

Abstract
One of the striking features of vascular endothelium, the single-cell-thick lining of the cardiovascular system, is its phenotypic plasticity. Various pathophysiologic factors, such as cytokines, growth factors, hormones, and metabolic products, can modulate its functional phenotype in health and disease. In addition to these humoral stimuli, endothelial cells respond to their biomechanical environment, although the functional implications of this biomechanical paradigm of activation have not been fully explored. Here we describe a high-throughput genomic analysis of modulation of gene expression observed in cultured human endothelial cells exposed to two well defined biomechanical stimuli—a steady laminar shear stress and a turbulent shear stress of equivalent spatial and temporal average intensity. Comparison of the transcriptional activity of 11,397 unique genes revealed distinctive patterns of up- and down-regulation associated with each type of stimulus. Cluster analyses of transcriptional profiling data were coupled with other molecular and cell biological techniques to examine whether these global patterns of biomechanical activation are translated into distinct functional phenotypes. Confocal immunofluorescence microscopy of structural and contractile proteins revealed the formation of a complex apical cytoskeleton in response to laminar shear stress. Cell cycle analysis documented different effects of laminar and turbulent shear stresses on cell proliferation. Thus, endothelial cells have the capacity to discriminate among specific biomechanical forces and to translate these input stimuli into distinctive phenotypes. The demonstration that hemodynamically derived stimuli can be strong modulators of endothelial gene expression has important implications for our understanding of the mechanisms of vascular homeostasis and atherogenesis.
From the moment the heart starts beating and blood flow is first established during vertebrate development, the cardiovascular system is constantly exposed to biomechanical stimulation. The pulsatile nature of blood flow generates a complex interplay of three distinct types of fluid mechanical forces: wall shear stresses, cyclic strains, and hydrostatic pressures (1). These hemodynamic factors not only influence the structure and function of the heart as a pump (2), but also act on the cells that comprise the walls of the distributive vascular network (3). Indeed, there is increasing evidence that biomechanical stimulation plays a key role in the maintenance of vascular integrity, as well as the development of vascular diseases. For example, the earliest atherosclerotic lesions in humans and experimental animals typically develop in the vicinity of branch points and areas of major curvature within the arterial vasculature—regions associated with complex flow disturbances, such as nonlaminar flow, flow reversal, and roaming stagnation points. In contrast, unbranched arterial geometries that are exposed to more uniform laminar flows appear relatively protected from lesion development (4, 5). This strikingly nonrandom, geometrically defined localization of atherosclerotic lesions strongly suggests an important influence of local hemodynamics on the underlying pathogenic mechanisms (6).
The endothelial lining of the heart and vasculature comprises a dynamic interface with the blood that acts as an integrator and transducer of both biomechanical and humoral stimuli (7). This single-cell-thick layer is able to rapidly sense changes in blood flow and respond by secreting or metabolizing potent vasoactive substances (e.g., nitric oxide) that contribute to pressure/flow homeostasis (8). In the face of chronic flow changes a more deliberate structural remodeling of the vessel wall also can occur via endothelium-dependent mechanisms (9, 10). Certain of these adaptive responses reflect changes in endothelial gene expression, and studies in in vitro model systems have confirmed that fluid shear stresses, comparable to those generated by the frictional force of blood flow on the endothelial lining in vivo, can directly influence transcriptional events in cultured endothelial monolayers (11). A central question in the field of vascular biology, currently, is how these mechanical forces are sensed by the cells of the blood vessel wall and then translated into pathophysiologically relevant phenotypic changes. Activation of various signaling cascades and transcription factor systems, as well as the identification of shear-stress-response elements in the promoters of several genes relevant to the atherosclerotic process (e.g., platelet-derived growth factor A, platelet-derived growth factor B, macrophage chemoattractant protein-1, vascular cell adhesion molecule 1) have helped to provide insight into the cellular mechanisms linking shear stress stimuli and genetic regulatory events (8, 12). The recent in vivo demonstration of enhanced activation of the NF-κB signaling pathway in geometrically defined, atherosclerosis-prone regions of the mouse aorta (13) lends further support to this biomechanical paradigm of endothelial activation (7).
In an effort to gain a more complete appreciation of the extent and biologic significance of endothelial activation by biomechanical stimuli, we have sought to apply high-throughput techniques for genomic analysis of phenotypic modulation (11, 14). Here we report the application of transcriptional profiling, using cDNA arrays, to assess the global patterns of gene expression in cultured human umbilical vein endothelial cells (HUVEC) exposed to two definably distinct biomechanical stimuli that have potential pathophysiological correlates. A physiologically relevant level of steady laminar shear stress (LSS, 10 dyn/cm2) and a turbulent (non-laminar) shear stress (TSS) of comparable spatial and temporal average amplitude each were compared with standard static (no flow) culture conditions. Cluster analyses of these transcriptional profiling data revealed distinctive patterns of gene expression, thus validating our working hypothesis that endothelial cells can differentially sense and transduce different biomechanical input stimuli. Further, we have applied various molecular and morphological approaches to document the impact of these transcriptional changes at the level of integrative cellular functions.
Materials and Methods
Cell Isolation and Culture.
HUVEC were isolated from several segments of normal term cords, pooled, and cultured in medium 199 (BioWhittaker) supplemented with 50 μg/ml endothelial cell growth supplement (Collaborative Research), 100 μg/ml heparin (Sigma), 100 units/ml penicillin-G + 100 μg/ml streptomycin (BioWhittaker), 2 mM l-Gln (GIBCO), and 20% FBS (BioWhittaker). Subculture 1 cells were plated at an initial density of ≈65,000 cells/cm2 on 0.1% gelatin-coated, culture-grade polystyrene plastic (Modern Plastics, Peabody, MA), or, for flow experiments, on specially designed 17.8-cm diameter plates (maxi-plates) fabricated from the same material. Cells derived from the same primary culture were used for each set of experimental comparisons (static vs. LSS vs. TSS). Each type of experiment was performed three times with different primary cell isolates.
Flow Apparatus.
Twenty-four hours after the initial plating, confluent HUVEC monolayers were exposed to well characterized hydrodynamically induced shear stresses in a cone and plate apparatus (15). For each shear experiment, the maxi-plate was transferred to the apparatus, and the entire volume of medium was replaced with fresh culture media. Fresh culture media also was replaced in the corresponding static cultures and maintained at 37°C in humidified 5% CO2/95% air. Culture medium in the shear apparatus was replenished during the experiment at an exchange rate of 0.5 ml/min, and the enclosed environment was maintained at 37°C in a humidified 5% CO2/95% air atmosphere. Endothelial cells were exposed to either steady LSS of 10 dyn/cm2 or TSS with an equivalent spatially and temporally averaged shear stress of 10 dyn/cm2 for 24 h. The rotation of the cone was ramped from 0 to maximum velocity over a 5-min interval. The modified Reynolds number, R̃, was used to determine the appropriate experimental conditions to induce laminar or turbulent flow (16). The parameter R̃ , described by Sdougas et al. (16), is a function of the local radius, the cone angle, the angular velocity of the cone, and the fluid kinematic viscosity of the culture media. It is predicted from this parameter that flow is laminar at R̃ ≪ 1 and turbulent for R̃ > 4. For LSS at 10 dyn/cm2, the apparatus was equipped with a 0.5° cone and rotated at a velocity of 100 rpm. In the case of turbulent flow, we used a 3° cone and a rotational velocity of 135 rpm. Because R̃ is proportional to the radial dimension, turbulent flow was established at radii ≥ 3.5 cm, which corresponded to a R̃ > 5 and represented an averaged spatial and temporal intensity of 10 dyn/cm2. Thus, for TSS experiments cells were harvested only from the outer portion of the culture plate (≥3.5 cm).
RNA Isolation and Array Hybridization.
After exposure to the appropriate stimulus, cells were rinsed twice with ice-cold PBS and scraped into Trizol (GIBCO). RNA was isolated by ethanol precipitation, and its quality was verified by the presence of intact ribosomal bands on a diagnostic gel. One microgram of total RNA was used to synthesize 33P-labeled probes by using Superscript II (GIBCO). Probes were purified by size-exclusion column chromatography (Bio-spin 6, Bio-Rad). Purified probes were denatured for 3 min at 100°C. A set of three cDNA arrays (GF200, GF204, and GF211, Research Genetics, Huntsville, AL), comprising a total of 14,836 elements, was used for hybridization for each experimental condition. Filters were prehybridized with MicroHyb hybridization solution (Research Genetics) containing Cot-1 (1 μg/ml) and Poly dA (1 μg/ml) for 6 h at 42°C in a rotator at 10 rpm. Probes were added to the hybridization solution, and the filters were hybridized for 16 h at 42°C at 10 rpm. Filters were washed twice with 2 × SSC, 1% SDS for 20 min at 50°C, 15 rpm and once with 0.5 × SSC, 1% SDS for 10 min at room temperature.
Quantification of Hybridized Arrays and Data Analysis.
Each filter was exposed to super resolution storage phosphor screens (Packard) for 1 h to determine the maximum intensity of radiolabeling. The screens were imaged with a Cyclone Storage Phosphor System (Packard) at 600 dpi by using the optiquant scanning software (Packard). In a given experiment, each filter was re-exposed for a time calculated to attain a comparable maximum intensity (at least 10,000) across the set. Images were imported into PATHWAYS 2.0 software (Research Genetics) to locate and quantify spot intensities. Data from pathways was transferred to a custom-designed database (Argus), programmed in visual basic, sql, and javascript, developed in our laboratory. Signal intensity on each filter was normalized so that the average intensity of all spots above background was equal to 2,000 (identical to the “normalize to all data” option in pathways). In preliminary analyses, we observed only small differences among different normalization methods, including selecting an unregulated control gene or normalizing to the median of the intensities of all spots. A distinctive design feature of our database program is the capability of comparing results for a given gene from multiple experiments simultaneously, facilitating the testing of reproducibility. An image of each spot also is displayed next to its numerical intensity, which allows rapid visual screening for hybridization artifacts. Details of the database structure, tools for analysis of gene expression, and other data-processing algorithms will be provided elsewhere (J.C., G. Weber, M.A.G., and G.G-C., unpublished work).
Criteria for Selecting Regulated Genes.
To quantify levels of gene regulation we divided the normalized intensity of a spot in one condition by the normalized intensity of the corresponding spot in the reference condition. This ratio represents the fold-change between the two conditions. For a gene to be included in the cluster analysis, it had to be regulated ≥2-fold with the normalized intensity of the more intense spot being greater than 1,500. When the arrays contained replicates of the same clone for a given gene, we only considered the ratios from the most intense pair of spots and all of the additional measurements at least half as intense. The ratio of the most intense pair of spots had to meet the 2-fold cutoff, and at least half of the remaining ratios had to meet a 1.5-fold cutoff. The ratios were exported to the program cluster (17) for hierarchical clustering. The confidence interval of a given ratio depends on many factors, such as the intensity of the imaged spots and the number of times a ratio was measured, and rigorous statistical techniques are being developed.
TaqMan Assays.
Purified, DNase-treated RNA was transcribed by using the TaqMan RT system (Applied Biosystems). A total of 37.5 ng of cDNA was used per PCR. For cyclin D1 a predeveloped TaqMan assay was used (Applied Biosystems). For cyclin B1 the following primers and TaqMan probe were used: forward primer, 5′-AAGGCGAAGATCAACATGGC-3′; reverse primer, 5′-GGCCTGCAGTTGTTCACTGA-3′; and TaqMan probe, 5′-CCTACGGCCCCTGCTGCAACCT-3′. Reactions were performed in a GeneAmp 5700 sequence detection system (Applied Biosystems).
Flow Cytometry.
Cells were rinsed with PBS, trypsinized, pelleted, and washed in FACS sample buffer (PBS without Ca2+/Mg2+ and 1 mg/ml glucose). After overnight fixation with 70% ethanol, cells were counted and ≈5 × 105 cells were aliquoted. After removal of the ethanol, propidium iodide staining solution (50 μg/ml final) with RNase (Roche, Indianapolis, IN) was added. A total of 1 × 105 cells were scanned in a FACSCalibur cytometer (Becton Dickinson), and data were analyzed in modfit lt. Fluorescence histograms were normalized so that the total area under the G0/G1, S, and G2/M curves was equal to 1, and the G0/G1 and G2/M peaks overlapped for all samples.
Immunofluorescence Microscopy.
Cells were rinsed with PBS at 37°C, fixed with 2% paraformaldehyde, and permeabilized with 0.1% Triton X-100 for 2 min. Cells then were incubated sequentially with antimyosin heavy chain antibody (1:25, rabbit polyclonal, Biomedical Technologies, Stoughton, MA) or antiplectin antibody (1:500, guinea pig polyclonal, Research Diagnostics, Flanders, NJ) for 1 h at room temperature, 2% goat serum in PBS, followed by FITC-conjugated goat anti-rabbit antibody (1:100, Jackson Laboratories) or FITC-conjugated goat anti-guinea pig antibody (1:150, Research Diagnostics). For F-actin staining, Oregon green 514 phalloidin (Molecular Probes) was used at 1:20. Cells were rinsed with PBS, and SYTOX orange (Molecular Probes) was added at a final concentration of 0.01 μM for 2 min. Cells were mounted in Gel-Mount (Biomeda, Foster City, CA).
Confocal Microscopic Analysis.
A Leica TCSNT confocal laser-scanning microscope acquired serial sections of each sample along the z axis, from the apical to the basal region of the cell, at 0.3-μm increments and a 0.2-μm pixel size resolution. Images were taken with a ×100 oil-immersion lens to generate an image stack of the immunostained protein and a corresponding stack of sytox-stained nuclei. To evaluate a comparable region within cells in different monolayers, the image of the center of the nucleus was selected as a reference point. To demonstrate the remodeling of cytoskeleton-associated proteins in the apical (supranuclear) region after flow, five sequential images of the immunostained protein, starting with the reference image and continuing toward the apex of the cell, were overlaid. This stacked image was merged with the corresponding reference image of the nuclei and represents a 1.5-μm thick section of each cell's apical region.
Results and Discussion
Endothelial Cells Exposed to Distinct Biomechanical Stimuli Exhibit Different Global Patterns of Gene Expression.
To examine the phenotypic plasticity of endothelial cells in the face of biomechanical activation, we used cDNA arrays to survey the transcriptional activity of a large number of genes in cultured HUVEC monolayers maintained under standard static (no flow) conditions compared with two distinct fluid dynamic conditions—steady laminar flow or turbulent flow. These flow regimens were adjusted to generate spatial and temporal averaged fluid shear stresses of comparable magnitudes (10 dyn/cm2) during an exposure period of 24 h. This particular time point was selected for comparison based on our previous experience with this in vitro model system, to allow various transient changes in transcriptional activation that primarily reflect the step-like transition from static (no flow) to a fluid dynamic culture condition to subside, and a more steady-state approximation of flow-dependent phenotype to emerge. Multiple arrays containing 11,397 unique human genes (Table 1) were hybridized with labeled cDNAs from the various experimental conditions. Of the genes represented in these arrays, 52.3% were named genes and 47.7% were expressed sequence tags, according to the Unigene database annotation (6/2000). When the normalized intensity data from two different experiments in which HUVEC were maintained under static (no flow) conditions were compared on a log-log plot (Fig. 1a), nearly all of the points fell along the unity line, indicating that gene expression levels overall were very similar in these two separate experiments (Fig. 1a). The total fraction of genes, in this relatively large sample of the human genome, that showed detectable levels of expression in cultured HUVEC (see Materials and Methods) was remarkably similar under the various experimental conditions examined: static vs. LSS, 23.6%; static vs. TSS, 21.2%; TSS vs. LSS, 21.5% (Table 1). In contrast, however, there were striking differences in the global patterns of gene regulation manifested in response to different stimuli (Fig. 1 b–d). For example, LSS stimulation elicited a broad pattern of up- and down-regulation of genes across all expression levels, when compared with static (no flow) conditions (Fig. 1b). On the other hand, TSS stimulation resulted in a global pattern of gene expression more reminiscent of static cultures (Fig. 1c vs. Fig. 1a). Direct comparison of LSS and TSS stimuli further illustrated the differences in global patterns of gene expression elicited by these distinct biomechanical stimuli (Fig. 1d). A quantitative tabulation of significant differences in gene expression under the experimental conditions examined is presented in Table 1. In general, LSS appeared to be a more potent stimulus (when applied to static monolayers) than TSS, resulting in significant (≥2-fold) changes in 205 genes compared with 86 genes, respectively. Interestingly, more genes were down-regulated by LSS and TSS than were up-regulated. Finally, when TSS and LSS conditions were directly compared, 100 genes showed significant differential regulation (68 up-regulated; 32 down-regulated, TSS/LSS), thus further indicating that cultured endothelial cells can discriminate between these distinct types of fluid mechanical stimulation. These transcriptional profiling data, analyzed in terms of global patterns of gene regulation, thus confirm and extend our working hypothesis that the endothelial cell is a mechanosensitive element in the blood vessel wall, whose phenotype can be modulated at the transcriptional level by fluid shear stresses (7, 11).
Table 1.
Quantitative summary of biomechanically induced changes in endothelial gene expression
| Condition | Expressed genes | Up-regulated genes | Down-regulated genes |
|---|---|---|---|
| Laminar/static | 2,684 (23.6%) | 65 (0.57%) | 140 (1.23%) |
| Turbulent/static | 2,419 (21.2%) | 12 (0.11%) | 74 (0.65%) |
| Turbulent/laminar | 2,445 (21.5%) | 68 (0.6%) | 32 (0.28%) |
Of 14,836 total array elements on the array set, there were 13,325 unique clones and 11,397 unique genes. Of these genes, 52.3% were named and 47.7% were expressed sequence tags. Percentages in the table are expressed as percent of unique genes.
Figure 1.
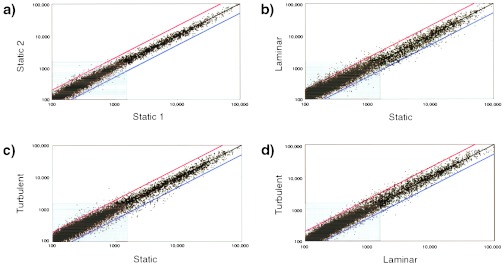
Global patterns of biomechanical activation of human endothelial genes revealed by transcriptional profiling. Scatterplots (log-log) of normalized intensities of each array element (13,325 unique clones) under the various experimental conditions examined: static 1 and static 2 (two separate no-flow experiments), TSS, and LSS. Differential expression of a given clone is reflected by deviation from the black diagonal line. Red diagonal defines ≥2-fold up-regulation; blue diagonal, ≥2-fold down-regulation. Gray box includes genes with levels of expression less than 1,500 (see Materials and Methods).
Categorization and Functional Annotation of Differentially Expressed Endothelial Cell Genes.
To begin to comprehend the endothelial phenotypes emerging in this model of biomechanical stimulation, we categorized the patterns of expression of known genes by different analytical methods. First, the genes that exhibited the greatest degree of change under the experimental conditions examined were grouped according to the direction (up or down) of their regulation. In Table 2, the 10 most regulated named genes are listed for each condition pair examined (LSS vs. static, TSS vs. static, TSS vs. LSS). Second, an average-linking hierarchical clustering algorithm (17) was applied to group named genes that were similarly regulated by each of the comparison conditions. As seen in Fig. 2, blocks of genes with similar color coding (connoting similar regulation across compared conditions) comprised qualitatively different categories. Some categories appear to be enriched for genes associated with particular functions. For example, many genes down-regulated by LSS (but less so with TSS) are known to be involved in the process of gene transcription, including Brahma (SWI/SNF matrix associated, subfamily a, member 2) and chromatin assembly factor 1, subunit B (p60). A rigorous analysis of such enrichment awaits a systematic classification of human transcripts into functional categories (18). It is notable that Fig. 2 contains examples of all possible combinations of up- and down-regulation among the three experimental conditions examined. For example, certain genes such as connective tissue growth factor were comparably up-regulated by both LSS and TSS, whereas others, such as CD164 (sialomucin), were comparably down-regulated by both stimuli, as compared with static control cultures. A subset of genes were found to be down-regulated by LSS and up-regulated by TSS (fibrogenic lymphokine, activating transcription factor 5), and vice versa (eukaryotic translation initiation factor 2, serine protease inhibitor Kazal type 2). These latter categories are particularly interesting because they may contain pathophysiologically relevant genes that would be hypothesized to contribute to biomechanically induced “atheroprotective” or “atheroprone” endothelial phenotypes in the in vivo setting (14). This entire data set can be found at a searchable database maintained by our laboratory at http://vessels.bwh.harvard.edu/papers/PNAS2001.
Table 2.
Endothelial-expressed genes showing greatest up- and down-regulation under various biomechanical conditions
| GenBank accession no. | Condition and gene description | GenBank accession no. | Condition and gene description |
|---|---|---|---|
| Laminar/static | ↓ Laminar/static | ||
| AA456008 | ALL1-fused gene from chromosome 1q | AA085749 | ATP binding protein |
| AA598794 | Connective tissue growth factor | AA479741 | TNFRSF1A modulator |
| AA448157 | Cytochrome P450, subfamily I | AA418118 | Cartilage paired-class homeoprotein 1 |
| AA487370 | Death-associated protein 6 | R54818 | Eukaryotic translation initiation factor 2B |
| H72187 | G protein, gamma 5 | AA457547 | Eukaryotic translation initiation factor 4 gamma, 3 |
| AA491206 | KIAA0217 protein | AA894648 | Homo sapiens clone 24583 mRNA sequence |
| AA436142 | Sparc/osteonectin | H23075 | Hydroxyacyl-Coenzyme A dehydrogenase/3-KCAT |
| T60235 | Spectrin, alpha, non-erythrocytic 1 (alpha-fodrin) | AA994205 | Peptide transporter 3 |
| AA478436 | Actin-dependent regulator of chromatin | AI022531 | Protein tyrosine phosphatase, receptor type |
| AA598758 | Tumor rejection antigen (gp96) 1 | T86027 | YDD19 protein |
| Turbulent/static | ↓ Turbulent/static | ||
| H28952 | ADP-ribosylation factor 4-like | AA477082 | TNFRSF1A modulator |
| N51018 | Biglycan | AA421296 | CD68 antigen |
| AA598794 | Connective tissue growth factor | W49715 | C-terminal binding protein 2 |
| AA975710 | DKFZP564F0923 protein | H68922 | Integrin, alpha 1 |
| H11003 | Endothelin 1 | AA417654 | Fibroblast growth factor receptor 3 |
| AA906997 | Homo sapiens mRNA; cDNA DKFZp762L137 | AA128153 | Interleukin 1 receptor-like 1 |
| AA630328 | Lectin, galactoside-binding, soluble, 3 (galectin 3) | T95668 | KIAA0631 protein |
| N54794 | Plasminogen activator inhibitor, type I | AA446251 | Laminin, beta 1 |
| H47015 | Ribosomal protein L34 | AA460330 | LIM and senescent cell antigen-like domains 1 |
| H65066 | Visinin-like 1 | AA460756 | Retinoblastoma-binding protein 2 |
| Turbulent/laminar | ↓ Turbulent/laminar | ||
| AA479741 | TNFRSF1A modulator | W49715 | C-terminal binding protein 2 |
| AA451684 | CD1D antigen, d polypeptide | AA448301 | Eukaryotic translation initiation factor 2 |
| T59055 | Exportin 1 (CRM1, yeast, homolog) | AA459208 | Follicular lymphoma variant translocation 1 |
| AA419620 | Fibroblast growth factor receptor 3 | T82817 | FOS-like antigen-1 |
| W80632 | Human BRCA2 region, mRNA sequence CG006 | AA431832 | Granulin |
| AA281731 | Natural killer-tumor recognition sequence | R63735 | Hypothetical protein |
| AA425655 | O-linked N-acetylglucosamine (GlcNAc) transferase | T95668 | KIAA0631 protein |
| AA488622 | Signal transducing adaptor molecule | AA447115 | Stromal cell-derived factor 1 |
| AA876165 | Spastic paraplegia 7, paraplegia | AA431611 | Thyroid hormone receptor interactor 7 |
| H15112 | Uracil-DNA glycosylase | AA497031 | TPA inducible protein |
Complete data set available in a searchable format at http://vessels.bwh.harvard.edu/papers/PNAS2001.
Figure 2.

Hierarchical clustering analysis of genes responding to biomechanical stimulation in cultured endothelial cells. Named genes whose mRNA levels showed significant up- or down-regulation (≥2-fold) and whose level of expression was ≥1,500 under laminar vs. static or turbulent vs. static conditions were selected. These 143 genes were clustered hierarchically into groups on the basis of the patterning of their expression profiles, by the procedure of Eisen et. al. (17). Each column represents a single experiment, and each row represents a single gene. For each gene, the ratio of mRNA levels for LSS/static conditions and TSS/static conditions, is denoted by a color code. Blue squares represent lower than static levels of gene expression in the LSS or TSS samples (ratios less than 1); white squares represent genes equally expressed (ratios near 1); red squares represent higher than static levels of gene expression (ratios greater than 1). Color saturation reflects the magnitude of the log/ratio (see color scale). Full cluster diagram with complete names and accession numbers is available at http://vessels.bwh.harvard.edu/papers/PNAS2001.
As a third approach, we subjectively grouped highly regulated genes, which have known or putative functions in mechanosignaling, vascular response-to-injury reactions, and atherogenesis (Table 3). We validated the regulation of several of these genes at the mRNA level by using quantitative real-time PCR (TaqMan), and their translation into protein by using Western blot analysis (Table 3). This list contains 28 genes of diverse functions, including genes involved in lipid metabolism (apolipoprotein E, long chain fatty acid-CoA ligase 3, megalin); the purinoreceptor, P2X4, which mediates fluid shear stress-dependent activation of calcium influx in endothelial cells (19); as well as isoforms of α, β and γ subunits of G proteins. The coordinated up-regulation of trimeric G proteins is interesting because these molecules have been implicated in mechanosignaling in response to fluid shear stress (20, 21). Certain genes involved in vascular responses to injury also showed significant regulation, including fibrogenic lymphokine (fibrosin), thioredoxin reductase, and matrix-gla protein. Thioredoxin reductase is recognized to be an important modulator of the redox state of the cell (22), whereas matrix-gla protein is a negative regulator of vascular calcification (23). Other genes whose regulated expression also may be relevant to vascular pathophysiology include connective tissue growth factor (CTGF), a pleiotropic growth factor, which can act on endothelial cells, smooth muscle cells, and fibroblasts (24); and CYP1B1, a cytochrome P450 enzyme that catalyzes estradiol hydroxylation and activates exogenous chemicals (25). Finally, we observed significant biomechanical regulation of various genes encoding extracellular matrix components, matrix receptors, and matrix remodeling enzymes, such as fibronectin, secreted protein acidic and rich in cysteine (SPARC), laminin β1, α1 integrin, thrombospondin 4, and matrix metalloproteinase-1. This cluster is consistent with the dynamic role of matrix biology in vascular morphogenic and response-to-injury processes, including hemostasis/thrombosis, angiogenesis, atherogenesis, and hypertension (26, 27).
Table 3.
Biomechanically responsive genes involved in mechanosignaling, response to injury, and atherogenesis
| Accession no. (GenBank) | Gene name (UniGene) | Laminar/static | Turbulent/static | Function |
|---|---|---|---|---|
| AA252968 | Annexin VIII | 2.2 | 1.9 | Anticoagulant |
| AA478589 | Apolipoprotein E* | 2.1 | ‡ | Lipid metabolism |
| AA598561 | CD164 antigen, sialomucin | −2.4 | −2.6 | Adhesion receptor |
| AA451684 | CD1D antigen, d polypeptide | −1.9 | 1.5 | Lipid-antigen presentation |
| AA136271 | CD58 antigen, (lymphocyte function-associated antigen 3) | −2.2 | −1.7 | T cell recognition |
| H65052 | Coagulation factor II (prothrombin) | −2.4 | ‡ | Pro-coagulant |
| AA598794 | Connective tissue growth factor† | 4.1 | 5.7 | Pleotropic functions |
| AA448157 | Cytochrome P450, subfamily I (dioxin-inducible), B1* | 2.7 | ‡ | Estradiol hydroxylation |
| W31074 | Fatty-acid-Coenzyme A ligase, long-chain 3 | −2.3 | −1.9 | Fatty acid metabolism |
| AA719257 | Fibrogenic lymphokine† | −1.2 | 1.7 | Fibrosis |
| R62612 | Fibronectin 1 | 2.4 | ‡ | Extracellular matrix |
| R43581 | G protein, alpha stimulating activity polypeptide 1 | 2.6 | 2.0 | GPCR signaling |
| AA487912 | G protein, beta polypeptide 1 | 2.5 | ‡ | GPCR signaling |
| H72187 | G protein, gamma 5 | 4.6 | ‡ | GPCR signaling |
| H68922 | Integrin, alpha 1 | −1.9 | −4.0 | Extracellular matrix receptor |
| AA446251 | Laminin, beta 1 | −1.4 | −3.0 | Extracellular matrix |
| AA630328 | Lectin, galactoside-binding, soluble, 3 (galectin 3) | 1.2 | 2.5 | RAGE, pleiotropic |
| AA630104 | Lipase A, lysosomal acid, cholesterol esterase | −2.1 | −1.0 | Cholesterol metabolism |
| R76808 | Low density lipoprotein-related protein 2 | −1.1 | −2.4 | Lipoprotein transport |
| AA143331 | Matrix metalloproteinase-1† | 1.7 | 1.2 | Extracellular matrix |
| AA155913 | Matrix-gla protein† | 1.8 | 0.9 | Vascular calcification |
| R51346 | Methionine aminopeptidase; elF-2-associated p67 | −2.3 | 1.0 | Angiogenesis |
| R60722 | Purinergic receptor P2X, ligand-gated ion channel, 4* | −2.3 | ‡ | Purinergic receptor |
| R96668 | Small inducible cytokine subfamily A (Cys-Cys), member 15 | −2.3 | ‡ | Chemokine |
| AA436142 | Sparc/osteonectin | 2.8 | ‡ | Extracellular matrix |
| AA453335 | Thioredoxin reductase* | 4.1 | ‡ | Redox |
| AA437064 | Thrombospondin 4 | −1.3 | −2.1 | Extracellular matrix |
| R76436 | Thromboxane A synthase 1 (cytochrome P450, subfamily V) | 1.3 | −1.6 | Vasoconstriction, procoagulant |
Verified by Western blot.
Verified by TaqMan assay.
Not detected.
These various approaches to analyzing the transcriptional profiling data emerging from our initial studies of cultured human endothelial cells under three well defined conditions—a quasi-physiological level of shear stress, a comparable spatial and temporal averaged level of TSS, and standard, static (no flow) culture, thus amply support the premise that the regulation of gene expression in this cell type is selectively and differentially sensitive to biomechanical stimulation. Next, we examined whether certain of these patterns of gene expression observed at the mRNA level are actually translated into changes in functional phenotype at the level of the intact cell. In particular, we focused on the modulation of cytoskeletal architecture and proliferative state—two aspects of endothelial cell biology that have long been appreciated to be responsive to biomechanical stimulation (8).
Remodeling of the Endothelial Cell Cytoskeleton Under Flow.
A number of genes with known cytoskeleton-related functions were observed to be significantly regulated in HUVEC exposed to steady LSS stimulation for 24 h. In Fig. 3a, 12 examples have been ordered according to their extent of up-regulation (red color-coded) or down-regulation (blue color-coded) by LSS versus static (no flow) conditions. Several of the most strikingly up-regulated genes are directly involved with structural and contractile properties of the cellular cytoskeleton (e.g., beta-actin, myosin heavy chain, plectin, vimentin, alpha-spectrin, actin depolymerizing factor), strongly suggesting that an active remodeling of cytoskeletal elements is induced by steady LSS biomechanical stimulation. Interestingly, TSS stimulation (which characteristically does not result in extensive cell shape change or alignment; ref. 28), resulted in less extensive regulation of this cytoskeletal gene cluster (data not shown). To examine the cell biological implications of this transcriptional program we performed confocal immunofluorescence microscopy to visualize the rearrangement of certain of these structural proteins in LSS-stimulated cells. Fig. 3b illustrates optical sections of the apical (supranuclear) compartment of confluent HUVEC under static (no flow) and LSS (10 dyn/cm2, 24 h) conditions, after immunostaining for F-actin, myosin heavy chain, and plectin. Accompanying the pronounced cell shape change from polygonal (static, Left) to fusiform and aligned (LSS, Right), the development of robust actin- and myosin-containing filamentous elements is evident in the apical region of the LSS stimulated cells. These “apical stress fibers,” which also have been described in vivo in aortic endothelial cells (29, 30), presumably reflect an adaptive response to the imposed uni-directional fluid shear stress, and thus represent a functional phenotypic correlate of cytoskeletal gene activation. Plectin, a versatile cytoskeletal linker protein that binds to actin, alpha-spectrin, and vimentin, and regulates actin dynamics, also showed a similar pattern of induction and subcellular localization under LSS stimulation, thus suggesting a previously unappreciated role in strengthening the endothelial cytoskeleton in the face of biomechanical loading. Interestingly, plectin-null mice exhibit bleeding (31), but the contribution of altered structural integrity of the endothelium to this phenotype remains to be explored. Finally, in addition to the structural implications of this shear-stress-induced cytoskeletal remodeling, the coordinated and sustained up-regulation of certain of these cytoskeletal proteins, and their organization in the vicinity of the apical cell membrane, a primary locus of biomechanical loading (8), points to their potential role in mechanosensing and/or mechanotransduction (8, 32).
Figure 3.

Changes in cytoskeletal elements of endothelial cells exposed to LSS. (a) Cytoskeleton-related genes showing significant up- or down-regulation in response to LSS stimulation. (b) Fluorescence confocal micrographs of the apical (supranuclear) region of HUVEC stained green for F-actin (Top), myosin heavy chain (Middle), and plectin (Bottom). Cells were counterstained with SYTOX (red) to identify nuclei. (Left) HUVEC under static (no flow) conditions; (Right), HUVEC exposed to LSS (10 dyn/cm2 for 24 h).
LSS and TSS Differentially Regulate Endothelial Cell Cycle.
Several genes known to be involved in cell cycle progression were differentially regulated by LSS and TSS (Fig. 4a). Validation of the levels of expression of cyclins B1 and D1 by TaqMan analysis (5′ nuclease fluorigenic quantitative PCR assay) confirmed the trends observed in the array data (Fig. 4b). Because cyclin D1 and cyclin B1 are markers for early G1 and G2/M, respectively, we used flow cytometry to compare the overall cell cycle activity under static, LSS and TSS conditions. As seen in Fig. 4c, exposure of HUVEC to LSS (10 dyn/cm2, 24 h) results in an increase in the G0/G1 population (from 81.4 ± 1.8%, static, to 94.1 ± 0.5%, LSS, n = 3, P ≤ 0.01) and a concomitant decrease in S (from 11.3 ± 1.8%, static, to 1.7 ± 0.1%, LSS, n = 3, P ≤ 0.01) and G2/M (from 7.5 ± 1.5%, static, to 4.0 ± 0.5%, LSS, n = 3, P = 0.025). Thus, exposure of HUVEC to LSS results in decreased proliferation, even in the presence of continuously replenished 20% serum and growth factors. In contrast, exposure of HUVEC to TSS resulted in only a slight increase of cells in G0/G1, compared with static conditions (from 81.4 ± 1.8%, static, to 86.7 ± 3.8%, TSS, n = 3, P = 0.07). We observed no change or an increase of cells in G2/M (from 7.5 ± 1.5%, static, to 11.4 ± 3.5%, TSS, n = 3, P ≥ 0.05), but a significant decrease in S phase (from 11.3 ± 1.8%, static to 2.0 ± 0.5%, TSS, n = 3, P ≤ 0.01). Notably, this latter observation suggests a temporal or mechanistic uncoupling of the S and G2 phases of the cell cycle machinery under TSS conditions. Further experiments are necessary to distinguish between these two possibilities. The increase in mRNA levels of cyclin D1 under LSS conditions is in agreement with the increase in the G0/G1 population observed by FACS analysis, suggesting a block in early G1. In contrast, TSS caused only a slight increase in cyclin D1 transcript levels, consistent with the small G0/G1 increase observed in the FACS data.
Figure 4.
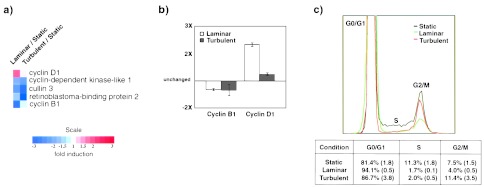
Cell cycle dynamics in biomechanically stimulated endothelial cells. (a) Cell cycle-related genes regulated by LSS or TSS compared with static conditions. (b) Relative mRNA levels of cyclin B1 and cyclin D1 as measured by TaqMan assay. β2-microglobulin, a gene not regulated by these stimuli, was used for normalization. Measurements were done in duplicate by using mRNA samples from three independent experiments. Bars represent means ± SE. (c) (Upper) Representative propidium iodide DNA-staining profile of HUVEC under static (no flow), LSS, or TSS (10 dyn/cm2 for 24 h). (Lower) Quantitative analysis of FACS data for indicated phases of the cell cycle. Percentages represent means from three independent experiments (SE in parentheses).
Previous studies have shown that exposure of endothelial cells to LSS decreases cell proliferation via the up-regulation of p21cip1 (33, 34), p53, and GADD45 (34). p21cip1 and p53 proteins are known regulators of progression through G1 (35), and their increased activity may be the cause of the G1 block observed here. The observations regarding the levels of cyclin B1 and the cell cycle status in LSS could be explained by at least two different mechanisms. First, cyclin B1 levels may be down-regulated because there are fewer cells in G2/M as seen in the FACS analysis. Second, the cells in G2/M could be expressing lower levels of cyclin B1. The latter possibility is supported by the recent observations that p53 expression decreases mRNA and protein levels of cyclin B1 and attenuates the activity of the cyclin B1 promoter (36). Although these mechanistic explanations are in agreement with our current knowledge about the cell cycle, it remains to be seen whether other biomechanically regulated genes (e.g., retinoblastoma-binding protein2, Fig. 4a), also participate significantly in this complex process. Nonetheless, it is clear from these data that the cell cycle machinery in human endothelial cells is differentially regulated in response to LSS and TSS stimulation. Importantly, these in vitro observations are consistent with previous in vivo reports of different mitotic activities in various anatomical regions of the aorta, in particular increased endothelial cell turnover in the arch and branch points, where the spatial- and time-varying characteristics of blood flow are more complex (37).
In conclusion, the distinctive global patterns of gene expression and resultant phenotypes observed in this study constitute the strongest evidence, to date, that vascular endothelial cells can discriminate among different types of biomechanical stimuli. Further analysis of this transcriptional profiling dataset (available in a searchable format at http://vessels.bwh.harvard.edu/papers/PNAS2001) should help to characterize the distinct features of this novel paradigm of endothelial activation. This experimental approach is being applied to other paradigms of endothelial cell gene regulation, in vitro and in vivo. A systematic comparison of these various transcriptional programs associated with both adaptive and dysfunctional endothelial phenotypes promises to provide new fundamental insights into the role of the endothelial lining of the cardiovascular system in health and disease.
Acknowledgments
We thank G. Weber (Decision Systems Group, Brigham and Women's Hospital) for help in data analysis and web site design, K. Case and V. Davis for primary isolation of HUVEC, C. F. Dewey (Fluid Mechanics Laboratory, Massachusetts Institute of Technology) for his longstanding collaboration, and R. Fenner and P. Morley (Massachusetts Institute of Technology) for design and fabrication of the flow apparatus. This study was supported primarily by grants from the National Heart Lung and Blood Institute, National Institutes of Health (P50-HL56985; R37-HL511509).
Abbreviations
- HUVEC
human umbilical vein endothelial cells
- LSS
laminar shear stress
- TSS
turbulent shear stress
- dyn
dynes
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 29, 1997.
References
- 1.Dewey C F. Adv Exp Med Biol. 1979;115:55–89. [Google Scholar]
- 2.Sadoshima J, Izumo S. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 3.Davies P F, Barbee K A, Volin M V, Robotewskyj A, Chen J, Joseph L, Griem M L, Wernick M N, Jacobs E, Polacek D C, et al. Annu Rev Physiol. 1997;59:527–549. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- 4.Gimbrone M A, Jr, Topper J N, Nagel T, Anderson K R, García-Cardeña G. Ann NY Acad Sci. 2000;902:230–239. doi: 10.1111/j.1749-6632.2000.tb06318.x. [DOI] [PubMed] [Google Scholar]
- 5.Traub O, Berk B C. Arterioscler Thromb Vasc Biol. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 6.Gessner F B. Circ Res. 1973;33:259–266. [Google Scholar]
- 7.Gimbrone M A, Jr, Nagel T, Topper J N. J Clin Invest. 1997;100:S61–S65. [PubMed] [Google Scholar]
- 8.Davies P F. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langille B L, O'Donnell F. Science. 1986;231:405–407. doi: 10.1126/science.3941904. [DOI] [PubMed] [Google Scholar]
- 10.Gibbons G H, Dzau V J. N Engl J Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 11.Topper J N, Gimbrone M A., Jr Mol Med Today. 1999;5:40–46. doi: 10.1016/s1357-4310(98)01372-0. [DOI] [PubMed] [Google Scholar]
- 12.Resnick N, Gimbrone M A., Jr FASEB J. 1995;9:874–882. doi: 10.1096/fasebj.9.10.7615157. [DOI] [PubMed] [Google Scholar]
- 13.Hajra L, Evans A I, Chen M, Hyduk S J, Collins T, Cybulsky M I. Proc Natl Acad Sci USA. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topper J N, Cai J, Falb D, Gimbrone M A., Jr Proc Natl Acad Sci USA. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bussolari S R, Dewey C F, Jr, Gimbrone M A., Jr Rev Sci Instrum. 1982;53:1851–1854. doi: 10.1063/1.1136909. [DOI] [PubMed] [Google Scholar]
- 16.Sdougas H P, Bussolari S R, Dewey C F., Jr J Fluid Mechanics. 1984;138:379–404. [Google Scholar]
- 17.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mewes W H, Frishman D, Gruber C, Geier B, Haase D, Kaps A, Lemcke K, Mannhaupt G, Pfeiffer F, Schuller C, et al. Nucleic Acids Res. 2000;28:37–40. doi: 10.1093/nar/28.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto K, Korenaga R, Kamiya A, Ando J. Circ Res. 2000;87:385–391. doi: 10.1161/01.res.87.5.385. [DOI] [PubMed] [Google Scholar]
- 20.Gudi S R, Clark C B, Frangos J A. Circ Res. 1996;79:834–839. doi: 10.1161/01.res.79.4.834. [DOI] [PubMed] [Google Scholar]
- 21.Redmond E M, Cahill P A, Sitzmann J V. Arterioscler Thromb Vasc Biol. 1998;18:75–83. doi: 10.1161/01.atv.18.1.75. [DOI] [PubMed] [Google Scholar]
- 22.Anema S M, Walker S W, Howie A F, Arthur J R, Nicol F, Beckett G J. Biochem J. 1999;342:111–117. [PMC free article] [PubMed] [Google Scholar]
- 23.Luo G, Ducy P, McKee M D, Pinero G J, Loyer E, Behringer R R, Karsenty G. Nature (London) 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 24.Moussad E E, Brigstock D R. Mol Genet Metab. 2000;71:276–292. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 25.Hanna I H, Dawling S, Roodi N, Guengerich F P, Parl F F. Cancer Res. 2000;60:3440–3444. [PubMed] [Google Scholar]
- 26.Vu T H, Werb Z. Genes Dev. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 27.Libby P, Tanaka H. Prog Cardiovasc Dis. 1997;40:97–106. doi: 10.1016/s0033-0620(97)80002-3. [DOI] [PubMed] [Google Scholar]
- 28.Davies P F, Remuzzi A, Gordon E J, Dewey C F, Gimbrone M A., Jr Proc Natl Acad Sci USA. 1986;83:2114–2117. doi: 10.1073/pnas.83.7.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White E W, Gimbrone M A, Jr, Fujiwara K. J Cell Biol. 1983;97:416–424. doi: 10.1083/jcb.97.2.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong A J T, Pollard T D, Herman I M. Science. 1983;219:867–869. doi: 10.1126/science.6681677. [DOI] [PubMed] [Google Scholar]
- 31.Andra K, Lassmann H, Bittner R, Shorny S, Fassler R, Propst F, Wiche G. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ingber D E. Cell. 1993;75:1249–1252. doi: 10.1016/0092-8674(93)90612-t. [DOI] [PubMed] [Google Scholar]
- 33.Akimoto S, Mitsumata M, Sasaguri T, Yoshida Y. Circ Res. 2000;86:185–190. doi: 10.1161/01.res.86.2.185. [DOI] [PubMed] [Google Scholar]
- 34.Lin K, Hsu P P, Chen B P, Yuan S, Usami S, Shyy J Y, Li Y S, Chien S. Proc Natl Acad Sci USA. 2000;97:9385–9389. doi: 10.1073/pnas.170282597. . (First Published August 1, 2000; 10.1073/pnas.170282597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puri P L, Maclachlan T K, Levrero M, Giordano A. In: The Molecular Basis of Cell Cycle and Growth Control. Stein G S, Baserga R, Giordano A, Denhardt D T, editors. New York: Wiley; 1999. pp. 15–79. [Google Scholar]
- 36.Innocente S A, Abrahamson J L A, Cogswell J P, Lee J M. Proc Natl Acad Sci USA. 1999;96:2147–2152. doi: 10.1073/pnas.96.5.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caplan B A, Schwartz C J. Atherosclerosis. 1973;17:401–417. doi: 10.1016/0021-9150(73)90031-2. [DOI] [PubMed] [Google Scholar]