to manufacturer’s protocol. Transfections for Western blotting analysis and activity assays were carried out in 25 cm2 flasks (Corning) with 2 μg of DNA/flask. In preparation for confocal microscopy experiments, cells were seeded on 4-well borosilicate cover-slips (Lab-Tek) and transfected with 0.2 μg of DNA/well. At 24 h post-transfection, cells were stressed with 80 μM cumene hydroperoxide (Sigma-Aldrich) for 2 h at 37 °C. Media not containing stressors was added in parallel to “unstressed” samples and those cells were kept at 37 °C for 2 h, just as was done with the “stressed” cells.
PAFAH-II Western Blot Analysis
After generation of oxidative stress, cells were washed twice with ice-cold PBS buffer (Invitrogen), resuspended in 1 ml PBS buffer and cell density was determined by measuring the OD600nm. Cells were spun down at 4,000 rpm, resuspended and sonicated in the appropriate volume of lysis buffer (20 mM Tris-Cl, pH 8.0, containing 150 mM NaCl and 10% glycerol) to ensure equal cell densities. Sonicates were mixed with equal volumes of 2x SDS-PAGE loading buffer containing 50 mM DTT and resolved on a 12% Tris-Glycine gel, along with Magic Mark XP Western MW standard (Invitrogen). Proteins were transferred to a HyBond ECL nitrocellulose membrane (GE Healthcare) at 4 °C overnight at 60 mA. The membrane was washed in TBS-T buffer (20 mM Tris-Cl, pH 7.6, containing 137 mM NaCl and 0.1% Tween-20, Sigma-Aldrich) and blocked in 5% non-fat dry milk in TBS-T for 1 h. After three washes, the membrane was incubated with either green fluorescent protein (GFP) antibody (Abcam, suitable for detecting both CFP and YFP) or PAFAH-II antibody (Proteintech Group) at a 1:1,000 dilution for 1 h, followed by three washes and treatment with an anti-chicken–HRP or anti-rabbit-HRP linked secondary antibody (Cell Signaling Technology) at a 1:2,000 dilution for 30 min, followed by 5 washes in TBS-T buffer. All blotting steps were carried out at room temperature. The membrane was treated with ECL Western blotting substrate (Pierce) for 5 min and imaged using a luminescence filter on an ImmunoChem™8800 Imager (AlphaInnotech) and a 5 min exposure. Densitometry analyses of all Western blots were carried out using ImageJ 141o software.
PAFAH-II Activity Assays
The activity of PAFAH-II samples were determined with a PAF Acetylhydrolase Assay Kit (Cayman Chemical), which uses the unnatural substrate 2-thio PAF. A 1 ml assay mixture contained 100 mM Tris-Cl, pH 7.6, 0.1 mM DTNB, 185 μM 2-thio PAF and the reaction was initiated with the addition of 10–50 μl of PAFAH-II. The reaction was monitored continuously at 412 nm for 5 min. The absorbance of 5-thio-2-nitrobenzoic acid was recorded in mAbs/min. To measure the total PAFAH-II activity, HEK293 cell lysates were prepared as described above. Lysate of untransfected cells were used to obtain a baseline reading. PAFAH-II activity in cell lysates was also measured using either soluble or insoluble fractions that were separated by centrifugation at 5,000 g for 15 min at 4 °C. Activity of recombinant GST-WT-PAFAH-II and GST-Triple-mutant-PAFAH-II (both without a myristoyl group) were measured in a similar fashion and normalized to the starting enzyme concentrations determined by BCA assay (Pierce) and SDS-PAGE gel analysis.
Confocal Microscopy and Colocalization of Fluorescent Protein Fusions
Cells were imaged using a LSM DUO confocal microscope using the LSM 510 META scanhead with the majority of the images generated using a 60x Plan-Apochromat (numerical aperture 1.4) oil immersion objective lens. Some preliminary data was gathered with a 40x Plan-Neofluar (numerical aperture 1.3) oil immersion objective lens on a Zeiss LSM 510 NLO.
To quantify the number of cells displaying PAFAH-II membrane localization morphology, ten random frames of cells expressing each construct were imaged during three independent experiments. During each confocal experiment, 45–50 cells were imaged for each construct and the total percentage of cells with membrane-bound morphology was calculated.
To determine whether PAFAH-II localized to the ER and Golgi, the ER and Golgi Organelle Lights (Invitrogen) kits with a red fluorescent protein (RFP) tag were used. These kits utilize a baculovirus suspension that contains a fluorescently tagged organelle-specific protein construct. The infection with the Organelle Lights was done according to the manufacturer’s protocol when seeded cells reached 70% confluence. At 24 h post-infection with Organelle Lights, cells were transfected with PAFAH-II DNA constructs as described above. 24 h post-transfection, oxidative stress was induced and cells were imaged. The 458 nm, 514 nm and 561 nm laser lines were used with the BP 465–510 emission filter for the CFP tag, the BP 520–555 emission filter for the YFP tag and the LP 575 emission filter for the RFP tag, respectively.
RESULTS
Homology Modeling and Membrane Orientation of PAFAH-II
A homology model of PAFAH-II was built using the program MODELLER and the pPAFAH crystal structure as a template (13). The structure of pPAFAH is the only one to date with sufficient homology (43% identity, 62% similarity) to make a reliable model of PAFAH-II. The generation of an accurate alignment is a critical step in producing valid homology models when using a single template of moderate homology. We initially attempted to build a model of PAFAH-II based on a simple pair-wise sequence alignment. This model and subsequent attempts using alternate “sequence only” alignments were not successful in giving us a reliable model. This issue was indicated by a lack of convergence in energy when performing energy minimization after initial generation of the model. When a structurally-influenced alignment (Figure S1 of the Supporting Information) was performed using the SALIGN function of the program MODELLER, we were able to generate models which converged upon energy minimization. Twenty independent modeling runs were performed in this fashion. Each of the resulting 20 models were highly similar, with the greatest variation in two surface loops, which are not predicted to be near the membrane-binding surface. The final minimized PAFAH-II model gave a backbone RMSD of 0.81 Å compared to the pPAFAH template structure.
To examine the potential interactions of PAFAH-II at the membrane, the Orientations of Proteins in Membranes (OPM) database computational approach (17, 19) was used. The spatial arrangement of our PAFAH-II homology model was predicted with respect to the hydrocarbon core of the lipid bilayer with an interaction energy of −7 kcal/mol. It is important to point out that the membrane boundaries of PAFAH-II were computed from its unmyristoylated form. The data predicts three regions to be in or near the membrane (Figure 2): two helices rich in hydrophobic residues (residues 326–332 and 71–81) and the N-terminus (positioned near helix 326–332). Five hydrophobic residues are predicted to be embedded into the membrane: L327, I328 and F331 from one helix, and L76 and L79 from the other helix. The fact that the N-terminus is predicted to be near the membrane led us to hypothesize that the myristoyl group is critical for membrane binding in addition to the hydrophobic residues.
Figure 2.

Unmyristoylated PAFAH-II homology model and its predicted membrane binding orientation. White spheres represent the hydrophilic and hydrophobic interface of the phospholipid bilayer from a prediction performed by A. Lomize (17, 19). Shown in magenta (oxygen atoms red and nitrogen atoms blue) is the PAFAH-II catalytic triad (S236, D259 and H314) based on the conserved active site residues of pPAFAH. The N-terminus, shown in blue, is the site of myristoylation in vivo. The C-terminus is shown in red. Helices predicted to embed into the membrane are shown with the hydrophobic residues L76 and L79 shown in yellow, and the L327, I328, and F331 residues shown in green.
Myristoyl Group and Hydrophobic Patch Drive PAFAH-II Membrane Anchoring
To determine the effect of the myristoyl group on the ability of PAFAH-II to become membrane-anchored, the distribution in mammalian cells of wild-type (WT) PAFAH-II and an unmyristoylated G2A-mutant was examined. Confocal microscopy images show that WT-PAFAH-II-YFP was distributed in both the cytosol and cytoplasmic membranes of the cell prior to oxidative stress. Upon generation of oxidative stress, WT-PAFAH-II concentration in the cytoplasm-facing membranes increased (Figure 3). On the other hand, before and after oxidative stress, unmyristoylated G2A-PAFAH-II-YFP was seen throughout the cytosol and in the nucleus to a smaller extent (Figure 3). These results indicate that the myristoyl group of PAFAH-II plays a critical role in its localization to the membranes in response to stress.
Figure 3.
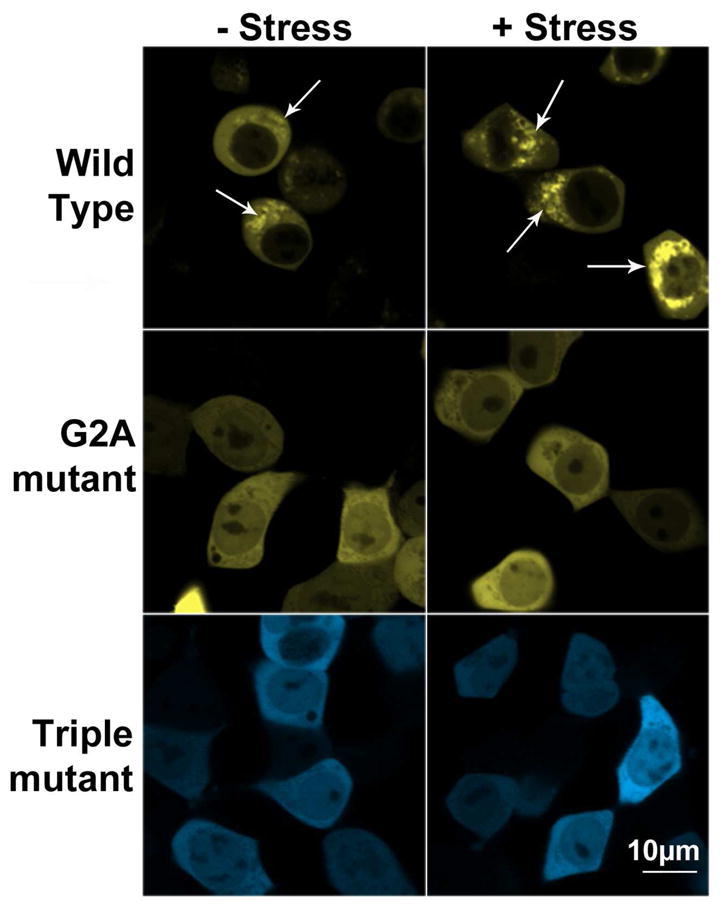
Confocal microscopy of HEK293 cells expressing PAFAH-II fusions before and after stress. Representative images showing WT-PAFAH-II-YFP, G2A-PAFAH-II-YFP, or Triple-mutant-PAFAH-II-CFP cellular location. Left column images are of unstressed cells, right column images are of cells stressed with 80 μM cumene hydroperoxide for 2 h. WT-PAFAH-II-YFP is evenly distributed in the cytoplasm and cytoplasmic membranes prior to stress and becomes more heavily membrane bound after stress (white arrows). Both mutants were completely cytoplasmic (and nucleoplasmic) prior to and after stress.
Additionally, three of the five predicted membrane binding hydrophobic residues (L327, I328, and F331) were mutated to serine to characterize the role of the predicted membrane-binding interface. Unlike the wild-type enzyme, the Triple-mutant-PAFAH-II-CFP fusion was found to be cytosolic and to a smaller degree, nucleoplasmic, prior to stress (Figure 3). Furthermore, it failed to localize to cytoplasmic membranes once treated with cumene hydroperoxide, much like the behavior of the G2A-mutant (Figure 3). Localization experiments with single hydrophobic patch PAFAH-II mutants (L327S, I328S and F331S) also gave similar results as the triple mutant and were cytosolic and partially nuclear (Figure S2 of the Supporting Information). To examine whether the fluorescent protein tags influenced the trafficking observed between the wild-type (YFP tag) and the Triple-mutant (CFP tag), the location of non-fused YFP and CFP proteins were observed by confocal microscopy. The data confirm that both CFP and YFP tags behave identically and remain evenly distributed in the cytosol, both under stressed and unstressed conditions (data not shown). In addition, the G2A-mutant was cloned to have CFP as the C-terminal fusion. Localization experiments with that construct show the same distribution as was seen with the G2A-PAFAH-II-YFP construct (data not shown).
To show that the confocal images depicting the localization of PAFAH-II in HEK293 cells (Figure 3) were representative of the whole population of cells, statistical analysis of PAFAH-II localization in a selection of random cells was preformed (see Experimental Procedures). For the WT-PAFAH-II construct, membrane-bound phenotype was present in 94.2 ± 5.4 % of the total cells. For the G2A-mutant, only 3.5 ± 1.2% of total cells had a detectable membrane bound fraction of PAFAH-II, while 8.5 ± 2.1% of total cells expressing the Triple-mutant, displayed a detectable membrane-bound enzyme.
Moreover, total exogenous PAFAH-II expression was tested to demonstrate that accumulation of the WT-PAFAH-II in the cytoplasmic membranes was a direct result of enzyme trafficking, rather than an increase in protein expression levels. Western blot analysis using GFP antibodies, of all three PAFAH-II constructs clearly showed that expression levels remained similar before and after stress (Figure 4A, B). It is also important to point out that no detectable amount of endogenous PAFAH-II was observed in non-transfected HEK293 cells prior to or after treatment with the oxidative stressor when probed with an antibody against PAFAH-II (Figure 4C).
Figure 4.

PAFAH-II expression levels. (A) Representative GFP Western blot of HEK293 lysates expressing PAFAH-II constructs before and after treatment with 80 μM cumene hydroperoxide as an oxidative stressor. GFP antibodies are suitable for detection of both YFP and CFP proteins. (B) PAFAH-II construct expression in HEK293 cell lysates before and after stress, measured by relative luminescence intensity and subsequent densitometry analysis from four independent GFP Western blots. (C) Using a PAFAH-II antibody, HEK293 cells were shown to not have any detectable levels of endogenous PAFAH-II before or after stress, as seen in the “No DNA” sample (expected MW: 44 kDa). Cells expressing WT-PAFAH-II-YFP construct were used as a positive control.
Effect of PAFAH-II Mutations on Activity
To compare the activity of PAFAH-II constructs, activity assays with a physiologically relevant substrate (2-thio-PAF) were conducted. PAFAH-II activity was measured on whole cell lysates, as well as soluble and insoluble fractions. Activity assays on whole cell lysates demonstrate that WT-PAFAH-II and G2A-PAFAH-II have comparable activity after normalization of PAFAH-II concentration by Western blot. However, the activity of the Triple-mutant-PAFAH-II was reduced two fold (Figure 5A). Similar results were obtained from activity assays on the soluble fraction (data not shown). Activity assays on the insoluble fraction showed that WT-PAFAH-II was the only construct with measurable activity, both before and after stress. The WT-PAFAH-II activity in the insoluble fraction increased upon treatment with the oxidative stressor (Figure 5B). Addition of various concentrations of detergent in assay buffer did not increase insoluble PAFAH-II activity (data not shown).
Figure 5.

PAFAH-II activity assays. Relative enzyme activity (mAbs/min) was determined using the substrate 2-thio-PAF. (A) Enzymatic activity of PAFAH-II constructs in whole HEK293 cell lysates is reported from three independent experiments. (B) Relative enzymatic activity of WT-PAFAH-II-YFP from unstressed and stressed HEK293 cells are shown from the insoluble fractions of three independent experiments. The asterisk indicates a statistically significant increase in activity between unstressed and stressed samples (student t-test, p=0.028). Activity of G2A-PAFAH-II-YFP and Triple-mutant-PAFAH-II-CFP was undetectable in the insoluble fraction (data not shown). (C) Enzymatic activity of unmyristoylated, GST-tagged WT and Triple-mutant-PAFAH-II expressed and purified from E. coli are compared to show the effect of the triple mutation on enzyme activity.
To test whether the hydrophobic patch mutations were the direct cause of the activity difference, we performed activity assays on two E. coli expressed GST-fusion constructs: GST-WT-PAFAH-II and GST-Triple-mutant-PAFAH-II. Since both of these constructs were expressed in E. coli, both were unmyristoylated. Comparison of the GST-WT-PAFAH-II and GST-Triple-mutant-PAFAH-II at equal concentrations (as judged by BSA and SDS-PAGE gel analysis) showed that 2-thio-PAF hydrolysis was more than 5-fold lower for the Triple-mutant as compared to the wild-type (Figure 5C). Similar results were seen when the GST tag was cleaved from both constructs (data not shown). These results indicate that absence of the hydrophobic residues was the sole reason for a decreased activity of the triple mutant in vivo.
WT-PAFAH-II Localizes to the ER and Golgi
Confocal images showed that WT-PAFAH-II-YFP accumulated in organelles enveloping the nucleus. These structures were reminiscent of either membranes of the ER or Golgi apparatus. To further characterize PAFAH-II localization, RFP fluorescently tagged ER and Golgi resident proteins, calreticulin and N-acetylgalactosaminyl-transferase-2, respectively, were separately used in colocalization studies with our fluorescent protein fusions of PAFAH-II. For both ER and Golgi experiments, partial colocalization of these organelles with the WT-PAFAH-II-YFP was observed prior to oxidative stress. A dramatic increase in colocalization signal upon oxidative stress indicated that the WT-PAFAH-II traffics to the ER and Golgi membranes (Figures 6 and 7). In contrast, the G2A -mutant and the Triple-mutant were unable to localize to the membranes of the ER and Golgi, and remained cytosolic. The WT-PAFAH-II colocalization images chosen in Figures 6 and 7 were representative of about half of the cell population. The other half (in both ER and Golgi colocalization experiments) only had partial PAFAH-II localization in ER or Golgi, pointing to the fact that the enzyme that was not localized in a labeled organelle (e.g., Golgi) was found in the organelle that was unlabeled in those experiments (e.g., ER) (Figure S3 of the Supporting Information). These data indicate that PAFAH-II traffics to both the ER and Golgi, and does not have a detectable preference for one organelle over the other.
Figure 6.

ER colocalization studies. Confocal images of HEK293 cells infected with an ER marker (calreticulin-RFP) and subsequently transfected with WT-PAFAH-II-YFP, G2A-PAFAH-II-YFP, or Triple-mutant-PAFAH-II-CFP. Cell images are of unstressed cells and cells stressed with 80 μM cumene hydroperoxide for 2 h. WT-PAFAH-II-YFP co-localizes with ER marker-RFP (white arrows). Both YFP/CFP (labeled as PAFAH-II) and RFP (labeled as ER) channels are shown separately, as well as a merge image.
Figure 7.

Golgi colocalization studies. Confocal images of HEK293 cells infected with a Golgi marker (N-acetylgalactosaminyltransferase-2-RFP) and subsequently transfected with WT-PAFAH-II-YFP, G2A-PAFAH-II-YFP, or Triple-mutant-PAFAH-II-CFP. Cell images are of unstressed cells and cells stressed with 80 μM cumene hydroperoxide for 2 h. WT-PAFAH-II-YFP co-localizes with Golgi marker-RFP (white arrows). Both YFP/CFP (labeled as PAFAH-II) and RFP (labeled as ER) channels are shown separately, as well as a merge image.
DISCUSSION
Significant efforts have been made to elucidate the physiological substrate and to understand the function and larger role of PAFAH-II. However, less focus has been placed on the structural features involved in PAFAH-II binding to membranes in response to oxidative stress and on its precise cellular location. Here we show that PAFAH-II must have both the myristoyl group and intact hydrophobic residues in helix 327–331 to order to associate with the membrane in response to oxidative stress within human embryonic kidney cells (Figure 3). In addition, our work shows that following oxidative stress this enzyme associates with the membranes of the ER and Golgi (Figures 6 and 7). This information will help in elucidating the physiological substrate for this enzyme and pinpoint more specific roles that PAFAH-II plays in the health of cells and disease.
Enzyme functional studies are often advanced when reliable structural models of the protein are available. Despite our success in solving the structure of pPAFAH (13), PAFAH-II has proven to be a difficult enzyme to crystallize; thus, homology modeling has been necessary to provide a working model. Although an earlier homology model of PAFAH-II was generated using a lipase structure from Streptomyces exfoliatus (20, 21) as a template, this alignment produced an imperfect model, due to <10% sequence identity between the two enzymes. In contrast, the new homology model of PAFAH-II presented in this study provides a reliable and validated structural model as a starting point to assess enzyme function. Using the pPAFAH structure as a template was the first key step in achieving a reliable model of PAFAH-II. The pPAFAH template is a closer homologue of PAFAH-II, with 43% sequence identity and 62% similarity. Additionally, the modeling approach that utilized a secondary structure influenced sequence alignment (SALIGN module of MODELLER) gave a robust model that was stable to geometry optimization refinement. Finally, the experiments presented in this study have begun to test the structural model of PAFAH-II, as well as validated our prediction of the enzyme’s association with a membrane surface.
To date, several functional studies of PAFAH-II have revealed the role it plays in oxidative stress survival by scavenging oxidatively-fragmented phospholipids (2, 3, 10). We have expanded on these initial studies by providing a model of structural aspects of PAFAH-II that are responsible for the enzyme association with the membrane during oxidative stress. Three regions of PAFAH-II were predicted to be in close proximity to the membrane using our homology model and the OPM database methods (Figure 2): the N-terminus and two α-helices rich in hydrophobic residues (residues L76 and L79 in the first helix and residues L327, I328 and F331 in the second helix). The accuracy of our model is supported by the fact that both unmyristoylated PAFAH-II and pPAFAH are predicted to bind the membrane via homologous regions, with a nearly identical orientation and penetration depth. However, it is intriguing to note that PAFAH-II is predicted to bind to membranes less favorably than pPAFAH (interaction energy of −7 kcal/mol and −14 kcal/mol, respectively). We believe that this difference in binding energies is due, in part, to the fact that the myristoyl group of PAFAH-II was not included in the OPM analysis since it was not part of the homology model. Future work is directed at predicting the change of interaction energy including the N-terminal myristoylation.
Since PAFAH-II is myristoylated, the proximity of the N-terminus to the membrane surface led us to hypothesize the possible involvement of the myristoyl group in membrane anchoring. However, work on other myristoylated proteins has demonstrated that the presence of the myristoyl group is not sufficient for stable membrane binding and that some other structural features are necessary (12). Thus, we examined the role of both the myristoyl group and the hydrophobic helix containing L327, I328 and F331 in PAFAH-II membrane localization. As will be discussed below, the localization studies unequivocally show that the absence of either the myristoyl group or the three hydrophobic residues was sufficient to prevent ROS mediated localization of PAFAH-II to the cytoplasmic membranes of cells. To achieve this goal, we expressed six different constructs of PAFAH-II in mammalian cells: wild-type, G2A (unmyristoylated) mutant, the hydrophobic patch Triple-mutant (L327S/I328S/F331S), as well as single mutations of these three hydrophobic residues. Based on our confocal data, the WT-PAFAH-II was found to be both membrane-bound and cytoplasmic under unstressed conditions (Figure 3). This result is consistent with the previous fractionation studies and immunoblot analyses that demonstrated a nearly equal presence of cytoplasmic and membrane associated PAFAH-II in unstressed cells (2,9). A clearly visible increase in the amounts of membrane bound PAFAH-II upon oxidative stress is also consistent with the previous results.
In contrast to the wild-type enzyme, the ability to become membrane associated was fully abrogated in the unmyristoylated G2A-mutant, the myristoylated Triple-mutant (Figure 3), as well as the myristoylated single mutants (L327S, I328S and F331S, Supporting Figure S2). We conclude that both the myristoyl group and the hydrophobic residues are necessary for proper enzyme association with the membrane.
Our quantitative comparison of expression levels (Figure 4) for each construct pre- and post-stress has convincingly shown that total PAFAH-II levels do not change in response to oxidative stress. Therefore, accumulation of myristoylated WT-PAFAH-II in the cytoplasmic membranes upon oxidative stress was solely due to changes in enzyme location and was not due to an increase in PAFAH-II expression levels. We also compared the enzymatic function of our PAFAH-II variants by assessing their ability to catalyze the hydrolysis of the substrate 2-thio-PAF. Our results showed that the activity of the G2A-mutant was indistinguishable from that of the WT-PAFAH-II in whole cell lysate (Figure 5A) or the soluble fraction (data not shown). Previous reports had suggested that the myristoyl group was not important for membrane association since unmyristoylated PAFAH-II had full activity in the soluble fractions (22). Our combined approach that monitored both PAFAH-II activity and localization, demonstrated that the myristoyl group is not needed for catalytic activity, but is absolutely necessary for enzyme association with the membrane where it is believed to function.
Interestingly, the activity of the Triple-mutant in whole cell lysates (Figure 5A) and in the soluble fraction (data not shown) is 2-fold lower compared to the wild-type and the G2A-mutant. This difference of activity is likely due to differential enzyme partitioning of the wild-type vs. the Triple-mutant enzyme into aggregated substrate used in our thio-PAF assay. In order to further investigate why the Triple-mutant has a decreased activity, we also compared the activity of the wild-type and Triple-mutant-PAFAH-II, expressed and purified from E. coli. Since myristoylation does not naturally occur in prokaryotes, neither of these proteins contained a myristoyl group, which allowed us to directly compare the effect of only the Triple-mutant on catalysis, and do so with enzymes that were purified to homogeneity. The activity of the E. coli expressed Triple-mutant was 5-fold lower than that of the wild-type (Figure 5C), thereby demonstrating that the hydrophobic mutations were the cause of the activity decrease observed in the mammalian cells. However, the activity decrease of the Triple-mutant was emphasized when the myristoyl group was not present, as was the case for E. coli expressed protein. This suggests that both the myristoyl group and hydrophobic patch are important for properly positioning the enzyme so it can access its substrate.
It is noteworthy that only the WT-PAFAH-II had measurable activity in the insoluble fraction (Figure 5B). These results, yet again, confirm that only myristoylated PAFAH-II with an intact hydrophobic patch is able to associate with the membrane. Also, the activity of WT-PAFAH-II increased in the insoluble fraction upon oxidative stress, confirming that PAFAH-II becomes more heavily membrane bound upon oxidative stress conditions. It is surprising that the increase in activity, though statistically significant, is small. However, the small magnitude of this increase may be due to the reversibility of the oxidative stress response. Moreover, this small increase and the rather low PAFAH-II activity in the insoluble fraction is certainly compounded by difficulties in measuring protein activity of membrane-associated proteins with this particular assay.
Until now, the precise cellular location of PAFAH-II in the cell was not known. Immunostaining experiments in MDBK cells displayed punctate patterns of PAFAH-II location and were hypothesized to be in the ER. However, ER-specific labeling was not previously carried out (10). In our present work, we monitored the location of fluorescently labeled PAFAH-II in relation to RFP-tagged ER and Golgi resident proteins. Our co-localization experiments show that WT-PAFAH-II resides both in the cytosol and in the cytoplasm-facing membranes of the ER and the Golgi apparatus before and upon oxidative stress (Figures 6 and 7). Both ER and Golgi are key cellular sites for lipid synthesis and processing, and it seems appropriate that PAFAH-II would become more concentrated in these cellular membranes after oxidative stress, where the enzyme could then function to scavenge oxidatively-damaged phospholipids.
Knowing the precise location of PAFAH-II in the cell allows for better understanding of its substrates. Even though PAFAH-II has been shown to hydrolyze PAF, a growing number of reports suggest that oxidized and fragmented phospholipids are the most relevant physiological substrates of PAFAH-II (1, 7, 11). Interestingly, alkylacetylglycerol choline phosphotransferase, an enzyme involved in the final step of PAF synthesis, is located in the outer leaflet of the ER (23). The results of our localization studies place PAFAH-II in the outer leaflet membranes of the ER, both before and after oxidative stress. Therefore, it is reasonable to argue that PAF may be a physiologically important substrate. ER localization would allow PAFAH-II to access newly synthesized PAF molecules and to potentially serve as a control mechanism in keeping PAF levels in check.
While structural requirements for PAFAH-II binding to the membrane have been examined here, the importance of the second hydrophobic helix (containing L76 and L79) has yet to be determined. Additionally, the overall mechanism that explains why PAFAH-II is found both in the cytosol and in the membranes is still unknown. A myristoyl-switch regulatory mechanism could allow the exposure of the myristoyl group upon oxidative stress. Other myristoyl-switch proteins are known to become membrane-bound due to binding of small molecules or calcium to the protein (i.e. ARFs and recoverin) or due to post-translational modifications (nitrosylation, glutathionylation, etc), leading to the exposure of the myristoyl group (24–26). We are currently probing differences between cytoplasmic and membrane bound PAFAH-II to potentially identify a trigger that controls how the myristoylated PAFAH-II transforms from a soluble to a membrane-bound protein.
In summary, we generated a PAFAH-II homology model that was used to develop a prediction of PAFAH-II membrane association and to understand the structural features responsible for this interaction. Mutagenesis and localization experiments in mammalian cells allowed us to test our model and to confirm that both the myristoyl group and the hydrophobic patch residues are required for PAFAH-II association with cytosolic facing membranes. Finally, it has been demonstrated by both localization results and enzymatic assay that WT-PAFAH-II is localized in the cytosol, as well as in the outer leaflet membranes of the ER and Golgi apparatus, and the enzyme becomes more concentrated in those membranes upon oxidative stress.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Jeffrey Caplan at the University of Delaware Core Imaging Facility for his help with confocal image data collection and analysis. The authors are thankful to Dr. Prabha Srinivasan and Dr. Damien Thévenin for helpful discussions and editing of this manuscript, as well as Dr. John Koh and Dr. Daniel Simmons for use of their lab space and equipment. We also thank Dr. Andrei Lomize for predicting the orientation of PAFAH-II on a bilayer using methods of the OPM database. The authors are grateful to Sam Linton for generation of the triple mutation of PAFAH-II.
Funding Sources: This work was supported by NIH grants 5P20RR015588 from the National Center for Research Resources and 5R01HL084366 from the National Heart, Lung, and Blood Institute to BJB. Support for Bioimaging work was also provided in part by NIH NCRR IDeA Network of Biomedical Research Excellence (INBRE) grant 2P20RR0164720.
ABBREVIATIONS
- CFP
cyan fluorescent protein
- DOPE
discrete optimized potential energy
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- OPM
orientations of proteins in membranes
- PAFAH-II
platelet-activating factor acetylhydrolase type-II
- PAF
platelet-activating factor
- pPAFAH
plasma PAFAH
- ROS
reactive oxygen species
- RFP
red fluorescent protein
- Triple-mutant
triple mutation L327S/I328S/F331S of the membrane binding surface of PAFAH-II
- YFP
yellow fluorescent protein
- WT
wild-type
Footnotes
Figure S1. Amino acid sequence alignment of PAFAH-II and pPAFAH. Figure S2: Localization of hydrophobic patch single mutants of PAFAH-II. Figure S3: PAFAH-II partial localization in the ER and Golgi. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rice SQ, Southan C, Boyd HF, Terrett JA, MacPhee CH, Moores K, Gloger IS, Tew DG. Expression, purification and characterization of a human serine-dependent phospholipase A2 with high specificity for oxidized phospholipids and platelet activating factor. Biochem J. 1998;330(Pt 3):1309–1315. doi: 10.1042/bj3301309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marques M, Pei Y, Southall MD, Johnston JM, Arai H, Aoki J, Inoue T, Seltmann H, Zouboulis CC, Travers JB. Identification of platelet-activating factor acetylhydrolase II in human skin. J Invest Dermatol. 2002;119:913–919. doi: 10.1046/j.1523-1747.2002.01859.x. [DOI] [PubMed] [Google Scholar]
- 3.Hattori K, Adachi H, Matsuzawa A, Yamamoto K, Tsujimoto M, Aoki J, Hattori M, Arai H, Inoue K. cDNA cloning and expression of intracellular platelet-activating factor (PAF) acetylhydrolase II. Its homology with plasma PAF acetylhydrolase. J Biol Chem. 1996;271:33032–33038. doi: 10.1074/jbc.271.51.33032. [DOI] [PubMed] [Google Scholar]
- 4.Hattori K, Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Purification and characterization of platelet-activating factor acetylhydrolase II from bovine liver cytosol. J Biol Chem. 1995;270:22308–22313. doi: 10.1074/jbc.270.38.22308. [DOI] [PubMed] [Google Scholar]
- 5.Tjoelker LW, Stafforini DM. Platelet-activating factor acetylhydrolases in health and disease. Biochim Biophys Acta. 2000;1488:102–123. doi: 10.1016/s1388-1981(00)00114-1. [DOI] [PubMed] [Google Scholar]
- 6.Stafforini DM, McIntyre TM, Zimmerman GA, Prescott SM. Platelet-activating factor acetylhydrolases. J Biol Chem. 1997;272:17895–17898. doi: 10.1074/jbc.272.29.17895. [DOI] [PubMed] [Google Scholar]
- 7.Kono N, Inoue T, Yoshida Y, Sato H, Matsusue T, Itabe H, Niki E, Aoki J, Arai H. Protection against oxidative stress-induced hepatic injury by intracellular type II platelet-activating factor acetylhydrolase by metabolism of oxidized phospholipids in vivo. J Biol Chem. 2008;283:1628–1636. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- 8.Pegorier S, Stengel D, Durand H, Croset M, Ninio E. Oxidized phospholipid: POVPC binds to platelet-activating-factor receptor on human macrophages. Implications in atherosclerosis. Atherosclerosis. 2006;188:433–443. doi: 10.1016/j.atherosclerosis.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Arai H. Platelet-activating factor acetylhydrolase. Prostaglandins Other Lipid Mediat. 2002;68–69:83–94. doi: 10.1016/s0090-6980(02)00023-0. [DOI] [PubMed] [Google Scholar]
- 10.Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. Protection against oxidative stress-induced cell death by intracellular platelet-activating factor-acetylhydrolase II. J Biol Chem. 1997;272:32315–32320. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- 11.Foulks JM, Weyrich AS, Zimmerman GA, McIntyre TM. A yeast PAF acetylhydrolase ortholog suppresses oxidative death. Free Radic Biol Med. 2008;45:434–442. doi: 10.1016/j.freeradbiomed.2008.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 13.Samanta U, Bahnson BJ. Crystal structure of human plasma platelet-activating factor acetylhydrolase: structural implication to lipoprotein binding and catalysis. J Biol Chem. 2008;283:31617–31624. doi: 10.1074/jbc.M804750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 15.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 17.Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 18.Rohman M, Harrison-Lavoie KJ. Separation of copurifying GroEL from glutathione-S-transferase fusion proteins. Protein Expr Purif. 2000;20:45–47. doi: 10.1006/prep.2000.1271. [DOI] [PubMed] [Google Scholar]
- 19.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. Positioning of proteins in membranes: a computational approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei Y, Swenson L, Castro C, Derewenda U, Minor W, Arai H, Aoki J, Inoue K, Servin-Gonzalez L, Derewenda ZS. Structure of a microbial homologue of mammalian platelet-activating factor acetylhydrolases: Streptomyces exfoliatus lipase at 1.9 A resolution. Structure. 1998;6:511–519. doi: 10.1016/s0969-2126(98)00052-5. [DOI] [PubMed] [Google Scholar]
- 21.Derewenda ZS, Ho YS. PAF-acetylhydrolases. Biochim Biophys Acta. 1999;1441:229–236. doi: 10.1016/s1388-1981(99)00158-4. [DOI] [PubMed] [Google Scholar]
- 22.Bae K, Longobardi L, Karasawa K, Malone B, Inoue T, Aoki J, Arai H, Inoue K, Lee T. Platelet-activating factor (PAF)-dependent transacetylase and its relationship with PAF acetylhydrolases. J Biol Chem. 2000;275:26704–26709. doi: 10.1074/jbc.M003951200. [DOI] [PubMed] [Google Scholar]
- 23.Snyder F. CDP-choline:alkylacetylglycerol cholinephosphotransferase catalyzes the final step in the de novo synthesis of platelet-activating factor. Biochim Biophys Acta. 1997;1348:111–116. doi: 10.1016/s0005-2760(97)00109-4. [DOI] [PubMed] [Google Scholar]
- 24.Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-myristoylation. J Biol Chem. 2001;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- 25.MacRitchie AN, Gardner AA, Prescott SM, Stafforini DM. Molecular basis for susceptibility of plasma platelet-activating factor acetylhydrolase to oxidative inactivation. FASEB J. 2007;21:1164–1176. doi: 10.1096/fj.06-6743com. [DOI] [PubMed] [Google Scholar]
- 26.Ames JB, Ishima R, Tanaka T, Gordon JI, Stryer L, Ikura M. Molecular mechanics of calcium-myristoyl switches. Nature. 1997;389:198–202. doi: 10.1038/38310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.