

Abstract
Edman degradation remains the primary method for determining the sequence of proteins. In this study, accelerator mass spectrometry was used to determine the N-terminal sequence of glutathione S-transferase at the attomole level with zeptomole precision using a tracer of 14C. The transgenic transferase was labeled by growing transformed Escherichia coli on [14C]glucose and purified by microaffinity chromatography. An internal standard of peptides on a solid phase synthesized to release approximately equal amounts of all known amino acids with each cycle were found to increase yield of gas phase sequencing reactions and subsequent semimicrobore HPLC as did a lactoglobulin carrier. This method is applicable to the sequencing of proteins from cell culture and illustrates a path to more general methods for determining N-terminal sequences with high sensitivity.
Characterization and identification of proteins remains critically important in the biological sciences (1). In high throughput biology, proteomics involves separation of complex biological mixtures by two-dimensional (2D) gel electrophoresis, characterization of all spots by high sensitivity tools including mass spectrometry and Edman degradation sequencing, and assignment of biological function based on powerful database searches and protein profiling patterns that reflect the cellular gene regulation (2). This trend will be accelerated as genomic sequences become available with increasing speed. Several mass spectral techniques including “peptide fingerprinting” by matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF; ref. 3) and “sequence tag search” by nano-electrospray ionization (ESI) have become the essential tools for rapid and sensitive screening of known proteins (4). However, if a protein of interest is not found in the database, amino acid sequence information must be obtained for further biological and biochemical studies. Primary sequence data can be obtained by direct N-terminal sequence analysis of the protein or from internal peptide fragments. Although mass spectral techniques can generate sequence information at the attomole level (5, 6), de novo sequencing by MS/MS is difficult, is limited to peptides less than 20 to 25 residues long, and sometimes gives ambiguous sequence callings (e.g., isomeric residues, Leu/Ile, and isobaric amino acids, Gln/Lys). Edman degradation is still the standard and most extensively used method for sequencing an unknown protein and has the advantage of providing easily interpreted long amino acid sequences. Thus, the combination of Edman chemistry and mass spectrometry generates a powerful and versatile approach to identify proteins.
Extensive research has led to progressively more sensitive Edman sequence analysis. The first automated “spinning cup” sequencer analyzed nanomole quantities of proteins or peptides in the 1970s (7), and gas phase sequencers in the 1980s (8) and 1990s (9) provided much improved sensitivity at the low-picomole and high-femtomole level, respectively. The major sensitivity improvement of gas phase or liquid pulse Edman sequencers resulted from the introduction of the RP-HPLC for phenylthiohydantoin (PTH)-amino acid detection (10). A 2-mm-diameter C-18 column can detect low-picomole PTH-amino acids, whereas a capillary (0.3–0.5 mm in diameter) column HPLC can push the sensitivity to femtomole levels (≈300–500). Although the sensitivity gap between Edman and mass spectrometry has narrowed considerably in recent years, neither technique is able to overcome the subfemtomole barrier for unknown proteins, and both need improvement to sequence very low abundance proteins expressed in the cell.
Accelerator mass spectrometry (AMS) is a tandem spectrometry tool that counts rare atoms by eliminating molecular isobars in high energy collisions, followed by identification of individual ions by energy loss quantitation. It has been extensively used in archaeological studies and earth sciences (11). AMS can accurately measure <106 atoms [1 attomole (amol)] of 14C in a sample containing ≈1 mg of total carbon and can analyze hundreds of samples per day. Unlike liquid scintillation counting (LSC), which counts decay events of a radioisotope, AMS is a mass spectrometric technique that detects individual 14C nuclei. Over the past decade, AMS has been applied to life science studies and has increased precision and sensitivity of detection for 3H and 14C by several magnitudes (12–18). In this study, we describe an increase in Edman sequencing sensitivity by introducing AMS as a PTH-amino acid detection system.
Materials and Methods
Preparation of Labeled Protein.
Uniformly labeled [14C]glutathione S-transferase (GST) was prepared as follows. The DNA plasmid containing mouse GST Yc (19), provided by T. Bammler (University of Washington, Seattle), was used for transformation of BL21(DE3) cells from Escherichia coli. The bacteria harboring the GST vector were grown for 26 h at 37°C with shaking, in M9 minimal medium containing ampicillin (50 μg/ml). One microliter of this cell culture was transferred into 200 μl of M9 medium containing 85 μCi (1 Ci = 37 GBq) d-[U-14C]glucose (323 mCi/mmol; Amersham Pharmacia) with 350 μg of nonlabeled d-glucose. The culture was incubated at 37°C with shaking until the OD600 reached 0.5–1.0. Production of recombinant GST was then induced by the addition of isopropyl β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. The bacteria were grown for an additional 12 h at 37°C with shaking and harvested by centrifugation at 2,000 g for 5 min at 4°C. Cells were resuspended in 100 μl of cell wall disrupting reagent BugBuster (Novagen) with the nuclease Benzonase (Novagen) to reduce viscosity. Cells were gently shaken at room temperature for 20 min, and the suspension was centrifuged at 16,000 × g for 20 min at 4°C. The supernatant was applied to glutathione Sepharose 4B beads (75 μl bed volume; Sigma) in Eppendorf tubes, which were equilibrated in buffer A (50 mM Tris⋅HCl, pH 7.4/200 mM NaCl/0.5 mM DTT), and incubated for 35 min at 4°C. The suspension was centrifuged at 500 g for 5 min at 4°C, and the supernatant was discarded. The pellet was washed four times with 1 ml of buffer A. The [14C]GST was eluted three times with 50 μl of buffer B (200 mM Tris⋅HCl, pH 9.0/50 mM reduced glutathione) at 4°C and eluents were pooled. The buffer was replaced with PBS containing 0.5 mM DTT by using Microcon 10 (Millipore). The concentration of uniformly labeled [14C]GST was determined by SDS/PAGE analysis with silver-staining using nonlabeled GST as a standard. The concentration of the standard GST was determined by amino acid analysis. Scion Image (Scion, Frederick, MD) was used for this quantitative analysis. A Wallac 1409 LSC (Wallac, Gaithersburg, MD) was used to determine the specific activity.
Random Peptide-Bead Synthesis.
Peptide beads were designed to release equimolar amounts of each of the natural amino acids with each Edman cycle. Fluorenylmethoxycarbonyl-amino acid mixtures (Fmoc, 5 eq) were coupled with TentaGel NH2 resin (0.26 mmol/g; Rapp Polymere, Tübingen, Germany) using the 1,3-diisopropylcarbodiimide/1-hydroxybenzotriazole (5 eq) method (20). Fmoc groups were removed with a 25% piperidine/dimethylformamide (DMF) solution and a step-wise coupling reaction was carried out until 10-mer peptide-beads were obtained. After Fmoc removal, the random peptide-beads were washed with DMF, methanol, and dichloromethane, and dried under vacuum.
Protein Sequencing.
Automated protein sequencing was performed on a 477A sequencer equipped with a 120A HPLC system (PE Applied Biosystems). All reagents and solvents used for the sequencer were obtained from PE Applied Biosystems and checked for 14C content by AMS before use. The solution of [14C]GST was diluted to concentrations of 10–1,000 amol/μl with 0.1% TFA in water containing β-lactoglobulin (2.5 pmol/μl). The concentration of each diluted [14C]GST solution was determined with 14C measurement by AMS. Polybrene (3.0 mg) was applied to a glass filter on the reaction chamber and subjected to three precycles. Before the addition of [14C]GST, β-lactoglobulin (25 pmol) and peptide-beads (4–7 beads) were loaded on the glass filter and dried under argon gas. The sequencer program was modified so that a known amount of standard PTH-amino acids were delivered to the conversion flask each cycle before the transfer of anilinothiazolinone amino acids to the flask in addition to the amino acids from the peptide beads and lactoglobulin. HPLC fractions were collected every 30 or 60 sec into borosilicate glass culture tubes (6 × 50 mm), which were pyrolyzed before use to remove any residual carbon.
AMS Sample Preparation and Measurement.
Each sample was collected in a small glass tube and 50 μl of carbon carrier solution (40.0 mg/ml tributyrin in methanol) was added before drying in a vacuum centrifuge to yield 1.19 mg of carrier C. Three tributyrin carrier blanks were prepared with each set of fractions. The samples were graphitized for AMS as described by Vogel (21). Typical AMS measurement times were 3 min/sample, with a counting precision of 1.4–2.0% and a SD among 3–7 measurements of 1–3%. The 14C/13C ratios of unknowns were normalized to measurements of four identically prepared standards of known isotope concentration (Australian National University Sucrose; ref. 22).
Results and Discussion
Sequencing of Low Femtomole (fmol) Quantities of 14C-Labeled GST.
Intrinsically labeled [14C]GST was prepared by using an E. coli expression system and purified by affinity chromatography. The specific radioactivity of this protein was determined by LSC to be 3.04 Ci/mmol. This corresponds to a 14C/C ratio of 0.0426 mol of 14C per mol of total C for the GST. Generally, sequence information of ten residues is sufficient to obtain a good DNA primer for PCR analysis and to search protein databases. A sequence analysis of 1.76 fmol of [14C]GST was carried out for ten cycles in the presence of 50 pmol of β-lactoglobulin and four peptide-beads as carriers. Minute quantities of protein (≈1 fmol) are susceptible to nonspecific absorptive losses during sample handling and sequencing so we used β-lactoglobulin to prevent this loss. Peptide-beads with a random mixture of sequences were also applied to the reaction chamber. The PTH-amino acids generated from the peptide-beads after each Edman degradation were detected by UV monitoring, but they were invisible to AMS. Because the peptide-beads could not be washed out from the reaction chamber, we could check the efficiency of the Edman degradation reaction from each cycle. The fractions collected every 1 min were measured by AMS. Because this HPLC eluent has sodium acetate as a buffer, which does not evaporate, it increases the carbon content, lowering the isotope ratio of carrier Rcarrier + buffer by the amount shown in (Eq. 1). Thus, the 14C content of each HPLC fraction (14Cfraction) was calculated with Eq. 2.
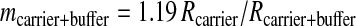 |
1 |
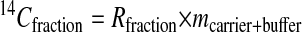 |
2 |
In these equations, the mass mcarrier + buffer was expressed in mg of C, and the isotope ratios Rcarrier, Rfraction were expressed in mol of 14C/mg of C. Fig. 1 shows the 14C level of HPLC fractions for ten cycles. Each PTH-amino acid was easily identified by comparing its retention time with those of standard PTH-amino acids that were derived from both peptide-beads and lactoglobulin solutions prepared in the conversion flask, facilitating the identification of PTH-amino acids. Chromatograms of 14C for ten cycles measured by AMS showed no peaks other than those for PTH-amino acids. After the fourth cycle the chromatogram becomes complicated because proline was not completely cleaved under these conditions. Eq. 3 was used to calculate the yield of each PTH-amino acid.
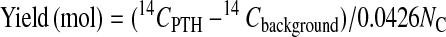 |
3 |
In this equation, 14CPTH, 14Cbackground, and NC represent the 14C amount of PTH-amino acid and background from HPLC samples measured by AMS (mol) and the number of carbon atoms in the molecular formula of each amino acid, respectively. Because the 14C content of each PTH-amino acid is proportional to NC, a yield of PTH-amino acid is calculated from dividing (14CPTH − 14Cbackground) by NC and 14C/C ratio of [14C]GST, 0.0426. The repetitive yield of the ten cycle sequence analyses was 88% (Fig. 2). This result indicates that protein and peptide-beads as carriers prevent not only nonspecific adsorption, but also washout from the reaction chamber. At high sensitivity, many peaks other than PTH-amino acids appear in the HPLC chromatograms, which increase background levels. Most current protein sequencers use capillary HPLC and a UV monitoring system that can detect 100 fmol levels of PTH-amino acids. AMS quantification combined extremely high sensitivity (amol levels of 14C) with high selectivity because 14C-labeled materials were generated only from the labeled protein in this method. The resulting low background level permitted selective and sensitive identification of PTH-amino acids.
Figure 1.

Amount of 14C measured by AMS in HPLC fractions of 1.76 fmol [14C]GST (N-terminal sequence: NH2-Ala-Gly-Lys-Pro-Val-Leu-His-Tyr-Phe-Asp) following sequence analysis. The position of PTH-amino acids assigned for cycles 1 through 10 are indicated by the arrows. The values in parentheses are yields calculated as described in the text. Samples were collected every 60 sec, and each sample was measured by AMS at least three times. The SDs among measurements ranged from 0.56–3.68 amol of 14C. HPLC fraction data in subsequent figures also is based on three to seven replicates with SDs of 1–3%. The amounts of 14C of background of each cycle ranged from 9.86 to 11.9 amol, and the SDs from 0.39 to 2.47 amol. Some data points are missing because of difficulty in sample preparation for AMS.
Figure 2.

Repetitive cycle yield of 1.76 fmol of [14C]GST with Edman-AMS sequencing. The repetitive yield was calculated from the slope of linear regression analysis for the plot of PTH-amino acid yields excluding cycles 7 (His) and 10 (Asp) (open circles).
Sequencing of 450 amol of [14C]GST.
Fig. 3 shows a typical UV trace and AMS measurement of sequencing analysis of 450 amol of [14C]GST for five cycles. A series of HPLC elution fractions bracketing the corresponding PTH-amino acid of each cycle were collected at 30 sec intervals. AMS analysis of these fractions shows in Fig. 3 that the PTH-amino acid peaks were significantly elevated above the background levels. The sensitivity of microsequencing may be limited not only by the detection system, but also by incomplete reaction of Edman degradation (23), however in this case the initial yield of this sequencing reaction (28%) is a limitation of this sequencer because reaction conditions were the same as for 100 pmol reactions due to the presence of carrier.
Figure 3.

(A) Typical UV trace of random peptide-bead internal standard (7 beads) and carrier protein, β-lactoglobulin (50 pmol) co-chromatographing with AMS detection. (B) Amount of 14C measured by AMS in HPLC fractions collected every 30 sec manually of 453 amol [14C]GST sequence analysis. The values in parentheses are yields calculated as described in the text.
The Sensitivity of Edman-AMS Sequencing.
We attempted to sequence 260 and 102 amol of [14C]GST. Fig. 4 shows results of PTH-amino acid fractions for cycles one and three of five measured by AMS. In the 260-amol analysis, the background levels were consistent and PTH-amino acid peaks were easily recognized. Likewise, the analysis for 102 amol of GST showed recognizable peaks. Although the sensitivity of this method depends on the specific activity of the labeled protein, 102 amol level sequencing, which is about 1,000 times more sensitive than the current Edman sequencer, could be carried out by using AMS as a detection system with even low 14C incorporation. AMS provides not only sensitive detection of 14C, but also accurate quantification of 14C amounts. Fig. 5 shows the relationship between the amount of protein applied to sequencer and initial yields of PTH-Ala calculated from 14C contents. There is a linear relationship over three orders of magnitude, indicating that sequence information obtained by the AMS detection system is highly quantitative, and that the 1–3% SD of the 3–7 replicates per sample demonstrates high precision. This result indicates that the sensitivity of protein sequencing is limited primarily by the detection system rather than Edman degradation reaction.
Figure 4.

Amount of 14C measured by AMS in HPLC fractions of 260 and 102 amol [14C]GST sequence analysis collected as described above. The values in parentheses are yields calculated as described in the text.
Figure 5.

Relationship between the amount of [14C]GST applied to the sequencer and the amount of PTH-Ala in cycle 1.
The improvement in the sensitivity of Edman degradation has had a major influence on the techniques used to purify proteins. For example recent improvements in the sensitivity of Edman technology have allowed biochemists to move from multiple steps of low yield and low resolution preparative purification techniques, such as gel permeation and ion exchange chromatography, to one or two very high yield steps by using technologies such as microbore HPLC and electrophoresis, which were previously considered only analytical methods. This process also allows for the use of much smaller amounts of protein as starting material and finer resolution of tissue samples. These AMS methods open the door to the use of still higher resolution techniques that are difficult to scale up to the pico- or femtomole level. This attomole sensitivity in turn will allow biochemistry to be done on tiny tissue samples, such as those from laser microdissection (24). This technology can be applied to the labeling of proteins in very small volumes with 14C followed by high resolution separation of the component proteins.
At present this AMS technique is restricted to analyzing 14C-labeled proteins. However, we are exploring an alternate and more general technology in which unlabeled proteins are reacted with 14C-labeled PITC or other chemistries to yield 14C-labeled amino acids. Such extrinsic methods require scaling down the size of Edman reactions to have reasonable rates of reaction and low background, as well as technologies to remove 14C-labeled impurities. Alternate approaches, such as the combination of capillary electrophoresis and thermo-optical absorbance, are reaching attomole detection (25). Other sensitive detection methods, such as laser induced fluorescence (26), might also be used in attomole Edman sequencing. These techniques, as well as AMS detection, will benefit from the use of microfluidics allowing sample volumes to be reduced dramatically.
At this time AMS sequencing involves the use of very large, complex, and expensive equipment and over 30 h of bench time to analyze a single run. In addition, current AMS technology requires isolated solid samples before measurement that can limit throughput for complex analyses. However, a variety of biological applications are driving the development of accelerator mass spectrometers (for nuclei such as 3H and 14C) that are small, of reduced cost, and have high throughput. We predict that the use of AMS in biological sciences, including the analysis of biopolymers, will increase dramatically in the near future.
Chemical labels using rare long-lived isotopes provide distinctive and specific signals that are quantifiable in a much larger background of unlabeled peptides and proteins. The approach described in this study provides a promising technology for attomole level protein sequencing. Such highly sensitive Edman methods are likely to complement the increasingly sensitive mass spectral methods used to provide some sequence information on femtomole levels and accurate mass on even subfemtomole concentrations (27, 28).
Acknowledgments
This work was supported by funds from the University of California (UC Presidents Campus Laboratory Collaboration 95–103 and Intra-University Agreement B29 1419, Lawrence Livermore National Laboratory–University of California, Davis), and the National Institute of Environmental Health Sciences (NIEHS) Superfund Basic Research Program (P42 ES04699). Additional support was provided by NIEHS Grants R01 ES02710 and P30 ES05707. This work was performed in part under the auspices of the U.S. Department of Energy by University of California Lawrence Livermore National Laboratory under Contract No. W-7405-Eng-48.
Abbreviations
- PTH
phenylthiohydantoin
- AMS
accelerator mass spectrometry
- LSC
liquid scintillation counting
- GST
glutathione S-transferase
- Fmoc
9-fluorenylmethoxycarbonyl
References
- 1.Fernandez J, Gharahdaghi F, Mische S M. Electrophoresis. 1998;19:1036–1045. doi: 10.1002/elps.1150190619. [DOI] [PubMed] [Google Scholar]
- 2.Blackstock W P, Weir M P. Trends Biotechnol. 1999;17:121–127. doi: 10.1016/s0167-7799(98)01245-1. [DOI] [PubMed] [Google Scholar]
- 3.Henzel W J, Billeci T M, Stults J T, Wong S C, Grimley C, Watanabe C. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilm M, Mann M. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 5.Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Nature (London) 1996;379:466–469. doi: 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- 6.McCormack A L, Schieltz D M, Goode B, Yang S, Barnes G, Drubin D, Yates J R. Anal Chem. 1997;9:767–776. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 7.Edman P, Begg G. Eur J Biochem. 1967;1:80–91. doi: 10.1007/978-3-662-25813-2_14. [DOI] [PubMed] [Google Scholar]
- 8.Hewick R M, Hunkapiller M W, Hood L E, Dreyer W J. J Biol Chem. 1981;256:7990–7997. [PubMed] [Google Scholar]
- 9.Lavin A E, Merewether L A, Clogston C L, Rohde M F. In: Techniques in Protein Chemistry VIII. Marshak D R, editor. San Diego: Academic; 1997. pp. 57–67. [Google Scholar]
- 10.Zimmerman C L, Appella E, Pisano J J. Anal Biochem. 1977;77:569–573. doi: 10.1016/0003-2697(77)90276-7. [DOI] [PubMed] [Google Scholar]
- 11.Vogel J S, Turteltaub K W, Finkel R, Nelson D E. Anal Chem. 1995;67:353a–359a. doi: 10.1021/ac00107a001. [DOI] [PubMed] [Google Scholar]
- 12.Buchholz B A, Fultz E, Haack K W, Vogel J S, Gilman S D, Gee S J, Hammock B D, Hui X, Wester R C, Maibach H I. Anal Chem. 1999;71:3519–3526. doi: 10.1021/ac990152g. [DOI] [PubMed] [Google Scholar]
- 13.Buchholz B A, Arjomand A, Dueker S R, Schneider P D, Clifford A J, Vogel J S. Anal Biochem. 1999;269:348–352. doi: 10.1006/abio.1999.4041. [DOI] [PubMed] [Google Scholar]
- 14.Creek M R, Mani C, Vogel J S, Turteltaub K W. Carcinogenesis. 1997;18:2421–2427. doi: 10.1093/carcin/18.12.2421. [DOI] [PubMed] [Google Scholar]
- 15.Dingley K H, Roberts M L, Velsko C A, Turteltaub K W. Chem Res Toxicol. 1998;10:1217–1222. doi: 10.1021/tx9801458. [DOI] [PubMed] [Google Scholar]
- 16.Gilman S D, Gee S J, Hammock B D, Vogel J S, Haack K, Buchholz B A, Freeman S, Wester R C, Hui X Y, Maibach H I. Anal Chem. 1998;70:3463–3469. doi: 10.1021/ac971383v. [DOI] [PubMed] [Google Scholar]
- 17.Shan G, Huang W, Gee S J, Buchholz B A, Vogel J S, Hammock B D. Proc Natl Acad Sci USA. 2000;97:2445–2449. doi: 10.1073/pnas.040575997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turteltaub K W, Felton J S, Gledhill B L, Vogel J S, Southon J R, Caffee M W, Finkel R C, Nelson D E, Proctor I D, Davis J C. Proc Natl Acad Sci USA. 1990;87:5288–5292. doi: 10.1073/pnas.87.14.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buetler T M, Eaton D L. Cancer Res. 1992;52:314–318. [PubMed] [Google Scholar]
- 20.Dooley C T, Houghten R A. In: Methods in Molecular Biology: Combinatorial Peptide Library Protocols. Cabilly S, editor. Vol. 87. Totowa, NJ: Humana Press; 1998. pp. 13–24. [Google Scholar]
- 21.Vogel J S. Radiocarbon. 1992;34:344–350. [Google Scholar]
- 22.Polach H A. In: Radiocarbon Dating: Proceedings of the Ninth International Conference Los Angeles and La Jolla 1976. Berger R, Suess H E, editors. Berkeley, CA: Univ. of California Press; 1979. pp. 115–124. [Google Scholar]
- 23.Matsudaira P. In: A Practical Guide to Protein and Peptide Purification for Microsequencing. Matsudaira P, editor. San Diego: Academic; 1993. pp. 1–13. [Google Scholar]
- 24.Ornstein D K, Gillespie J W, Paweletz C P, Duray P H, Gerring J, Vocke C D, Topalian S L, Bostwick D G, Linehan W M, Petricoin E F, III, et al. Electrophoresis. 2000;21:2235–2242. doi: 10.1002/1522-2683(20000601)21:11<2235::AID-ELPS2235>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 25.Xing-Fang L, Waldron K C, Black J, Lewis D, Ireland I, Dovichi N J. Talanta. 1997;44:401–411. doi: 10.1016/s0039-9140(96)02076-0. [DOI] [PubMed] [Google Scholar]
- 26.Eggertson M J, Craig D B. Biomed Chromatogr. 2000;14:156–159. doi: 10.1002/1099-0801(200005)14:3<156::AID-BMC942>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Martin S E, Shabanowitz J, Hunt D F, Marto J A. Anal Chem. 2000;72:4266–4274. doi: 10.1021/ac000497v. [DOI] [PubMed] [Google Scholar]
- 28.Pierce R A, Field E D, den Haan J M M, Caldwell J A, White F M, Marto J A, Wang W, Frost L M, Blokland E, Reinhardus C, et al. J Immunol. 1999;163:6360–6364. [PubMed] [Google Scholar]