

Background: NHERF tethers the PTHR at the cell membrane. Upon PTHR activation and internalization, the fate of NHERF is uncertain.
Results: NHERF interaction with the PTHR promotes ezrin association and delays arrestin recruitment to the receptor.
Conclusion: NHERF engages dynamically with the PTHR, regulating its association and dissociation with protein partners.
Significance: NHERF association with the PTHR coordinates spatiotemporal receptor signaling and action.
Keywords: Arrestin, Epithelial Cell, Ezrin, Fluorescence Resonance Energy Transfer (FRET), G Protein-coupled Receptors (GPCR), Parathyroid Hormone, Receptor Endocytosis, Scaffold Proteins, NHERF, PDZ Proteins
Abstract
Na/H exchanger regulatory factor-1 (NHERF1) is a cytoplasmic PDZ (postsynaptic density 95/disc large/zona occludens) protein that assembles macromolecular complexes and determines the localization, trafficking, and signaling of select G protein-coupled receptors and other membrane-delimited proteins. The parathyroid hormone receptor (PTHR), which regulates mineral ion homeostasis and bone turnover, is a G protein-coupled receptor harboring a PDZ-binding motif that enables association with NHERF1 and tethering to the actin cytoskeleton. NHERF1 interactions with the PTHR modify its trafficking and signaling. Here, we characterized by live cell imaging the mechanism whereby NHERF1 coordinates the interactions of multiple proteins, as well as the fate of NHERF1 itself upon receptor activation. Upon PTHR stimulation, NHERF1 rapidly dissociates from the receptor and induces receptor aggregation in long lasting clusters that are enriched with the actin-binding protein ezrin and with clathrin. After NHERF1 dissociates from the PTHR, ezrin then directly interacts with the PTHR to stabilize the PTHR at the cell membrane. Recruitment of β-arrestins to the PTHR is delayed until NHERF1 dissociates from the receptor, which is then trafficked to clathrin for internalization. The ability of NHERF to interact dynamically with the PTHR and cognate adapter proteins regulates receptor trafficking and signaling in a spatially and temporally coordinated manner.
Introduction
The Na+/H+ exchanger regulatory factor-1 (NHERF1),2 known also as the EBP50 (50-kDa ezrin-binding protein), is a cytoplasmic scaffolding protein implicated in protein targeting and in the assembly of multi-protein complexes (1–4). NHERF1 belongs to a family of adaptor proteins consisting of four members. All of them possess tandem PDZ (postsynaptic density 95/disc large/zona occludens) domains: two in NHERF1 and NHERF2 and four in NHERF3 and NHERF4 (5). PDZ domains are globular structures of ∼90 residues that mediate protein-protein interactions by binding to other PDZ domains or, more commonly by recognizing short amino acid motifs at the carboxyl terminus of target proteins (6, 7). The Type I motif takes the form (D/E)(S/T)XΦ, where X is promiscuous residue, and Φ is a hydrophobic residue such as Leu, Ile, Val, or Met (8). In addition to the tandem PDZ domains, NHERF1 and NHERF2 contain a carboxyl-terminal ezrin-binding domain that engages ezrin, radixin, or moesin through their respective amino-terminal 4.1 protein-ezrin-radixin-moesin (FERM) domain and enables tethering to the actin cytoskeleton through their carboxyl terminus (1, 2, 5, 9). NHERF1 is differentially expressed in mammalian tissues, with particularly high levels found in polarized epithelial cells (10). NHERF1 recruits receptors, ion transporters, Gαq, phospholipase C, RabGAP EPI64, and other adaptor proteins to plasma membranes, regulating their distribution, signaling, and trafficking (2). G protein-coupled receptors (GPCRs), including the parathyroid hormone type-1 receptor (PTHR), are primary NHERF1 targets (4, 11, 12).
Acting through the PTHR, PTH and the PTH-related protein regulate mineral ion homeostasis and bone turnover, respectively. The PTHR possesses a PDZ recognition domain (ETVM) at its intracellular carboxyl terminus that binds NHERF1 PDZ1 with greater affinity than PDZ2 (13, 14). This interaction and the tethering of NHERF1 to the actin cytoskeleton through the ezrin-binding domain forms a complex that stabilizes the PTHR at the cell membrane, thereby delaying endocytosis and desensitization (15–17). NHERF1-null mice (18) and patients (19) harboring NHERF1 polymorphisms or coding region mutations display mineral ion wasting and a bone phenotype, consistent with an important biological role of NHERF1/PTHR interactions. Upon activation by PTH, the PTHR recruits β-arrestins, traffics to clathrin, and is endocytosed in early endosomes and eventually recycled to the membrane or targeted for degradation (20–22). However, the fate of NHERF1 and the subsequent events triggered by the activation of the PTHR remain unknown. In contrast to the static model of PTHR-NHERF1 interaction previously proposed (23), we now show dynamic interactions between NHERF1 and the PTHR that determine and coordinate the series of events preceding receptor internalization. Upon PTH stimulation, NHERF1 rapidly dissociates from the PTHR, resulting in receptor redistribution in membrane-retained clusters. Although NHERF is no longer linked to the receptor, by promoting association of the PTHR to an ezrin-actin cytoskeletal complex and by delaying β-arrestin1/2 recruitment, NHERF1 decreases internalization of the PTHR clusters.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
Rat osteosarcoma (ROS) 17/2.8 cells were cultured in DMEM/F-12 50/50 supplemented with 10% FBS (Cellgro, Mediatech, Herndon, VA), 100 units/ml penicillin, and 100 μg/ml streptomycin. ROS-T6-N4 cells that stably express NHERF1 regulated by a tetracycline repressor protein were generated as described (16) and selected with zeocin (0.4 mg/ml). The cells were maintained at 37 °C in a humidified atmosphere of 5% CO2. For transient transfections, ROS, ROS-T6-N4, and HEK 293 cells were grown on 25-mm coverslips 12 h prior to transfection with FuGENE 6 (Roche Applied Science) in complete medium. After 24 or 48 h, coverslips in HEPES/BSA buffer (HEPES buffer containing 0.1% (w/v) BSA) were transferred to an Attofluor chamber (Invitrogen) for live cell imaging or for immunofluorescence experiments.
cDNA Constructs
For CFPNHERF and YFPNHERF cDNA, the carboxyl-terminal HA epitope of pcDNA3.1+ NHERF1-HA (15) was deleted by PCR amplification using the forward primer (AAA TCT CGA GCT AGC GCG GAC GCA GCG GCC) with an XhoI restriction site and the reverse primer (AAA GGA TCC TCA GAG GTT GCT GAA GAG TTC) with a BamHI restriction site. The purified PCR fragment was cut by XhoI and BamHI, and a 1.1-kb fragment without the epitope was subcloned into an mCerulean 3-C1 vector (CFPNHERF) or a Venus-C1 vector (YFPNHERF). The fidelity of the plasmids was confirmed by sequencing (ABI PRISM 377; Applied Biosystems, Foster City, CA) and subsequent sequence alignment (NCBI Blast) with human NHERF1.
For PTHRCFPETVM, the truncated form of the receptor HA-PTHR 480-STOP (24) was used as a parent vector to insert BamHI, XhoI, and Xba1 restriction sites at the carboxyl terminus of the receptor by site-directed mutagenesis and loop-in PCR (25). mCerulean 3-C1 was cut with BamHI and XhoI, and the mCerulean insert was subcloned into the carboxyl terminus of the HA-PTHR 480-STOP (Scheme 1). An insert containing the sequence for the terminal 22 amino acids of the full-length PTHR, which includes the PDZ recognition motif, ETVM, was subcloned between XhoI and Xba1 in the HA-PTHR 480-STOP vector. The fidelity of the plasmids was confirmed by sequencing, and the localization of PTHRCFP and the interaction with NHERF1 was tested by live cell imaging.
SCHEME 1.

The PTHRCFPETVM chimera includes the PTHR(1–480), a cerulean fluorescent protein, and the carboxyl-terminal 22 amino acids of the PTHR containing the NHERF-binding domain ETVM (18 aa + ETVM). This construct signals and traffics indistinguishably from full-length PTHR.
mCherryNHERF and GFPPTHR were generously provided by Dr. A. C. Newton (University of California) and Dr. C. Silve (INSERM, Paris, France), respectively. Membrane-targeted CFP (myristoylation-palmitoylation CFP) (26), HA-PTHR (15), GFPNHERF (27), mCerulean 3-C1 (28), Tomatoβ-Arrestin1, CFPβ-Arrestin1, YFPβ-Arrestin2 (29), PTHR-ETVA, with a mutation of the carboxyl-terminal amino acid of PTHR from methionine to alanine (M593A) (30), a dominant negative form of dynamin, K44A-dynamin-pcDNA3.1 (31), and the EPAC biosensor (32) were previously reported.
Fluorescence Recovery after Photobleaching (FRAP)
FRAP measurements were performed with a Nikon A1 microscope on circled areas <2.5-μm diameter at the plasma membrane adjacent to the coverslip. The bleaching time was 5-ms with 488- and 514-nm laser lines, and the fluorescence recovery was recorded for 5 min. Recovery curves were calculated for each value according to the following equation,
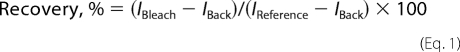 |
where IBleach is the fluorescence intensity of the bleached spot, IBack is the fluorescence intensity of the background, and IReference is the fluorescence intensity of control regions in other cells or regions far removed from the target cell. The data were fit to an exponential decay and plotted using Prism (GraphPad, San Diego, CA) to calculate the half-life (t½) for each condition. The mobile fraction (%) (17) was calculated as follows,
where Ipost is the average fluorescence intensity value of the plateau after bleaching, IBl is the fluorescence intensity immediately recorded after bleaching, and Ipre is the average fluorescence intensity of the plateau before bleaching.
The diffusion coefficient (D) was calculated from the Stokes-Einstein equation for two-dimensional diffusion (D = r2/4t½), where r is the radius of the bleached target and t½ is the half-life of the fluorescence recovery.
Receptor Immobilization by Antibody Cross-linking
Immobilization of GFPPTHR at the cell membrane was performed as described (33). Briefly, medium was removed, and the cells were washed three times with buffer containing 150 mm NaCl, 10 mm Na-HEPES, 12.8 mm d-glucose, 2.5 mm KCl, 0.5 mm MgCl2, and 0.5 mm CaCl2, pH 8.0, at room temperature. For antibody cross-linking, the cells were incubated sequentially for 5 min each (separated by three buffer washes) in 1:200 anti-GFP rabbit IgG (Invitrogen) and biotin-XX goat anti-rabbit antiserum (Invitrogen).
Total Internal Reflection Microscopy (TIRF)
TIRF microscopy was performed using a Nikon A1 equipped with an X60 TIRF objective controlled by NIS Elements (Nikon). Images were recorded at 30-s intervals for 20 min. The collected data were exported to Image J (34). Fluorescence protein colocalization and average PTHR puncta size were analyzed using the JACoP plug-in (35) and particle size analysis, respectively.
Fluorescence Resonance Energy Transfer (FRET)
Interactions between PTHR and β-arrestins in the presence or absence of NHERF1 were measured as an increase of FRET between YFPβ-Arrestin2 and PTHRCFP. Time lapse movies were recorded with a Nikon A1s confocal microscope attached to a Ti-E inverted base using a 40× 1.30 NA plan-apo objective. The FRET signal was calculated as the normalized ratio of the YFP and CFP emission (FYFP/FCFP) (36).
Donor Dequenching after Acceptor Photobleaching
FRET between CFP and YFP was determined using a modification of a previously described protocol (37). Briefly, CFP fluorescence was recorded (457-nm excitation) followed by 1-min of YFP bleaching (514-nm ∼100% bleach intensity), and the increase of CFP fluorescence intensity after bleaching of the acceptor was measured for 2 min. Direct bleaching of CFP was determined in cells expressing only CFPβ-arrestin and was negligible. A CFP protein with a myristoylation/palmitoylation modification (26) to target the cell membrane was used with YFPNHERF to calculate nonspecific increases of FRET after photobleaching of the acceptor. FRET efficiency was calculated as follows,
 |
where Donor+Acceptor and Donor−Acceptor refer to fluorescence measurement in the presence and absence of acceptor, respectively.
Immunofluorescence
HEK 293 cells were cultured on poly-d-lysine glass coverslips, transfected with GFPPTHR-ETVM or GFPPTHR-ETVA plasmids, and allowed to grow for 48 h until 80% confluent. 100 nm hPTH(1–34) (Bachem, Torrance, CA) was added for the indicated times, and the cells were washed in PBS, fixed for 10 min in 2% paraformaldehyde in PBS, and permeabilized with 0.1% Triton X-100 in PBS for 20 min. Nonspecific binding was blocked with 5% goat serum in PBS for 1 h. The cells were then incubated with monoclonal anti-ezrin antibody (Invitrogen) (1:1000) for 2 h. After washing with PBS, the cells were incubated with anti-mouse Alexa 546 (Invitrogen) (1:2000) for 1 h, and the slides were mounted with aqueous mounting medium for TIRF evaluation. All of the incubations were performed at room temperature.
Colocalization Analysis
TIRF and confocal images from combinations of β-arrestins, NHERF1, and PTHR expressed at the cell membrane were analyzed in a pair-wise manner using Image J. Subcellular areas from different cells where compared using the Pearson correlation coefficient (ρ) (38), which is defined here as the ratio of the covariance of the red and green color images divided by the product of the standard deviation of the normalized image intensities, was calculated with the JACoP plug-in (34). The values of this coefficient range from −1, indicating the complete absence of overlap between pixels from the two images, to +1, indicating perfect image correlation. Pearson's correlation coefficient accounts only for the similarity of shapes between the two images and does not depend upon image pixel intensity values.
RESULTS
Modulation of PTHR Distribution and Mobility by NHERF1
We first characterized the cell surface distribution of PTHR and NHERF1 by TIRF and confocal microscopy in ROS cells that do not express NHERF (30). ROS cells coexpressing GFPPTHR and mCherryNHERF1 exhibited extensive colocalization at the cell membrane (Figs. 1A and 2B) and closely adjacent regions with a Pearson's correlation coefficient of 0.80 ± 0.01. Under resting conditions, PTHR and NHERF1 are thus membrane-delimited and colocalize in ROS cells.
FIGURE 1.

Dynamics of NHERF1 and PTHR trafficking at the plasma membrane. A, cell surface distribution of NHERF1 and PTHR. ROS cells were transfected with mCherryNHERF and GFPPTHR and examined by TIRF microscopy 24 h after transfection. Colocalization of NHERF1 and PTHR in different subcellular regions was estimated with Pearson's correlation coefficient of eight cells from independent experiments. B–E, PTHR and NHERF1 mobility was determined FRAP. At time 0, circular spots 2 μm in diameter were bleached using a 561- or 488-nm laser (for mCherryNHERF and GFPPTHR, respectively) at 50% power for 5 ms. FRAP was measured over 5 min (1 min shown). B, PTHR FRAP calculated in the presence or absence of NHERF1. C, NHERF1 FRAP calculated in the presence or absence of PTHR. D, GFPPTHR was immobilized at the membrane as described under “Experimental Procedures,” and NHERF1 and PTHR FRAP were calculated. Half-time recovery values were analyzed for each FRAP curve. E, half-time recovery values from the FRAP analysis were used to calculate the diffusion coefficients of PTHR or PTHR-ETVA ± NHERF1 or cytochalasin D (Cyto. D) and NHERF1 ± nonimmobilized or immobilized PTHR. The data were collected from 8–17 cells from three independent experiments. *, p ≤ 0.05 versus corresponding control.
FIGURE 2.

Effects of PTH-activated PTHR on NHERF1 mobility and β-arrestin localization. A, ROS cells were cotransfected with mCherryNHERF and GFPPTHR. After 24 h, half-time recovery values were calculated from FRAP curves at different time intervals following challenge with 100 nm hPTH(1–34). The data shown represent the average ± S.E. from at least three independent experiments. B and C, ROS cells cotransfected with GFPPTHR, mCherryNHERF (B), or mCherryNHERF and YFPβ-Arrestin2 (C) were examined at 0, 5, 10, 20, and 30 min after exposure to 100 nm hPTH(1–34) by live cell confocal imaging microscopy. The red, green, and yellow channels were separated using spectral unmixing software (NIS-ElementsTM; Nikon). Representative images of seven cells from three independent experiments are displayed.
Next, we determined the mobility and dynamics of PTHR and NHERF1 by FRAP of 2-μm diameter areas on the basal membrane of ROS cells. GFPPTHR fluorescence recovery in the absence of NHERF1 was rapid (t½ = 16.3 ± 2.9 s) (Fig. 1B), and the mobile PTHR fraction was high (67%). By contrast, in the presence of NHERF1, GFPPTHR fluorescence recovery was substantially slowed (t½ = 36.1 ± 9.0 s), and the mobile fraction of PTHR was reduced (28%) (Fig. 1B), demonstrating that binding to NHERF1 diminishes PTHR mobility.
Conditions were reversed, and we measured the mobility of mCherryNHERF in the presence and absence of PTHR.mCherryNHERF exhibited slower fluorescence recovery in the presence of PTHR (t½ = 18.5 ± 2.9 s) compared with its absence (t½ = 8.5 ± 2.3 s) (Fig. 1C). Notably, the mobile fractions were similar under both conditions (90% without PTHR; 89% with PTHR). Furthermore, similar NHERF1 FRAP recovery values were obtained when the PTHR was fully immobilized by antibody cross-linking at the cell membrane (Fig. 1D). PTHR diffusion was slower in cells expressing NHERF1, and NHERF1 diffusion decreased in the presence of either mobile (“free”) or immobile PTHR (Fig. 1E). However, PTHR immobilization did not further decrease NHERF1 diffusion compared with “free” PTHR (Fig. 1E). In addition, a PTHR mutant that does not interact with NHERF1 (PTHR-ETVA) showed high diffusion coefficient values both in the presence or absence of NHERF1 (Fig. 1E). Together, these results are consistent with the view that the interaction between PTHR with a functional PDZ-binding domain and NHERF1 reduces the diffusion of both proteins without binding to an immobile complex.
We then asked whether an intact actin cytoskeleton is required for NHERF1 to tether the receptor at the cell membrane. Incubation with cytochalasin D, an inhibitor of actin polymerization, increased the PTHR diffusion coefficient despite the coexpression with NHERF1 (Fig. 1E). These data confirm a previous report (17) suggesting that NHERF1-mediated PTHR immobilization at the cell surface involves interactions with the actin cytoskeleton.
Ligand-induced Dissociation of NHERF1 and PTHR
We next characterized NHERF1-PTHR interactions upon activation of the receptor by measuring mCherryNHERF mobility following PTH stimulation. FRAP curves were generated at discrete times after the addition of the agonist, and t½ values were derived from each curve. Over the immediate time frame of 1–240 s, NHERF1 fluorescence recovery after PTHR stimulation was significantly shorter (Table 1), corresponding to more rapid recovery and higher mobility of NHERF1 when the receptor was activated compared with nonstimulated conditions. Interestingly, the t½ values of mCherryNHERF after PTH stimulation were similar to those obtained for mCherryNHERF in the absence of PTHR (t½ = 8.5 ± 2.3 s), suggesting rapid release of NHERF1 from the receptor upon PTH activation.
TABLE 1.
NHERF1 mobility increases upon PTHR stimulation by PTH
ROS cells were transfected with GFPPTHR and mCherryNHERF. Photobleaching of NHERF1 in discrete areas colocalizing with PTHR was performed at different time points after PTH(1–34) stimulation. Fluorescence intensity for each bleached area was recorded, and the data were exported to GraphPad Prism 5.0. FRAP curves and their corresponding t½ values were obtained for each condition, fitting NHERF1 FRAP data to a one-phase decay function. The data were collected from five independent experiments.
| Time after hPTH(1–34) stimulation |
|||||
|---|---|---|---|---|---|
| 0 s | 10 s | 40 s | 80 s | 240 s | |
| FRAP t½ (s) | 17.47 ± 1.83 | 9.37 ± 1.04a | 7.50 ± 1.00a | 6.51 ± 0.97a | 9.37 ± 0.81a |
a p ≤ 0.01 versus 0 s after PTH stimulation.
The NHERF1 diffusion coefficients were calculated from the t½ FRAP values of mCherryNHERF with or without stimulated PTHR. NHERF1 diffusion near the cell membrane rapidly increased after PTHR stimulation by PTH (Fig. 2A), showing that NHERF1 mobility increases following activation of the PTHR and remains elevated throughout the period of study (240 s).
To define the itinerary of NHERF1 upon PTHR stimulation by PTH, we analyzed the subcellular distribution of NHERF1 at different focal planes in live cells by confocal microscopy. After stimulation with PTH, NHERF1 remained adjacent to the cell membrane (Fig. 2, B and C). Although PTHR redistributed largely together with β-arrestin2 in cytosolic compartments during 10 min of PTH stimulation, a considerable amount of PTHR remained at the cell surface (Fig. 2, B and C).
The dynamic interactions of NHERF1 and PTHR at the plasma membrane and adjoining regions were then characterized using TIRF microscopy. In the absence of NHERF1, GFPPTHR rapidly internalized to the cytoplasm (Fig. 3, A and B). In contrast, coexpression of mCherryNHERF with GFPPTHR delayed and decreased receptor internalization (Fig. 3, A and B), consistent with earlier observations (17, 30). Moreover, upon PTH stimulation, cell surface GFPPTHR, when coexpressed with NHERF1, exhibited a greater redistribution from localized areas to disperse puncta across the cell membrane, whereas mCherryNHERF did not redistribute (Fig. 3A and supplemental Fig. S1), supporting the view that NHERF1 dissociates from PTHR upon PTH stimulation and persistently resides at the cell membrane and adjacent regions.
FIGURE 3.

NHERF1 effects on PTH-activated PTHR cell surface distribution. ROS cells were transfected with GFPPTHR in the absence or presence of mCherryNHERF, and the distribution of PTHR at the cell membrane after PTH stimulation was analyzed by TIRF microscopy. A, PTHR localization in the absence and presence of NHERF1 and before and after the addition of 100 nm hPTH(1–34). The insets show the representative area containing PTHR particles. Bar, 1 μm. B, PTH-induced PTHR internalization determined from the corresponding TIRF images. C, GFPPTHR fluorescence intensity within circular areas (≤1 μm diameter) from measurements of 20–40 cell surface regions from TIRF images. D, GFPPTHR particle average size calculated with Image J from 35 cell surface regions from TIRF images. Cells coexpressing mCherryNHERF and GFPPTHR were transfected with dominant negative K44A-dynamin (K44A) or empty vector. *, p ≤ 0.05 versus −NHERF1; a, p ≤ 0.05 versus −NHERF1 + K44A. E and F, HEK 293 cells were transfected with GFPPTHR-ETVM or GFPPTHR-ETVA. Ezrin immunofluorescence after a 5-min stimulation with 100 nm hPTH(1–34) was determined as described under “Experimental Procedures.” Ezrin colocalization with PTHR-ETVM or PTHR-ETVA is shown in E. The images shown in F are representative of three independent determinations.
PTHR Clustering
To characterize the behavior of the GFPPTHR puncta at the cell surface, we analyzed the initial changes of fluorescence intensity after PTH stimulation in the absence or presence of NHERF1. Both conditions revealed a peak of clustered fluorescence intensity after PTH stimulation of greater magnitude in the presence of NHERF1 (area under curve = 4,279) than in its absence (area under curve = 2,259) (Fig. 3C). These GFPPTHR particles subsequently colocalized with dsREDclathrin-coated pits (Pearson's correlation coefficient = 0.87 ± 0.02). The GFPPTHR puncta appeared and disappeared as clathrin-coated pits are formed and undergo scission from the plasma membrane or adjacent regions and internalize to the cytoplasm. In addition to their greater fluorescence intensity, the average size of the GFPPTHR particles increased in cells expressing NHERF1 (Fig. 3D). Furthermore, NHERF1 attenuated the post-peak decrease in GFPPTHR puncta fluorescence intensity (Fig. 3C). These results suggest that NHERF1 promotes persistent PTHR clustering after PTH stimulation.
Next, we inquired whether the increased size of the PTHR clusters induced by NHERF1 was due to inhibition of receptor internalization or to PTHR particle-directed rearrangement. We used a dominant negative form of dynamin (K44A-dynamin), a GTPase that regulates the formation of clathrin-coated vesicles (39), to inhibit receptor internalization (30). K44A-dynamin significantly increased the size of GFPPTHR puncta in the absence of NHERF1. However, K44A-dynamin did not affect the size of GFPPTHR particles in cells expressing NHERF1 (Fig. 3D). Moreover, inhibition of PTHR internalization by K44A-dynamin did not fully recapitulate the increase in particle size induced by NHERF1. These results suggest that receptor clustering at the cell membrane promoted by NHERF1 is achieved by a combination of inhibition of receptor internalization and by PTHR particle rearrangement. This interpretation is supported by receptor binding studies with [125I]PTH in ROS cells showing that the amount of endogenous receptor at the cell surface when expressing NHERF1 and/or K44A-dynamin is the same after PTH as in nonstimulated conditions (data not shown), consistent with the view that NHERF1 promotes receptor aggregation in membrane clusters and does not increase the number of cell surface receptors. We hypothesized that PTHR clusters are retained at the cell membrane by a NHERF1-mediated mechanism that involves the tethering of receptors to the actin cytoskeleton. However, NHERF1 did not colocalize with the PTHR clusters after PTH stimulation (supplemental Fig. S1). This observation is compatible with the finding that NHERF1 promptly dissociates from the PTHR upon ligand activation. We therefore determined whether ezrin, a cytoskeleton adaptor protein that binds NHERF1 but can directly interact with PTHR (40), acts to scaffold the PTHR and the actin cytoskeleton after dissociation of NHERF1 from the PTHR. HEK 293 cells expressing endogenous NHERF1 and ezrin were transfected with GFPPTHR-ETVM, which interacts with NHERF1 or GFPPTHR-ETVA that does not. Using TIRF immunofluorescence we observed that upon stimulation with PTH, ezrin colocalized with GFPPTHR-ETVM clusters at the cell membrane but not with the GFPPTHR-ETVA clusters (Fig. 3, E and F), suggesting that NHERF1 promotes ezrin-PTHR interactions.
NHERF1 Does Not Stably Interact with β-Arrestins
Given that PTHR associates dynamically with NHERF1 and that PTH promotes recruitment of β-arrestins to the activated receptor (20, 30, 41), we used TIRF to test whether NHERF1 and β-arrestin interact at the plasma membrane or the closely adjacent cytoplasm. In nonstimulated ROS cells overexpressing the PTHR, we observed no detectable colocalization of YFPNHERF and Tomatoβ-arrestin1 (Fig. 4A). Furthermore, after PTH stimulation, Tomatoβ-arrestin1 was recruited to the membrane but did not affect the pattern of YFPNHERF distribution at the cell surface (Fig. 4A). Similar results were obtained using GFPNHERF with Tomatoβ-arrestin1 and mCherryNHERF with YFPβ-arrestin2 (data not shown).
FIGURE 4.

Interactions of NHERF1 and β-arrestin. A, ROS cells were cotransfected with YFPNHERF, Tomatoβ-Arrestin1, and HA-PTHR for 24 h. After the addition of 100 nm PTH, the cells were examined by TIRF microscopy using the Nikon Perfect Focus System®. Note the arrestin recruitment to the edges of the cell, where PTHR is more abundant. Representative images from six independent experiments are shown. B, efficiency of CFPmembrane (Mem.) protein, PTHRCFPETVM, or CFPβ-arrestin with YFPNHERF, PTH(1–34)-stimulated PTHRCFPETVM with YFPβ-arrestin, and CFPEPACYFP FRET pairs were analyzed using donor dequenching after acceptor photobleaching as described under “Experimental Procedures.” *, p ≤ 0.05 versus membrane protein-NHERF pair; **, p ≤ 0.01 versus membrane protein-NHERF pair; †, p ≤ 0.05 versus β-arrestin-NHERF pair; ††, p ≤ 0.01 versus β-arrestin-NHERF pair. C, ROS-T6-N4 cells were cotransfected with PTHRCFP and YFPβ-Arrestin2 and 0, 10, or 100 ng/ml tetracycline was added for 48 h to induce NHERF1 expression. Top panel, immunoblot showing differences in NHERF1 induction with 0, 10, or 100 ng/ml tetracycline. Bottom panel, changes in FRET were recorded after the addition of PTH. The data shown correspond to ≥8–10 cells from three independent experiments.
To confirm that NHERF1 and β-arrestin do not interact, we performed donor dequenching after acceptor photobleaching using a FRET pair consisting of CFPβ-arrestin as the donor and YFPNHERF as the acceptor. FRET efficiency did not increase after YFP photobleaching (Fig. 4B), suggesting that within the limit of detection, YFPNHERF and CFPβ-arrestin do not interact. A CFPmembrane-targeted YFPNHERF FRET pair was used to exclude nonspecific increases of CFP fluorescence after YFP photobleaching (Fig. 4B). Positive controls using the intramolecular FRET-based cAMP biosensor EPAC1CFP/YFP, PTHRCFPETVM-YFPNHERF, or PTH(1–34)-stimulated PTHRCFPETVM-β-Arrestin2YFP pairs displayed increased FRET efficiency after YFP photobleaching (Fig. 4B). Supplemental Fig. S2 shows the increase in CFP fluorescence intensity after acceptor photobleaching for the EPAC biosensor and the PTHRCFPETVM-YFPNHERF or PTH(1–34)-stimulated PTHRCFPETVM-β-ArrestinYFP pairs compared with CFPβ-arrestin-YFPNHERF and CFPmembrane-targeted YFPNHERF FRET pairs.
We characterized the kinetics of PTHR, NHERF1, and β-Arrestin interactions after receptor activation. ROS cells harboring inducible NHERF1 (ROS-T6) were transiently transfected with PTHRCFPETVM and YFPβ-Arrestin2 cDNA constructs. Recruitment of β-Arrestin2 to the receptor upon PTH stimulation was confirmed by confocal microscopy (supplemental Fig. S3). FRET analysis of β-Arrestin2 association with the receptor after PTH stimulation in the absence of NHERF1 showed a similar magnitude of response but slower kinetics (t½ = 58 ± 5 s) (Fig. 4C) than in the presence of modest (tetracycline, 10 ng/ml; t½ = 80 ± 11 s) or maximal (tetracycline, 100 ng/ml; t½ = 247 ± 21 s) NHERF1 induction (16), suggesting that NHERF1 delays the recruitment of β-arrestins to PTHR. No interaction between NHERF1 and β-Arrestin was detected by FRET in ROS cells.
To characterize the interrelated patterns of NHERF1, PTHR, and β-arrestin distribution at the cell membrane after PTHR activation, we analyzed the colocalization of GFPPTHR and mCherryNHERF or GFPPTHR and Tomatoβ-arrestin1 by TIRF microscopy. Upon stimulation with PTH, we observed a decrease in the Pearson correlation coefficient, corresponding to the amount of PTHR colocalizing with NHERF1, consistent with a redistribution of the receptor away from NHERF1 (Fig. 5, A and C, and Table 2). As expected, β-arrestin1 subsequently colocalized with PTHR in response to PTH (Fig. 5B and Table 2 and supplemental Fig. S4). No colocalization between NHERF1 and β-arrestin1 was observed (Fig. 5C and supplemental Fig. 4). The half-time of the rapid dissociation of the PTHR-NHERF1 complex (t½ = 4.99 min) compared with the slower formation of the PTHR-β-Arrestin1 complex (t½ = 10.83 min) suggests that the PTHR dissociates from NHERF1 before engaging β-Arrestin1 and does not form a stable ternary complex of the three proteins.
FIGURE 5.

Effects of PTH on NHERF1, β-Arrestin1, and PTHR colocalization. ROS cells were cotransfected with GFPPTHR and mCherryNHERF, or GFPPTHR, Tomatoβ-Arrestin1, and HA-NHERF1 for 24 h and were examined by TIRF microscopy. A, NHERF1 and PTHR colocalization are shown before and 15 min after the addition of 100 nm hPTH(1–34). B, β-Arrestin1 and PTHR colocalization are shown before and 15 min after the addition of 100 nm hPTH(1–34). C, analysis of time-dependent PTHR colocalization (ρ) with NHERF1 and β-Arrestin1 after PTH stimulation. Representative details of captured images are shown. The data represent measurements from three independent experiments.
TABLE 2.
PTHR colocalization with NHERF1 before PTH and β-arrestin1 after stimulation by PTH
ROS cells were cotransfected with GFPPTHR and mCherryNHERF, or GFPPTHR, Tomatoβ-Arrestin1 and HA-NHERF1 for 24 h and were examined by TIRF microscopy. NHERF or β-Arrestin1 colocalization with PTHR was measured by Pearson's correlation coefficient.
| Time after PTH stimulation (min) | Pearson's correlation coefficient |
|
|---|---|---|
| NHERF/PTHR | β-Arrestin1/PTHR | |
| 0 | 0.85 ± 0.04 | 0.45 ± 0.11 |
| 2.5 | 0.71 ± 0.03 | 0.52 ± 0.09 |
| 5 | 0.53 ± 0.08a | 0.59 ± 0.11 |
| 10 | 0.36 ± 0.11a | 0.71 ± 0.07a |
| 15 | 0.32 ± 0.11a | 0.78 ± 0.05a |
a p ≤ 0.05 versus 0 s after PTH stimulation.
DISCUSSION
Several scaffold proteins regulate GPCR signaling and trafficking (42–44). NHERF1 mediates the assembly in functional complexes of proteins possessing PDZ recognition ligands (4). However, the mechanism whereby NHERF1 promotes the interactions of different proteins with the same receptor is unknown, as is the behavior of NHERF1 itself upon receptor activation and internalization. Here, we show that NHERF1 establishes dynamic interactions with the PTHR within membrane microdomains, suggesting that NHERF1 promotes the recruitment of specific signaling proteins in a temporally and spatially coordinated manner at different steps of receptor activation and internalization. We chose the PTHR as a model to characterize the fate of NHERF1 because the phenotype of NHERF1-null mice (18) and humans bearing NHERF1 polymorphisms or mutations (19) involves renal mineral ion wasting, osteopenia, and osteomalacia, suggesting that PTHR interactions with NHERF1 modulate vital physiological functions.
Several transporters and receptors harboring PDZ-binding domains localize to apical membranes in epithelial cells because of the interaction of these motifs with NHERF1 (3). Moreover, NHERF1 interactions with cytoskeletal binding proteins (ezrin, radixin, and moesin) promote the tethering of PDZ-containing transporters and receptors, including the PTHR, to the actin cytoskeleton (17, 45). Reduced PTHR mobility in the presence of NHERF1, as reported here, presumably arises from the assembly of a PTHR-NHERF1-ezrin ternary complex. Supporting this interpretation, our results show that NHERF1 reduces PTHR diffusion only when the receptor contains an intact ETVM motif and the actin cytoskeleton is not compromised. Moreover, the NHERF1 diffusion coefficient is reduced in the presence of the PTHR. Engagement of FERM with the ezrin-binding domain induces conformational changes in NHERF1 structure by a long range, interdomain allosteric mechanism (46). This allosteric action may promote formation of the PTHR-NHERF1-ezrin-actin complex. Binding of the ezrin FERM domain to the carboxyl terminus of NHERF1 increases the binding affinity of both NHERF1 PDZ1 and PDZ2 domains (46).
Although NHERF1 mobility diminished in the presence of the PTHR, complete immobilization of the PTHR did not further decrease NHERF1 diffusion. This finding suggests that NHERF1-PTHR interactions are transient. PDZ domains bind their ligands with modest affinities (Kd = ∼1 μm), which makes them appropriate for reversible and adaptable interactions (47). The terminal methionine of the PTHR carboxyl-terminal PDZ motif, ETVM, has been hypothesized to impart a lower affinity for NHERF1 PDZ domains (40). Receptors lacking PDZ-interacting motifs and cells expressing the PTHR, but not NHERF1, nevertheless localize to apical cell membranes (13, 48), implying that direct NHERF1 interactions do not fully account for receptor targeting to apical cell membranes. Thus, it is likely that other proteins participate in apical targeting of certain membrane-delimited receptors, including the PTHR. The PTHR interacts directly with the cytoskeletal adaptor protein ezrin, which was suggested to subserve this adapter function (40). Ezrin-PTHR interactions, however, seem not to be sufficient to direct apical PTHR localization insofar as distal kidney cells, which lack NHERF1 and express the PTHR and abundant ezrin, but the receptor localizes entirely to basolateral membranes (49). Nonetheless, ezrin may play a role in PTHR clustering.
It has been proposed that a ternary complex comprised of the PTHR, NHERF1, and ezrin is formed by the interaction of the FERM domain of ezrin with the intracellular carboxyl-terminal tail of the PTHR (40). The sodium-hydrogen exchanger-3 (NHE-3), which like the PTHR possesses an atypical PDZ recognition motif, STHM, also binds directly to ezrin (50). Furthermore, NHE-3 binds both to NHERF1 and ezrin to form a ternary complex (51). The presence of these low affinity, atypical motifs in the PTHR and NHE-3 in conjunction with their ability to form ternary complexes raises the possibility that NHERF1 facilitates receptor/transporter and ezrin-actin cytoskeleton interactions rather than forming a stable component of the complex. A similar process may account for the signaling switch of the PTHR between Gαq and Gαs induced by NHERF1 (12, 14). The ability of NHERF1 to engage the ezrin-actin cytoskeleton and also Gαq while concurrently interacting with the PTHR is consistent with the view that dynamic exchange occurs between effector proteins and NHERF1. Hence, NHERF1 may mediate ezrin and Gαq interactions with the PTHR by transiently and reversibly binding these proteins and increasing their proximity. The presence of specific proteins with the ability to interact in a discrete subcellular environment may accelerate and prioritize directed signaling pathways. The acceleration of signaling may be the most important contribution of a multi-PDZ scaffold, in which receptors, G proteins, and effectors are expressed at low abundance (52).
The present results show that the mobility of NHERF1 rapidly increases after PTHR stimulation. We postulate that upon agonist binding, the receptor undergoes a conformational change that weakens its affinity for NHERF1 and accounts for the rapid dissociation of NHERF1 from the receptor, even when the receptor is irreversibly immobilized at the plasma membrane. Consistent with such an argument, the t½ for recovery of NHERF1 after FRAP in the absence of PTHR is similar to that for NHERF1 in the presence of the PTH-stimulated PTHR. This finding suggests rapid dissociation of NHERF1 from the receptor upon PTH stimulation. This conclusion is also supported by the prompt redistribution of the PTHR away from areas of colocalization with NHERF1 upon PTH addition.
Although NHERF1 dynamically interacts with the PTHR, it nonetheless delays ligand-induced PTHR internalization (15, 17). In this setting, we propose that ezrin mediates NHERF1 interaction with the cytoskeleton, thereby retarding PTHR endocytosis by transiently stabilizing the receptor in a multi-protein complex at the cell membrane. In this setting, NHERF1 interferes with β-arrestin binding to the PTHR, thus enhancing PTHR membrane retention (16). The formation of the PTHR-NHERF1-ezrin complex may sterically interfere with β-arrestin binding to the PTHR. Our results show that NHERF1 does not interact with β-arrestins under resting or PTHR-stimulated conditions. The absence of direct interactions of NHERF1 with β-arrestin is entirely consistent with the lack of a PDZ recognition motif in mammalian β-arrestins. Further, in the absence of NHERF1, the interaction of the PTHR with β-arrestin2 was rapid and plateaued by 80 s, whereas in the presence of NHERF1, the interaction between the PTHR and β-arrestin2 was delayed by 380 s and not complete for 8 min. Moreover, the t½ values of NHERF1-PTHR and PTHR-β-arrestin colocalization suggest antecedent dissociation of NHERF1 from the receptor with delayed recruitment of β-arrestins. Thus, trafficking of β-arrestins to the receptor and consequent receptor internalization may be impeded, whereas NHERF1 or the ezrin-actin cytoskeleton complex is bound to the receptor. However, a ternary complex of PTHR-NHERF1 and β-Arrestin has been described (23). Such an interaction may transiently occur when β-arrestins are recruited to the receptor and NHERF1 is being released. This brief interaction could be stabilized by cross-linking agents (23). In our hands, after PTH stimulation, full-length HA-tagged PTHR showed negligible coimmunoprecipitation with β-arrestin in the presence of NHERF compared with strong interaction in the absence of NHERF (16). Further, the formation of a PTHR-NHERF1-β-Arrestin ternary complex was undetectable in the absence of a cross-linking agent. Alternatively, PTHR-NHERF1-β-Arrestin complexes may form in CHO cells, which do not express ezrin.
The present results show that NHERF1 not only decreases PTHR internalization upon PTH stimulation but also favors receptor rearrangement in long lasting cell surface clusters. PDZ-mediated interactions can cluster GPCR cargo to the actin cytoskeleton (53). According to this model, the maturation of clathrin-coated pits is interrupted when PDZ-containing GPCRs are present in the cargo, resulting in delayed recruitment of dynamin and subsequent inhibition of clathrin-coated pit scission. Thus, NHERF1 tethers the PTHR to the actin cytoskeleton under basal conditions, whereas ezrin links stimulated receptors to clathrin-coated pits. According to this view, under resting conditions NHERF1 and ezrin stabilize the PTHR at the plasma membrane. Upon PTHR activation, NHERF1 dissociates from the PTHR, and ezrin tethers the receptor to the actin cytoskeleton, stabilizing PTHR clusters at the cell surface and delaying the scission of clathrin-coated pits. The dynamic interactions of NHERF1 with the receptor and the ability of NHERF1 to engage multiple adapter and trafficking proteins in a spatially and temporally coordinated manner is a key mechanism modulating PTHR function. Similar behavior is likely to extend to the interactions between NHERF1 and other GPCRs and transporters possessing PDZ-binding motifs.
Supplementary Material
Acknowledgments
We thank Dr. Guillermo Romero for constructive suggestions throughout the course of this work and Sarah Richards for designing and generating the described constructs.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK06998, R01DK054171 (to P. A. F.), U54RR022241 (to S. C. W.), and R01DK087688 (to J.-P. V.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- NHERF
- Na/H exchange regulatory factor
- PTH
- parathyroid hormone
- PTHR
- type 1 PTH receptor
- GPCR
- G protein-coupled receptor
- TIRF
- total internal reflected fluorescence
- FRAP
- fluorescence recovery after photobleaching
- ROS
- rat osteosarcoma cells
- EPAC
- exchange protein activated by cAMP
- FERM
- band 4.1, ezrin, radixin, moesin homology domain.
REFERENCES
- 1. Bretscher A., Edwards K., Fehon R. G. (2002) Nat. Rev. Mol. Cell Biol. 3, 586–599 [DOI] [PubMed] [Google Scholar]
- 2. Voltz J. W., Weinman E. J., Shenolikar S. (2001) Oncogene 20, 6309–6314 [DOI] [PubMed] [Google Scholar]
- 3. Shenolikar S., Weinman E. J. (2001) Am. J. Physiol. Renal Physiol. 280, F389–F395 [DOI] [PubMed] [Google Scholar]
- 4. Weinman E. J., Hall R. A., Friedman P. A., Liu-Chen L. Y., Shenolikar S. (2006) Annu. Rev. Physiol. 68, 491–505 [DOI] [PubMed] [Google Scholar]
- 5. Seidler U., Singh A. K., Cinar A., Chen M., Hillesheim J., Hogema B., Riederer B. (2009) Ann. N.Y. Acad. Sci. 1165, 249–260 [DOI] [PubMed] [Google Scholar]
- 6. Hung A. Y., Sheng M. (2002) J. Biol. Chem. 277, 5699–5702 [DOI] [PubMed] [Google Scholar]
- 7. Stiffler M. A., Chen J. R., Grantcharova V. P., Lei Y., Fuchs D., Allen J. E., Zaslavskaia L. A., MacBeath G. (2007) Science 317, 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Songyang Z., Fanning A. S., Fu C., Xu J., Marfatia S. M., Chishti A. H., Crompton A., Chan A. C., Anderson J. M., Cantley L. C. (1997) Science 275, 73–77 [DOI] [PubMed] [Google Scholar]
- 9. Fehon R. G., McClatchey A. I., Bretscher A. (2010) Nat. Rev. Mol. Cell Biol. 11, 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weinman E. J., Steplock D., Shenolikar S. (1993) J. Clin. Invest. 92, 1781–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall R. A., Premont R. T., Chow C. W., Blitzer J. T., Pitcher J. A., Claing A., Stoffel R. H., Barak L. S., Shenolikar S., Weinman E. J., Grinstein S., Lefkowitz R. J. (1998) Nature 392, 626–630 [DOI] [PubMed] [Google Scholar]
- 12. Mahon M. J., Donowitz M., Yun C. C., Segre G. V. (2002) Nature 417, 858–861 [DOI] [PubMed] [Google Scholar]
- 13. Mahon M. J., Segre G. V. (2004) J. Biol. Chem. 279, 23550–23558 [DOI] [PubMed] [Google Scholar]
- 14. Wang B., Ardura J. A., Romero G., Yang Y., Hall R. A., Friedman P. A. (2010) J. Biol. Chem. 285, 26976–26986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang B., Bisello A., Yang Y., Romero G. G., Friedman P. A. (2007) J. Biol. Chem. 282, 36214–36222 [DOI] [PubMed] [Google Scholar]
- 16. Wang B., Yang Y., Abou-Samra A. B., Friedman P. A. (2009) Mol. Pharmacol. 75, 1189–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wheeler D., Sneddon W. B., Wang B., Friedman P. A., Romero G. (2007) J. Biol. Chem. 282, 25076–25087 [DOI] [PubMed] [Google Scholar]
- 18. Shenolikar S., Voltz J. W., Minkoff C. M., Wade J. B., Weinman E. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11470–11475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karim Z., Gérard B., Bakouh N., Alili R., Leroy C., Beck L., Silve C., Planelles G., Urena-Torres P., Grandchamp B., Friedlander G., Prié D. (2008) N. Engl. J. Med. 359, 1128–1135 [DOI] [PubMed] [Google Scholar]
- 20. Ferrari S. L., Behar V., Chorev M., Rosenblatt M., Bisello A. (1999) J. Biol. Chem. 274, 29968–29975 [DOI] [PubMed] [Google Scholar]
- 21. Conway B. R., Minor L. K., Xu J. Z., D'Andrea M. R., Ghosh R. N., Demarest K. T. (2001) J. Cell Physiol. 189, 341–355 [DOI] [PubMed] [Google Scholar]
- 22. Chorev M. (2002) Receptors Channels 8, 219–242 [PubMed] [Google Scholar]
- 23. Klenk C., Vetter T., Zürn A., Vilardaga J. P., Friedman P. A., Wang B., Lohse M. J. (2010) J. Biol. Chem. 285, 30355–30362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sneddon W. B., Magyar C. E., Willick G. E., Syme C. A., Galbiati F., Bisello A., Friedman P. A. (2004) Endocrinology 145, 2815–2823 [DOI] [PubMed] [Google Scholar]
- 25. Geiser M., Cèbe R., Drewello D., Schmitz R. (2001) BioTechniques 31, 88–90, 92 [DOI] [PubMed] [Google Scholar]
- 26. Galperin E., Sorkin A. (2003) J. Cell Sci. 116, 4799–4810 [DOI] [PubMed] [Google Scholar]
- 27. Cunningham R., Steplock D., E. X., Biswas R. S., Wang F., Shenolikar S., Weinman E. J. (2006) Am. J. Physiol. Renal Physiol. 291, F896–F901 [DOI] [PubMed] [Google Scholar]
- 28. Rizzo M. A., Springer G. H., Granada B., Piston D. W. (2004) Nat. Biotechnol. 22, 445–449 [DOI] [PubMed] [Google Scholar]
- 29. Violin J. D., Ren X. R., Lefkowitz R. J. (2006) J. Biol. Chem. 281, 20577–20588 [DOI] [PubMed] [Google Scholar]
- 30. Sneddon W. B., Syme C. A., Bisello A., Magyar C. E., Rochdi M. D., Parent J. L., Weinman E. J., Abou-Samra A. B., Friedman P. A. (2003) J. Biol. Chem. 278, 43787–43796 [DOI] [PubMed] [Google Scholar]
- 31. Altschuler Y., Barbas S. M., Terlecky L. J., Tang K., Hardy S., Mostov K. E., Schmid S. L. (1998) J. Cell Biol. 143, 1871–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nikolaev V. O., Bünemann M., Hein L., Hannawacker A., Lohse M. J. (2004) J. Biol. Chem. 279, 37215–37218 [DOI] [PubMed] [Google Scholar]
- 33. Fonseca J. M., Lambert N. A. (2009) Mol. Pharmacol. 75, 1296–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abramoff M. D., Magelhaes P. J., Ram S. J. (2004) Biophotonics. Int. 11, 36–42 [Google Scholar]
- 35. Bolte S., Cordelières F. P. (2006) J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 36. Vilardaga J. P., Nikolaev V. O., Lorenz K., Ferrandon S., Zhuang Z., Lohse M. J. (2008) Nat. Chem. Biol. 4, 126–131 [DOI] [PubMed] [Google Scholar]
- 37. Bünemann M., Frank M., Lohse M. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 16077–16082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manders E. M., Verbeek F. J., Aten J. A. (1993) J. Microsc. 169, 375–382 [DOI] [PubMed] [Google Scholar]
- 39. Zhang J., Ferguson S. S., Barak L. S., Ménard L., Caron M. G. (1996) J. Biol. Chem. 271, 18302–18305 [DOI] [PubMed] [Google Scholar]
- 40. Mahon M. J. (2009) Mol. Endocrinol. 23, 1691–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vilardaga J. P., Frank M., Krasel C., Dees C., Nissenson R. A., Lohse M. J. (2001) J. Biol. Chem. 276, 33435–33443 [DOI] [PubMed] [Google Scholar]
- 42. Jeleń F., Oleksy A., Smietana K., Otlewski J. (2003) Acta Biochim. Pol. 50, 985–1017 [PubMed] [Google Scholar]
- 43. Bockaert J., Fagni L., Dumuis A., Marin P. (2004) Pharmacol. Ther. 103, 203–221 [DOI] [PubMed] [Google Scholar]
- 44. Lee H. J., Zheng J. J. (2010) Cell Commun. Signal 8, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lau A. G., Hall R. A. (2001) Biochemistry 40, 8572–8580 [DOI] [PubMed] [Google Scholar]
- 46. Li J., Callaway D. J., Bu Z. (2009) J. Mol. Biol. 392, 166–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harris B. Z., Lim W. A. (2001) J. Cell Sci. 114, 3219–3231 [DOI] [PubMed] [Google Scholar]
- 48. Capuano P., Bacic D., Roos M., Gisler S. M., Stange G., Biber J., Kaissling B., Weinman E. J., Shenolikar S., Wagner C. A., Murer H. (2007) Am. J. Physiol. Cell Physiol. 292, C927–C934 [DOI] [PubMed] [Google Scholar]
- 49. Amizuka N., Lee H. S., Kwan M. Y., Arazani A., Warshawsky H., Hendy G. N., Ozawa H., White J. H., Goltzman D. (1997) Endocrinology 138, 469–481 [DOI] [PubMed] [Google Scholar]
- 50. Cha B., Tse M., Yun C., Kovbasnjuk O., Mohan S., Hubbard A., Arpin M., Donowitz M. (2006) Mol. Biol. Cell 17, 2661–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Donowitz M., Mohan S., Zhu C. X., Chen T. E., Lin R., Cha B., Zachos N. C., Murtazina R., Sarker R., Li X. (2009) J. Exp. Biol. 212, 1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ranganathan R., Ross E. M. (1997) Curr. Biol. 7, R770–R773 [DOI] [PubMed] [Google Scholar]
- 53. Puthenveedu M. A., von Zastrow M. (2006) Cell 127, 113–124 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.