

Abstract
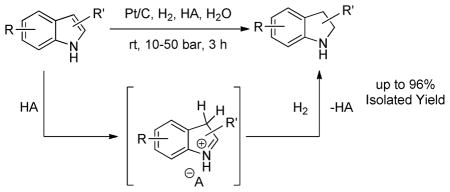
An environmentally benign procedure for the hydrogenation of unprotected indoles is described. The hydrogenation reaction is catalyzed by Pt/C and activated by p-toluenesulfonic acid in water as a solvent. The efficacy of the method is illustrated by the hydrogenation of a variety of substituted indoles to their corresponding indolines which were obtained in excellent yields.
Hydrogenation of indole is a difficult task due to the highly resonance stabilized aromatic nucleus.1 In addition, the hydrogenated product, a cyclic secondary amine, could poison the metal catalyst and hinder the progress of the reaction2 and the product indoline can undergo further hydrogenation to form byproducts up to octahydroindole. Over the years, extended efforts have been devoted to the regioselective hydrogenation of indoles to indolines. The most environmentally benign tool for this reaction is heterogeneous catalytic hydrogenation.3 However, challenges for the use of this truly green tool for the selective hydrogenation of indole still remain.4 One of the most effective methods for this transformation utilizes NaBH3CN, which gives the desired indolines in good yields. This method has its major drawbacks; it uses superstoichiometric amounts (up to 3 eq.) of the hydride reagent and the reaction generates large amounts of toxic byproducts such as cyanides.5 Other reagents used for this transformation include triethylamine/borane,6 triethylsilane/trifluoroacetic acid,7 or PhSiH3.8 All of these methods use stoichiometric amounts of reagents and generate extensive amount of waste. The reduction of indoles to indolines was also reported by catalytic hydrogenation. The use of hydrogen has advantages over H-transfer reagents; most importantly that no waste is generated after the reaction from hydrogen. The major challenge in catalytic hydrogenation of indoles is that it often proceeds further than the indoline stage or occurs at positions other than the desired 2,3-double bond.9 Homogeneous catalytic methods for the hydrogenation of N-protected indoles using hydrogen involve the use of Rh,10 Ru11 and Ir12 complexes.
Heterogeneous catalysts are easy to store and handle, usually are not moisture sensitive and are easy to separate and recycle.13 Despite some progress in selective hydrogenation of indoles to indolines by heterogeneous catalysis, the available examples are mostly limited to N-protected indoles.14 The only available report, to the best of our knowledge, for the hydrogenation of unprotected indole by catalytic heterogeneous hydrogenation using hydrogen gas requires very harsh conditions such as hydrogen pressure of 150 bar and temperature of 227 °C giving moderate yield.15
Under acidic conditions, indole can be protonated at the C-3 position generating an iminium ion with disrupted aromaticity.16 This iminium ion could be efficiently hydrogenated by homogeneous catalytic hydrogenation to obtain asymmetric indolines using the Pd(OCOCF3)2 catalyst.17 The method appeared to be efficient; however, it was limited to 2-substituted indoles that are less prone to polymerization. Thus, while the application of a co-acid is a reasonable idea, it also raises further problems. As indole is a highly activated aromatic compound, even weak electrophiles initiate polymerization that results in significant amount of byproducts.18 The other problem is overhydrogenation, mainly to octahydroindole.
The topic has undoubtedly attracted significant attention due to the importance of the indoline moiety that can be found in a number of naturally occurring, biologically active and pharmaceutically important compounds.19 Aside from the above discussed hydrogenations,10–12,14,15,17 indolines can be synthesized by direct ring closure through amination reactions.20
Continuing our efforts towards developing sustainable organic transformations, herein we report a robust methodology for highly regioselective, Brønsted acid-induced heterogeneous catalytic hydrogenation of unprotected indoles (Scheme 1). The reaction is catalyzed by the inexpensive and commercially available Pt/C catalyst and occurs under mild conditions such as moderate hydrogen pressure, at room temperature, generally requires short reaction times and most importantly utilizes water as a solvent.
Scheme 1.

Synthetic strategy for the catalytic heterogeneous hydrogenation of indoles in water.
Hydrogenation of indole was selected as a model reaction. The reaction was carried out in ethanol using a Pt/C catalyst and hydrogen gas. The reaction revealed significant selectivity issues such as over hydrogenation to octahydroindole and polymerization. As our goal was to develop a green reduction to produce indolines from unprotected indoles, we decided to further explore this reaction. We adopted the use of the acid additive even though that method had failed for unprotected indoles, since protonation appears to be a reasonable strategy to disrupt the aromatic system of indole. In order to determine optimum conditions, a detailed investigation of the effect of various experimental variables was undertaken. The results are presented in Table 1.
Table 1.
The synthesis of indoline from indole under various experimental conditions
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | HA | solvent | H2 (bar) | time (h) | conversion (%) | selectivity (%) |
| 1 | Pt/C | - | EtOH | 50 | 2 | 2 | 100/0 |
| 2 | Pt/C | TFA | EtOH | 50 | 2 | 62 | 77/23 |
| 3 | Pt/C | TFA | MeOH | 50 | 2 | 66 | 76/24 |
| 4 | Pt/C | TFA | Toluene | 50 | 2 | 53 | 38/62 |
| 5 | Pt/C | TFA | Hexane | 50 | 2 | 61 | 13/87 |
| 6 | Pt/C | TFA | CF3CH2OH | 50 | 2 | 100 | 71/29 |
| 7 | Pt/C | TFA | H2O | 50 | 2 | 67 | 100/0 |
| 8 | Pt/C | CSA | H2O | 50 | 2 | 100 | 81/19 |
| 9 | Pt/C | p-TSA | H2O | 50 | 2 | 100 | 87/13 |
| 10 | Pt/C | HCOOH | H2O | 50 | 2 | 52 | 85/15 |
| 11 | Pt/C | CF3SO3H | H2O | 50 | 2 | 78 | 100/0 |
| 12 | Pt/Al2O3 | p-TSA | H2O | 50 | 2 | 88 | 100/0 |
| 13 | Pd/C | p-TSA | H2O | 50 | 2 | 56 | 100/0 |
| 14 | Pt/C | p-TSA | H2O | 10 | 2 | 36 | 100/0 |
| 15 | Pt/C | p-TSA | H2O | 20 | 2 | 56 | 100/0 |
| 16 | Pt/C | p-TSA | H2O | 30 | 2 | 78 | 100/0 |
| 17 | Pt/C | p-TSA | H2O | 40 | 2 | 82 | 100/0 |
| 18 | Pt/C | p-TSA | H2O | 30 | 3 | 100 | 100/0 |
| 19 | Pt/C | - | H2O | 30 | 3 | 3 | 100/0 |
Conversions and selectivities were determined by GC. Major byproducts include octahydroindole, dimer and trimer of indole and their corresponding hydrogenated products. TFA = trifluoroacetic acid. CSA = camphorsulfonic acid. p-TSA = p-toluenesulfonic acid.
The reactions were carried out on a 1mmol scale.
When carried out without an acid additive, the reaction yielded the product in traces (Entry 1). Using 1 eq. of trifluoroacetic acid (TFA), however, a substantial amount of the desired product was observed with octahydroindole and polymerized byproducts. When carried out in hexane, the reaction resulted in 61% conversion and 13% selectivity for the desired product. A similar phenomenon was observed in toluene, although the selectivity (38%) was higher than in hexane. The conversion and selectivity increased in ethanol (62% conversion, 77% selectivity) and methanol (66% conversion, 76% selectivity). Thus, it was concluded that the selectivity of product increased with increasing nucleophilicity of the solvent (Entries 2–5). It appears that the decreasing nucleophilicity of the medium increases the acidity of the system, thus promoting polymerization.
Therefore, in order to minimize the polymerization, it was envisioned that further increasing the nucleophilicity of the medium would alleviate the acidity of the Brønsted acid, thereby minimize the possibility of polymerization.
One obvious choice to increase solvent nucleophilicity was the selection of water as a solvent. When the reaction was carried out in aqueous medium, polymerization was completely hindered and only indoline and indole were observed (Entry 7). Then, it was decided to test different Brønsted acid additives. When camphorsulfonic acid (CSA) and p-toluenesulfonic acid (p-TSA) were used as additives (Entries 8 and 9), complete conversion was observed and the selectivity of indoline increased substantially with octahydroindole being the only byproduct. It appears that there is an acidity optimum for the reaction. The selectivity decreased when a weaker acid (formic acid) or a stronger acid (triflic acid) were used as additives (Entries 10 and 11). The use of different catalysts such as Pt/Al2O3 (Entry 12) or Pd/C (Entry 13) increased the selectivity but these were less active than the Pt/C catalyst and resulted in incomplete conversion of indole. After determining the optimum solvent and acid additive, attention was focused on optimizing the hydrogen pressure and reaction time. It was observed that the reaction proceeded with 100% selectivity even at 10 bar pressure, however, with a relatively low rate (Entry 14, 36% yield in 2 h). The optimum hydrogen pressure of 30 bar gave 100 % selectivity for indoline and quantitative conversion of indole (Entry 18). To confirm the effect of the acid additive, a reaction was carried out under the optimized conditions but without using p-TSA. As indicated, there was only 3% product formation and indole was recovered after the reaction (Entry 19). This observation confirmed the role of the acid additive. Even though the reaction is catalytic with respect to p-TSA, 1.2 equivalent of p-TSA was required to ensure short reaction times. As the reaction progresses, the indoline formed tends to decrease the concentration of free p-TSA in the solution by the formation of an ammonium ion and the reaction gets sluggish if less than 1 equivalent of p-TSA is used. This side reaction however, has a rather positive effect on the overall process; the formation of an ammonium ion occupies the lone pair of the secondary amine, therefore completely suppressing its catalyst poisoning effect that greatly inhibited the heterogeneous catalytic hydrogenation earlier. Using the optimized conditions, the method was applied to a variety of indoles with different substituents on the carbocyclic as well as the heterocyclic ring in order to study the steric and electronic effects on the outcome of the reaction. The results are summarized in Scheme 2.
Scheme 2.

Scope of the Pt/C catalyzed hydrogenation of substituted indoles in water
Determined by 1H NMR
A variety of indoles gave indolines in good yields with excellent selectivities. C-5 electron donating substituents such as 5-methyl and 5-methoxy gave the desired products in excellent yields (96 % and 95 % respectively). 5-fluoroindole also afforded the desired product in excellent yield (93 %). However, a 5-chloro substituent was not completely tolerated and the product (46 %) was obtained along with the dehalogenated derivative (42 %) as well as octahydroindole (12 %). A less active Pt/Al2O3 catalyst, however, was found to effectively catalyze the reaction with high selectivity and gave the desired product in 72 % yield. Increased catalyst loading and hydrogen pressure were required for 7-ethyl and 2-methylindole most likely because the alkyl group adjacent to the nitrogen hindered adsorption on the surface of the catalyst and resulted in a rate decrease. The rate of hydrogenation was comparatively low in the case of methyl indole-5-carboxylate probably due to the electron withdrawing nature of the substituent and also required increased catalyst loading. In the case of ethyl indole-2-carboxylate the substrate was completely deactivated. Selectivity was a major problem for N-methylindole since it was easily over reduced to the octahydro product, thus both catalyst loading and hydrogen pressure were decreased. 2,3-Disubstituted indoles required higher pressure and catalyst loading (Scheme 2). Both hexahydrocyclopentaindole and hexahydrocarbazole exclusively formed the cis products whereas 2,3-dimethylindoline was observed as a mixture of cis and trans isomers in 6:1 ratio.21 Tryptamine which contains two nitrogen atoms required 2.2 eq. p-TSA to give 2,3-dihydrotryptamine in good isolated yield (68%).
In the reaction mechanism the acid additive has a crucial role; it temporarily disrupts the aromaticity of the heterocyclic ring of the indole forming an iminium ion (Fig 1). This step occurs by p-TSA protonating the most nucleophilic C-3 position of indole. Once the iminium ion is formed, it dissolves in the aqueous medium and the solvation in water prevents further nucleophilic attack by another indole molecule thus preventing polymerization. The attack is prevented in part due to phase separation between the water soluble iminium ion and the insoluble indole. In addition, the iminium ion is solvated by water molecules that further eliminates the steric availability for subsequent attack by excess indole. The iminium ion quickly undergoes hydrogenation to form indoline. The methodology described here is a highly green synthesis design.22 The major advantages are as follows:
Figure 1.
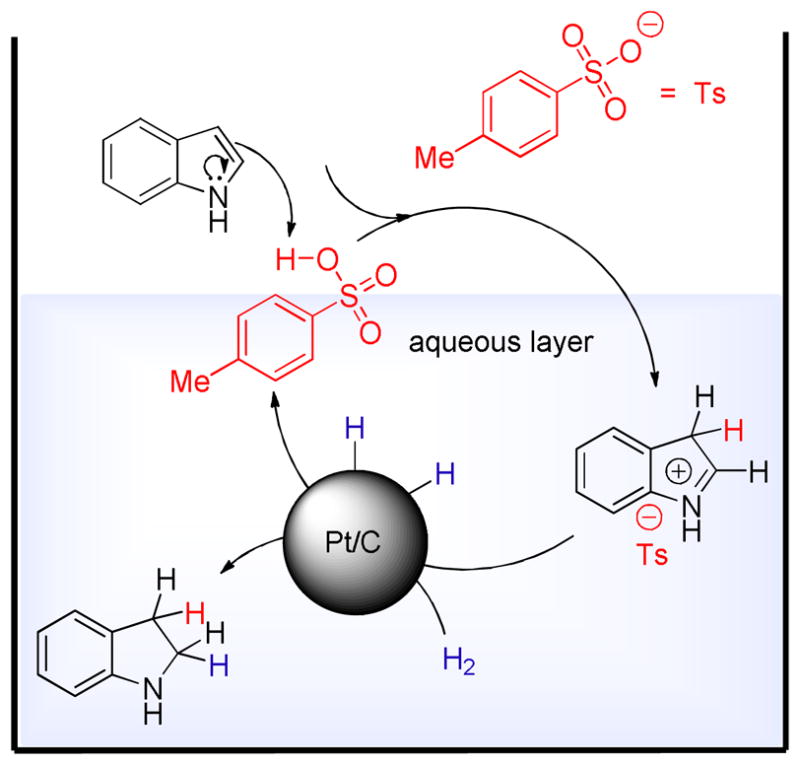
Mechanism for acid-induced Pt/C-catalyzed hydrogenation of indole to indoline in water.
(i) The reaction is carried out on unprotected indoles, which eliminates the protection and deprotection steps required in all previous attempts and the losses and waste formation associated with them.; (ii) The reaction is carried out in water, the most benign, readily available and inexpensive solvent.; (iii) The use of a heterogeneous Pt/C catalyst allows easy catalyst recovery and makes use of a continuous flow process possible.; (iv) Reactions were carried out at room temperature in short reaction times and the temperatures and hydrogen pressures in this process are significantly lower than those previously reported in heterogeneous systems.; (v) The atom economy is near 100 % in the reaction as both starting materials (indole and hydrogen) are completely used up and there is no byproduct formation.; (vi) The acid additive is not consumed in the reaction and p-TSA can be recovered and reused.; (vii) With all the above in consideration, the reaction does not generate waste.
In summary, a heterogeneous catalytic hydrogenation methodology for the direct synthesis of indolines from unprotected indoles in water was developed. In the presence of a readily available Brønsted acid such as p-toluenesulfonic acid, a broad variety of unprotected indoles were hydrogenated to corresponding indolines by a heterogeneous Pt/C catalyst. The reactions were carried out at room temperature at a moderate hydrogen pressure. Products were obtained in excellent yields in short reaction times. The process, with its highly effective, inexpensive and selective design provides a green solution to a long standing challenge, the selective hydrogenation of indoles to indolines.
Supplementary Material
Acknowledgments
Financial support provided by University of Massachusetts Boston and National Institute of Health (R-15 AG025777-03A1) is gratefully acknowledged.
Footnotes
Supporting Information Available General procedure along with spectroscopic data of all products.
References
- 1.Bird CW. Tetrahedron. 1992;48:335. [Google Scholar]
- 2.Maxted EB, Biggs MS. J Chem Soc. 1957:3844. [Google Scholar]
- 3.Nishimura S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis. Wiley; New York: 2001. [Google Scholar]
- 4.Zhou YG. Acc Chem Res. 2007;40:1357. doi: 10.1021/ar700094b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Gribble GW, Lord PD, Skotnicki J, Dietz SE, Eaton JT, Johnson J. J Am Chem Soc. 1974;96:7812. [Google Scholar]; (b) Gribble GW, Hoffman JH. Synthesis. 1977:859. [Google Scholar]
- 6.Berger JG. Synthesis. 1974. p. 508. [Google Scholar]
- 7.Lanzilotti AE, Littell R, Fanshawe WJ, McKenzie TC, Lovell FM. J Org Chem. 1979;44:4809. [Google Scholar]
- 8.Tan M, Zhang Y. Tetrahedron Lett. 2009;50:4912. doi: 10.1016/j.tetlet.2009.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson B. Chem Rev. 1969;69:785. doi: 10.1021/cr60262a003. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kuwano R, Sato K, Kurokawa T, Karube D, Ito Y. J Am Chem Soc. 2000;122:7614. [Google Scholar]; (b) Kuwano R, Kaneda K, Ito T, Sato K, Kurokawa T, Ito T. Org Lett. 2004;6:2213. doi: 10.1021/ol049317k. [DOI] [PubMed] [Google Scholar]
- 11.Kuwano R, Kashiwabara M. Org Lett. 2006;8:2653. doi: 10.1021/ol061039x. [DOI] [PubMed] [Google Scholar]
- 12.Baeza A, Pfaltz A. Chem Eur J. 2010;16:2036. doi: 10.1002/chem.200903105. [DOI] [PubMed] [Google Scholar]
- 13.(a) Horvath I, editor. Encyclopedia of Catalysis. Wiley; New York: 2003. [Google Scholar]; (b) Kulkarni A, Török B. Curr Org Synth. 2011;8:187. [Google Scholar]
- 14.(a) Gilchrist TL, Graham K. Tetrahedron. 1997;53:791. [Google Scholar]; (b) Chandrasekhar S, Basu D, Reddy CR. Synthesis. 2007. p. 1509. [Google Scholar]
- 15.Shaw JE, Stapp PR. J Heterocyclic Chem. 1987;24:1477. [Google Scholar]
- 16.(a) Chen CB, Wang XF, Cao YJ, Cheng HG, Xiao WJ. J Org Chem. 2009;74:3532. doi: 10.1021/jo900104r. [DOI] [PubMed] [Google Scholar]; (b) Gilchrist TL. Heterocyclic Chemistry. 3. Pearson: Essex; 1997. p. 240. [Google Scholar]
- 17.(a) Wang DS, Chen QA, Li W, Yu CB, Zhou YG, Zhang X. J Am Chem Soc. 2010;132:8909. doi: 10.1021/ja103668q. [DOI] [PubMed] [Google Scholar]; (b) Wang DS, Tang J, Zhou YG, Chen MW, Yu CB, Duan Y, Jiang GF. Chem Sci. 2011;2:803. [Google Scholar]
- 18.Ishii H, Murakami K, Sakurada E, Hosoya K, Murakami Y. J Chem Soc, Perkin Trans 1. 1988:2377. [Google Scholar]
- 19.(a) Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. Chem Rev. 1997;97:787. doi: 10.1021/cr960095g. [DOI] [PubMed] [Google Scholar]; (b) Dounay ZAB, Overman LE, Wrobleski AD. J Am Chem Soc. 2005;127:10186. doi: 10.1021/ja0533895. [DOI] [PubMed] [Google Scholar]; (c) Natesh R, Schwager SLU, Evans HR, Sturrock ED, Acharya KR. Biochemistry. 2004;43:8718. doi: 10.1021/bi049480n. [DOI] [PubMed] [Google Scholar]; (d) Gruenfeld N, Stanton JL, Yuan AM, Ebetino FH, Browne JJ, Gude C, Huebner CF. J Med Chem. 1983;26:1277. doi: 10.1021/jm00363a012. [DOI] [PubMed] [Google Scholar]
- 20.(a) Anas S, Kagan HB. Tetrahedron: Asymmetry. 2009;20:2193. [Google Scholar]; (b) Liu D, Zhao G, Xiang L. Eur J Org Chem. 2010:3975. [Google Scholar]; (c) Viswanathan R, Prabhakaran EN, Plotkin MA, Johnston JN. J Am Chem Soc. 2003;125:163. doi: 10.1021/ja0284308. [DOI] [PubMed] [Google Scholar]
- 21.Please see the supporting information for details.
- 22.Anastas PT, Warner JC. Green Chemistry: Theory and Practice. Oxford University Press; Oxford: 1998. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.