

Abstract
Neurotoxin receptor site-3 at voltage-gated Na+ channels is recognized by various peptide toxin inhibitors of channel inactivation. Despite extensive studies of the effects of these toxins, their mode of interaction with the channel remained to be described at the molecular level. To identify channel constituents that interact with the toxins, we exploited the opposing preferences of LqhαIT and Lqh2 scorpion α-toxins for insect and mammalian brain Na+ channels. Construction of the DIV/S1-S2, DIV/S3-S4, DI/S5-SS1, and DI/SS2-S6 external loops of the rat brain rNav1.2a channel (highly sensitive to Lqh2) in the background of the Drosophila DmNav1 channel (highly sensitive to LqhαIT), and examination of toxin activity on the channel chimera expressed in Xenopus oocytes revealed a substantial decrease in LqhαIT effect, whereas Lqh2 was as effective as at rNav1.2a. Further substitutions of individual loops and specific residues followed by examination of gain or loss in Lqh2 and LqhαIT activities highlighted the importance of DI/S5-S6 (pore module) and the C-terminal region of DIV/S3 (gating module) of rNav1.2a for Lqh2 action and selectivity. In contrast, a single substitution of Glu-1613 to Asp at DIV/S3-S4 converted rNav1.2a to high sensitivity toward LqhαIT. Comparison of depolarization-driven dissociation of Lqh2 and mutant derivatives off their binding site at rNav1.2a mutant channels has suggested that the toxin core domain interacts with the gating module of DIV. These results constitute the first step in better understanding of the way scorpion α-toxins interact with voltage-gated Na+-channels at the molecular level.
Keywords: Mutant, Neurotoxin, Protein Structure, Sodium Channels, Sodium Transport, Voltage-gated Sodium Channel, Mutagenesis, Scorpion Alpha-toxins, Sea Anemone Toxin, Specificity
Introduction
Voltage-gated sodium channels (Nav)3 play a central role in excitability (1) and are targeted by a large variety of toxins used for prey and defense (2). Among a number of binding sites (3), neurotoxin receptor site-3 has been defined pharmacologically as the channel site of interaction with peptide toxins from scorpions, sea anemones, and spiders capable of inhibiting the channel inactivation process. Despite their similar effect and ability to compete for binding (3–5), these toxins differ tremendously in structure (6, 7), thus raising questions as to commonalities and differences in the molecular structure of receptor site-3.
Navs are composed of a pore-forming α-subunit (∼260 kDa) that in mammals is associated with one or two β-subunits and in insects with the TipE accessory subunit (1, 8–12). The α-subunit consists of four homologous domains (DI-DIV), each composed of six transmembrane segments (S1–S6) connected by intra- and extracellular loops. A key feature of Navs is the voltage-dependent activation enabled by the gating module formed by transmembrane segments S1–S4 in each of the four domains. The positively-charged S4 segments respond to changes in membrane potential and move outwards across the membrane electric field, leading to opening of the channel pore and transient increase in sodium conductance that is followed by fast inactivation (1, 13). The fast inactivation is coupled to the movement of the voltage sensor in DIV, which triggers the occlusion of the inner side of the channel pore by the intracellular loop that connects DIII and DIV (inactivation loop; 5, 14–16). Site-3 toxins have been shown to impede the movement of the voltage sensor in DIV, thereby inhibiting fast inactivation (17, 18).
Receptor site-3 has been localized at low resolution to the extracellular loops in domains I and IV using a photoaffinity-labeled scorpion α-toxin (Lqq5 from Leiurus quinquestriatus quinquestriatus) and antibodies directed to specific regions of the external loops in domains I (S5–S6) and IV (S3–S4 and S5–S6) of the rat brain channel rNav1.2 (19, 20). Channel chimeras between rNav1.2 and the cardiac channel subtype rNav1.5 (21, 22) suggested a role for DIV/S3-S4 loop in the interaction of rNav1.2a brain channel with Lqq5. Of particular interest was the result of charge inversion at position 1613 (E1613R), which decreased the affinity for Lqq5 by 62-fold (21). Mutagenesis of nearby residues in the rNav1.2a channel and the equivalent loop of the skeletal muscle Nav (rNav1.4) suggested a putative role for other residues of DIV/S3-S4 (Asp-1428, Lys-1432, Tyr-1433, Phe-1434, and Val-1435) in the interaction of the sodium channel with scorpion α-toxins (21, 23, 24; Fig. 1). Despite these results, the channel components that constitute receptor site-3 have not been fully determined.
FIGURE 1.

Sodium channel topology. Top, transmembrane folding diagram of the α-subunit of a voltage-gated sodium channel. Cylinders represent transmembrane α-helical segments. The gating module (segments S1–S4) of each domain resides near the pore module region (segments S5–S6) of the next domain in a clockwise orientation. Boldface lines represent the external and internal loops that connect the transmembrane segments. The pore loop (SS1–SS2) is colored red, and the inactivation ball is indicated by the IFM motif. Site-3 has been thus far assigned to the extracellular loops S5–S6 of DI and DIV and DIV/S3-S4 (19–21, 23, 24). The position of Glu-1613 is highlighted by a blue circle. Bottom, sequence alignment of DIV/S3-S4 extracellular loop (in italics) of the mammalian brain channel (rNav1.2a), mammalian skeletal muscle channel (rNav1.4), heart channel (hNav1.5) and Drosophila channel (DmNav1). Residue substitutions that affected the activity of scorpion α-toxins are in blue (Glu-1613 at rNav1.2a DIV/S3-S4 (21)) and in red (Lys-1432, Tyr-1433, Phe-1434, and Val-1435 of rNav1.4 (23, 24)).
Scorpion α-toxins constitute a family of structurally related polypeptides of 61–67 amino acids reticulated by four conserved disulfide bridges. Despite the similarity in sequence and three-dimensional structure, these toxins exhibit substantial differences in preference for mammalian and insect Navs, and on this basis are subdivided into subgroups (reviewed in Ref. 3) (i) α-toxins highly active on mammals and very weak on insects, such as Aah2 from Androctonus australis hector, Lqh2 from Leiurus quinquestriatus hebraeus (EC50 = 13.6 ± 1.4 nm and 6920 ± 1420 nm on rNav1.2a and the Drosophila DmNav1 channel, respectively, expressed in Xenopus oocytes; Ref. 25), and Lqq5 from L. quinquestriatus quinquestriatus (KD = 1.71 ± 1.1 nm, as determined on rNav1.2a expressed in tsA-201 cells; Ref. 21); (ii) α-toxins that are highly active on insects and very weak at mammalian brain Navs, such as LqhαIT (EC50 = 0.36 ± 0.04 nm on DmNav1, and 17800 ± 357 nm on rNav1.2a; Ref. 25); and (iii) α-like toxins that are active both in mammalian brain and on insects.
The pharmacology, electrophysiological effects, structures, and bioactive surfaces (see Fig. 2) of scorpion α-toxins have been studied extensively (3). Their functional surface is bipartite and is divided between two domains: a core domain that involves short loops that connect the conserved secondary structure elements of the molecule core and an NC domain composed of the five-residue turn (residues 8–12) and the C-terminal segment (see Fig. 2A; Refs. 26–29). The difference in amino acid composition and spatial arrangement of the NC domain was suggested to dictate the variations in preference among α-toxins for distinct Navs.
FIGURE 2.

Comparison of Lqh2 and LqhαIT. Top, bioactive surfaces of the two toxins. Lqh2 modeling was based on the reported structure of the almost identical toxin Aah2 (Protein Data Bank code 1AHO), and LqhαIT structure was determined (Protein Data Bank code 2ASC). The gray ribbons indicate the backbone structures covered by a semitransparent molecular surface of the toxins. Bioactive residues are shown as sticks (26, 29, 40). Residues of the core domain are colored magenta, and residues of the NC domain are colored blue. Bottom, sequence alignment of the two toxins. The bioactive surface of scorpion α-toxins consists of the conserved core domain (residues in magenta) and the diverse NC domain (residues in blue). Lqh2 and LqhαIT are similar in structure and share ∼70% sequence similarity, yet they exhibit opposing preferences for the mammalian brain and insect Navs (Table 1).
Here, we used two α-toxins, Lqh2 and LqhαIT, which vary greatly in preference for insect and mammalian brain Nav, to analyze channel constituents involved in this selective recognition. We chose Nav1.2 for construction of chimeric Nav channels because it is more sensitive to Lqh2 and much less sensitive to LqhαIT than cardiac or skeletal muscle NaV channels. Construction of rNav1.2a external loops in the background of DmNav1 as well as point mutagenesis revealed the role of DI/S5-S6 and DIV/S3 in the selective recognition by Lqh2. Comparison of depolarization-induced dissociation of Lqh2 and its mutant derivatives from various channel mutants suggested that the core domain of Lqh2 interacts with the voltage-sensing module in DIV of the channel.
EXPERIMENTAL PROCEDURES
Toxin Production and Modification
Lqh2 was produced in Escherichia coli strain BL21 as described (29). The toxin derivatives bear a His6 tag and a thrombin cleavage site at their N termini that does not hamper their activity (25).
Channel Modification
The cDNA encoding the DmNav1 sodium channel of Drosophila melanogaster cloned in pAlter expression vector (Promega) was digested by XbaI, ApaI, and NotI, dividing the entire sequence to two fragments, one encoding domains I and II (3650 bp) and the other encoding domains III and IV (2890 bp). The two fragments were cloned into pBluescript (Stratagene), and the resulting plasmids were used in all further steps of PCR-driven mutagenesis and construction of channel mutants. Mutagenized DNA fragments were back inserted to the original plasmid and the DNA sequence was verified prior to RNA production for injection into Xenopus laevis oocytes. The cDNA encoding the rNav1.2a rat brain sodium channel, cloned in the expression vector pCDM8 (Invitrogen), was used in a similar way for mutagenesis, and BstEII and BspMI restriction sites were used for back insertion to the original plasmid.
Expression of Navs in Oocytes and Two-electrode Voltage Clamp Experiments
cRNAs encoding the α-subunit of each channel and the auxiliary β1 and TipE subunits were transcribed in vitro using T7 RNA polymerase and the mMESSAGE mMACHINETM system (Ambion, Austin, TX) and injected into Xenopus oocytes as described previously (30). Currents were measured 1–3 days after injection using a two-electrode voltage clamp and a Gene Clamp 500 amplifier (Axon Instruments, Union City, CA). Data were sampled at 10 kHz and filtered at 5 kHz. Data acquisition was controlled by a Macintosh PPC 7100/80 computer, equipped with an ITC-16 analog/digital converter (Instrutech Corp., Port Washington, NY), utilizing Synapse (Synergistic Systems). Capacitance transients and leak currents were removed by subtracting a scaled control trace utilizing a P/6 protocol (31). Bath solution contained the following: 96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2, 5 mm HEPES, pH 7.5. Toxins were diluted with bath solution containing 1 mg/ml bovine serum albumin and applied directly to the bath in the final desired concentration. To avoid application artifacts, 1 mg/ml bovine serum albumin solution was applied prior to toxin addition. For the G-V analysis, mean conductance (G) was calculated from the peak current-voltage relations using the equation G = I/(V − Vrev), where I is the peak current, V is the membrane potential, and Vrev is the reversal potential. The normalized conductance-voltage relations were fit with either a one- or two-component Boltzmann distribution according to Equation 1,
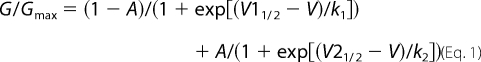 |
where V1½ and V2½ are the respective membrane potentials for two populations of channels for which the mean conductance is half maximal, k1 and k2 are their respective slopes, and A defines the proportion of the second population (amplitude) with respect to the total. For fits in which only one population of channels was apparent, A was set to zero. The voltage dependence of steady-state fast inactivation was described using a single Boltzmann distribution as shown in Equation 2,
where I is the peak current obtained at the depolarizing test step, Imax is the current without a preceding conditioning step, V is the membrane potential of the conditioning step, Vh is the membrane potential at which half-maximal inactivation is achieved, k is the slope factor, α0 is the remaining normalized peak current at highly depolarizing conditioning potentials, and α1 is the normalized amplitude (32).
Dose-response Curves of α-Toxin Effect on Fast Inactivation
Currents were elicited by a 50-ms depolarization to −20 mV from a −80 mV holding potential in the presence of increasing toxin concentrations. At each toxin concentration, the currents were allowed to reach a steady-state level prior to the final measurement. The dose dependence for toxin-induced removal of fast inactivation is calculated by plotting the ratio of the steady-state current remaining 50 ms after depolarization (Iss) to the peak current (Ipeak) as a function of toxin concentration, normalized to the maximal effect set to 1, and fitted with the Hill equation, where H is the Hill coefficient, [toxin] is the toxin concentration, and a0 is the offset measured prior to toxin application. The a1 − a0 amplitude provides the maximal effect obtained at saturating toxin concentrations. EC50 is the toxin concentration at which half-maximal inhibition of fast inactivation is obtained. To reduce variability, H was set to 1 in all calculations.
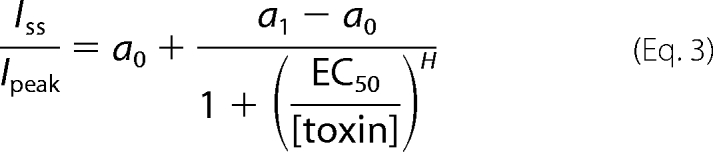 |
Determination of Voltage-dependent Dissociation of Toxin
Voltage-dependent toxin dissociation was measured with a two-pulse protocol. Conditioning dissociation pulses between −20 mV to +105 mV were applied from a −100 mV holding potential, following 50 ms at −100 mV for channels recovery from fast inactivation. Sodium currents were then elicited with a 50-ms test pulse to −20 mV. The experiments were conducted at saturating toxin concentrations, and a 30-s interval between test pulses ascertained maximal toxin rebinding. The extent of removal of fast inactivation represented by the ratio Iss/Ipeak was plotted as a function of the conditioning voltage and was fitted with the Boltzmann distribution described in Equation 4,
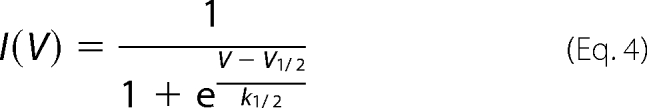 |
where V½ is the half-maximal dissociation voltage, and k½ is the corresponding slope factor.
RESULTS
The extreme difference in potency of Lqh2 and LqhαIT on rNav1.2a rat brain and DmNav1 Drosophila voltage-gated sodium channels is correlated with differences in their bioactive surfaces as well as differences in their channel receptor sites. Whereas the toxins have been thoroughly dissected and their bioactive surfaces documented (Fig. 2) (26, 29), their channel receptor sites are described incompletely. Based on previous reports suggesting that receptor site-3 is associated with channel external loops of domains IV and I (19, 21, 23, 24, 33), our approach to identify channel constituents that determine toxin recognition was to first uncover the extracellular loops involved with toxin selectivity and then use this information to characterize toxin interaction with receptor site-3.
Construction of rNav1.2a External Loops in DmNav1 and Analysis of Sensitivity to Lqh2 and LqhαIT
Stepwise construction of the four external loops DIV/S1-S2, DIV/S3-S4, DI/S5-SS1, and DI/SS2-S6 from rNav1.2a in the background of DmNav1 (see sequences in supplemental Fig. 1) provided channel chimeras, which we named by indicating the parent channel in full type and the substituted segments or amino acid residues as superscripts (supplemental Table 1). The voltage-dependent activation and inactivation properties of these chimeras varied only slightly from those of DmNav1 (supplemental Table 1). However, the sensitivity of these chimeric channels to the two toxins varied greatly (Table 1). Most dramatic was the decrease in sensitivity of the final chimera DmNav1rNav1.2a(DI/S5-SS1+SS2-S6,DIV/S1-S2+S3-S4) to LqhαIT by nearly 3 orders of magnitude from an EC50 of 0.36 ± 0.04 nm for DmNav1 to an EC50 of 241 ± 47 nm for the chimera (Table 1). On the other hand, Lqh2 activity increased from an EC50 value of 6920 ± 1420 nm for DmNav1 to an EC50 value of 26.7 ± 3.1 nm for the chimera (Table 1 and Fig. 3A). This suggested that the mammalian receptor site-3 of Lqh2 has been nearly fully constructed in the background of the insect channel. Therefore, this chimera was named DmNav1rNav1.2a(site-3 face).
TABLE 1.
Effect of Lqh2 and LqhαIT on channel mutants
The channels were clamped at a holding potential of −80 mV, and currents were elicited by depolarization to −20 mV in the presence of increasing toxin concentrations. At each toxin concentration, the currents were allowed to reach a steady-state level prior to the final measurement. Determination of dose-dependent effect of the toxin (removal of fast inactivation) is described in detail under “Experimental Procedures”, and the EC50 values provided are mean ± S.E., where n stands for the number of cells analyzed. No effect denotes lack of effect on channel inactivation in the presence of 5 μm toxin.
| Channel derivative | EC50 of Lqh2 | EC50 of LqhαIT |
|---|---|---|
| nm | nm | |
| DmNav1 unmodified | 6920 ± 1420 | 0.36 ± 0.04 |
| DmNav1D1701E | >5000 (n = 3) | 50 ± 4.1 (n = 3) |
| DmNav1D1701R | No effect | No effect |
| DmNav1rNav1.2a(DIV/S1-S2) | 1530 ± 433 (n = 4) | 3.2 ± 0.6 (n = 5) |
| DmNav1rNav1.2a(DIV/S1-S2+S3-S4) | 120 ± 12 (n = 5) | 57 ± 17 (n = 3) |
| DmNav1rNav1.2a(DIV/S3-MFLA) | 495 ± 113 (n = 3) | 1.4 ± 0.1 (n = 3) |
| DmNav1rNav1.2a(DI/S5-SS1) | >2500 (n = 3) | 0.65 ± 0.1 (n = 3) |
| DmNav1rNav1.2a(DI/SS2-S6) | >5000 (n = 3) | 0.54 ± 0.06 (n = 2) |
| DmNav1rNav1.2a(D1/S5-SS1+SS2-S6) | 123 ± 26 (n = 3) | 0.48 ± 0.12 (n = 3) |
| DmNav1rNav1.2a(site-3 face)a | 26.7 ± 3.1 (n = 5) | 241 ± 47 (n = 4) |
| DmNav1rNav1.2a(site-3 face-E1613D) | 22.5 ± 1.9 (n = 3) | 0.37 ± 0.03 (n = 4) |
| rNav1.2aE1613D | 14.5 ± 5.7 (n = 4) | 18.8 ± 3.3 (n = 4) |
| rNav1.2a unmodified | 13.6 ± 1.4 (n = 3) | 17,800 ± 357 (n = 5) |
a Site-3 face denotes DI/S5-SS1+SS2-S6, DIV/S1-S2+S3-S4 exchanged in DmNav1 by their rNav1.2a equivalents.
FIGURE 3.

Lqh2 and LqhαIT activities on modified channels. A, dose response curve of Lqh2 on the channel chimera DmNav1rNav1.2a(site-3 face) expressed in Xenopus oocytes. Each point represents mean ± S.E. of at least n = 3. Inset, representative traces of Lqh2 activity on the channel chimera. The oocytes were clamped at −80 mV, and currents were elicited by 50 ms depolarization to −20 mV from a −80 mV holding potential in the presence of increasing toxin concentrations. At each toxin concentration, the currents were allowed to reach a steady-state level prior to the final measurement (see “Experimental Procedures”). B, dose response curve of LqhαIT activity on the channel mutant rNav1.2aE1613D. Inset, representative traces of LqhαIT activity on the channel mutant expressed in Xenopus oocytes using a similar protocol as described in A. A further increase in toxin concentration did not yield further inhibition of channel inactivation.
To clarify the contribution of each loop to toxin recognition, we first examined the S1–S2 and S3–S4 external loops of the gating-module in DIV. Because DmNav1rNav1.2a(DIV/S3-S4) did not express in oocytes, we examined the sensitivity of DmNav1rNav1.2a(DIV/S1-S2) and DmNav1rNav1.2a(DIV/S1-S2+S3-S4) to Lqh2 and LqhαIT. The activity of Lqh2 on DmNav1rNav1.2a(DIV/S1-S2+S3-S4) was 58-fold higher compared with its activity on the unmodified DmNav1 and 4.5-fold weaker than that at DmNav1rNav1.2a(Site-3 face), whereas the activity of LqhαIT on this chimera decreased 159-fold (Table 1). The swap of DIV/S1-S2 (chimera DmNav1rNav1.2a(DIV/S1-S2)) improved Lqh2 activity by only 4.5-fold (Table 1), highlighting the greater significance of loop DIV/S3-S4 in determining the specificity of Lqh2 binding.
We next analyzed the external loops of DI pore module. The sensitivity of DmNav1rNav1.2(DI/S5-SS1+SS2-S6) to Lqh2 increased 56-fold compared with the sensitivity of DmNav1 to this toxin, whereas these substitutions had barely an effect on LqhαIT activity. However, the sensitivity of DmNav1rNav1.2(DI/S5-SS1) and DmNav1rNav1.2(DI/SS2-S6) to Lqh2 was as poor as that of DmNav1, and the LqhαIT effect on these channel chimeras hardly changed (Table 1). Thus, single loop substitutions were not very effective suggesting that there may be a cooperative effect of the two extracellular loops in the pore module on the ability of Lqh2 to interact with the brain sodium channel. Overall, these results indicated that both the gating module of DIV and the pore module of DI were required to form a complete Lqh2 receptor site in DmNav1.
Substitution of Residues in DIV External Loop That Differ between rNav1.2a and DmNav1
The substantial role of DIV/S3-S4 in channel sensitivity to the two α-toxins (Table 1) as well as previous reports on changes in activity of α-toxins upon substitution of Glu-1613 in rNav1.2a (21) and its Asp-1428 and Asp-1701 equivalents in rNav1.4 (23, 33) and DmNav1 (7) suggested that differences in this loop between the two channels might be involved with the varying potency of α-toxins. Hence, we examined the effect of reciprocal exchange of Glu-1613 at rNav1.2a and Asp-1701 at DmNav1 on channel sensitivity to Lqh2 and LqhαIT. Whereas the substitution D1701E at DmNav1 markedly reduced LqhαIT activity (139-fold decrease; Table 1), this channel mutant remained insensitive to Lqh2. In sharp contrast was the effect of the reciprocal substitution in rNav1.2a (E1613D), which improved LqhαIT affinity by ∼1000-fold, closely resembling the potency of Lqh2 at rNav1.2a (EC50 = 18.8 ± 3.3 nm; Table 1; Fig. 3B). Following this striking result, we analyzed the Glu-to-Asp substitution in the context of the chimera DmNav1rNav1.2a(site-3 face). We found that the activity of LqhαIT on DmNav1rNav1.2a(site-3 face-E1613D) was restored (EC50 = 0.37 ± 0.03 nm) and was similar to the activity of the toxin at DmNav1, whereas Lqh2 activity persisted (Table 1; Fig. 3A). Thus, Glu-1613 at DIV/S3-S4 is a key factor that hinders LqhαIT interaction with receptor site-3 of the rat brain channel rNav1.2a.
A prominent difference in this channel region is the amino acid composition of the distal part of DIV/S3 (positions 1697–1700 in DmNav1; Leu-Val-Leu-Ser versus positions 1609–1612 in rNav1.2a; Met-Phe-Leu-Ala; Fig. 1). Substitution of this amino acid stretch by its rNav1.2a equivalent resulted in a channel mutant, DmNav1rNav1.2a(DIV/S3-MFLA), whose sensitivity to Lqh2 increased 14-fold, with only a minor effect in its sensitivity to LqhαIT (Table 1), compared with DmNav1. This suggested that this region in DIV gating module of rNav1.2a plays a role in Lqh2 interaction with its receptor.
Analysis of Voltage-dependent Dissociation of Lqh2 Mutants from Nav1.2a
The swap of the Lqh2 receptor at rNav1.2a onto DmNav1 indicated an important role of the gating module of DIV and pore module of DI in toxin recognition. On the toxin side, the bioactive surface of Lqh2 has recently been shown to be composed of two domains: the core domain (Phe-15, Arg-18, Trp-38, and Asn-44) and the NC domain (Lys-2, Thr-57, Lys-58; Fig. 2A; Ref. 29). On these grounds and the fact that binding of scorpion α-toxins is voltage-dependent (1, 34–37), which suggests toxin binding at the mobile voltage-sensing region, we analyzed which of the toxin bioactive domains interacts with the DIV gating module of rNav1.2a. This analysis was based on the assumption that the dissociation of toxin mutants upon depolarization would vary from that of the unmodified toxin if the substitutions affect a site of interaction with the channel gating module.
We first analyzed the voltage-dependent dissociation of Lqh2 at a saturating concentration from rNav1.2a expressed in Xenopus oocytes by applying a series of depolarizing prepulses to +105 mV of variable durations (see “Experimental Procedures”). However, under this protocol with prepulse duration of up to 200 ms, the toxin had barely dissociated, as indicated by the persistent inhibition of inactivation following the test pulse (Fig. 4A). Considerable dissociation of Lqh2 was obtained following a prepulse duration of at least 500 ms, and the apparent toxin effect was nearly abolished after 2 s (not shown). Because depolarizing prepulses exceeding 200 ms promote channel entrance into slow inactivation (38), we limited the assays with saturating toxin concentrations to a 200-ms depolarizing prepulse (Fig. 4A, inset). The same protocol was used to analyze Lqh2 mutants K2A, F15A, N44A, and T57A. This analysis revealed that the voltage-dependent dissociation of Lqh2 substituted at the core domain was markedly enhanced, especially for F15A and N44A. The V½ for dissociation of mutants Lqh2F15A and Lqh2N44A was 82.5 ± 0.6 and 53 ± 2.7 mV, respectively, compared with 129 ± 13 mV for Lqh2 (Table 2), and a complete dissociation was observed following a depolarizing prepulse to +105 mV (Fig. 4A). On the other hand, the voltage-dependent dissociation of toxin mutants Lqh2K2A and Lqh2T57A of the NC domain (Fig. 2; Ref. 29) was similar to that of the unmodified toxin, despite a large increase in their EC50 values (Fig. 4B and Table 2). These results have suggested that the core domain of Lqh2 interacts with a channel region undergoing modification upon depolarization.
FIGURE 4.

Effects of substitutions in Lqh2 on activity and voltage-induced dissociation off rNav1.2a and its F1610A and E1613R mutants. A, dissociation of Lqh2 and derivatives off rNav1.2a upon depolarizing prepulses. Toxin effect was monitored by a test pulse to −20 mV as a function of 200 ms varying depolarizing prepulses (between −20 and +105 mV; see inset and “Experimental Procedures”). Note the difference between core domain to NC domain toxin mutants. B, dose-dependent effect of Lqh2 mutants on rNav1.2a (filled symbols) and channel mutant rNav1.2aE1613R (open symbols). C, dissociation of Lqh2 and derivatives off rNav1.2aE1613R channel mutant upon depolarizing prepulses. D, dissociation of Lqh2 and derivatives off rNav1.2aF1610A channel mutant upon depolarizing prepulses. Symbols in C and D are as described in B. The voltage protocol in C and D is identical to that described in A.
TABLE 2.
Effects of Lqh2 mutants and their voltage-induced dissociation off rNav1.2a and the F1610A and E1613R channel mutants
The EC50 values for rNav1.2a are from Kahn et al. (29). The voltage at which 50% of the toxin dissociated (V½) was calculated from the slopes presented at Fig. 4, A, C, and D. The values provided are mean ± S.E. of three to six measurements (n). ND, not determined (when the EC50 values could not be calculated because a saturating effect could not be reached).
| Channel | Toxin |
|||||
|---|---|---|---|---|---|---|
| WT | K2A | F15A | W38A | N44A | T57A | |
| rNav1.2a | ||||||
| EC50 (nm) | 13.6 ± 1.4 | 179 ± 22 | 72 ± 3.7 | 1180 ± 207 | 394 ± 43 | 1220 ± 40 |
| V½ (mV) | 129 ± 13 | >130 | 82.5 ± 0.6 | 111 ± 2 | 53 ± 2.7 | >130 |
| F1610A | ||||||
| EC50 (nm) | 63 ± 3.6 | 864 ± 39 | 2540 ± 160 | 3880 ± 76 | ND | 1820 ± 307 |
| V½ (mV) | 93.8 ± 7.2 | ND | 49 ± 3.8 | ND | ND | 89 ± 6.4 |
| E1613R | ||||||
| EC50 (nm) | 15.7 ± 1.6 | ND | 1640 ± 91 | ND | 3430 ± 234 | 1760 ± 203 |
| V½ (mV) | 64.4 ± 4.4 | ND | 6.0 ± 0.3 | ND | −10.7 ± 2a | 50 ± 1.6 |
a N44A dissociation was best fit with a two-component Boltzmann distribution with a major component of 0.69, V½ = −10.7 ± 2 mV, and a minor component, V½ = 45 ± 1 mV.
Effects of Channel Substitutions on Voltage-dependent Dissociation of Lqh2
Because an E1613R substitution in rNav1.2a has been shown to enhance the dissociation of the α-toxin Lqq5 (21), we examined the effects of charge neutralization and inversion at this position (E1613N and E1613R) on Lqh2 voltage-dependent dissociation (Tables 2 and 3). Notably, Lqh2 voltage-dependent dissociation off rNav1.2aE1613N and rNav1.2aE1613R channel mutants was markedly facilitated compared with its dissociation off the unmodified channel. The dissociation off rNav1.2aE1613R was more prominent as it began at +40 mV and was complete following a prepulse to +105 mV (Fig. 4, A and C, and Table 2). However, despite the substantial shift in V½ for both channel mutants, the EC50 values of Lqh2 barely changed (Tables 2 and 3). This result suggested that substitution of Glu-1613 at DIV/S3-S4 impairs an interaction of Lqh2 with an activated and/or fast-inactivated channel state.
TABLE 3.
Lqh2 activity and voltage-dependent dissociation off rNav1.2a and mutants
Determination of the dose-dependent effect of the toxin (removal of fast inactivation) is described in detail under “Experimental Procedures”, and the EC50 values provided are mean ± S.E. of at least three measurements (n). V½, the voltage at which 50% of the toxin dissociated off the channel.
| Channel | Toxin (Lqh2) |
|
|---|---|---|
| EC50 | V½ | |
| nm | mV | |
| rNav1.2a | 13.6 ± 1.4 | 129 ± 13 |
| T1560A | 55 ± 4.2 | >105 |
| M1609A | 22 ± 2.9 | >105 |
| L1611A | 12.8 ± 1.8 | >105 |
| E1613N | 11.4 ± 1.8 | 101 ± 1.7 |
| Y1618A | 16.3 ± 2.2 | >105 |
| F1619A | 17.4 ± 1.7 | >105 |
| V1620A | 14.2 ± 0.9 | >105 |
Based on the results of the dissociation assays, we examined the effects of Lqh2 and its bioactive surface mutants on a number of channel mutants modified at the DIV extracellular loop S1–S2, the distal region of S3 and the beginning of S3–S4 (Table 3). These channel determinants were selected on the basis of the swap experiments, which suggested that they determine the selectivity of Lqh2, and on unpublished results of the effects of point mutations at DIV/S1-S2 on Lqh2 activity (39). Of all channel mutants examined, prominent changes in Lqh2 EC50 values were obtained only for T1560A (DIV/S1-S2) and F1610A (distal region of DIV/S3; Tables 2 and 3).
Voltage-dependent Dissociation of Toxin Mutants from Channel Mutants
Analysis of the voltage-dependent dissociation of Lqh2 mutants Lqh2F15A and Lqh2N44A off rNav1.2aE1613R revealed substantial enhancement, as indicated by the prominent shifts in V½ to more negative membrane potentials (V½ values of 6.0 ± 0.3 mV and −10.7 ± 2 mV, respectively, compared with 64.4 ± 4.4 mV for Lqh2; Fig. 4C; Table 2). In light of this result, we sought other residues that putatively interact with Lqh2. As replacement of the Leu-Val-Leu-Ser sequence at the C-terminal end of DIV/S3 in DmNav1 by its rNav1.2a equivalent, Met-Phe-Leu-Ala, resulted in gain of Lqh2 function at the chimeric channel, DmNav1rNav1.2a(DIV/S3-MFLA) (Table 1), we examined the effect of substitutions M1609A and F1610A at rNav1.2a on Lqh2 activity. Whereas substitution M1609A had no effect (Table 3), the substitution F1610A resulted in 5-fold increase in the EC50 of Lqh2 (Table 2). Accordingly, we analyzed the effect of F1610A substitution on the voltage-dependent dissociation of Lqh2 and its mutant derivatives. As shown in Fig. 4D and Table 2, the dissociation of Lqh2 off rNav1.2aF1610A was markedly facilitated and observed at a lower membrane potential compared with the toxin dissociation off the unmodified channel. Furthermore, whereas the dissociation of the NC-domain toxin mutant, Lqh2T57A, off rNav1.2aF1610A resembled that of the unmodified toxin, the dissociation of the core domain toxin mutant Lqh2F15A off rNav1.2aF1610A was markedly facilitated (Fig. 4D; Table 2). These results suggested that upon interaction, the core domain of Lqh2 is in close proximity to the distal region of S3 at DIV in the rat brain sodium channel.
DISCUSSION
Determination of receptor sites of Nav modifiers is complex due to the lack of channel structure and their conformational rearrangements during gating. The experimental approach in the present study was to first identify channel regions involved in toxin selectivity and then dissect the relevant regions in search for specific residues associated with the receptor site. Although receptor site-3 is not necessarily constituted from components that differ between the brain and insect sodium channels, the successful swap and gain of high activity of Lqh2 at the chimeric channel DmNav1rNav1.2a(site-3 face) has indicated that the external loops of the gating module of domain IV and pore module of domain I in rNav1.2a play an important role in toxin selectivity and that they are spatially arranged in the chimeric channel as in rNav1.2a. The inverse experiment to construct rNav1.2a such that it acquires high sensitivity to LqhαIT surprisingly did not require swap of external loops from DmNav1. Instead, a single conservative substitution, E1613D, converted the brain channel to high sensitivity toward LqhαIT. In the skeletal muscle channel Nav1.4 and cardiac channel Nav1.5, the position equivalent to Glu-1613 is occupied by an Asp residue, and both channels are sensitive to LqhαIT (32, 36). Moreover, substitution at this position in rNav1.4 (D1428E) decreased the effect of LqhαIT (23). These observations are in concert with the gain of function of LqhαIT at the E1613D mutant of rNav1.2a and indicate that Glu-1613 at DIV/S3-S4 of rNav1.2a is in close proximity to the surface of interaction with LqhαIT. The gain of rNav1.2a sensitivity to LqhαIT upon a single substitution demonstrates that the brain channel bears a receptor site for LqhαIT, and the primary reason for lack of LqhαIT activity is the hindrance caused by Glu-1613. Furthermore, these results also suggest that receptor sites 3 at both the brain and insect channels are similar although not identical.
The interaction of DIV/S3-S4 external loop with scorpion α-toxins has been demonstrated in previous studies (21, 23, 40, 41), and it was proposed that the positively charged S4 segment moves outwards upon depolarization and is capable of removing the bound toxin from its receptor site (1, 5, 15, 16, 21, 34). Perturbation of this movement by the bound toxin then inhibits the subsequent conformational change required for fast inactivation. However, in contrast to the large detrimental effect of E1613R substitution at rNav1.2a on Lqq5 activity (21), this substitution had comparatively little effect on Lqh2 activity except when assayed in the toxin dissociation protocol (Fig. 4A; Table 2). This difference in effect may be attributed to the difference between the toxins but also to the experimental system employed for channel expression, such that DIV/S3-S4 may not be displayed identically when expressed in mammalian cells versus Xenopus oocytes. Indeed, we find a large effect of the E1613R mutation on Lqh2 action for Nav1.2 channels expressed in the human embryonic kidney cell line tsA-201 (39). Nevertheless, the results obtained with Lqq5 and Lqh2 applied onto rNav1.2a expressed in mammalian cells (21, 23, 39) and the results regarding toxin dissociation obtained in this study corroborate the suggestion that Glu-1613 in rNav1.2a is in very close proximity to the interacting surface of scorpion α-toxins with this brain sodium channel.
On the basis of the bipartite bioactive surface of Lqh2 (29) and the successful swap of its receptor, we assume that Lqh2 interacts with the channel such that one of the two functional domains at the toxin surface recognizes the gating module at DIV and the other toxin domain interacts with the pore module of DI. The involvement of the distal part of DIV/S3 in this interaction might take place within a crevice in the membrane-channel interface that enables access of the toxin core domain, a scenario that resembles the interaction of a scorpion β-toxin with DII of rNav1.2a (42).
Our suggestion that the core domain of Lqh2 interacts with DIV gating module is based on the findings of large changes in the depolarization-induced dissociation of the core domain toxin mutants compared with the unmodified toxin (Fig. 4A), as well as on the enhanced dissociation of Lqh2 from the channel mutants modified at this channel region, rNav1.2aE1613R and rNav1.2aF1610A (Table 2; Fig. 4, C and D). We used these data to construct an initial model of the putative interaction of Lqh2 with rNav1.2a by employing the three-dimensional structure of the potassium channel Kv1.2 (43) and assuming that the intersegmental region of both channel types would be similar (Fig. 5). In this initial model, Phe-15 of the toxin is in close proximity to Phe-1610 in DIV/S3 and to Glu-1613 in DIV/S3-S4, whereas Asn-44 of the toxin is in close proximity to Glu-1613, in agreement with our conclusion that the toxin core domain interacts with the voltage-sensing module of the channel. Although at this stage we are unable to determine which of the toxin domains interacts first with the channel, from the toxin unbinding experiments it seems that, upon depolarization, dissociation of the toxin off the channel is initiated at the core domain.
FIGURE 5.

Model of Lqh2 interaction with rNav1.2 resting state. The external loops DIV/S1-S2 and S3-S4 of rNav1.2a were constructed on the structural model of Kv1.2 in its resting state (43) using the Swiss-PdbViewer. The internal loops and the gating modules of DI, DII, and DIII were removed. Shown in black are the remaining pore modules of DII, DIII, and DIV, whereas the DI pore module is shown in light gray. Due to the substantial difference in size, the DI/S5-SS1 external loop is omitted (indicated by the orange dashes). DIV/S1 is shown in green, DIV/S2 is shown in blue, DIV/S3 is shown in light orange, and DIV/S4 is shown in dark orange. Lqh2 modeling relied on its close resemblance to Aah2 (29; Protein Data Bank code 1AHO). Phe-15 and Asn-44 are bioactive residues of Lqh2 that are in close proximity to Phe-1610 and Glu-1613 of the channel, respectively (colored sticks according to their chemical nature). Docking of Lqh2 core domain at DIV gating module was performed using DockingServer. Although the residues of the toxin NC domain that may interact with residues at DI/S5-SS1 and DIV/S1-S2 have not been clarified yet, further modeling was performed manually to show this proximity, while avoiding side chain clashes. The final figure was drawn using PyMOL.
In summary, this study reveals sodium channel determinants involved in scorpion α-toxin selectivity as well as illuminates for the first time the α-toxin domain that interacts with the channel voltage sensor, which enables initial modeling of its docking at the channel. Further mutagenesis and double-mutant cycle analysis, including residues that are spatially conserved in sodium channels, are required to identify the individual amino acid residues in the DI/S5-S6 and DIV/S1-S2 loops that participate in toxin binding.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant 1U01 NS058039-01 (to W. A. C. and M. G.). This work was also supported by United States-Israel Binational Agricultural Research and Development Grant IS-4313-10 (to M. G. and D. G.) and by Israeli Science Foundation Grant 107/08 (to M. G. and D. G.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Fig. 1.
- Nav(s)
- voltage-gated sodium channel(s).
REFERENCES
- 1. Catterall W. A. (2000) Neuron 26, 13–25 [DOI] [PubMed] [Google Scholar]
- 2. Zlotkin E. (1999) Annu. Rev. Entomol 44, 429–455 [DOI] [PubMed] [Google Scholar]
- 3. Gordon D., Karbat I., Ilan N., Cohen L., Kahn R., Gilles N., Dong K., Stühmer W., Tytgat J., Gurevitz M. (2007) Toxicon 49, 452–472 [DOI] [PubMed] [Google Scholar]
- 4. Cestèle S., Catterall W. A. (2000) Biochimie 82, 883–892 [DOI] [PubMed] [Google Scholar]
- 5. Ulbricht W. (2005) Physiol. Rev. 85, 1271–1301 [DOI] [PubMed] [Google Scholar]
- 6. Gilles N., Harrison G., Karbat I., Gurevitz M., Nicholson G. M., Gordon D. (2002) Eur. J. Biochem. 269, 1500–1510 [DOI] [PubMed] [Google Scholar]
- 7. Moran Y., Kahn R., Cohen L., Gur M., Karbat I., Gordon D., Gurevitz M. (2007) Biochem. J. 406, 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng G., Deák P., Chopra M., Hall L. M. (1995) Cell 82, 1001–1011 [DOI] [PubMed] [Google Scholar]
- 9. Warmke J. W., Reenan R. A., Wang P., Qian S., Arena J. P., Wang J., Wunderler D., Liu K., Kaczorowski G. J., Van der Ploeg L. H., Ganetzky B., Cohen C. J. (1997) J. Gen. Physiol. 110, 119–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qu Y., Curtis R., Lawson D., Gilbride K., Ge P., DiStefano P. S., Silos-Santiago I., Catterall W. A., Scheuer T. (2001) Mol. Cell Neurosci. 18, 570–580 [DOI] [PubMed] [Google Scholar]
- 11. Yu F. H., Westenbroek R. E., Silos-Santiago I., McCormick K. A., Lawson D., Ge P., Ferriera H., Lilly J., DiStefano P. S., Catterall W. A., Scheuer T., Curtis R. (2003) J. Neurosci. 23, 7577–7585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Derst C., Walther C., Veh R. W., Wicher D., Heinemann S. H. (2006) Biochem. Biophys. Res. Commun. 339, 939–948 [DOI] [PubMed] [Google Scholar]
- 13. Tombola F., Pathak M. M., Isacoff E. Y. (2006) Annu. Rev. Cell Dev. Biol. 22, 23–52 [DOI] [PubMed] [Google Scholar]
- 14. West J. W., Patton D. E., Scheuer T., Wang Y., Goldin A. L., Catterall W. A. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang Y. C., Kuo C. C. (2003) J. Neurosci. 23, 4922–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldin A. L. (2003) Curr. Opin. Neurobiol. 13, 284–290 [DOI] [PubMed] [Google Scholar]
- 17. Sheets M. F., Kyle J. W., Kallen R. G., Hanck D. A. (1999) Biophys. J. 77, 747–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campos F. V., Chanda B., Beirão P. S., Bezanilla F. (2008) J. Gen. Physiol. 132, 251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomsen W. J., Catterall W. A. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 10161–10165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tejedor F. J., Catterall W. A. (1990) Cell Mol. Neurobiol. 10, 257–265 [DOI] [PubMed] [Google Scholar]
- 21. Rogers J. C., Qu Y., Tanada T. N., Scheuer T., Catterall W. A. (1996) J. Biol. Chem. 271, 15950–15962 [DOI] [PubMed] [Google Scholar]
- 22. Qu Y., Rogers J. C., Chen S. F., McCormick K. A., Scheuer T., Catterall W. A. (1999) J. Biol. Chem. 274, 32647–32654 [DOI] [PubMed] [Google Scholar]
- 23. Leipold E., Lu S., Gordon D., Hansel A., Heinemann S. H. (2004) Mol. Pharmacol. 65, 685–691 [DOI] [PubMed] [Google Scholar]
- 24. Leipold E., Hansel A., Olivera B. M., Terlau H., Heinemann S. H. (2005) FEBS Lett. 579, 3881–3884 [DOI] [PubMed] [Google Scholar]
- 25. Weinberger H., Moran Y., Gordon D., Turkov M., Kahn R., Gurevitz M. (2010) Mol. Biol. Evol. 27, 1025–1034 [DOI] [PubMed] [Google Scholar]
- 26. Karbat I., Frolow F., Froy O., Gilles N., Cohen L., Turkov M., Gordon D., Gurevitz M. (2004) J. Biol. Chem. 279, 31679–31686 [DOI] [PubMed] [Google Scholar]
- 27. Karbat I., Kahn R., Cohen L., Ilan N., Gilles N., Corzo G., Froy O., Gur M., Albrecht G., Heinemann S. H., Gordon D., Gurevitz M. (2007) FEBS J. 274, 1918–1931 [DOI] [PubMed] [Google Scholar]
- 28. Liu L. H., Bosmans F., Maertens C., Zhu R. H., Wang D. C., Tytgat J. (2005) FASEB J. 19, 594–596 [DOI] [PubMed] [Google Scholar]
- 29. Kahn R., Karbat I., Ilan N., Cohen L., Sokolov S., Catterall W. A., Gordon D., Gurevitz M. (2009) J. Biol. Chem. 284, 20684–20691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shichor I., Zlotkin E., Ilan N., Chikashvili D., Stuhmer W., Gordon D., Lotan I. (2002) J. Neurosci. 22, 4364–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wallner M., Weigl L., Meera P., Lotan I. (1993) FEBS Lett. 336, 535–539 [DOI] [PubMed] [Google Scholar]
- 32. Chen H., Heinemann S. H. (2001) J. Gen. Physiol. 117, 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gordon D., Kallen R. G., Heinemann S. H. (2004) in Neurotox '03: Neurotoxicological Targets from Functional Genomics & Proteomics (Beadle D., Mellor I. R., Usherwood P. N. eds) pp. 59–68, Society of Chemical Industry, London [Google Scholar]
- 34. Catterall W. A. (1979) J. Gen. Physiol. 74, 375–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Strichartz G. R., Wang G. K. (1986) J. Gen. Physiol. 88, 413–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen H., Gordon D., Heinemann S. H. (2000) Pflugers Arch. 439, 423–432 [DOI] [PubMed] [Google Scholar]
- 37. Gilles N., Leipold E., Chen H., Heinemann S. H., Gordon D. (2001) Biochemistry 40, 14576–14584 [DOI] [PubMed] [Google Scholar]
- 38. Featherstone D. E., Richmond J. E., Ruben P. C. (1996) Biophys. J. 71, 3098–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang J., Yarov-Yarovoy V., Kahn R., Gordon D., Gurevitz M., Scheuer T., Catterall W. A. (2011) Biophys. J. 100, 422a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schnur E., Turkov M., Kahn R., Gordon D., Gurevitz M., Anglister J. (2008) Biochemistry 47, 911–921 [DOI] [PubMed] [Google Scholar]
- 41. Bosmans F., Martin-Eauclaire M. F., Swartz K. J. (2008) Nature 456, 202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cestèle S., Yarov-Yarovoy V., Qu Y., Sampieri F., Scheuer T., Catterall W. A. (2006) J. Biol. Chem. 281, 21332–21344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pathak M. M., Yarov-Yarovoy V., Agarwal G., Roux B., Barth P., Kohout S., Tombola F., Isacoff E. Y. (2007) Neuron 56, 124–140 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.