

Abstract
Histone–lysine acetylation is a vital chromatin post-translational modification involved in the epigenetic regulation of gene transcription. Bromodomains bind acetylated lysines, acting as readers of the histone-acetylation code. Competitive inhibitors of this interaction have antiproliferative and anti-inflammatory properties. With 57 distinct bromodomains known, the discovery of subtype-selective inhibitors of the histone–bromodomain interaction is of great importance. We have identified the 3,5-dimethylisoxazole moiety as a novel acetyl-lysine bioisostere, which displaces acetylated histone-mimicking peptides from bromodomains. Using X-ray crystallographic analysis, we have determined the interactions responsible for the activity and selectivity of 4-substituted 3,5-dimethylisoxazoles against a selection of phylogenetically diverse bromodomains. By exploiting these interactions, we have developed compound 4d, which has IC50 values of <5 μM for the bromodomain-containing proteins BRD2(1) and BRD4(1). These compounds are promising leads for the further development of selective probes for the bromodomain and extra C-terminal domain (BET) family and CREBBP bromodomains.
Introduction
Genetic information is encoded in DNA by the specific linear sequence of its bases. The expression of this information is regulated by protein scaffolds that are capable of reading the complex code of post-translational modifications that occur on chromatin. Acetylation of histones lysine residues is one such essential component in the epigenetic regulation of gene expression.(1) Additionally, recent proteomics studies have demonstrated that lysine acetylation occurs in over 1750 cellular proteins involved in diverse roles such as cell cycle, splicing, nuclear transport, and actin nucleation.(2) Lysine acetylation on histones and other proteins is effected by the dynamic interplay of acetyltransferase (HAT) and deacetylase (HDAC) enzymes, which is analogous to the regulation of serine, threonine, and tyrosine phosphorylation by kinases and phosphatases.
Bromodomains are a family of conserved ∼110 amino acid modules that bind selectively to acetylated lysines present in proteins, notably histones,(3) and are thereby thought to participate in deciphering the histone code.(4) Bromodomains have been classified into several distinct subgroups according to the function of their parent protein: (i) histone acetyltransferases (HATs), including CREBBP, GCN5, PCAF, and TAFII250; (ii) in components of ATP-dependent chromatin-remodeling complexes such as Swi2/Snf2; and (iii) the BET (bromodomain and extra C-terminal domain) family, a class of transcriptional regulators carrying tandem bromodomains and an extra terminal domain.(5)
Although the biological roles of most bromodomains in the human genome remain elusive, those that have been characterized are fundamental. For example, the BET bromodomain-containing protein (BRD) 4 plays a key role in several cellular processes, including mitosis.(6) Expression levels of BRD4 correlate with breast cancer survival rates,(7) and in a subset of malignant squamous carcinomas, the N-terminal bromodomains of BRD4 are fused in frame to the NUT gene giving rise to extremely aggressive tumor growth.(8) Knockdown experiments have implicated BRD4 in the transcriptional regulation of viruses such as HIV(9) and EBV,(10) as well as the degradation of HPV.(11) BRD4 was also shown to be required for transcriptional coactivation of NF-κB, regulating the transcription of P-TEFb-dependent pro-inflammatory target genes.(12) A second BET-family protein, the testis-specific BRDT, is essential for male germ cell differentiation in selective domain knockout mice that were viable but sterile.(13)
The development of small molecule inhibitors of bromodomain binding to histones and other acetyl-lysine-containing proteins is in its infancy. Weakly potent small molecule ligands of the cAMP response element-binding protein (CREB) binding protein (CREBBP) bromodomain, discovered by NMR screening of acetyl-lysine (KAc) mimics, have been shown to modulate p53 stability and function in response to DNA damage in cells.(14) More recently, a 4-hydroxyphenylazobenzenesulfonic acid derivative was shown to bind to the CREBBP bromodomain, with KD = 19 μM, and consequently blocks p53-dependent apoptosis in cardiomyocytes under DNA-damage stress.(15)
Ligands have also been developed for the BET family of bromodomains. A benzimidazole-derived BRD2(1) ligand with KD = 28 μM reduced the transcription of BRD2-dependent genes.(16) Antiproliferative effects against a number of cell lines have been claimed in a patent for a series of thienotriazolodiazepine BRD2/3/4 blockers with nanomolar in vitro peptide displacement activity.(17) The structurally related (+)-JQ1 (Figure 1A), recently characterized as a BET-family selective chemical probe, shows specific antiproliferative effects in BRD4-dependent cell lines and patient-derived xenograft models.(18) In addition, a recently identified BET bromodomain blocker (I-BET, Figure 1B) suppressed lipopolysaccharide-inducible genes in macrophages and showed anti-inflammatory effects in mice.(19) This work demonstrates the feasibility of inhibiting KAc binding to bromodomains with small molecules that occupy the well-defined KAc-binding pocket. It is possible that targeting this pocket might prove more tractable than developing inhibitors of catalytic activity in certain enzymes. In addition, small molecule inhibition of bromodomain–protein interactions might elicit subtly different pharmacological responses to the inhibition of the catalytic domain of the same protein.(20)
Figure 1.

(A) Structure of the BET probe (+)-JQ1.(18) (B) Structure of the BET probe I-BET.(19a) (C) Structure of dihydroquinazolinone-containing 3,5-dimethylisoxazole derivative 1. (D) The concept employed in the design of the 3,5-dimethylisoxazole-based bromodomain inhibitors.
The findings outlined above demonstrate clearly the need for systematic generation of high affinity subfamily selective bromodomain-binding small molecules that can be used as chemical probes to explore further the roles of individual bromodomains. These molecules will provide complementary data to studies using knockdown, peptide and antibody approaches, and will enable prediction of the likely phenotypes of eventual small molecule drugs. As part of our ongoing goal to generate cell-penetrant chemical probes for bromodomains, we have previously developed bromodomain–histone peptide binding assays to allow the identification of fragment and small molecule ligands.(21) Herein, we describe the use of these assays, together with a structure-based design approach, to identify and develop the structure–activity relationships (SAR) of a novel series of 3,5-dimethylisoxazole-containing bromodomain–histone binding inhibitors.
We demonstrate that the 3,5-dimethylisoxazole moiety is an effective KAc mimic and show how differential substitution of the 3,5-dimethylisoxazole core gives derivatives that bind with some selectivity and useful affinity to either the first bromodomain of human BRD4 [BRD4(1)] or the bromodomain of human CREBBP.
Results and Discussion
In the course of our work, we discovered that the solvent N-methyl pyrrolidinone (NMP) interacts with the KAc-binding pocket of a range of bromodomains, showing weak affinity but, because of its low molecular weight, high ligand efficiency (0.30–0.53, see Supporting Information, Table S1), with IC50 values ranging from 34 mM for the bromodomain adjacent to zinc finger domain 2B (BAZ2B) to 2.3 mM for CREBBP.(21) During subsequent studies on methyl-bearing heterocycles, we purchased and tested the dihydroquinazolinone-containing 3,5-dimethylisoxazole derivative 1 (Figure 1C) and observed unexpectedly low IC50 values of ∼3 μM against the first bromodomain of BRD2 [BRD2(1)] and ∼7 μM against BRD4(1). Docking studies suggested that two binding modes were possible for this compound, with either the dihydroquinazolinone or 3,5-dimethylisoxazole moiety acting as a KAc bioisostere (data not shown). To explore the possibility that the 3,5-dimethylisoxazole might occupy the KAc-binding pocket, we obtained an X-ray crystal structure of the complex between human BRD4(1) and 1.
The X-ray crystal structure of the BRD4(1) complex was solved at 1.98 Å (Figure 2A, PDB ID: 3SVF). It demonstrated that the 3,5-dimethylisoxazole was indeed acting as a KAc bioisostere, accepting a hydrogen bond from the conserved N residue within the binding pocket [N140 in BRD4(1)], which mimics the key interaction made with the carbonyl oxygen of KAc. Additionally, we observed additional unexplained electron density at the 4-position of the dihydroquinazolinone that was interpreted as a substitution of one hydrogen atom by an ethylene glycol unit (Figure 2B). As dihydroquinazolinones are liable to benzylic oxidation under laboratory conditions (Supporting Information, Figure S1),(22) we hypothesized that this product 2 might arise from oxidation at this position to form the corresponding iminium species, followed by the addition of ethylene glycol, which is present in the crystallization buffer (see Supporting Information, Figure S1 for mass spectrometry analysis). Interestingly, only one enantiomer was observed in the X-ray crystal structure; the ethylene glycol unit attached to the stereogenic center occupies a hydrophobic groove formed by W81 and P82 from the ZA loop and I146 and M149 from helix C. Occupation of this site by the 4-chlorophenyl substituent of (+)-JQ1 and I-BET (Figure 1A,B) is believed to contribute to the selectivity of the these compounds for the BET subfamily over other bromodomains since a similar groove is not available in other bromodomains (Figure 2C).(18) In addition, we observed that a sample of 1 displayed significant cytotoxicity in the HeLa human cell line, while (+)-JQ1 showed no cytotoxicity over the same concentration range (Supporting Information, Figure S2). Therefore, the development of analogues that do not possess a readily oxidized benzylic methylene unit, but that could form interactions with this groove, was pursued. The mono- and di-meta-substituted 3,5-dimethyl-4-phenylisoxazole scaffolds (3a–d and 4a–d) were chosen to allow extension from the aromatic core in two directions (Figure 1B). Initial docking studies suggested that a single meta-substituent would occupy a position similar to that of the ethylene glycol moiety of 2 (Supporting Information, Figure S3A). A second meta-substituent would be directed into a groove (referred to by Nicodeme et al.(19a) and Chung et al.(19b) as the ZA channel) defined by W81 on one side and L92 on the other (Supporting Information, Figure S3A). Consequently, the 3,5-dimethylisoxazole derivatives 3a–d and 4a–d were selected as target molecules that contain a range of substituents to enable exploration of the KAc-binding pocket of the bromodomains.
Figure 2.

(A) The X-ray crystal structure of 2 (carbon = yellow, PDB ID, 3SVF) bound to human BRD4(1). The conserved N140 residue is highlighted in green. (B) 2Fo – Fc map at 2σ of the observed ligand 2. (C) Overlay of crystal structures of 2 (carbon = yellow) and (+)-JQ1 (carbon = orange, PDB ID: 3MXF) bound to human BRD4(1), showing that the ethylene glycol unit binds in the same region as the chlorophenyl moiety of (+)-JQ1. The images were generated and structure alignment conducted using PyMOL.
The syntheses of 3a–d and 4a–d are shown in Schemes 1–3. Initial work focused on the direct arylation of 3,5-dimethylisoxazole using Pd-catalyzed C–H bond activation.(23) While this reaction proceeded well with bromobenzene 5, lower yields were obtained on reaction with 6 and 10 (Supporting Information, Scheme S1). Furthermore, the reaction has been reported to be less successful with electron-rich aryl bromides.(23) Conversely, Molander et al. have demonstrated that potassium heteroaryltrifluoroborates react efficiently with both electron-rich and electron-deficient aryl halides, with low catalyst loadings and minimal protodeboronation.(24) Therefore, we prepared potassium (3,5-dimethylisoxazol-4-yl)trifluoroborate 8 from (3,5-dimethylisoxazol-4-yl)boronic acid.(24) Coupling of 8 to commercially available aryl bromides 5 and 6, and aryl bromide 7 (easily prepared from 3-bromophenol,(25)Supporting Information, Scheme S2) afforded 3,5-dimethyl-4-phenylisoxazole 3a and the 3′-substituted 3,5-dimethyl-4-phenylisoxazoles 3b and 3c in excellent yields (Scheme 1). Although the coupling reactions did proceed when conducted with (3,5-dimethylisoxazol-4-yl)boronic acid (data not shown), the yields observed were lower than those in the route described above.
Scheme 1. Synthesis of the 3′-Substituted 3,5-Dimethyl-4-phenylisoxazole Derivatives 3a–c.

Conditions: (a) R = H: Na2CO3, Pd(OAc)2, RuPhos, EtOH, 85 °C, 4 h, 85%. R = Ac: Na2CO3, Pd(OAc)2, RuPhos, EtOH, 85 °C, 13 h, 86%. R = OEt: Na2CO3, Pd(OAc)2, RuPhos, EtOH, 85 °C, 2 h, 87%.
Scheme 3. Synthesis of the Secondary Alcohols 3d and 4d.

Conditions: (a) NaBH4, MeOH, rt, 2 h, R = H: 77%. R = OEt: 81%.
The 3′,5′-disubstituted 3,5-dimethyl-4-phenylisoxazoles 4a–c were prepared from 3-bromo-5-hydroxybenzoic acid 9 (Scheme 2). Alkylation of 9 afforded the ethyl ester 10, which was coupled to 8, yielding 4a. Subsequent LiOH-mediated hydrolysis furnished the carboxylic acid 4b. Alternatively, the ethyl ester of 10 was converted to the acetyl group via the Weinreb amide 12 in three steps; coupling, under the same conditions employed above, yielded the isoxazole 4c. The methyl ketones 3b and 4c were converted to the racemic secondary alcohols 3d and 4d by reduction of the ketone with NaBH4 (Scheme 3).
Scheme 2. Synthesis of the 3′,5′-Substituted 3,5-Dimethyl-4-phenylisoxazole Derivatives 4a–c.

Conditions: (a) EtBr, K2CO3, DMF, 100 °C (microwave), 15 min, 96%; (b) 8, Na2CO3, Pd(OAc)2, RuPhos, EtOH, 100 °C, 48 h, 70%; (c) LiOH, THF, H2O, rt, 16 h, 83%; (d) LiOH, THF, H2O, rt, 23 h, 95%; (e) NHMe(OMe)·HCl, HBTU, iPr2NEt, DMF, rt, 14 h, 80%; (f) MeMgBr, THF, rt, 15 h, 86%; (g) 8, Na2CO3, Pd(OAc)2, RuPhos, EtOH, 85 °C, 64 h, 67%.
To enable accurate comparison with our new compounds, a sample of 1 was resynthesized (Supporting Information, Scheme S3). Biological evaluation of 1 was conducted using freshly prepared DMSO solutions to minimize the possibility of oxidation (see Supporting Information for details).
To determine whether the compounds inhibited bromodomain–histone binding, we utilized a peptide displacement assay based on AlphaScreen technology.(21) For the first bromodomain of BRD2 [BRD2(1)] and BRD4(1), a histone H4 peptide acetylated at K5, 8, 12, and 16 was employed, while for CREBBP an H3 peptide acetylated at K56 was used. IC50 values for compounds 1, 3a–d, and 4a–d are shown in Table 1 (dose–response curves for 1, compounds 4b and 4d in BRD4(1), BRD2(1), and CREBBP are shown in Supporting Information, Figure S4). With the exception of 3a, all compounds inhibited BRD4(1) and BRD2(1) with IC50 ≤ 50 μM, the most potent compounds showing low micromolar activity. Introduction of an ethoxy substituent at R2 increases potency against BRD4(1) and BRD2(1). A variety of substituents at R1 are well tolerated, with the exception of R1 = CO2H in which the positioning of a negatively charged substituent in a hydrophobic site is likely to disfavor binding. IC50 values were generally higher against CREBBP, except for 4b (R1 = CO2H) where the IC50 for CREBBP was lower than that for BRD4(1) and similar to that for BRD2(1). Gratifyingly, 4b and 4d did not show any cytotoxicity in a human cell line (Supporting Information, Figure S2).
Table 1. Effect of 3′- and 5′-Substituents on Competitive Displacement of Histone Peptides from the BRD4(1) and BRD2(1), and CREBBP Bromodomaina.


Protein and peptide concentration: 50 nM. Protein and peptide concentration: 100 nM. Heat map shows relative IC50 values; red indicates low IC50 values, and green indicates high IC50 values. Ranges in parentheses represent 95% confidence intervals resulting from sigmoidal curve fitting to duplicate data. n.d. = not determined.
To investigate the selectivity of these compounds against other bromodomains, we employed a binding assay based on protein stability shift (Table 2).(26) Of the compounds that produced substantial changes in melting temperature (ΔTmobs), most displayed selectivity for BET bromodomains. However, 4b–d also produce an appreciable ΔTmobs with CREBBP, in agreement with the IC50 values. Indeed, 4b showed selectivity, albeit modest, for CREBBP over the BET bromodomains.
Table 2. ΔTmobs against Selected Bromodomainsa.

Compound concentration, 100 μM; protein concentration, 2 μM. Heat map shows relative ΔTmobs; red indicates large ΔTmobs, and green indicates small ΔTmobs. Experiments were carried out in duplicate; average values are reported.
In order to confirm that the dimethylisoxazole moiety was acting as a KAc mimic and to elucidate structure–activity profiles for these compounds, we obtained cocrystal structures of BRD4(1) with 4d and the CREBBP bromodomain with 4b (Figures 3 and 4).
Figure 3.

(A) X-ray crystal structure of 4d (carbon = yellow) bound to human BRD4(1) (PDB ID: 3SVG). The conserved N140 residue is highlighted in green. Black dashed lines represent hydrogen bonds between 4d, structured water molecules (red spheres), and interacting residues (labeled). Image generated using PyMOL. (B) A schematic representation of the key interactions formed between 4d and human BRD4(1).
Figure 4.

(A) X-ray crystal structure of 4b (carbon = yellow) bound to the bromodomain of human CREBBP (PDB ID: 3SVH). The conserved N1168 residue is highlighted in green. Black dashed lines represent hydrogen bonds among 4b, structured water molecules (red spheres), and interacting residues (labeled). Image generated using PyMOL. (B) Schematic representation of the key interactions formed between 4b and the bromodomain of human CREBBP.
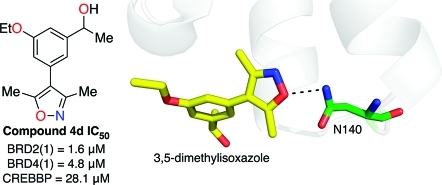
The costructure of 4d bound to BRD4(1) was obtained at 1.68 Å resolution (PDB ID: 3SVG). Globally, the ligand-bound and apo structures (PDB ID: 2OSS) are very similar, with a rmsd of 0.17. This structure confirms that 4d binds with the dimethylisoxazole oriented into the KAc-binding pocket as anticipated by our docking studies (Supporting Information, Figure S3B). It is predicted that the isoxazole oxygen atom forms the expected hydrogen bond with the amine group of the conserved N140 (green, Figure 3A). The isoxazole nitrogen atom interacts with the phenol group of Y97 via the structured water molecule A (Figure 3B).
Although 4d was synthesized as a racemate, the electron density for this compound implies that the (R)-enantiomer crystallizes preferentially with BRD4(1). The secondary alcohol forms a network of hydrogen bonds with water molecules B–E, which interact with D144, D145, and the carbonyl oxygen atom of N140. The methyl group attached to the secondary alcohol binds within the hydrophobic pocket (pocket 2, Figure 3B) that is defined by I146, M149, and W81, the latter forming part of the WPF shelf (described by Nicodeme et al. as W81, P82, and F83).(19a) This is the same pocket that binds the ethylene glycol unit of 2 (Supporting Information, Figure S3C) and the chlorophenyl moieties of I-BET and (+)-JQ1 (Supporting Information, Figure S3D). The ethyl ether binds within the ZA channel (pocket 3, Figure 3B) that accommodates the thiophene moiety of (+)-JQ1 and the methylated phenol of I-BET. Therefore, analysis of the X-ray crystal structure of 4d in complex with BRD4(1) shows that this compound is forming interactions in many of the same regions as (+)-JQ1 and I-BET, perhaps explaining how a compound with relatively low molecular weight (261) still shows good affinity for BRD4(1) and reasonable selectivity for the BET subfamily of bromodomains. Indeed, 4d has ligand efficiencies of 0.39 for BRD4(1) and 0.43 for BRD2(1), which compare favorably with the value of 0.32 for (+)-JQ1 at BRD4(1) (see Supporting Information, Table S1 for details of ligand efficiency calculations). These ligand efficiencies suggest that compound 4d is a promising lead for the development of BET bromodomain ligands that display potencies similar to those of (+)-JQ1 and I-BET but with lower molecular mass than those of these compounds.
Identification of CREBBP bromodomain inhibitors has so far proved challenging, with only two studies identifying small-molecule ligands for this bromodomain.14,15 Therefore, given that compound 4b shows some selectivity for CREBBP bromodomain over other subfamilies, the costructure of 4b bound to the CREBBP bromodomain was obtained at 1.80 Å resolution (PDB ID: 3SVH). Compound 4b, which possesses a carboxylic acid group in place of the secondary alcohol of 4d, binds to the CREBBP bromodomain in an orientation similar to that adopted by 4d when binding to BRD4(1). The dimethylisoxazole moiety again acts as a KAc mimic and occupies the KAc-binding pocket. Likewise, the isoxazole oxygen atom forms a hydrogen bond with the amine group of N1168 (equivalent to N140 in BRD4(1), green, Figure 4A). The isoxazole nitrogen atom also interacts with the phenol group of Y1125 [equivalent to Y97 in BRD4(1)] via the structured water molecule A. The dihedral angle between the 3,5-dimethylisoxazole and the phenyl ring of compounds 2, 4b, and 4d differs in the crystal structures obtained. However, this angle is observed as 50° in the case of 2 [BRD4(1)], 45° in the case of 4d [BRD4(1)] and 39° in the case of 4b (CREBBP), which are all similar to the angle of 40° in the predicted lowest energy conformation (Supporting Information, Figure S5), suggesting that these compounds can bind in the KAc-binding pocket without the introduction of significant strain. The oxygen atom of the ethoxy group in 4b forms a hydrogen bond with structured water G, which interacts with the turn-forming residues 1110–1113. Interestingly, this structured water molecule is also present in BRD4(1), but the orientation of the ethoxy group in compound 4d is such that it does not form a similar hydrogen bond, at least within crystals. Residue W81, which plays a key role in defining the binding site in BRD4(1), is not present in CREBBP. The equivalent amino acid is L1109, which although also hydrophobic, is oriented away from the binding site, providing more space in this region. R1173 provides a basic rim to the binding pocket, which is not present in BRD4. Although these residues are not in direct contact with the carboxylic acid of 4b, it is possible that a weak electrostatic interaction might explain, at least in part, the preferential binding of this compound to CREBBP over the BET bromodomains. It should be noted that CREBBP crystallized as a dimer, with a second molecule of 4b bound at the dimer interface. This phenomenon is thought to result from the relatively high concentration of 4b used in obtaining the structure and is unlikely to be responsible for this compound’s ability to prevent the CREBBP–KAc interaction.
In summary, we have discovered an entirely new chemical class of bromodomain ligands comprising low-molecular weight 4-phenyl-3,5-dimethylisoxazole derivatives. Using structure-based approaches, we have demonstrated that the unstable dihydroquinazolinone moiety of lead 1 can be replaced with a single aromatic ring without significant loss of affinity. Most of the resultant 4-phenyl-3,5-dimethylisoxazoles are selective ligands for the BET family of bromodomains. However, compounds 4b–d produced a marked increase in Tmobs for CREBBP, suggesting that 3,5-dimethylisoxazoles are also useful leads for selective inhibitors of CREBBP-KAc interactions, which have proved elusive to date. The costructures that we have obtained demonstrate that the 3,5-dimethylisoxazole moiety acts as a KAc bioisostere in two classes of bromodomains, corroborating the binding data and IC50 values presented. As few chemical classes of small-molecule inhibitors of bromodomain–histone interactions have been described previously,15−20 this work represents a significant advance in the SAR of these important proteins.
In conclusion, we have exploited the serendipitous discovery of a KAc mimic to develop a novel class of compounds that inhibit bromodomain–histone interactions. Crystallographic analysis and rational design have provided insight into the structural requirements for potent, selective BRD4(1) and CREBBP bromodomain ligands. These compounds are invaluable leads in the continuing search for chemical probes of epigenetic targets.
Experimental Section
General Experimental
1H NMR spectra were recorded on Bruker DPX400 or DQX400 (400 MHz) or Bruker AVII 500 (500 MHz) using deuterochloroform (unless indicated otherwise) as a reference for the internal deuterium lock. The chemical shift data for each signal are given as δH in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δH (TMS) = 0.00 ppm. The multiplicity of each signal is indicated by s (singlet); br s (broad singlet); d (doublet); t (triplet); q (quartet); dd (doublet of doublets); ddd (doublet of doublet of doublets); or m (multiplet). The number of protons (n) for a given resonance signal is indicated by nH. Coupling constants (J) are quoted in Hz and are recorded to the nearest 0.1 Hz. Identical proton coupling constants (J) are averaged in each spectrum and reported to the nearest 0.1 Hz. The coupling constants are determined by analysis using Bruker TopSpin software.
13C NMR spectra were recorded on Bruker DQX400 (100 MHz) or Bruker AVII 500 (125 MHz) spectrometers with broadband proton decoupling and a internal deuterium lock. The chemical shift data for each signal are given as δC in units of parts per million (ppm) relative to tetramethylsilane (TMS) where δC (TMS) = 0.00 ppm.
19F NMR spectra were recorded on Bruker AVII 500 (470 MHz) using a broadband proton decoupling pulse sequence and deuterium internal lock. The chemical shift data for each signal are given as δF in units of parts per million (ppm).
11B NMR spectra were recorded on a Bruker DRX500 (160 MHz). The chemical shift data for each signal are given as δB in units of parts per million (ppm).
Mass spectra were acquired on a VG platform spectrometer. Electrospray ionization spectra were obtained on Micromass LCT Premier and Bruker MicroTOF spectrometers, operating in positive or negative mode, from solutions of MeOH. m/z values are reported in Daltons and followed by their percentage abundance in parentheses.
Melting points were determined using a Kofler hot stage microscope and are uncorrected.
Compound purity for all tested compounds was determined by elemental analysis, obtained at the Elemental Analysis Service, London Metropolitan University, London. Elemental analysis was carried out in duplicate; average values are reported in Supporting Information. For all tested compounds, experimentally determined hydrogen, carbon, and nitrogen composition was within ±0.4% of the expected value, implying a purity of >95%.
Synthetic Procedures and Characterization for Compounds 3a–d, 4a–d
3,5-Dimethyl-4-phenylisoxazole 3a(27)
Method A
To a dry 2–5 mL microwave vial were added 8 (108 mg, 532 μmol), Pd(OAc)2 (2 mg, 8 μmol), RuPhos (7 mg, 15 μmol), and anhydrous Na2CO3 (106 mg, 1.00 mmol). The vial was sealed and purged with argon (3 × evacuate/fill). Bromobenzene 5 (79 mg, 53 μL, 496 μmol) and degassed EtOH (2.8 mL) were added by syringe, and the mixture was heated at 85 °C for 4 h. The mixture was cooled to rt and filtered through a thin pad of silica (eluent CH2Cl2) and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 6 → 30% Et2O/petroleum ether) gave 3a as a colorless oil which crystallized slowly on standing (73 mg, 85%); mp 37–38 °C (lit. 42–43 °C);(27)1H NMR (400 MHz, CDCl3) 2.29 (s, 3H), 2.42 (s, 3H), 7.24–7.29 (m, 2H), 7.34–7.40 (m, 1H), 7.42–7.48 (m, 2H); m/z (ES+) 174 ([M + H]+, 73), 196 ([M + Na]+, 100), 369 ([2M + Na]+, 19). These data are in good agreement with the literature values.(27)
Method B
To a dry 2–5 mL microwave vial were added PdCl2 (0.5 mg, 3 μmol) and KOAc (98 mg, 1.0 mmol). The vial was sealed and purged with argon (3× evacuate/fill). Degassed DMAc (2.5 mL), bromobenzene 5 (53 μL, 79 mg, 503 μmol), and 3,5-dimethylisoxazole (74 μL, 73 mg, 752 μmol) were added, and the reaction was heated at 130 °C for 27 h. The mixture was filtered through Celite (eluent Et2O), and the filtrate was washed with water (40 mL) and brine (40 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (gradient elution, gradient 3 → 30% Et2O/petroleum ether) gave 3a as a pale yellow solid (60 mg, 69%).
1–3-(3,5-Dimethylisoxazol-4-yl)phenyl)ethanone 3b(23)
Method A
To a dry 2–5 mL microwave vial were added 8 (108 mg, 532 μmol), Pd(OAc)2 (2 mg, 8 μmol), RuPhos (7 mg, 15 μmol), and anhydrous Na2CO3 (106 mg, 1.00 mmol). The vial was sealed and purged with argon (3× evacuate/fill). 1-(3-Bromophenyl)ethanone 6 (100 mg, 66 μL, 500 μmol) and degassed EtOH (3.8 mL) were added by syringe, and the mixture was heated at 85 °C for 13 h. The mixture was cooled to rt and filtered through a thin pad of silica (eluent 25% MeOH/EtOAc) and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 6 → 50% EtOAc/petroleum ether) gave 3b as a pale yellow solid (93 mg, 86%); mp 107–110 °C (50% Et2O/40–60 °C petroleum ether); 1H NMR (400 MHz, CDCl3) 2.29 (s, 3H), 2.43 (s, 3H), 2.65 (s, 3H), 7.44–7.49 (m, 1H), 7.53–7.60 (m, 1H), 7.85–7.89 (m, 1H), 7.93–7.98 (m, 1H); m/z (ES+) 216 ([M + H]+, 54), 238 ([M + Na]+, 100), 453 ([2M + Na]+, 65). These data are in good agreement with the literature values.(23)
Method B
To a dry 2–5 mL microwave vial were added PdCl2 (0.5 mg, 3 μmol) and KOAc (98 mg, 1.0 mmol). The vial was sealed and purged with argon (3× evacuate/fill). Degassed DMAc (2.5 mL), 1-(3-bromophenyl)ethanone 6 (66 μL, 100 mg, 502 μmol), and 3,5-dimethylisoxazole (74 μL, 73 mg, 752 μmol) were added, and the reaction was heated at 130 °C for 20 h. The mixture was filtered through Celite (eluent EtOAc), and the filtrate was washed with water (2 × 25 mL) and brine (25 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography (gradient elution, gradient 6 → 50% EtOAc/petroleum ether) gave 3b as a pale yellow solid (55 mg, 51%).
4-(3-Ethoxyphenyl)-3,5-dimethylisoxazole 3c
To a dry 2–5 mL microwave vial were added 8 (108 mg, 532 μmol), Pd(OAc)2 (2 mg, 8 μmol), RuPhos (7 mg, 15 μmol), and anhydrous Na2CO3 (106 mg, 1.00 mmol). The vial was sealed and purged with argon (3× evacuate/fill). Compound 7 in degassed EtOH (178 μM, 2.8 mL, 499 μmol) was added by syringe, and the mixture was heated at 85 °C for 2 h. The mixture was cooled to rt and filtered through a thin pad of silica (eluent CH2Cl2) and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 5 → 40% Et2O/petroleum ether) gave 3c as a pale yellow solid (94 mg, 87%); mp 51–52 °C (15% Et2O/petroleum ether); 1H NMR (400 MHz, CDCl3) 1.44 (t, J = 7.0 Hz, 3H), 2.27 (s, 3H), 2.41 (s, 3H), 4.06 (q, J = 7.0, 2H), 6.78 (dd, J = 2.4, 1.6 Hz, 1H), 6.82 (ddd, J = 7.7, 1.6, 0.9 Hz, 1H), 6.89 (ddd, J = 8.2, 2.4, 0.9 Hz, 1H), 7.33 (dd, J = 8.2, 7.7 Hz, 1H); HRMS m/z (ES+) found [M + Na]+ 240.0994, C13H15NNaO2 requires M+ 240.0995; m/z (ES+) 218 ([M + H]+, 47), 240 ([M + Na]+, 100), 457 ([2M + Na]+, 99).
(RS)-1-(3-(3,5-Dimethylisoxazol-4-yl)phenyl)ethanol 3d
To a solution of 3b (100 mg, 465 μmol) in anhydrous MeOH (2 mL) at 0 °C was added NaBH4 (18 mg, 476 μmol). The mixture was warmed to rt and stirred for 2 h, then quenched with saturated aqueous NH4Cl (10 mL). The solution was extracted with CH2Cl2 (3 × 10 mL), and the combined organic layers were washed with water (30 mL) and brine (30 mL), then dried (MgSO4), filtered, and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 8 → 70% EtOAc/40–60 °C petroleum ether) gave 3d as a colorless solid (78 mg, 77%); mp 72–73 °C (40% EtOAc/40–60 °C petroleum ether); 1H NMR (400 MHz, CDCl3) 1.53 (d, J = 6.4 Hz, 3H), 2.26 (s, 3H), 2.30 (br s, 1H), 2.40 (s, 3H), 4.95 (q, J = 6.4 Hz, 1H), 7.13–7.17 (m, 1H), 7.26–7.29 (m, 1H), 7.34–7.44 (m, 2H); HRMS m/z (ES+) found [M + Na]+ 240.1002, C13H15NNaO3 requires M+ 240.0995; m/z (ES+) 218 ([M + H]+, 26), 240 ([M + Na]+, 81), 457 ([2M + Na]+, 100).
Ethyl 3-(3,5-dimethylisoxazol-4-yl)-5-ethoxybenzoate 4a
Method A
To a dry 10–20 mL microwave vial were added 8 (408 mg, 2.01 mmol), 10 (500 mg, 1.83 mmol), Pd(OAc)2 (4 mg, 16 μmol), RuPhos (26 mg, 56 μmol), and anhydrous Na2CO3 (388 mg, 3.66 mmol). The vial was sealed and purged with argon (3× evacuate/fill). Degassed EtOH (10 mL) was added by syringe, and the mixture was heated at 100 °C for 48 h. The mixture was cooled to rt and filtered through a thin pad of silica (eluent CH2Cl2) and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 6 → 50% Et2O/petroleum ether) gave 4a as a colorless solid (370 mg, 70%); mp 102–105 °C (CH2Cl2); 1H NMR (400 MHz, CDCl3) 1.40 (t, J = 7.1 Hz, 3H), 1.46 (t, J = 7.0 Hz, 3H), 2.28 (s, 3H), 2.42 (s, 3H), 4.11 (q, J = 7.0 Hz, 2H), 4.40 (q, J = 7.1 Hz, 2H), 6.96 (dd, J = 2.3, 1.7 Hz, 1H), 7.52 (dd, J = 1.7, 1.4 Hz, 1H), 7.55 (dd, J = 2.3, 1.4 Hz, 1H); HRMS m/z (ES+) found [M + Na]+ 312.1204, C16H19NNaO4 requires M+ 312.1206; m/z (ES+) 290 ([M + H]+, 29), 312 ([M + Na]+, 50), 602 ([2M + Na]+, 100).
Method B
To a dry 0.5–2 mL microwave vial were added PdCl2 (0.5 mg, 3 μmol), KOAc (36 mg, 367 μmol), and 10 (28 mg, 103 μmol). The vial was sealed and purged with argon (3× evacuate/fill). Degassed DMAc (1 mL) and 3,5-dimethylisoxazole (15 μL, 15 mg, 154 μmol) were added, and the reaction was heated at 130 °C for 44 h. The mixture was filtered through Celite (eluent EtOAc), and the filtrate was washed with water (2 × 30 mL) and brine (30 mL), dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (gradient elution, gradient 5 → 60% EtOAc/petroleum ether) gave 4a as a colorless solid (25 mg, 44%).
3-(3,5-Dimethylisoxazol-4-yl)-5-ethoxybenzoic acid 4b
To a solution of 4a (300 mg, 1.04 mmol) in THF (5 mL) were added water (2.5 mL) and LiOH (37 mg, 1.56 mmol), and the mixture was stirred for 16 h at rt. NaOH (1 M, 10 mL) was then added, and the mixture was washed with Et2O (3 × 10 mL). The solution was acidified to pH 2 with HCl (1 M) and extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were washed with brine (100 mL), dried (MgSO4), filtered, and concentrated in vacuo. Repeated recrystallization from cyclohexane/CHCl3 (2:1) gave 4b as a colorless crystalline solid (225 mg, 83%); mp 191–193 °C (cyclohexane/CHCl3, 2:1); 1H NMR (500 MHz, CDCl3) 1.48 (t, J = 7.0 Hz, 3H), 2.31 (s, 3H), 2.45 (s, 3H), 4.14 (q, J = 7.0 Hz, 2H), 7.03–7.05 (m, 1H), 7.60–7.63 (m, 2H); HRMS m/z (ES+) found [M + Na]+ 284.0886, C14H15NNaO4 requires M+ 284.0893; m/z (ES+) 262 ([M + H]+, 32), 284 ([M + Na]+, 100), 545 ([2M + Na]+, 75).
1-(3-(3,5-Dimethylisoxazol-4-yl)-5-ethoxyphenyl)ethanone 4c
To a dry 2–5 mL microwave vial were added 8 (88 mg, 433 μmol), 13 (100 mg, 411 μmol), Pd(OAc)2 (1 mg, 4 μmol), RuPhos (6 mg, 13 μmol), and anhydrous Na2CO3 (87 mg, 821 μmol). The vial was sealed and purged with argon (3× evacuate/fill). Degassed EtOH (2.3 mL) was added by syringe, and the mixture was heated at 85 °C for 64 h. The mixture was cooled to rt and filtered through a thin pad of silica (eluent CH2Cl2) and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 6 → 50% EtOAc/petroleum ether) gave 4c as a pale yellow solid (71 mg, 67%); mp 104–106 °C (CH2Cl2); 1H NMR (500 MHz, CDCl3) 1.47 (t, J = 7.0 Hz, 3H), 2.29 (s, 3H), 2.43 (s, 3H), 2.62 (s, 3H), 4.13 (q, J = 7.0 Hz, 2H), 6.98 (dd, J = 2.2, 1.5 Hz, 1H), 7.41 (dd, J = 1.5, 1.5 Hz, 1H), 7.46 (dd, J = 2.2, 1.5 Hz, 1H); HRMS m/z (ES+) found [M + Na]+ 282.1098, C15H17NNaO3 requires M+ 282.1101; m/z (ES+) 260 ([M + H]+, 35), 282 ([M + Na]+, 62), 541 ([2M + Na]+, 100).
(RS)-1-(3-(3,5-Dimethylisoxazol-4-yl)-5-ethoxyphenyl)ethanol 4d
To a solution of 4c (30 mg, 123 μmol) in anhydrous MeOH (1 mL) at 0 °C was added NaBH4 (6 mg, 159 μmol). The mixture was warmed to rt and stirred for 2 h, then quenched with saturated aqueous NH4Cl (10 mL). The solution was extracted with CH2Cl2 (3 × 10 mL), and the combined organic layers were washed with water (30 mL) and brine (30 mL), then dried (MgSO4), filtered, and concentrated in vacuo. Purification by silica gel column chromatography (gradient elution, gradient 10 → 100% EtOAc/40–60 °C petroleum ether) gave 4d as a pale yellow viscous oil (26 mg, 81%); 1H NMR (400 MHz, CDCl3) 1.44 (t, J = 7.0 Hz, 3H), 1.52 (d, J = 6.4 Hz, 3H), 2.14 (br s, 1H), 2.27 (s, 3H), 2.41 (s, 3H), 4.07 (q, J = 7.0 Hz, 2H), 4.91 (q, J = 6.4 Hz, 1H), 6.66–6.69 (m, 1H), 6.81–6.84 (m, 1H), 6.92–6.95 (m, 1H); HRMS m/z (ES+) found [M + Na]+ 284.1246, C15H15NNaO3 requires M+ 284.1257; m/z (ES+) 262 ([M + H]+, 21), 284 ([M + Na]+, 47), 545 ([2M + Na]+, 100).
Computational Procedures
Docking Procedures for Structure-Aided Design
Docking of compounds was carried out using the standard protocol(28) implemented in ICM-Pro 3.6-1 (Molsoft LLC). The costructure of 2 bound to BRD4(1) (PDB ID: 3SVF) was used to define the receptor; only water molecules interacting with 2 were included.
Conformational Analysis of Lead Compounds
Conformational analysis was performed for the two cocrystallized compounds (4b and 4d) using MacroModel (version 9.8) in Schrodinger Suite 2010 from Schrodinger LLC. The dihedral angle between the two ring systems was subjected to coordination scan with 1 degree of increment. The chemical structures from Ligprep were used as the starting structure. The default option was used, including a force field of OPLS2005, a solvent of water, and a PRCG minimization protocol with a convergence gradient of 0.05. The relative energy in kcal/mol of each minimized structure was then plotted versus the dihedral angle.
Biological Evaluation of Compounds
Protein Expression and Purification
Proteins were cloned, expressed, and purified as previously described.(18)
Peptides
H4Ac4 peptide (BRD2 and BRD4 assays, H2N-SGRGK(Ac)GGK(Ac)GLGK(Ac)-GGAK(Ac)RHRK(Biotin)-COOH) and H3K56Ac peptide (CREBBP assay, H2N-ALREIRRYQK(Ac)STELLIRKLK(Biotin)-COOH) were synthesized by Tufts University Core Facility.
Thermal Stability Shift Assay
Thermal melting experiments were carried out using the Mx3005p real-time PCR machine (Agilent) and employing a protein concentration of 2 μM and an inhibitor concentration of 100 mM. Buffer conditions were 10 mM HEPES, pH 7.5, 500 mM NaCl and 1:1000 dilution of SyproOrange (Invitrogen, CA). The assay and data evaluation were carried out as described previously.(29) Experiments were performed in duplicate; average values are reported.
AlphaScreen Peptide Displacement Assay
Bromodomain AlphaScreen assays were carried out as previously described.18,21 All experiments were carried out in duplicate on the same plate. Interexperimental IC50 values generated for individual compounds typically varied over an ∼3-fold range. However, rank order remained highly consistent between repeat experiments, with Spearman rank order correlations for 3a–d and 4a–d >0.90 for repeats of each assay. Thus, for the purposes of SAR, data in Table 1 is given from a single assay, with intraexperimental variability expressed as 95% confidence intervals resulting from sigmoidal curve fitting to duplicate data.
Cytotoxicity Assay
HeLa cells were subcultured in DMEM supplemented with l-glutamine and 10% fetal calf serum. 20000 cells per well were seeded into a 96 well plate. After 24 h of incubation at 37 °C, the medium was changed, and compounds were applied at final concentrations of 10, 1, 0.1, and 0.01 μM. The cells were incubated for 24 or 48 h, after which time the medium was changed, and MTT solution (5 mg/mL) was added to the plate. After 2 h of incubation at 37 °C, the medium was replaced with 200 μL of DMSO to solubilize the formazan crystals. After a further 15 min, absorbance was measured at 550 nm using a spectrophotometer. Experiments were performed in triplicate on the same plate.
Acknowledgments
We thank Cancer Research U.K. for a studentship (to D.S.H.). S.J.C. thanks St. Hugh’s College, Oxford, for research support. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Foundation for Strategic Research, and the Wellcome Trust. We are grateful to Timothy Rooney for the preparation of 6-bromo-3-methyl-3,4-dihydroquinazolin-2(1H)-one and to Dr. Rod Chalk and Dr. James Wickens for MS studies. We thank Dr. Paul Brennan for useful discussions.
Glossary
Abbreviation Used
- BET
bromodomain and extra terminal domain
- BRD
bromodomain-containing protein
- BRD2(1)/(2)
first/second bromodomain of BRD2
- BRD4(1)/(2)
first/second bromodomain of BRD4
- BRDT
bromodomain, testis-specific
- CREB
cAMP response element-binding protein
- CREBBP
CREB binding protein
- DMAc
N,N-dimethylacetamide
- EBV
Epstein–Barr virus
- GCN5
general control of amino acid synthesis, yeast, homologue-like 2
- HAT
histone acetyltransferase
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HDAC
histone deacetylase
- KAc
acetyl-lysine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylltetrazolium bromide
- NUT
nuclear protein in testis
- P-TEFb
positive transcription elongation factor b
- PB1(5)
fifth bromodomain of PB1 (BAF180), a unique subunit of the polybromo (PBAF) complex
- PCAF
P300/CREBBP-associated factor
- RuPhos
2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl
- TAFII250
TATA box binding protein-associated factor, 250 kDa
Supporting Information Available
Supporting figures, schemes, and tables, crystallographic information, further general experimental details, further characterization for compounds 3a–d, 4a–d, and synthesis and characterization of compounds 1, 7, 8, and 10–13. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Astex Therapeutics, 436 Cambridge Science Park, Cambridge, CB4 0QA, U.K.
Accession Codes
† Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession codes 3SVF (2), 3SVG (4d), and 3SVH (4b).
Supplementary Material
References
- Turner B. M. Histone acetylation and an epigenetic code. BioEssays 2000, 22, 836–845. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [DOI] [PubMed] [Google Scholar]
- a Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M. M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [DOI] [PubMed] [Google Scholar]; b Ornaghi P.; Ballario P.; Lena A. M.; Gonzalez A.; Filetici P. The bromodomain of Gcn5p interacts in vitro with specific residues in the N terminus of histone H4. J. Mol. Biol. 1999, 287, 1–7. [DOI] [PubMed] [Google Scholar]; c Hudson B. P.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Solution structure and acetyl-lysine binding activity of the GCN5 bromodomain. J. Mol. Biol. 2000, 304, 355–370. [DOI] [PubMed] [Google Scholar]; d Jacobson R. H.; Ladurner A. G.; King D. S.; Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [DOI] [PubMed] [Google Scholar]; e Owen D. J.; Ornaghi P.; Yang J. C.; Lowe N.; Evans P. R.; Ballario P.; Neuhaus D.; Filetici P.; Travers A. A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Strahl B. D.; Allis C. D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]; b Winston F.; Allis C. D. The bromodomain: a chromatin-targeting module?. Nat. Struct. Biol. 1999, 6, 601–604. [DOI] [PubMed] [Google Scholar]; c Jenuwein T.; Allis C. D. Translating the histone code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]; d Agalioti T.; Chen G.; Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002, 111, 381–392. [DOI] [PubMed] [Google Scholar]; e Loyola A.; Almouzni G. Bromodomains in living cells participate in deciphering the histone code. Trends Cell Biol. 2004, 14, 279–281. [DOI] [PubMed] [Google Scholar]
- Florence B.; Faller D. V. You bet-cha: a novel family of transcriptional regulators. Front. Biosci. 2001, 6, D1008–1018. [DOI] [PubMed] [Google Scholar]
- Yang Z.; He N.; Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28, 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford N. P.; Alsarraj J.; Lukes L.; Walker R. C.; Officewala J. S.; Yang H. H.; Lee M. P.; Ozato K.; Hunter K. W. Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 6380–6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a French C. A.; Kutok J. L.; Faquin W. C.; Toretsky J. A.; Antonescu C. R.; Griffin C. A.; Nose V.; Vargas S. O.; Moschovi M.; Tzortzatou-Stathopoulou F.; Miyoshi I.; Perez-Atayde A. R.; Aster J. C.; Fletcher J. A. Midline carcinoma of children and young adults with NUT rearrangement. J. Clin. Oncol. 2004, 22, 4135–4139. [DOI] [PubMed] [Google Scholar]; b Haruki N.; Kawaguchi K. S.; Eichenberger S.; Massion P. P.; Gonzalez A.; Gazdar A. F.; Minna J. D.; Carbone D. P.; Dang T. P. Cloned fusion product from a rare t(15;19)(q13.2;p13.1) inhibit S phase in vitro. J. Med. Genet. 2005, 42, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Huang K.; Jung K. J.; Cho W. K.; Klase Z.; Kashanchi F.; Pise-Masison C. A.; Brady J. N. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 2009, 83, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.; Wang S.; Nguyen T.; Shire K.; Frappier L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 2008, 82, 12009–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon D.; Joubert S.; Senechal H.; Fradet-Turcotte A.; Torre S.; Archambault J. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Yang X. D.; Zhou M. M.; Ozato K.; Chen L. F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang E.; Nickerson H. D.; Wen D.; Wang X.; Wolgemuth D. J. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507–3515. [DOI] [PubMed] [Google Scholar]
- Sachchidanand; Resnick-Silverman L.; Yan S.; Mutjaba S.; Liu W. J.; Zeng L.; Manfredi J. J.; Zhou M. M. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006, 13, 81–90. [DOI] [PubMed] [Google Scholar]
- Borah J. C.; Mujtaba S.; Karakikes I.; Zeng L.; Muller M.; Patel J.; Moshkina N.; Morohashi K.; Zhang W.; Gerona-Navarro G.; Hajjar R. J.; Zhou M. M. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 2011, 4, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Umehara T.; Sasaki K.; Nakamura Y.; Nishino N.; Terada T.; Shirouzu M.; Padmanabhan B.; Yokoyama S.; Ito A.; Yoshida M. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem. Biol. 2011, 18, 495–507. [DOI] [PubMed] [Google Scholar]
- Miyoshi S.; Ooike S.; Iwata K.; Hikawa H.; Sugahara K. Antitumor Agent. Int. Pat. Appl. WO 2009/084693, 2009.
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chung C.-w.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M. G.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clément C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [DOI] [PubMed] [Google Scholar]
- Bowers E. M.; Yan G.; Mukherjee C.; Orry A.; Wang L.; Holbert M. A.; Crump N. T.; Hazzalin C. A.; Liszczak G.; Yuan H.; Larocca C.; Saldanha S. A.; Abagyan R.; Sun Y.; Meyers D. J.; Marmorstein R.; Mahadevan L. C.; Alani R. M.; Cole P. A. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. D.. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. Biosyst. 2011, DOI: 10.1039/C1MB05099K. [DOI] [PubMed] [Google Scholar]
- a Burgey C. S.; Stump C. A.; Nguyen D. N.; Deng J. Z.; Quigley A. G.; Norton B. R.; Bell I. M.; Mosser S. D.; Salvatore C. A.; Rutledge R. Z.; Kane S. A.; Koblan K. S.; Vacca J. P.; Graham S. L.; Williams T. M. Benzodiazepine calcitonin gene-related peptide (CGRP) receptor antagonists: optimization of the 4-substituted piperidine. Bioorg. Med. Chem. Lett. 2006, 16, 5052–5056. [DOI] [PubMed] [Google Scholar]; b Shaw A. W.; Paone D. V.; Nguyen D. N.; Stump C. A.; Burgey C. S.; Mosser S. D.; Salvatore C. A.; Rutledge R. Z.; Kane S. A.; Koblan K. S.; Graham S. L.; Vacca J. P.; Williams T. M. Caprolactams as potent CGRP receptor antagonists for the treatment of migraine. Bioorg. Med. Chem. Lett. 2007, 17, 4795–4798. [DOI] [PubMed] [Google Scholar]
- Fall Y.; Reynaud C.; Doucet H.; Santelli M. Ligand-free-palladium-catalyzed direct 4-arylation of isoxazoles using aryl bromides. Eur. J. Org. Chem. 2009, 2009, 4041–4050. [Google Scholar]
- Molander G. A.; Canturk B.; Kennedy L. E. Scope of the Suzuki-Miyaura cross-coupling reactions of potassium heteroaryltrifluoroborates. J. Org. Chem. 2008, 74, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarju J.; Danks T. N.; Wagner G. Rapid microwave-assisted synthesis of phenyl ethers under mildly basic and nonaqueous conditions. Tetrahedron Lett. 2004, 45, 7675–7677. [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protocol. 2007, 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- Li X.; Du Y.; Liang Z.; Li X.; Pan Y.; Zhao K. Simple conversion of enamines to 2H-azirines and their rearrangements under thermal conditions. Org. Lett. 2009, 11, 2643–2646. [DOI] [PubMed] [Google Scholar]
- Abagyan R.; Totrov M.; Kuznetsov D. ICM: A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar]
- Bullock A. N.; Debreczeni J. É.; Fedorov O. Y.; Nelson A.; Marsden B. D.; Knapp S. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in Moloney murine leukemia virus (PIM-1) kinase. J. Med. Chem. 2005, 48, 7604–7614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.