

Abstract
Although members of a virus family produce similar gene products, those products may have quite different functions. Simian virus 40 (SV40) large T antigen (LT), for example, targets p53 directly, but murine polyomavirus LT does not. SV40 small T antigen (SVST) has received considerable attention because of its ability to contribute to transformation of human cells. Here, we show that there are major differences between SVST and polyomavirus small T antigen (POLST) in their effects on differentiation, transformation, and cell survival. Both SVST and POLST induce cell cycle progression. However, POLST also inhibits differentiation of 3T3-L1 preadipocytes and C2C12 myoblasts. Additionally, POLST induces apoptosis of mouse embryo fibroblasts. SVST reduces the proapoptotic transcriptional activity of FOXO1 through phosphorylation. On the other hand, SVST complements large T antigen and Ras for the transformation of human mammary epithelial cells (HMECs), but POLST does not. Mechanistically, the differences between SVST and POLST may lie in utilization of protein phosphatase 2A (PP2A). POLST binds both Aα and Aβ scaffolding subunits of PP2A while SVST binds only Aα. Knockdown of Aβ could mimic POLST-induced apoptosis. The two small T antigens can target different proteins for dephosphorylation. POLST binds and dephosphorylates substrates, such as lipins, that SVST does not.
INTRODUCTION
DNA tumor viruses of the polyomavirus family have helped us understand important cellular processes, particularly those associated with the signal transduction of cell cycle regulation and transformation. For example, polyomavirus middle T antigen (PyMT) studies were crucial to the discovery of the importance of tyrosine phosphorylation (17) and phosphatidylinositol 3-kinase (PI3) kinase activity (70) in cellular signaling. Comparative studies of the individual members of the polyomavirus family have also been very informative. For example, p53 was discovered as a protein that associates with simian virus 40 (SV40) large T antigen (LT) (38, 39). Comparison of SV40 LT that binds p53 and transforms cells to polyomavirus LT (PyLT) that does neither of these focused attention on the role of p53 in cell cycle regulation.
All members of the polyomavirus family produce a small T antigen (ST) as one of the early gene products. Polyomavirus small T antigen (POLST) and SV40 small T antigen (SVST) share many structural similarities with each other (Fig. 1). Both have an N-terminal J domain with a conserved HPDKGG motif that can bind heat shock proteins, and both possess zinc-binding cysteine motifs. Both POLST and SVST bind and perturb protein phosphatase 2A (PP2A) (51). PP2A functions as a trimeric ABC complex, where the scaffolding A subunit binds a catalytic subunit (C) and some regulatory B subunits (30). STs bind to the A and C subunit complexes (51, 64), displacing or preventing B subunits from binding. Since SVST binds to regions of the A subunit involved in B binding (12, 14), the absence of B subunits in ST/PP2A complexes is not surprising.
Fig. 1.

Schematic representation of polyomavirus ST (POLST) and SV40 ST (SVST) protein structures with the main features indicated.
There is ample evidence suggesting that careful examination of ST is important. SVST contributes to the multioncoprotein-directed transformation of human cells (24, 59, 76). Transgenic SVST contributes to mammary gland carcinogenesis (22). POLST complements MT for both transformation (4, 45, 49) and tumor induction (3). POLST effects on the cell cycle are well documented. POLST promotes cell cycle progression (46) and complements LT for S-phase induction (5, 51). Array analysis showed that SVST (43) and POLST (35) have large effects on cellular mRNA levels. SVST can transactivate (19, 32, 40, 48, 53, 61, 68) or repress (67) various promoters. POLST activates the fos (46) and myc (36) promoters. POLST is known to promote changes in viral chromatin structure that may underlie altered transcriptional activity (16). DNA viruses are very much concerned with issues of cell survival. It is therefore not surprising that both small Ts affect survival. Both POLST and SVST can be antiapoptotic. POLST can protect against serum starvation-induced apoptosis (2) and resists the effects of p53-induced apoptosis (54). SVST opposes apoptosis induced by LT (37) or CD95 (20). Both POLST (2) and SVST (21) can also be proapoptotic under some circumstances.
Many ST functions are known to depend on their interactions with PP2A. Promotion of cell cycle by POLST (46), activation of promoters, such as myc (36), fos (46), cyclin A (60), or cyclin D (68), promotion of or protection from cell death (2), and transformation of human cells by SVST (24) all depend on PP2A. The fact that STs should work through PP2A is quite reasonable. PP2A, as one of the major cellular serine/threonine protein phosphatases, is involved in many processes, including transcription, translation, and replication (30, 31, 42, 47). It can function as a tumor suppressor (31, 69), and effects on PP2A are being increasingly connected to cancer (18). Decreasing the activity of PP2A toward myc, for example, has been associated with head and neck and colon cancer (33).
The consequences of ST binding to PP2A have been only partly worked out. SVST can inhibit PP2A activity in vitro (57, 74). In some situations, such as transformation of kidney cells, knockdown of specific PP2A B subunits mimics much of the effect of SVST in causing transformation, while their overexpression can reverse the effect of SVST (11). PP2A A subunit mutations that mimic haploinsufficiency are tumorigenic (10). All these results are consistent with an inhibition model. However, perturbation of PP2A function by SVST increases the activity of PP2A toward some substrates, such as histone H1 (74) or the androgen receptor (73). Such results suggest a more positive role.
Our interest in comparing POLST and SVST originated with the observation that POLST had a profound proapoptotic activity (2). This seemed at odds with the reported ability of SVST to promote human cell transformation (24, 59, 76). This work compares SVST and POLST side by side in several different systems and backgrounds: fibroblast survival assays, human mammary epithelial cell (HMEC) transformation, and 3T3-L1 preadipocyte or C2C12 myoblast differentiation. Our work shows that while the two small T antigens share sequence similarities and abilities, they also show a striking difference in effects on differentiation and the ability to promote cell death. In each case, the interaction with PP2A is critical. In examining ST interactions with PP2A, we see differences between POLST and SVST that could explain the altered biology.
MATERIALS AND METHODS
Retroviral infections.
Polyomavirus small T antigen, polyomavirus small T antigen defective in binding PP2A (POLST with the mutation W157S [POLST−], and SV40 small T antigen were cloned in Pinco vector (2) and pBABE-puro vector (44) (obtained from Addgene) at EcoRI restriction sites. For cloning in pBABE vector, small T antigens were tagged with a tandem FLAG and hemagglutinin (HA) tag at the C terminus (ST-FLAG-HA). Phoenix cells stably expressing packaging proteins were transiently transfected with control vector or small T antigen-containing vectors by the calcium phosphate method as described below (9). Briefly, 5 μg of the DNA was transfected into the cells that were grown in 60-mm plates and were 60 to 80% confluent. Next day, cells were washed with phosphate-buffered saline (PBS) and replaced with medium (Dulbecco's modified Eagle's medium [DMEM] containing 10% fetal calf serum). After 48 h, cells that were split a day before were infected with the viral supernatants obtained from the above transfected cells and supplemented with 8 μg of Polybrene/ml (Sigma) for at least 6 h. For isolation of ST-expressing cells, cells were selected and further grown in the presence of puromycin (2.5 μg/ml).
Transient transfections.
For transient transfections, we routinely used the N,N-bis(2-hydroethyl)-2-aminomethane sulfonic acid (BES)/CaCl2 method (9). Briefly, 100 mm plates of NIH 3T3 or HEK 293T cells were split a day before transfection. A total of about 6 to 10 μg of the DNA was mixed with 450 μl of water and then 500 μl of 2× BES (0.05 M BES, pH 6.95, 0.28 M NaCl, 0.015 M Na2HPO4). Finally, 50 μl of 2.5 M CaCl2 was added drop-wise, and the precipitate was allowed to form for 20 to 30 min. The precipitate was then added drop-wise on the plates and incubated overnight at 35°C. The plates were washed the next day with PBS, and then the medium was replaced with fresh medium. At 48 h posttransfection, the cells were harvested, and the extracts were used for the assays.
Western blotting.
Cells were washed with cold PBS, harvested, and resuspended in lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na3VO4) in the presence of protease inhibitors (leupeptin, pepstatin, and aprotinin at 1 μg/ml) and phosphatase inhibitors I and II (1:100 dilution; Sigma) for 30 min. Equal amounts of protein were loaded, as estimated by a Bradford assay (Bio-Rad Laboratories). Antibodies against Akt, phospho-Akt 473, and phospho-FOXO1/FOXO3a were from Cell Signaling. Anti-α-actin and FLAG were from Sigma. Anti-HA11 was from Covance. Anti-myosin heavy chain (MHC) was obtained from Developmental Studies Hybridoma Bank. For POLST and SVST, PN116 and pAb419 monoclonal antibodies were used, respectively. These were obtained from the Dana Farber Cancer Institute core facility (obtained from James DeCaprio), and they recognize the N-terminal domains.
DNA laddering.
For the assessment of chromosomal fragmentation occurring due to apoptosis, DNA was isolated as described previously (2). Briefly, NIH 3T3 cells were grown in 100-mm plates, harvested by scraping and centrifugation at 1,000 × g, and washed once with cold 1× PBS, and the cell pellets were then lysed in 0.4 ml of lysis buffer (10 mM Tris, pH 7.4, 25 mM EDTA, and 0.25% Triton X-100) on ice for 30 min. This was followed by centrifugation at 13,800 × g for 15 min, and the supernatant was treated with RNase A (200 μg/ml) at 37°C for 1 to 2 h, followed by incubation with proteinase K (100 μg/ml) at 56°C overnight. The mixtures were purified sequentially with phenol-chloroform and chloroform and then precipitated with 0.1 volume of 5 M NaCl and 2 volumes of ethanol at −20°C overnight. DNA extracts were pelleted after centrifugation, washed with 70% ethanol, dried, and resuspended in water. Equal amounts of the DNA (determined by spectrometry at 260/280 nm) were loaded and run in 2% agarose gel (30 to 50 V for 2 h), stained with ethidium bromide (1 μg/ml), and observed by UV transilluminator.
Cell cycle analysis by fluorescence-activated cell sorting (FACS).
NIH 3T3 cells were infected with control vector (Pinco) or small T antigen-containing retroviral vectors (Pinco-ST) as described above. Following infection, a homogenous population of cells expressing green fluorescent protein (GFP) was obtained by cell sorting. For cell cycle analysis, cells were grown to subconfluent density, washed once with PBS without Ca2+ and Mg2+ and then with PBS supplemented with 0.1% EDTA. The plates were incubated for 5 min at 37°C, and cells were dislodged by pipetting and collected in a 15-ml tube. To include apoptotic cells, loosely attached and floating cells were also collected at all stages. After centrifugation at 1,000 × g for 5 min, pellets were washed once with PBS supplemented with 1% serum and spun at 1,000 × g for 5 min again. The pellets were resuspended in 0.5 ml of PBS and fixed by the slow addition of 5 to 10 ml of ethanol while vortexing to prevent clumping. At this stage, the cells were stored at 4°C at least overnight in the dark. This was followed by centrifugation at 1,000 × g for 5 min, and cells were washed once with PBS–1% serum, spun again, resuspended in 200 to 500 μl of propidium iodide-RNase solution (50 μg/ml propidium iodide, 10 mM Tris, pH 7.5, 5 mM MgCl2, and 20 μg/ml RNase A), and incubated at 37°C for 30 min. Samples were then subjected to flow cytometry using a FACSCalibur and analyzed by ModFit.
Reporter assays.
Cells were passed 1 day prior to transfection so that they were 40 to 60% confluent next day. Typically, 3 μg of DNA was used for each 60-mm plate. The BES-calcium phosphate method was used for transfections as described above. Medium was changed the next day, and at 48 h posttransfection cells were harvested, and samples were used for luciferase assays. β-Galactosidase assays done on the same samples were used to normalize the luciferase values.
BrdU staining.
For measuring S-phase induction, a bromodeoxyuridine (BrdU) kit from Roche was used. Cells were grown overnight on coverslips in medium containing 0.2% serum. Next day, they were washed once with PBS and incubated for 1 h in standard cell culture medium (DMEM) that contained BrdU. Cells were washed three times with PBS, fixed in cold ethanol (−20°C), washed again three times with PBS, and followed by incubation with anti-BrdU antibody for 30 min at 37°C. Cells were again washed three times with PBS and incubated with tetramethyl rhodamine isocyanate (TRITC)-labeled secondary anti-mouse antibody at a dilution of 1:1,000 (Jackson Immunochemicals) for 30 min at 37°C. Cells were washed again three times and mounted on glass slides using Vectashield (Vector laboratories) that contained 4′,6′-diamidino-2-phenylindole (DAPI) as the mounting medium. BrdU-positive cells were observed by fluorescence microscopy at >620 nm, while DAPI-stained nuclei were observed at 420 nm. Pictures were taken at magnifications of ×10x and ×20. The percentage of cells undergoing DNA synthesis was calculated by counting BrdU-positive cells, which were a fraction of the total number of cells (as seen by DAPI) in a given field (number of BrdU-positive/total number of DAPI-positive cells × 100).
Soft-agar assay.
Soft-agar assays were done in 60-mm dishes in duplicates as described previously (78). A stock of 1.8% agar (Noble agar; Sigma) was autoclaved and stored at room temperature. The bottom agar was made by plating 4 ml of 0.6% agar in DMEM and left to solidify about 30 min. This was followed by adding 2 ml of top agar (0.3%) containing about 5 × 104 cells in minimal essential growth medium ([MEGM] DMEM/F-12 supplemented with 10 ng/ml human epidermal growth factor [hEGF], 0.5 μg/ml hydrocortisone, 10 μg/ml insulin, 1 ng/ml cholera toxin, 1% penicillin/streptomycin/fungizone [Invitrogen], and 0.5% fetal calf serum [FCS]) and left to solidify. The plates were fed with 500 μl of MEGM once or twice a week. The colonies (0.2 mm or bigger) were counted after about 6 weeks of growth.
Immunoprecipitation.
Cells were grown in 100-mm plates, washed one to two times with cold PBS, harvested, and lysed in 300 μl of extraction/lysis buffer in the presence of protease and phosphatase inhibitors for 30 min on ice. The extracts were spun down at 10,000 × g (13,000 rpm in a microcentrifuge) for 10 min. Sepharose beads were premixed with antibodies (protein G with monoclonal and protein A with polyclonal antibodies) for 30 min to 1 h. Equal amounts of the cell extracts (as determined by Bradford assay) were then mixed with the beads-antibody mixture, and mixing continued for at least 2 h at 4°C. This was followed by three washes with cold PBS and a final wash with cold water. The beads were mixed with dissociation buffer, boiled, and loaded on a gel, followed by Western blotting.
3T3-L1 cell differentiation.
Small T antigen- and control vector-expressing 3T3-L1 cells were obtained by retroviral infection using pBABE-puro vectors, as described above, and then selected with puromycin at a final concentration of 2.5 μg/ml. Cell lines were allowed to grow to confluence in DMEM containing 10% FCS. Two days later, this was replaced by induction medium consisting of 10% FCS in DMEM supplemented with 167 nM insulin, 1 mM dexamethasone, 0.5 mM isobutylmethylxanthine, and 5 mM troglitazone. After the second day, the medium was replaced with DMEM containing 10% FCS and 167 nM insulin for one more day. The cells were then allowed to fully differentiate in DMEM containing 10% FCS for 8 days with fresh medium added every 2 days.
For Oil Red O staining, the differentiated cells were fixed in 10% formalin for 10 min at room temperature and replaced with 10% formalin for 1 hour. The plates were washed with 60% isopropanol, dried, and stained with Oil Red O solution (3.5% Oil Red O in 60% isopropanol) for 10 min at room temperature. The plates were subsequently washed four times with water and dried, and pictures were taken at a magnification of ×10.
C2C12 differentiation.
ST- and control vector-expressing C2C12 cells were obtained by retroviral infection using pBABE-puro vectors as described above and then selected and grown in the presence of puromycin at a final concentration of 2.5 μg/ml. Cell lines were allowed to grow to confluence in DMEM containing 10% FCS. The medium was then replaced with DMEM containing 2% horse serum (HS) every 2 days. Cells lysates were collected 6 days postconfluence.
Knockdown of PP2A Aβ.
Lentivirus pLKO expressing short hairpin RNAs (shRNAs) against mouse PP2A Aβ (obtained from William Hahn) were used. Two of these hairpins, sh2 (CCCACAAAGTAAGAGAGCTTT) and sh4 (GCTGGGAAATTTCACTGGTTT), and a pLKO scrambled control at 3 μg were transfected into 293T cells along with packaging vectors PLP1 (4 μg), PLP2 (2.5 μg), and vesicular stomatitis virus glycoprotein 3 (VSVG3; 3 μg). At 48 h posttransfection the supernatant was collected, filtered, and added to 3T3-L1 cells at amounts that yielded similar amounts of infection based on measurement of puromycin resitance gene expression. Twenty-four hours later the infection medium was changed. At 48 h postinfection part of the cells were assayed by reverse transcription-PCR (RT-PCR) for Aβ knockdown and puromycin resistance, while 5% of each respective infection was plated to assay for growth for an additional 5 days. Pictures of the cells were then taken.
Knockdown was demonstrated by real-time PCR. RNA was isolated with an RNeasy Plus Mini Kit (Qiagen 74134) following the protocol supplied. Twenty microliters of cDNA was subsequently synthesized with 1 μg of RNA using an iScript cDNA synthesis kit by Bio-Rad. Real-time PCR was then set up with each sample containing 2 μl of the cDNA synthesis reaction mixture, 10 μl of LightCycler SYBR green I master mix (Roche 04707516001), primers at a final concentration of 0.5 μM, and water. The Aβ primers were TGGACCCCGACACGGAGTGG and GCGGACCATCGAAACCAGGAGC; the primers for the puromycin resistance gene (puromycin acetyltransferase, or PAC gene) were GGCCGAGTTGAGCGGTTCCC and CTCCACTCCGGGGAGCACGA. The reaction conditions consisted of 2 min at 95°C, followed by 45 cycles at 95°C, 60°C, and 72°C each for 10 s. Samples were assayed in triplicates.
RESULTS
Both polyomavirus and SV40 small T antigens promote cellular DNA synthesis.
Both polyomavirus and SV40 small T antigens are known to promote cellular DNA replication (15, 46). To compare the two small T antigens directly for their ability to induce S phase in the host cells, stable NIH 3T3 cells expressing either POLST or SVST were obtained by retroviral infection. To measure the ability of the STs to support ongoing cell cycle progression, a BrdU incorporation assay was carried out. After starvation overnight in medium containing 0.2% serum, cells were incubated for 1 h in medium containing BrdU. Cells were fixed and stained with anti-BrdU antibody to measure cells replicating DNA and stained with DAPI to determine the total cell number. Both SVST and POLST induced DNA synthesis although the effect of POLST was somewhat stronger (Fig. 2 A). Neither small T antigen is quite as strong as treatment with fresh 10% serum. Similar results were obtained with NIH 3T3 cells transiently cotransfected with either POLST or SVST with EF-GFP to identify transfected cells (data not shown). If the small T antigens are both promoting continued cell cycle progression, then genes associated with cell cycle progression should be stimulated. Cyclin D1 transcription is regulated as a function of cell cycle progression. The levels of cyclin D in stable cell lines expressing POLST or SVST were indeed elevated as well (Fig. 2B). The mechanism for this seems to be transcriptional. SVST is already known to activate cyclin D (68). Figure 2C shows that POLST also activates the cyclin D promoter. Together, reporter assays, BrdU incorporation experiments, and cyclin D expression in cell lines point to similar functions for the two small T antigens.
Fig. 2.
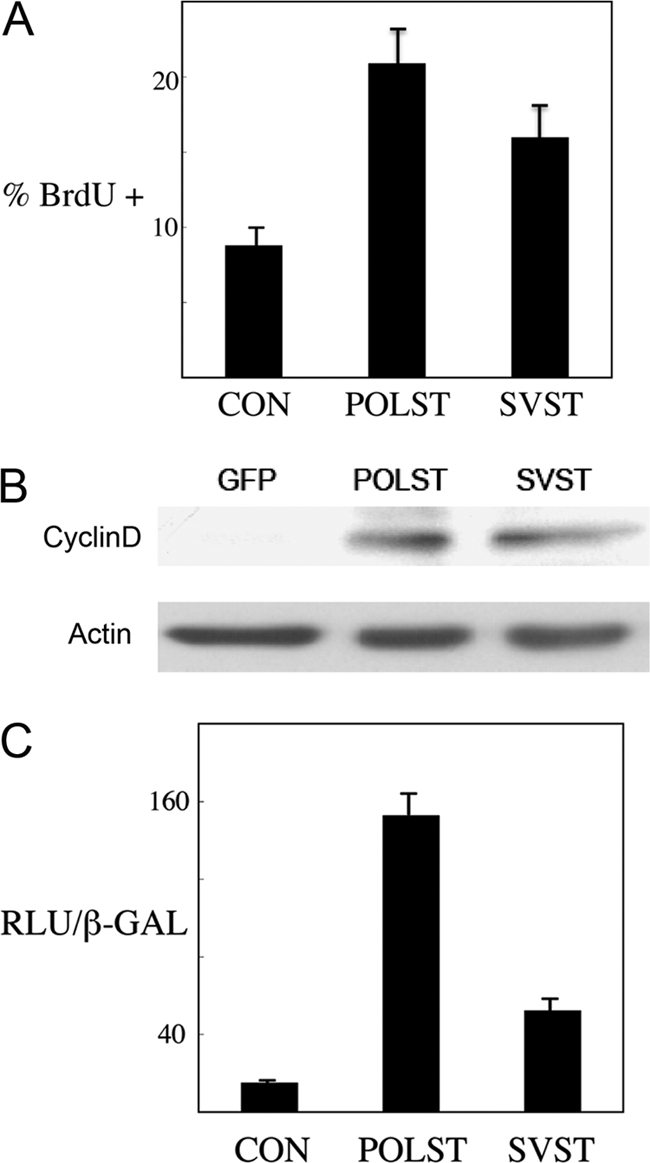
Cell cycle-related effects of STs. (A) Effect of STs on S-phase progression. NIH 3T3 cells expressing control vector (CON), POLST, or SVST, as obtained by retroviral infection, were grown overnight in medium containing 0.2% serum. Cells were then incubated in medium containing BrdU for 1 h and subjected to immunofluorescence. Percent cells undergoing S phase (% BrdU +) is measured by BrdU incorporation compared to the total number of cells, as measured by DAPI-stained nuclei. (B) After retroviral infection C2C12 myoblast cell pools were isolated that express GFP, POLST, or SVST. These respective cells were grown to around 60% confluence, and fresh medium containing 10% FCS was added to the cells at this point for 4 hours. Cell lysates were subsequently collected and analyzed by Western blotting for cyclin D expression using actin as a control. (C) Effect of STs on the cyclin D1 promoter. NIH 3T3 cells were cotransfected with cyclin D-luc reporter, small T antigens, or control vector and pCMV-β-Gal. The luciferase activity was normalized using β-galactosidase activity, and results are reported as the number of relative light units (RLU) × 10−3/β-galactosidase activity unit.
POLST blocks preadipocyte and myoblast differentiation while SVST does not.
The ability of the small T antigens to promote cell cycle progression caused us to wonder about their effects on differentiation. The ability of 3T3-L1 cells to differentiate into adipocytes (13, 25) or the ability of C2C12 myoblasts to differentiate into myotubes (41, 58) was blocked by SV40 LT or PyLT. These effects were dependent on their ability to promote cell cycle progression via association with the retinoblastoma (Rb) family of tumor suppressors.
3T3-L1 preadipocytes and C2C12 myoblast cell lines that expressed almost equal amounts of POLST, SVST, and W157S POLST were prepared by retroviral infection. W157S is a POLST mutant deficient in binding PP2A. These cells were compared to GFP controls for their ability to differentiate. Oil Red O, a traditional marker for adipocyte differentiation, was used to determine the extent of differentiation. POLST cells were defective in adipocyte differentiation, as indicated by the complete lack of Oil Red O staining (Fig. 3 A). The extent of myoblast differentiation was assessed by the expression of myosin heavy chain (MHC) in cells differentiated for 6 days (Fig. 3B). Wild-type POLST drastically reduced the expression of MHC. W157S POLST did not block differentiation in either system. This implicates PP2A as a POLST target in blocking differentiation. Though SVST also interacts with PP2A, it did not inhibit differentiation of either 3T3-L1 or C2C12 cells.
Fig. 3.
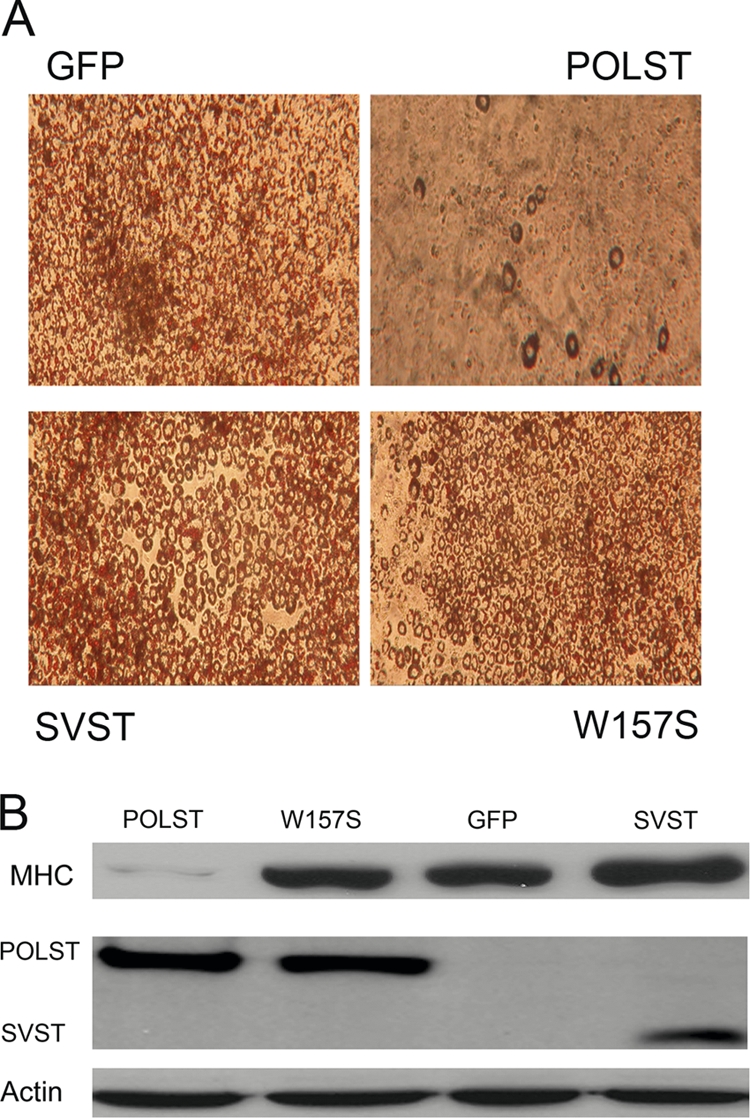
POLST inhibits differentiation of 3T3-L1 preadipocytes and C2C12 myoblasts. (A) After retroviral infection 3T3-L1 cell pools were isolated that expressed GFP, POLST, W157S POLST, or SVST. Subsequently, cells were allowed to differentiate. At 7 days postinduction, differentiating cells were fixed and stained with Oil Red O. (B) After retroviral infection C2C12 myoblast cell pools were isolated that expressed GFP, POLST, PP2A− W157S POLST, or SVST. Cell lysates were collected at 6 days postinduction of differentiation. Expression of MHC, a differentiation marker, was determined by Western blotting. PN116 antibody was used to detect the expression of POLST, while PAB419 antibody was used to detect the expression of SVST. α-Actin was used as a loading control.
SVST but not POLST promotes transformation of HMECs.
SVST was necessary for transformation of human mammary epithelial cells (HMECs) in combination with human telomerase reverse transcriptase (hTERT), SV40 large T antigen, and H-ras (24, 78). Soft-agar colonies were obtained when SVST was expressed in these cells but not in control HMECs expressing only hTERT, SV40 large T antigen, and H-ras. This function of SVST was dependent upon its ability to bind and inhibit PP2A. The activation of the PI3 kinase pathway leading to the activation of Akt and Rac seemed important for this effect (78). HMECs that expressed POLST or SVST at similar levels were generated by retroviral infection of the control HMECs (Fig. 4 C). Comparison of POLST- to SVST-expressing cells showed that POLST was not capable of inducing colonies to grow in soft agar (Fig. 4A and B). In this assay, as in differentiation assays, the two small T antigens gave quite different phenotypes.
Fig. 4.

Effect of STs on transformation measured by anchorage-independent growth. (A) HMECs that express hTERT, SV40 large T antigen, and H-ras were used as controls (CON) in soft-agar assays. POLST or SVST cells were then expressed. Colonies are shown after 6 weeks of growth. (B) Quantitative representation of the soft-agar assay. Colonies 0.2 mm or more in size were counted after 6 weeks of growth. (C) Cell extracts from HMECs were separated by PAGE and blotted with antibodies against POLST (PN116) or SVST (PAB419) to determine the relative expression levels of the STs. Total Akt was used as a loading control.
POLST but not SVST induced apoptosis in human and mouse cells.
On the basis of observations of HMECs over time in soft agar, it seemed as if POLST might have been stimulating a small amount of growth followed by loss of colonies. We have recently reported that POLST expression caused apoptosis in the host cells (2). It seemed possible that the differences in the transformation assay resulted from the effect of POLST on cell survival.
One simple test for an effect on survival is to ask whether control cells or those expressing POLST or SVST could grow out after retroviral infection and drug selection. NIH 3T3 cells were infected with GFP-control, POLST-expressing, or SVST-expressing retroviruses. The levels of infection were comparable, as judged by initial GFP and ST expression (data not shown). As shown in Fig. 5A, staining of cells with crystal violet showed that POLST-infected cells failed to grow out, as expected from the previous report. In contrast, both the control and SVST-expressing cells continued to grow and divide until they became confluent. To look at this more closely, infected 3T3 cells were examined in more detail. Following infection and expression of small T antigens, cells expressing POLST showed apoptotic features like rounding up and membrane blebbing, as well as condensed nuclear morphology (Fig. 5B). Further confirmation of apoptosis came from a distinct sub-G1 peak as seen in cell cycle histogram obtained by FACS analysis (Fig. 5C), as well as DNA laddering (Fig. 5D). As noted previously, these effects depended on the ability of POLST to associate with PP2A. By all of these criteria, SVST-expressing cells did not show any signs of apoptosis. This points to an important difference in the function of the small T antigens of the two different viruses.
Fig. 5.

Effect of STs on cell survival. NIH 3T3 cells expressing small T antigens were obtained by retroviral infection. (A) Cell survival following retroviral infection. Cells were infected with retrovirus containing only the puromycin resistance gene (pBABE-puro) as a control (CON) expressing POLST (pBABEpuro-POLST) or SVST (pBABEpuro-SVST). After selection in puromycin (2.5 μg/ml) for about 7 days, cells were fixed with ethanol, stained with 0.2% crystal violet, and washed (78). (B) Expression of STs was confirmed by GFP expression as seen in fluorescence microscopy, and the morphology of the cells expressing POLST indicated cell death. Nuclei were stained by DAPI and observed by fluorescence microscopy. (C) POLST induced apoptosis (APOP) as seen by FACS analysis. A sub-G1 peak was seen in the cell cycle histogram of POLST-expressing cells but not in control (CON) or SVST-expressing cells. FACS analysis showed that 37% of the infected cells with POLST had sub-G1 DNA content. (D) Apoptosis in POLST-expressing cells as indicated by DNA fragmentation. DNA was extracted from ST-expressing NIH 3T3 cells, separated on a 2% agarose gel, stained with ethidium bromide, and visualized on a UV transilluminator.
The signaling of the two small T antigens shows significant differences under the conditions of these experiments. Previous work has characterized some of the signaling associated with killing by POLST (2). In those earlier experiments, the effect of POLST on Akt seemed to be important because inhibition of Akt protected against cell death; the biological effect emanated from failure to activate phosphorylation of Akt S473.
Figure 6 A shows decreased phosphorylation of Akt S473 after coexpression of POLST, but not SVST, in transfection experiments in human 293T cells. A similar restriction in S473 phosphorylation of endogenous Akt can be seen in HMECs expressing POLST (Fig. 6B). In contrast, SVST-expressing HMEC cells had enhanced levels of S473 phosphorylation compared to controls. Similar results were obtained in 3T3-L1 (Fig. 6C) and in C2C12 myoblasts (Fig. 6D). FOXO1/FOXO3a transcription factors are well known substrates of Akt and are involved in cell survival. Fully activated Akt causes their phosphorylation and inactivation by cytoplasmic sequestration and degradation. Due to their role in apoptosis, we investigated the status of their phosphorylation in ST-expressing cells. Phosphorylation of FOXO1 and FOXO3a was observed only when SVST was expressed and not when POLST was expressed (Fig. 6E). Figure 6F shows a FOXO1 reporter assay using an insulin receptor substrate 1 (IRS-1)-luciferase (Luc) reporter construct as the target. Cotransfection of FOXO1 with the IRS-1 reporter shows activation. POLST had a small activating effect. SVST, on the other hand, clearly suppresses the ability of FOXO1 to activate. The presence of slower-migrating FOXO1 is consistent with the idea that FOXO1 phosphorylation is being affected (Fig. 6G). Both the phosphorylation and reporter assay results are consistent with the observed difference in killing between POLST and SVST. SVST repression of FOXO1 transcriptional activity was recently reported in another study (72). Phosphorylation of FOXO1 by SVST was mediated by inhibition of PP2A dephosphorylation activity.
Fig. 6.

ST effects on Akt signaling. (A) Effects of ST on Akt S473 phosphorylation. 293T cells were transiently cotransfected with HA-myr-Akt (HA-labeled myristoylated Akt) and pCMV-ST antigens. At 48 h posttransfection, cell extracts were blotted for phospho-Akt (S473) or HA (total Akt) as indicated. (B to D) Effects of overexpression of STs on endogenous Akt S473 phosphorylation. Extracts from control HMECs, 3T3-L1s, and C2C12s cells (CON) and those expressing either POLST or SVST were collected. Phosphorylation of endogenous Akt at S473 was detected by Western blotting. Total Akt was used as a loading control in each case. (E) Effects of overexpression of STs on FOXO1/FOXO3a phosphorylation. Extracts from control HMECs (CON) and those expressing either POLST or SVST were collected for Western blotting. Phospho-FOXO1/FOXO3a (T24/32) was detected with vinculin as a loading control. (F) Effects of STs on FOXO1 activation. 293T cells were transiently transfected with IRS-Luc, Flag-FOXO1, pCMV–β-Gal and either POLST or SVST. Extracts were obtained after 24 h and used for luciferase and β-Gal assays. The luciferase values (RLU × 10−3) were normalized by β-galactosidase values. Standard errors of the means are shown. (G) Flag-FOXO1 mobility was determined using anti-FLAG antibody after cotransfection with control vector, POLST, or SVST as described for panel F.
Differences in association of POLST and SVST with PP2A A subunit isoforms.
As noted above, POLST blocks differentiation in a PP2A-dependent manner under conditions where SVST does not. The killing observed with POLST depends on association with PP2A, but SVST that also associates with PP2A does not kill under the same conditions. SVST complements hTERT, h-Ras, and SV40 LT for HMEC transformation in a manner that depends on its association with PP2A, but POLST that associates with PP2A does not. This suggests that the two small T antigens deal with PP2A differently.
The A subunit of PP2A comes in two isoforms: Aα and Aβ. To determine whether the two small T antigens interacted in a similar or different way with PP2A A subunit isoforms, epitope-tagged PP2A Aα and Aβ isoforms were cotransfected with either POLST or SVST into NIH 3T3 cells. Immunoprecipitates of the PP2A isoforms were then blotted to determine the amount of associated ST (Fig. 7). While POLST interacted with both the Aα and Aβ isoforms of the A subunit, SVST showed little or no binding to the PP2A Aβ isoform. The same result was obtained in human 293T cells (data not shown).
Fig. 7.

Interaction of small T antigens with PP2A A isoforms. NIH 3T3 cells were cotransfected with EE-tagged PP2A Aα or Aβ and POLST or SVST. At 48 h after transfection, lysates were collected, and PP2A Aα or Aβ was immunoprecipitated (IP) with anti-EE antibody and blotted for small T antigens. Expression of small T antigens and PP2A was confirmed by Western blotting on whole-cell extracts (WCE).
This result suggests that POLST can exhibit unique functions through its interaction with PP2A Aβ. We therefore examined the effects of short hairpin knockdown of PP2A Aβ. De novo infection of 3T3-L1 cells was carried out with lentiviruses expressing a scrambled control or two different short hairpins to Aβ. Cells were assayed at 48 h postinfection for the amount of Aβ expression and the puromycin resistance gene (PAC gene) by real-time PCR. The equivalent levels of PAC expression ensured that the infections were equally efficient with different constructs. sh2 knocked down Aβ by 52%, while sh4 knocked down Aβ by 87% (Fig. 8). Cell death within 4 days was observed in response to infection by PP2A shRNA-containing viruses but not by the scrambled control. Importantly, the level of knockdown correlated inversely with cell survival. Therefore, death induced by knockdown of Aβ mimicked the effect of de novo infection of POLST. This suggests that POLST may indeed target Aβ as part of the apoptotic process.
Fig. 8.
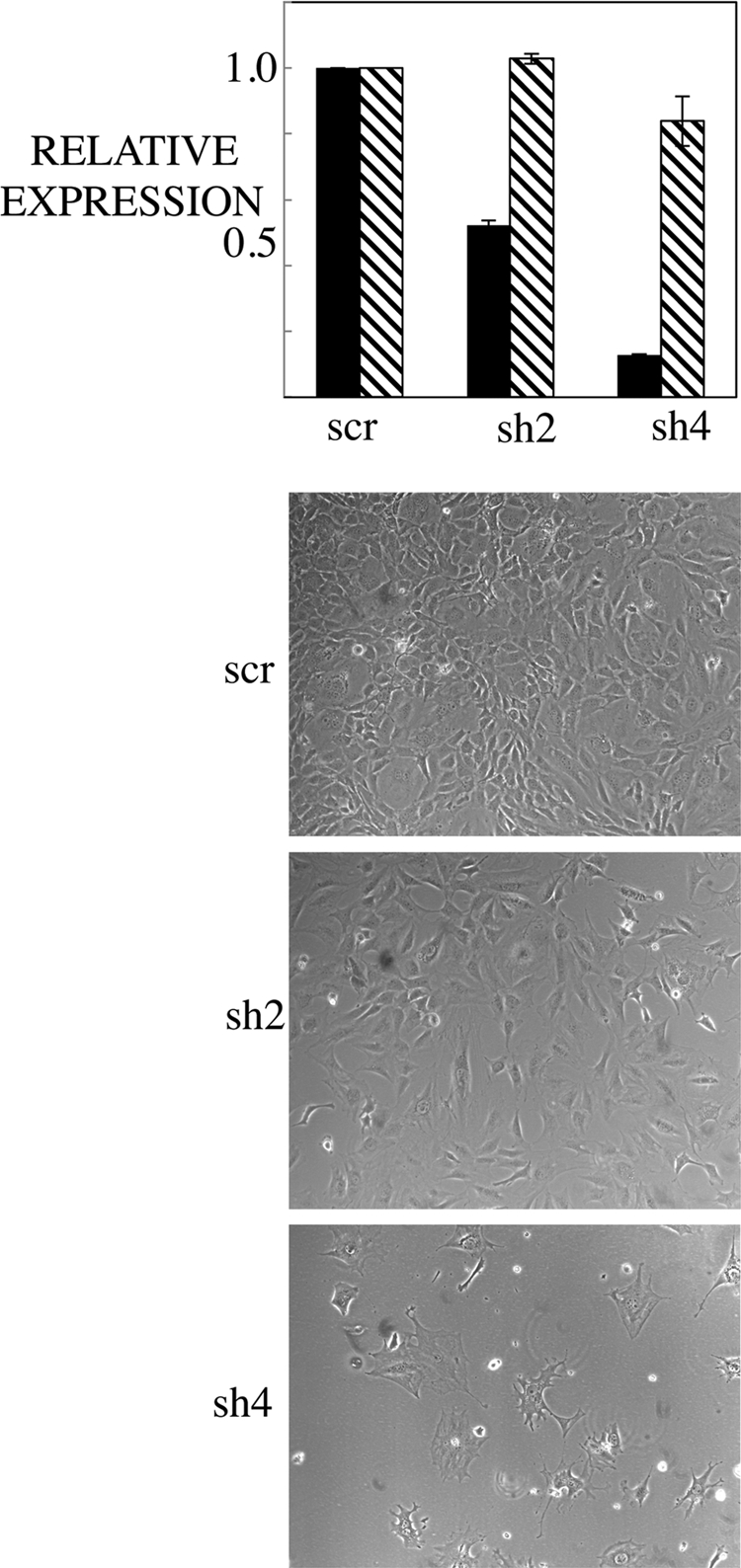
Short hairpin knockdown of PP2A Aβ induces cell death. 3T3-L1 cells were infected with pLKO control (scr, for scrambled) or sh2 Aβ or sh4 Aβ. At 48 h postinfection, mRNA was collected from the cells. RT-PCR was performed to assay for levels of Aβ (black bars) and the puromycin resistance gene (PAC gene; striped bars). Phase-contrast pictures were taken at 7 days postinfection (magnification, ×10).
POLST can interact with different cellular targets than SVST.
If the two small T antigens could interact differently with forms of PP2A, it seemed possible that that they might target different cellular proteins for dephosphorylation by PP2A. A proteomic screen was used to look for such proteins using POLST tagged for tandem affinity purification at the C terminus with FLAG and HA tags. Early results identified lipin 1 and lipin 2 as proteins that interacted with POLST. Immunoprecipitation of FLAG-tagged POLST or SVST showed that lipin 1 and lipin 2 associated with POLST and not SVST (Fig. 9 A). The electrophoretic mobility of lipin is known to be affected by phosphorylation, for example, in response to insulin signaling (27). As shown in Fig. 9B, expression of POLST increased the mobility of lipin while SVST had little effect. As expected, the ability of POLST to alter lipin mobility depended on its association with PP2A (data not shown). The results shown in Fig. 9 are consistent with the idea that POLST targets lipin as a substrate for PP2A dephosphorylation while SVST does not.
Fig. 9.
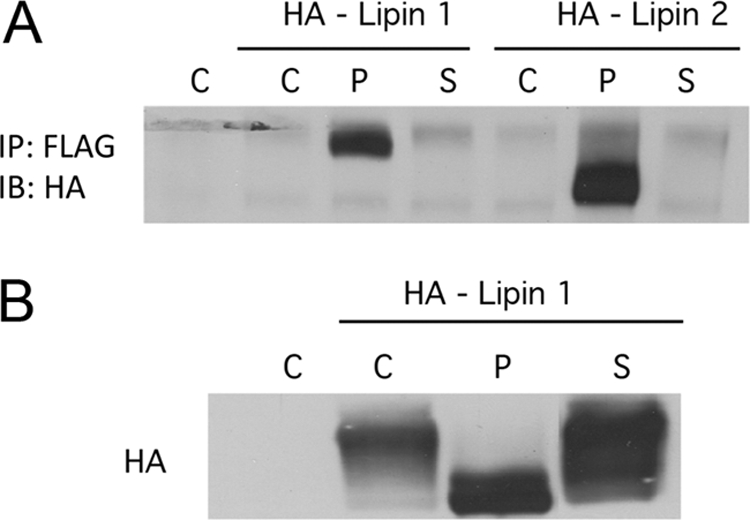
Interaction of STs with lipins. (A) Interaction of ST with both lipin 1 and lipin 2. 293T cells were cotransfected with either HA-lipin 1 or lipin 2 and empty vector (C), FLAG-POLST (P), or FLAG-SVST (S). STs were immunoprecipitated (IP) with anti-FLAG antibody, and the immunoprecipitates were blotted for lipins with anti-HA (IB). (B) The effect of STs on lipin 1 mobility. 293T cells were cotransfected with HA-lipin 1 and vector (C), POLST (P), or SVST (S). After approximately 40 h, cells extracts were collected and blotted for lipin using anti-HA antibody.
DISCUSSION
This work shows that while POLST and SVST may have common functions, there are important differences in their effects on cell differentiation and survival. This might have seemed surprising since the two viral proteins have similar structures. However, the idea that homologous proteins in a virus family can have different functions is hardly a new concept. Comparative studies on polyomavirus and SV40 large T antigens demonstrated the importance of p53 in controlling cell growth. Human papillomavirus (HPV) E7 protein contributes to transformation by binding Rb family members using an LXCXE motif. Bovine papillomavirus (BPV) E7 protein does not, but it may contribute to transformation via association with p600 (28). The differences between POLST and SVST appear to be related to the way they “handle” the same protein, the cellular phosphatase PP2A.
POLST, but not SVST, blocks differentiation of 3T3-L1 preadipocytes and C2C12 myoblasts. Previously, POLST has been shown to interfere with myeloid or monocytic differentiation of HL-60 cells (75). POLST blocks adipocyte and myoblast differentiation through PP2A since a POLST W157S mutant that does not bind PP2A fails to inhibit. While PP2A control of myoblast differentiation has not been previously reported, PP2A has previously been connected to adipocyte differentiation. PP2A inhibits C/EBP-α (65), which cooperates with peroxisome proliferator-activated receptor γ (PPARγ) to promote adipogenesis (26) and restricts cell cycle progression (65). PPARγ, a principal regulator of fat cell differentiation (62), reduces the amount of PP2A during cell cycle withdrawal (1).
Another prominent difference between POLST and SVST is seen in cell survival. In side-by-side comparisons in mouse NIH 3T3 cells or human mammary epithelial cells, POLST has a striking ability to induce cell death compared to SVST. This effect means that, unlike SVST, POLST supports transformation of human cells expressing hTERT, SV40 LT, and H-ras. However, it is important to remember that each small T antigen can be either proapoptotic or antiapoptotic under some conditions. For POLST, opposite effects are obtained depending on whether cells are growing in serum-containing medium or are serum starved (2). SVST can also be apoptotic (21), but SVST promoted survival in MDA-MB231 cells (29) or hepatocytes (20). The ability of POLST and SVST to regulate cell survival is connected to interaction with PP2A. For POLST to promote either cell death or cell survival, it must associate with PP2A (2). SVST's proapoptotic (21) or antiapoptotic (20) effects were lost when a mutant defective in PP2A binding was used.
The basis for the phenotypic difference between SVST and POLST may be rooted in effects on Akt (2). As shown previously, Akt inhibitors protect cells against POLST-mediated cell death. Differentiation of preadipocytes (34) or myoblasts (23, 71) has been shown to require Akt. SVST activates Akt in a number of cell types, such as keratinocytes (77), mammary cells (78), and 3T3-L1 cells (63). POLST reduced activation of Akt S473 phosphorylation in human (293T and HMECs) or mouse (3T3L1 or C2C12) cells. This result is reminiscent of the decrease in Akt phosphorylation when endothelin-1 blocks adipogenesis (6). Akt promotes survival by phosphorylating FOXO proteins, promoting cytoplasmic retention and inhibiting their activity (7, 8). SVST but not POLST promoted phosphorylation of FOXO proteins. Further, SVST but not POLST was able to suppress FOXO transcriptional activity.
POLST must associate with PP2A to block differentiation and regulate survival. Since SVST also associates with PP2A, it is important to understand how the interactions of SVST and POLST differ. Both SVST and POLST replace the B subunit, giving A-C-ST heterotrimers. One difference between POLST and SVST is that the two proteins can reach different substrates. The ability of POLST but not SVST to promote the dephosphorylation of lipin is a clear example. For lipin, POLST functions as a scaffold bringing together lipin with the PP2A phosphatase. This may be similar to how SVST increases the phosphatase activity of PP2A toward histone H1 (74) and the androgen receptor (73). Lipin is required for adipocyte differentiation (52), and insulin, one of the required ingredients for differentiation, promotes lipin phosphorylation (27). Dephosphorylation of lipin by POLST would therefore be expected to contribute to the block. We anticipate that additional specific substrates will be uncovered for both POLST and SVST.
A striking difference in interactions between PP2A and SVST or POLST is the ability of POLST to bind both the Aα and Aβ scaffolding subunits. This is not surprising, because PyMT, which contains 191 of the 195 residues of POLST, was shown to bind the Aβ subunit (79). From a structural point of view, it is surprising because Aα and Aβ are extremely similar in the regions that bind SVST (12, 14). Knowledge about the A isoform-specific function of PP2A is quite limited. Mutations in PP2A Aβ have been associated with lung and colon cancer (55, 66). The Aβ isoform seems to regulate RalA GTPases (56). There may be a direct connection between POLST binding of Aβ and particular outcomes since Aβ knockdown mimics POLST in promoting cell death. However, not all POLST functions are Aβ dependent. Coimmunoprecipitation experiments show that lipin binds both Aα/POLST and Aβ/POLST complexes (data not shown). POLST mutants that separate Aα and Aβ binding are needed to clarify roles for specific isoforms.
These studies once again show the value of studies on the comparative anatomy of different virus proteins. They point to the need for better mechanistic understanding of the roles of isoforms of PP2A in controlling phosphorylation of proteins such as Akt or lipin to produce different cellular outcomes.
ACKNOWLEDGMENTS
This work was supported by NIH grants to B.S.S. (CA34722 and CA50661) and T.M.R. (CA50661).
Footnotes
Published ahead of print on 10 August 2011.
REFERENCES
- 1. Altiok S., Xu M., Spiegelman B. M. 1997. PPARγ induces cell cycle withdrawal: inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes Dev. 11:1987–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrabi S., Gjoerup O. V., Kean J. A., Roberts T. M., Schaffhausen B. 2007. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 104:19011–19016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Asselin C., Gelinas C., Bastin M. 1983. Role of the three polyoma virus early proteins in tumorigenesis. Mol. Cell. Biol. 3:1451–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asselin C., Vass-Marengo J., Bastin M. 1986. Mutation in the polyomavirus genome that activates the properties of large T associated with neoplastic transformation. J. Virol. 57:165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berger H., Wintersberger E. 1986. Polyomavirus small T antigen enhances replication of viral genomes in 3T6 mouse fibroblasts. J. Virol. 60:768–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhattacharya I., Ullrich A. 2006. Endothelin-1 inhibits adipogenesis: role of phosphorylation of Akt and ERK1/2. FEBS Lett. 580:5765–5771 [DOI] [PubMed] [Google Scholar]
- 7. Brownawell A. M., Kops G. J., Macara I. G., Burgering B. M. 2001. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol. Cel.l Biol. 21:3534–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brunet A., et al. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868 [DOI] [PubMed] [Google Scholar]
- 9. Chen C. A., Okayama H. 1988. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques 6:632–637 [PubMed] [Google Scholar]
- 10. Chen W., Arroyo J. D., Timmons J. C., Possemato R., Hahn W. C. 2005. Cancer-associated PP2A Aα subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 65:8183–8192 [DOI] [PubMed] [Google Scholar]
- 11. Chen W., et al. 2004. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5:127–136 [DOI] [PubMed] [Google Scholar]
- 12. Chen Y., et al. 2007. Structural and biochemical insights into the regulation of protein phosphatase 2A by small t antigen of SV40. Nat. Struct. Mol. Biol. 14:527–534 [DOI] [PubMed] [Google Scholar]
- 13. Cherington V., Morgan B., Spiegelman M., Roberts T. M. 1986. Recombinant retroviruses that transduce individual polyoma tumor antigens: effects on growth and differentiation. Proc. Natl. Acad. Sci. U. S. A. 83:4307–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho U. S., et al. 2007. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 5:e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cicala C., et al. 1994. Simian virus 40 small-t antigen stimulates viral DNA replication in permissive monkey cells. J. Virol. 68:3138–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dahl J., Chen H. I., George M., Benjamin T. L. 2007. Polyomavirus small T antigen controls viral chromatin modifications through effects on kinetics of virus growth and cell cycle progression. J. Virol. 81:10064–10071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eckhart W., Hutchinson M. A., Hunter T. 1979. An activity phosphorylating tyrosine in polyoma T antigen immunoprecipitates. Cell 18:925–933 [DOI] [PubMed] [Google Scholar]
- 18. Eichhorn P. J., Creyghton M. P., Bernards R. 2009. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 1795:1–15 [DOI] [PubMed] [Google Scholar]
- 19. Garcia A., Cereghini S., Sontag E. 2000. Protein phosphatase 2A and phosphatidylinositol 3-kinase regulate the activity of Sp1-responsive promoters. J. Biol. Chem. 275:9385–9389 [DOI] [PubMed] [Google Scholar]
- 20. Gillet R., Cavard C., Grimber G., Briand P., Joulin V. 2001. Hepatic expression of SV40 small-T antigen blocks the in vivo CD95-mediated apoptosis. Biochem. Biophys. Res. Commun. 284:369–376 [DOI] [PubMed] [Google Scholar]
- 21. Gjoerup O., Zaveri D., Roberts T. M. 2001. Induction of p53-independent apoptosis by simian virus 40 small t antigen. J. Virol. 75:9142–9155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goetz F., et al. 2001. The SV40 small t-antigen prevents mammary gland differentiation and induces breast cancer formation in transgenic mice; truncated large T-antigen molecules harboring the intact p53 and pRb binding region do not have this effect. Oncogene 20:2325–2332 [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez I., et al. 2004. Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways in myogenesis. Mol. Cell. Biol. 24:3607–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hahn W. C., et al. 2002. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 22:2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Higgins C., Chatterjee S., Cherington V. 1996. The block of adipocyte differentiation by a C-terminally truncated, but not by full-length, simian virus 40 large tumor antigen is dependent on an intact retinoblastoma susceptibility protein family binding domain. J. Virol. 70:745–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu E., Tontonoz P., Spiegelman B. M. 1995. Transdifferentiation of myoblasts by the adipogenic transcription factors PPARγ and C/EBPα. Proc. Natl. Acad. Sci. U. S. A. 92:9856–9860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huffman T. A., Mothe-Satney I., Lawrence J. C., Jr 2002. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. U. S. A. 99:1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huh K. W., et al. 2005. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. U. S. A. 102:11492–11497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hui L., et al. 2005. mTOR-dependent suppression of protein phosphatase 2A is critical for phospholipase D survival signals in human breast cancer cells. J. Biol. Chem. 280:35829–35835 [DOI] [PubMed] [Google Scholar]
- 30. Janssens V., Goris J. 2001. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 353:417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janssens V., Goris J., Van Hoof C. 2005. PP2A: the expected tumor suppressor. Curr. Opin. Genet. Dev. 15:34–41 [DOI] [PubMed] [Google Scholar]
- 32. Johannessen M., Olsen P. A., Johansen B., Seternes O. M., Moens U. 2003. Activation of the coactivator four-and-a-half-LIM-only protein FHL2 and the c-fos promoter through inhibition of protein phosphatase 2A. Biochem. Pharmacol. 65:1317–1328 [DOI] [PubMed] [Google Scholar]
- 33. Junttila M. R., et al. 2007. CIP2A inhibits PP2A in human malignancies. Cell 130:51–62 [DOI] [PubMed] [Google Scholar]
- 34. Kim S. P., et al. 2010. Transcriptional activation of peroxisome proliferator-activated receptor-gamma requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem. Biophys. Res. Commun. 399:55–59 [DOI] [PubMed] [Google Scholar]
- 35. Klucky B., Koch B., Radolf M., Steinlein P., Wintersberger E. 2004. Polyomavirus tumorantigens have a profound effect on gene expression in mouse fibroblasts. Oncogene 23:4707–4721 [DOI] [PubMed] [Google Scholar]
- 36. Klucky B., Wintersberger E. 2007. Polyomavirus small T antigen transactivates genes by its ability to provoke the synthesis and the stabilization of MYC. Oncogene 26:6356–6360 [DOI] [PubMed] [Google Scholar]
- 37. Kolzau T., Hansen R. S., Zahra D., Reddel R. R., Braithwaite A. W. 1999. Inhibition of SV40 large T antigen induced apoptosis by small T antigen. Oncogene 18:5598–5603 [DOI] [PubMed] [Google Scholar]
- 38. Lane D. P., Crawford L. 1979. T antigen is bound to a host of protein in SV40-transformed cells. Nature 278:261–263 [DOI] [PubMed] [Google Scholar]
- 39. Linzer D. I., Maltzman W., Levine A. J. 1979. The SV40 A gene product is required for the production of a 54,000 MW cellular tumor antigen. Virology 98:308–318 [DOI] [PubMed] [Google Scholar]
- 40. Loeken M. R. 1992. Simian virus 40 small t antigen transactivates the adenovirus E2A promoter by using mechanisms distinct from those used by adenovirus E1A. J. Virol. 66:2551–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maione R, Fimia G. M., Holman P., Schaffhausen B., Amati P. 1994. Retinoblastoma antioncogene is involved in the inhibition of myogenesis by polyomavirus large T antigen. Cell Growth Differ. 5:231–237 [PubMed] [Google Scholar]
- 42. Millward T. A., Zolnierowicz S., Hemmings B. A. 1999. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 24:186–191 [DOI] [PubMed] [Google Scholar]
- 43. Moreno C. S., et al. 2004. Signaling and transcriptional changes critical for transformation of human cells by simian virus 40 small tumor antigen or protein phosphatase 2A B56γ knockdown. Cancer Res. 64:6978–6988 [DOI] [PubMed] [Google Scholar]
- 44. Morgenstern J. P., Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moule M. G., Collins C. H., McCormick F., Fried M. 2004. Role for PP2A in ARF signaling to p53. Proc. Natl. Acad. Sci. U. S. A. 101:14063–14066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mullane K. P., Ratnofsky M., Cullere X., Schaffhausen B. 1998. Signaling from polyomavirus middle T and small T defines different roles for protein phosphatase 2A. Mol. Cell. Biol. 18:7556–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mumby M. 2007. PP2A: unveiling a reluctant tumor suppressor. Cell 130:21–24 [DOI] [PubMed] [Google Scholar]
- 48. Mungre S., et al. 1994. Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small-t antigen in vitro decrease viral transformation. J. Virol. 68:1675–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Noda T., Satake M., Yamaguchi Y., Ito Y. 1987. Cooperation of middle and small T antigens of polyomavirus in transformation of established fibroblast and epithelial-like cell lines. J. Virol. 61:2253–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ogris E., Mudrak I., Wintersberger E. 1992. Polyomavirus large and small T antigens cooperate in induction of the S phase in serum-starved 3T3 mouse fibroblasts. J. Virol. 66:53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pallas D. C., et al. 1990. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60:167–176 [DOI] [PubMed] [Google Scholar]
- 52. Phan J., Peterfy M., Reue K. 2004. Lipin expression preceding peroxisome proliferator-activated receptor-gamma is critical for adipogenesis in vivo and in vitro. J. Biol. Chem. 279:29558–29564 [DOI] [PubMed] [Google Scholar]
- 53. Porras A., et al. 1996. A novel simian virus 40 early-region domain mediates transactivation of the cyclin A promoter by small-t antigen and is required for transformation in small-t antigen-dependent assays. J. Virol. 70:6902–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qian W., Wiman K. G. 2000. Polyoma virus middle T and small t antigens cooperate to antagonize p53-induced cell cycle arrest and apoptosis. Cell Growth Differ. 11:31–39 [PubMed] [Google Scholar]
- 55. Ruediger R., Pham H. T., Walter G. 2001. Alterations in protein phosphatase 2A subunit interaction in human carcinomas of the lung and colon with mutations in the A beta subunit gene. Oncogene 20:1892–1899 [DOI] [PubMed] [Google Scholar]
- 56. Sablina A. A., et al. 2007. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell 129:969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scheidtmann K. H., Mumby M. C., Rundell K., Walter G. 1991. Dephosphorylation of simian virus 40 large-T antigen and p53 protein by protein phosphatase 2A: inhibition by small-t antigen. Mol. Cell. Biol. 11:1996–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sheng Q., Love T. M., Schaffhausen B. 2000. J domain-independent regulation of the Rb family by polyomavirus large T antigen. J Virol 74:5280–5290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Skoczylas C., Fahrbach K. M., Rundell K. 2004. Cellular targets of the SV40 small-t antigen in human cell transformation. Cell Cycle 3:606–610 [PubMed] [Google Scholar]
- 60. Skoczylas C., Henglein B., Rundell K. 2005. PP2A-dependent transactivation of the cyclin A promoter by SV40 ST is mediated by a cell cycle-regulated E2F site. Virology 332:596–601 [DOI] [PubMed] [Google Scholar]
- 61. Sontag E., Sontag J. M., Garcia A. 1997. Protein phosphatase 2A is a critical regulator of protein kinase C zeta signaling targeted by SV40 small t to promote cell growth and NF-κB activation. EMBO J. 16:5662–5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tontonoz P., Hu E., Spiegelman B. M. 1994. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79:1147–1156 [DOI] [PubMed] [Google Scholar]
- 63. Ugi S., et al. 2004. Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24:8778–8789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Walter G., Ruediger R., Slaughter C., Mumby M. 1990. Association of protein phosphatase 2A with polyoma virus medium tumor antigen. Proc. Natl. Acad. Sci. U. S. A. 87:2521–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang G. L., Iakova P., Wilde M., Awad S., Timchenko N. A. 2004. Liver tumors escape negative control of proliferation via PI3K/Akt-mediated block of C/EBP alpha growth inhibitory activity. Genes Dev 18:912–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang S. S., et al. 1998. Alterations of the PPP2R1B gene in human lung and colon cancer. Science 282:284–287 [DOI] [PubMed] [Google Scholar]
- 67. Wang W. B., Bikel I., Marsilio E., Newsome D., Livingston D. 1994. Transrepression of RNA polymerase II promoters by the simian virus 40 small t antigen. J. Virol. 68:6180–6187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Watanabe G., et al. 1996. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc. Nat. Acad. Sci. 93:12861–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Westermarck J., Hahn W. C. 2008. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14:152–160 [DOI] [PubMed] [Google Scholar]
- 70. Whitman M., Kaplan D. R., Schaffhausen B., Cantley L., Roberts T. M. 1985. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 315:239–242 [DOI] [PubMed] [Google Scholar]
- 71. Wilson E. M., Rotwein P. 2007. Selective control of skeletal muscle differentiation by Akt1. J. Biol. Chem. 282:5106–5110 [DOI] [PubMed] [Google Scholar]
- 72. Yan L., et al. 2008. PP2A regulates the pro-apoptotic activity of FOXO1. J. Biol. Chem. 283:7411–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang C. S., et al. 2005. Simian virus 40 small t antigen mediates conformation-dependent transfer of protein phosphatase 2A onto the androgen receptor. Mol. Cell. Biol. 25:1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang S. I., et al. 1991. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol. Cell Biol. 11:1988–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yen A., Placanica L., Bloom S., Varvayanis S. 2001. Polyomavirus small t antigen prevents retinoic acid-induced retinoblastoma protein hypophosphorylation and redirects retinoic acid-induced G0 arrest and differentiation to apoptosis. J. Virol. 75:5302–5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yu J., Boyapati A., Rundell K. 2001. Critical role for SV40 small-t antigen in human cell transformation. Virology 290:192–198 [DOI] [PubMed] [Google Scholar]
- 77. Yuan H., Veldman T., Rundell K., Schlegel R. 2002. Simian virus 40 small tumor antigen activates AKT and telomerase and induces anchorage-independent growth of human epithelial cells. J. Virol. 76:10685–10691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhao J. J., et al. 2003. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3:483–495 [DOI] [PubMed] [Google Scholar]
- 79. Zhou J., Pham H. T., Ruediger R., Walter G. 2003. Characterization of the Aalpha and Abeta subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem. J. 369:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]