

Abstract
The large size of many novel therapeutics impairs their transport through the tumor extracellular matrix and thus limits their therapeutic effectiveness. We propose that extracellular matrix composition, structure, and distribution determine the transport properties in tumors. Furthermore, because the characteristics of the extracellular matrix largely depend on the tumor–host interactions, we postulate that diffusion of macromolecules will vary with tumor type as well as anatomical location. Diffusion coefficients of macromolecules and liposomes in tumors growing in cranial windows (CWs) and dorsal chambers (DCs) were measured by fluorescence recovery after photobleaching. For the same tumor types, diffusion of large molecules was significantly faster in CW than in DC tumors. The greater diffusional hindrance in DC tumors was correlated with higher levels of collagen type I and its organization into fibrils. For molecules with diameters comparable to the interfibrillar space the diffusion was 5- to 10-fold slower in DC than in CW tumors. The slower diffusion in DC tumors was associated with a higher density of host stromal cells that synthesize and organize collagen type I. Our results point to the necessity of developing site-specific drug carriers to improve the delivery of molecular medicine to solid tumors.
Blood-borne therapeutics must extravasate and penetrate the interstitial matrix to reach cancer cells in a tumor (1). We recently have shown that tumor–host interactions regulate transvascular transport in tumors (2), but how they affect tumor interstitial transport is not known. Because of uniformly elevated interstitial fluid pressure in solid tumors, convection in the tumor interstitium is negligible (3), and drug delivery through the extracellular matrix (ECM) relies on passive diffusive transport (4). Unfortunately, passive delivery becomes increasingly inefficient for larger particles. The success of novel cancer therapies that rely on large agents such as proteins, liposomes, nanoparticles, or gene vectors will hinge on their ability to penetrate the tumor interstitium (1, 5–7). It is thus vital to identify the ECM constituents and characteristics that restrict diffusion and to determine how these are affected by tumor type and site.
Different ECM components, including collagen, glycosaminoglycans, and proteoglycans such as decorin, form a complex structured gel (8). Resistance to interstitial flow has been strongly linked to glycosaminoglycans and especially hyaluronan (HA) (8–10). However, a recent in vivo study from our lab found an inverse correlation between collagen content of tumors and diffusion of IgG (11). Furthermore, in vitro experiments found that diffusion of albumin is weakly hindered in HA gels (10) but significantly hindered in collagen gels (12). Thus, we expect that tumor interstitial transport properties will depend on the volume, interaction, structure, and distribution of the matrix molecules and not simply on their overall levels (13). Furthermore, because the bulk of the matrix in many tumors is produced by stromal cells (14, 15), we hypothesize that the diffusion of macromolecules will depend on tumor–host interactions.
Here we present analysis of the combined effect of the ECM composition, structure, and distribution and the role of tumor–host interaction on diffusion in the tumor interstitium. Using the fluorescence recovery after photobleaching (FRAP) technique (11, 16, 17), we measured the diffusion coefficients of proteins, dextrans, and liposomes in two different human tumor xenografts implanted either in the dorsal chamber (DC) or cranial window (CW) in mice. Diffusion coefficients were related to the distribution and relative levels of collagen type I, decorin, and HA as determined from stained tissue sections. Collagen organization was characterized by transmission electron microscopy. We also estimated the effect of cellular geometry (tortuosity) on transport. The results provide critical data on the delivery of molecular medicine in solid tumors.
Materials and Methods
Fluorescent Tracers.
FITC-conjugated particles/molecules of various sizes were studied. In order of increasing size, these included lactalbumin and BSA (Molecular Probes), nonspecific IgG (Jackson ImmunoResearch), nonspecific IgM (Sigma), FITC-dextran 2,000,000 MW (Sigma), and liposomes. IgM was purchased unlabeled and then conjugated to FITC by using the Fluo EX-protein labeling kit (Molecular Probes). All other molecules were purchased in FITC-labeled form. Liposomes (150 nm in diameter—determined from the diffusion coefficients in solution by using Eq. 1) were prepared from dipamitoylphosphatidylcholine with 1 mol% of the fluorescent phospholipid carboxyfluorescein-dioleoyl phosphatidylethanolamine (18).
Animals and Tumors.
Human glioblastoma (U87) and melanoma (Mu89) were implanted in two different sites in severe combined immunodeficient mice as described: (i) on the s.c. tissue of the skin (DC) (19), and (ii) on the pial surface (CW) (2). The pial surface approximates an orthotopic site for U87 tumors whereas skin is orthotopic for Mu89. Tumors can be visualized directly in these preparations. Animals were used for experiments 3–4 weeks after tumor implantation.
Diffusion Measurements by FRAP.
Injection of tracer.
Small molecules (lactalbumin, BSA, and IgG) were injected i.v. via the tail vein. To ensure sufficient fluorescence and homogeneous distribution, molecules larger than IgG were introduced by direct intratumoral injection: 1 μl of fluorescent solution was infused through thin micropipettes (25–30 μm inner diameter) at constant pressure using a syringe pump (Harvard Apparatus) for 15–20 min. Diffusion was measured by FRAP 30 min after the end of micropipette injection. In preliminary studies, no statistical difference in the diffusion of IgG was found between i.v. or micropipette injections in the human sarcoma HSTS26T (high collagen content tumors; ref. 11) implanted in DC [D = (8.85 ± 0.8)⋅10−8 vs. (9.3 ± 0.7)⋅10−8 cm2⋅s−1 for micropipette and i.v. injection, respectively].
FRAP measurements.
The FRAP technique and method of analysis are described fully elsewhere (20). In brief, redistribution of fluorescent molecules in bleached tissue yields the effective diffusion coefficient, independently of convection (17). Unlike multiphoton FRAP (21), FRAP measurements are restricted to less than 100 μm from the tumor surface due to light scatter.
Hydrodynamic radius determination.
The hydrodynamic radius of the fluorescent molecules, RH, was determined from the diffusion coefficient, D0, in PBS solution at T = 26°C (299 K) using the Stokes–Einstein equation:
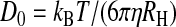 |
1 |
in which kB is Boltzmann constant, T is the temperature in K, and η is the viscosity of water (0.8705 cP at T = 299 K). Diffusion coefficients in solution then were scaled to T = 37°C by correcting for the effect of temperature on the viscosity.
Extracellular Space Organization.
Extracellular space organization was characterized in tissue sections embedded in the hydrophilic resin LR White (Ted Pella, Redding, CA). Tumors were fixed in 2.5% glutaraldehyde and 2.0% paraformaldehyde in PBS and embedded in the LR White resin (22). Toluidine blue-stained sections were photographed by using a color charge-coupled device camera mounted on a Nikon microscope.
Immunohistochemistry.
Rabbit antiserum against type I collagen (LF-67) (23) and against human (LF-136) (24, 25) and mouse (LF-113) (26) decorin were generously provided by Larry Fisher (National Institute of Dental Research, Bethesda, MD). LF-67, LF-136, and LF-113 were used at dilutions of 1:50, 1:500, and 1:1,000, respectively. Mouse anti-human collagen type IV (Dako) and rabbit anti-mouse collagen type IV (Chemicon) were used at dilutions 1:100 and 1:30, respectively. HA was detected with a HA biotinylated proteoglycan fragment (8 μg/ml), a generous gift of Charles Underhill (Georgetown University, Washington, DC).
Tumors were perfusion-fixed through the heart with 4% paraformaldehyde in PBS. The tissue was infiltrated with sucrose and embedded in OCT. For immunostaining, sections were blocked with rabbit or goat serum, incubated with the antibody overnight at 4°C and then with the appropriate secondary antibodies conjugated to Cy-5 (Jackson ImmunoResearch Laboratories). For HA staining, the sections were stained for 1 h with the biotinylated proteoglycan fragment diluted in 10% calf serum and incubated with Texas red-conjugated streptavidin (Jackson ImmunoResearch Laboratories). The cell nuclei were stained with the Alexis nuclear stain (Molecular Probes). Sections were photographed with a Leica TCS-NT4D confocal microscope. For quantification of the fraction of tissue occupied by collagen type I staining, photographs were taken with a custom-made two photon microscope based on a MRC 600 platform (Bio-Rad). Using a constant 10 mW of 720-nm light through a 0.9 numerical aperture water immersion lens, we generated image stacks of the histological slices, with 10–20 images per stack. A maximum intensity projection was performed on the image stacks to form a single image of the section, thereby ensuring that each pixel value represents the best colocalization of the excitation volume with the slice. Using a series of threshold pixel values, we determined what fraction of the pixels in a 50 × 100 μm window (oriented perpendicular to the tumor surface) were stained for collagen. The average pixel value of the collagen pixels was calculated as an indicator of collagen type I staining.
Electron Microscopy.
Organization of collagen bundles and interfibrillar spacing were characterized by electron microscopy. Tumors were fixed by immersion in 2.5% glutaraldehyde and 2.0% paraformaldehyde in PBS for 4–6 h. Small tumor pieces were washed overnight in PBS, dehydrated in ethanol, fixed in 1% osmium, and embedded in Polybed 812. Thin sections were stained with uranyl acetate and lead citrate and examined with a Phillips CM10 transmission electron microscope (Phillips Electronic Instruments, Mahwah, NJ) operating at 80 kV.
Results
Interstitial Diffusion Decreases with Increasing Molecular Size.
Fig. 1a presents diffusion coefficients obtained in Mu89 and U87 in both implantation sites. In the two tumors, the diffusion of larger molecules is significantly slower than that of smaller molecules. The decrease in diffusion with particle size in tumors is even greater than one would predict from pure solution data, due to the presence of cellular obstacles and matrix molecules. To examine these contributions, we introduce the concept of tortuosity.
Figure 1.

(a) Effective diffusion coefficients,
Deff, as a function of their experimental
hydrodynamic radius, RH. Diffusion
coefficients in PBS solution were measured at T =
26°C and scaled to 37°C according to the Stokes–Einstein equation.
Diffusion coefficients were measured in DC (filled symbols and dotted
lines) and CW (open symbols and continuous line) tumors.
(b) Interstitial diffusion coefficients in tumors
(Dint =
τ Deff) as a function of
hydrodynamic radius, RH, using the
experimentally obtained value τg =
1.19. The diffusion coefficients in solution
(D0) are pictured (▴) to
illustrate the ECM influence on retardation.
Deff) as a function of
hydrodynamic radius, RH, using the
experimentally obtained value τg =
1.19. The diffusion coefficients in solution
(D0) are pictured (▴) to
illustrate the ECM influence on retardation.
The increase in path length induced by physical obstacles and extracellular space connectivity is described by the tortuosity. The effective diffusion measured in tissues (27) is related to the tortuosity by Deff = (1/τ2)D0 (28). Geometric effects imposed by the organization of cells are likely to be the major hindrance to long-range diffusion of small molecules. Frictional effects assume greater importance as the size of diffusing particles increases to become comparable to the dimensions of channels through which they move. On this basis, we separate tortuosity into viscous (τv) and geometric (τg) contributions according to τ = τgτv (29) so that:
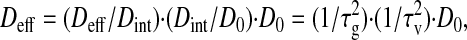 |
2 |
where Dint is the interstitial
diffusion coefficient and D0 the
diffusion coefficient in solution. The ratio
Deff/Dint
= 1/τ measures hindrance due to cellular
obstacles. The ratio
Dint/D0
= 1/τ
measures hindrance due to cellular
obstacles. The ratio
Dint/D0
= 1/τ measures hindrance within the ECM. The
geometric tortuosity may be estimated by using a sufficiently small
molecule for which viscous hindrance is negligible
(τv = 1) so that
Deff =
(1/τ
measures hindrance within the ECM. The
geometric tortuosity may be estimated by using a sufficiently small
molecule for which viscous hindrance is negligible
(τv = 1) so that
Deff =
(1/τ )D0. From
diffusion measurements of fluorescein
(RH = 0.4 nm) in U87 DC, the geometric
tortuosity was estimated at τg =
1.19. Fig. 1b presents the interstitial diffusion
coefficients in tumors (Dint =
τ
)D0. From
diffusion measurements of fluorescein
(RH = 0.4 nm) in U87 DC, the geometric
tortuosity was estimated at τg =
1.19. Fig. 1b presents the interstitial diffusion
coefficients in tumors (Dint =
τ Deff) as a
function of the hydrodynamic radius, illustrating that the reduction of
diffusion coefficient with particle size is greater in the ECM than in
solution.
Deff) as a
function of the hydrodynamic radius, illustrating that the reduction of
diffusion coefficient with particle size is greater in the ECM than in
solution.
Diffusion of Larger Particles Is Faster in CW Tumors Than in DC Tumors.
No statistical difference was observed in the diffusion coefficients of small molecules such as lactalbumin and BSA between the two tumor types and sites of implantation. However, the diffusion coefficients of larger molecules (particles equal in size and larger than IgG: RH ≥ 5.5 nm) were significantly decreased (P < 0.05) in DC as compared with CW tumors (Fig. 1). Fig. 1a illustrates the distinction between a “fast diffusion group” (CW tumors) and a “slow diffusion group” (DC tumors). The difference in diffusion increases with particle size and is striking for molecules such as dextran 2,000,000 MW. Fig. 1a also shows diffusion coefficients for liposomes (RH = 75 nm) in CW tumors. Diffusion coefficients of liposomes in DC tumors could not be assessed by FRAP due to prohibitively slow diffusion and inhomogeneous distribution of the particles.
The Capsule of DC Tumors Has a High Density of Fibroblast-Like Cells.
Significant differences in cellular content and ECM organization were found between DC and CW tumors. Typically, DC tumors were separated from the glass coverslip by a fibrous capsule composed of several layers of fibroblast-like cells, separated by ECM (Fig. 2 a and b). The ECM of the capsule was continuous with that of the underlying tumor cells. In Mu89, cellular nodules were surrounded by a thin layer of ECM and stromal cells, whereas in U87 single tumor cells or groups of tumor cells were separated by larger ECM spaces (Fig. 2 a and b). In contrast to DC tumors, at the outer edge of CW tumors, only one layer of fibroblast-like cells was observed in contact with the underlying tumor cells, which were separated from each other by narrow ECM spaces (Fig. 2 c and d).
Figure 2.
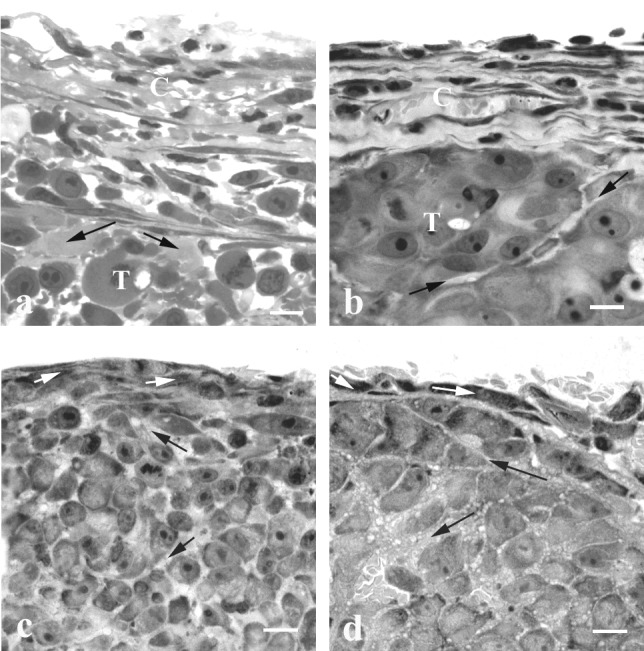
Light microscopy (LR White sections) of the peripheral region of DC and CW tumors. The capsule of U87 (a) and Mu89 (b) DC tumors is composed of several layers of fibroblast-like cells separated by ECM. Note the large intercellular spaces in U87 and the narrow space that separates two cellular nodules in Mu89. The connective tissue at the edge of U87 (c) and Mu89 (d) in the CW is composed of one fibroblast cell layer; the tumor cells are separated by narrow intercellular spaces. C, capsule; T, tumor; black arrows, ECM; white arrows, fibroblast-like cells. (Bar = 10 μm.)
DC Tumors Have High Levels of Collagen Type I and Fibrillar Collagen.
To compare the influence of tumor implantation site on the ECM, the distribution and staining intensity of collagen types I and IV, decorin, and HA were characterized. Collagen type I staining was abundant in DC tumors, approaching levels found in normal skin. In these tumors, collagen type I fibers were identified between the layers of fibroblast-like cells (Fig. 3a). In central regions of Mu89, tumor cell clusters were surrounded by thin layers of type I collagen, whereas in U87, tumor cell clusters or single cells were separated by wider spaces occupied by type I collagen. In comparison to DC tumors the staining occupied a smaller area in CW tumors (Fig. 3 a and b). Quantitative image analysis within the superficial 100 μm of Mu89 tumors revealed 36 ± 11% tissue area stained for collagen type I in the DC, as opposed to 12 ± 5% in the CW. The collagen type I staining also occupied a greater proportion of the ECM in DC than in CW tumors (Fig. 3 a and b). In CW tumors collagen type I was predominantly localized at the tumor edge with scattered staining between tumor cells (Fig. 3b). As expected, staining for collagen type IV was associated with tumor vessels in both sites (data not shown).
Figure 3.

Immunostaining for collagen type I (a and b) and decorin (c and d), and labeling for HA (e and f) in DC (a, c, and e) and CW (b, d, and f) tumors. Collagen type I occupies a greater area of the periphery in DC than in CW tumors. In both DC and CW tumors the decorin staining is restricted to the periphery of the tumor. HA staining is intense in the center of Mu89 in the DC, whereas in the periphery the staining is weak. (Bar = 100 μm.)
The collagen organization was characterized by electron microscopy. Fibrillar collagen was abundant in the capsule of DC tumors. Bundles of aligned and compact fibrils (interfibrillar spacing 20–42 nm) were found adjacent to bundles that were poorly organized with larger interfibrillar spaces (75–130 nm) (Fig. 4). In the center of U87 especially, fibrillar collagen was less abundant and poorly organized. This finding, coupled with the extensive collagen type I staining in the center of U87, suggests that the deposited collagen is poorly assembled. In CW tumors, collagen fibrils had no specific organization and appeared as isolated fibrils.
Figure 4.
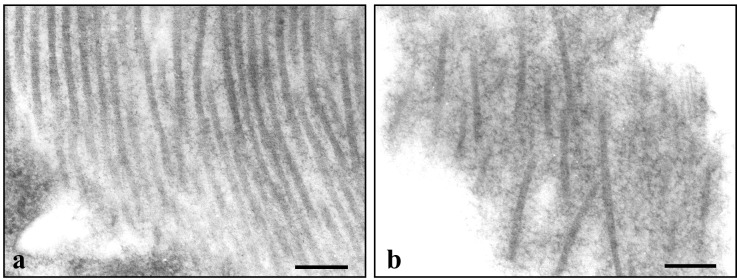
Electron microscopy of the organization of collagen fibrils in the capsule of U87 tumors in the DC. (a) The longitudinally oriented fibrils are parallel to one another with an interfibrillar spacing that varies from 20 to 42 nm. (b) The fibrils are poorly organized. The interfibrillar spacing varies between 75 and 130 nm. (Bar = 200 nm.)
Decorin Is Restricted to the Tumor Periphery.
Because decorin participates in the organization of fibrillar collagen, we characterized its distribution. Decorin was present between fibroblast-like cells in the capsule of DC tumors. However, in contrast to type I collagen, decorin immunostaining was not detected in the extracellular space separating tumor cells (Fig. 3c). In CW tumors, decorin staining was almost exclusively restricted to the tumor edge (Fig. 3d).
HA Staining Is Diffuse in CW Tumors but Associated with Tumor Cells in DC Tumors.
In comparison to the tumor center, HA staining was absent or significantly reduced in the capsule of DC tumors (Fig. 3e). In the center of Mu89 especially, tumor nodules were separated by intense HA staining. The staining intensity for HA was greater in skin than in DC tumors. In U87 and Mu89 in the CW, HA staining was distributed diffusely throughout the tumor. No obvious difference in the relative levels of HA was detected between tumor implantation sites.
The ECM Is of Host Origin.
The origin (tumor vs. host) of ECM components in the human tumor xenografts implanted in mice was determined by immunostaining. Staining of the ECM by antibodies against human decorin and collagen type IV was significantly weaker than for corresponding murine antibodies (data not shown), indicating that the ECM observed was primarily of host origin.
Discussion
Importance of Diffusion in Drug Design and Selection.
Our diffusion measurements provide necessary data for prediction of transport properties of therapeutic molecules over a wide range of molecular weights. Although no significant difference in diffusion coefficients was observed for small proteins (lactalbumin, albumin) between implantation sites, diffusion of larger molecules (IgM and dextran 2,000,000 MW) was 5- to 10-fold faster in CW tumors than in DC tumors.
Depending on tumor site, tumors fell into slow-diffusing (DC) and fast-diffusing (CW) groups, characterized by high and low collagen type I levels, respectively. The hindrance to diffusion of dextran 2,000,000 MW (RH = 19 nm) in DC tumors was comparable to that of liposomes (RH = 75 nm) in CW tumors (Fig. 1a). In DC tumors, diffusion of the same liposomes was prohibitively slow for measurement. A rough estimate based on extrapolation of the measured diffusion coefficients suggests that the diffusion of liposomes in DC tumors would be 1–2 orders of magnitude slower than in CW tumors. Thus, passive delivery of liposomes might be more feasible in low-collagen brain tumors than in high-collagen tumors. Our results emphasize that the delivery of larger particles will be highly influenced by the tumor site and possibly by other factors that influence ECM composition/structure.
Contributions of Geometric (Cellular) and Viscous (Matrix) Tortuosity to Diffusional Hindrance.
We estimated the geometric tortuosity in U87 DC tumors as τg = 1.19 ± 0.10. Our results compare well with previous Monte Carlo simulations, which predicted a tortuosity of 1.4 for three-dimensional radial diffusion through an array of evenly spaced cells (29, 30). The geometric tortuosity could vary with cellular arrangement and extracellular space connectivity. Complex cellular arrangements may differentially affect the transport of large vs. small particles, restricting large particles to wider intercellular paths. The matrix, its distribution, and organization further compound the hindrance via the viscous tortuosity, which, as shown in Fig. 1b, increases significantly for larger molecules and at higher collagen type I levels. Although the true tortuosity of long-range motion may indeed increase with particle size, the most likely explanation for the increased hindrance for larger particles is the increased viscous drag from solid obstacles (cells, matrix fibers) as the size of the diffusing particles becomes significant compared with intercellular or interfibrillar spacing (29, 31).
The Role of ECM Composition and Organization in Determining Transport.
Role of collagen.
Expanding on the results of Netti et al. (11), we find that collagen type I and its organization into fibrils have a significant role in limiting the diffusion of large molecules (e.g., IgG, IgM, and dextran 2,000,000 MW). Fibrillar collagen occupied a greater portion of the ECM in DC than in CW tumors. The narrow spacing (20–40 nm) between collagen fibrils will exclude or hinder (frictional interaction, steric hindrance) the migration of larger particles. The tortuous paths around compact collagen bundles or within loose bundles (interfibrillar spacing = 75–130 nm) also will hinder the diffusion of large molecules. Interestingly, the 5- to 10-fold difference in diffusion between CW and DC tumors was found for molecules with diameters approaching the interfibrillar spacing.
Role of proteoglycans.
The alignment and spacing of collagen fibrils is modulated by proteoglycans. The protein core of decorin binds to fibrils, and the dermatan/chondroitin sulfate side chains form complexes that bridge the interfibrillar space at intervals of 60–65 nm (32). Decorin knockout mice exhibit wider interfibrillar spaces in the skin, and inhibition of decorin synthesis by β-d xyloside induces large separations in the fibrillar collagen network of the corneal stroma (33, 34). Thus, the wider interfibrillar spaces in the center of U87 in the DC could be due to the reduced expression of decorin. However, the presence of occasional compact collagen bundles in the center of U87 and the tightly organized fibrillar collagen in the center of Mu89 where decorin expression is reduced suggest that other proteoglycans, possibly lumican (35, 36), may participate in the organization of fibrillar collagen in these tumors. It remains to be established whether the interaction between proteoglycans and fibrillar collagen limits the diffusion of macromolecules in tumors.
Role of HA.
The low levels or absence of HA staining in the capsule of DC tumors suggest that HA was not a contributor to transport hindrance in these tumors. In the tumor center, the higher levels of HA could potentially influence interstitial transport. Several studies have clearly demonstrated that HA impedes fluid flow in tissues (37, 38), whereas the effect of HA on the diffusion of macromolecules in normal or tumor tissues is yet to be determined. The degradation of HA in normal tissues with hyaluronidase either does not modify or even decreases the diffusion of albumin (38, 39). Indeed, we have observed that hyaluronidase decreases diffusion of IgG in tumors (unpublished results). Based on these results, it is possible that the swelling potential of intact HA increases the pore size between ECM molecules and thus actually facilitates diffusion.
Role of tumor site and tumor–host interactions.
Differences in the levels of collagen type I and decorin between DC and CW tumors reflect the greater recruitment of stromal cells (e.g., fibroblasts) in DC tumors. The greater accumulation of collagen type I and decorin in DC tumors was associated with a higher density of stromal cells. In general, stromal cells, and not neoplastic cells, synthesize these molecules in carcinomas (14, 40). Immunostaining also showed that decorin and collagen type IV in Mu89 and U87 were produced by host (murine) cells. In contrast, previous studies have shown that HA is produced by neoplastic cells as well as by stromal cells (41, 42). In vitro, paracrine interactions and direct cell–cell contact between tumor cells and fibroblasts can increase the fibroblast synthesis of collagen type I, HA, and decorin (40, 42, 43).
Conclusion.
The present study provides critical data on the diffusive transport of particles in tumors for a wide range of particle sizes. Tumors studied fell into slow vs. fast diffusion groups, corresponding to high vs. low collagen type I content, respectively, supporting a central role for fibrillar collagen in determining interstitial hindrance. The results demonstrate that diffusion of large molecules (IgG, IgM, dextran 2,000,000 MW, and liposomes) is much faster in CW than in DC tumors. The greater hindrance to diffusion in DC tumors was associated with a higher density of host stromal cells, which synthesize and organize collagen type I. These results also point to the necessity of site-specific drug carriers to improve drug delivery. Finally, our results underscore that efficient gene therapy will require a better integration of drug design and in vivo experimentation.
Acknowledgments
We thank Sylvie Roberge and Julia Kahn for their outstanding technical assistance and Mary McKee and the Massachusetts General Hospital Program in Membrane Biology for their help with the electron microscopy. This work was supported by an Outstanding Investigator Grant (R35-CA56591) and a Program Project Grant (P01-CA-80124) from the National Cancer Institute (to R.K.J.). S.R. and E.B.B. are supported by National Institutes of Health Fellowships (F32-CA83248 to S.R., T32-CA73479 to E.B.B.). T.D.M. is supported by a grant from the National Institutes of Health (5 T32 GM08334) Interdepartmental Biotechnology Program, Biotechnology Process Engineering Center at the Massachusetts Institute of Technology. Y.I. is supported by a fellowship from The Japan Health Sciences Foundation.
Abbreviations
- CW
cranial window
- DC
dorsal chamber
- ECM
extracellular matrix
- FRAP
fluorescence recovery after photobleaching
- HA
hyaluronan
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jain R K. Nat Med. 1998;4:655–657. doi: 10.1038/nm0698-655. [DOI] [PubMed] [Google Scholar]
- 2.Hobbs S K, Monsky W L, Yuan F, Roberts W G, Griffith L, Torchilin V P, Jain R K. Proc Natl Acad Sci USA. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boucher Y, Baxter L T, Jain R K. Cancer Res. 1990;50:4478–4484. [PubMed] [Google Scholar]
- 4.Netti P A, Hamberg L M, Babich J W, Kierstad D, Graham W, Hunter G H, Wolf G L, Fischman A, Boucher Y, Jain R K. Proc Natl Acad Sci USA. 1999;96:3137–3142. doi: 10.1073/pnas.96.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan F, Dellian M, Fukumura D, Leunig M, Berk D A, Torchilin V P, Jain R K. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 6.Jacobs A, Breakefield X O, Fraefel C. Neoplasia. 1999;1:402–416. doi: 10.1038/sj.neo.7900056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weyerbrock A, Oldfield E H. Curr Opin Oncol. 1999;11:168–173. doi: 10.1097/00001622-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Gribbon P M, Maroudas A, Parker K H, Winlove C P. In: Connective Tissue Biology: Integration and Reductionism. Reed R K, Rubin K, editors. London: Portland; 1998. pp. 95–124. [Google Scholar]
- 9.Levick J R. Q J Exp Physiol. 1987;72:409–437. doi: 10.1113/expphysiol.1987.sp003085. [DOI] [PubMed] [Google Scholar]
- 10.Comper W D, Laurent T C. Physiol Rev. 1978;58:255–315. doi: 10.1152/physrev.1978.58.1.255. [DOI] [PubMed] [Google Scholar]
- 11.Netti P A, Berk D A, Swartz M A, Grodzinsky A J, Jain R K. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 12.Shenoy V, Rosenblatt J. Macromolecules. 1995;28:8751–8758. [Google Scholar]
- 13.Ogston A G, Preston B N, Wells J D. Proc R Soc London Ser A. 1973;333:297–316. [Google Scholar]
- 14.Brown L F, Guidi A J, Schnitt S J, Van De Water L, Iruela-Arispe M L, Yeo T K, Tognazzi K, Dvorak H F. Clin Cancer Res. 1999;5:1041–1056. [PubMed] [Google Scholar]
- 15.Toole B P, Biswas C, Gross J. Proc Natl Acad Sci USA. 1979;76:6299–6303. doi: 10.1073/pnas.76.12.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chary S R, Jain R K. Proc Natl Acad Sci USA. 1989;86:5385–5389. doi: 10.1073/pnas.86.14.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berk D A, Yuan F, Leunig M, Jain R K. Proc Natl Acad Sci USA. 1997;94:1785–1790. doi: 10.1073/pnas.94.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szoka F, Jr, Papahadjopoulos D. Annu Rev Biophys Bioeng. 1980;9:467–508. doi: 10.1146/annurev.bb.09.060180.002343. [DOI] [PubMed] [Google Scholar]
- 19.Leunig M, Yuan F, Menger D, Boucher Y, Goetz A F, Messmer K, Jain R K. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 20.Berk D A, Yuan F, Leunig M, Jain R K. Biophys J. 1993;65:2428–2436. doi: 10.1016/S0006-3495(93)81326-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown E B, Wu E S, Zipfel W, Webb W W. Biophys J. 1999;77:2837–2849. doi: 10.1016/S0006-3495(99)77115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman G R. J Histochem. 1987;19:118. doi: 10.1007/BF01682756. [DOI] [PubMed] [Google Scholar]
- 23.Bernstein E F, Fisher L W, Li K, LeBaron RG, Tan E M, Uitto J. Lab Invest. 1995;72:662–669. [PubMed] [Google Scholar]
- 24.Bianco P, Fisher L W, Young M F, Termine J D, Robey P G. J Histochem Cytochem. 1990;38:1549–1563. doi: 10.1177/38.11.2212616. [DOI] [PubMed] [Google Scholar]
- 25.Fisher L W, Termine J D, Young M F. J Biol Chem. 1989;264:4571–4576. [PubMed] [Google Scholar]
- 26.Fisher L W, Stubbs J T, 3rd, Young M F. Acta Orthop Scand Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
- 27.el-Kareh A W, Braunstein S L, Secomb T W. Biophys J. 1993;64:1638–1646. doi: 10.1016/S0006-3495(93)81532-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicholson C, Phillips J M. J Physiol (London) 1981;321:225–257. doi: 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rusakov D A, Kullmann D M. Proc Natl Acad Sci USA. 1998;95:8975–8980. doi: 10.1073/pnas.95.15.8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K C, Nicholson C. Proc Natl Acad Sci USA. 2000;97:8306–8311. doi: 10.1073/pnas.150338197. . (First Published July 11, 2000; 10.1073/pnas.150338197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips R J. Biophys J. 2000;79:3350–3353. doi: 10.1016/S0006-3495(00)76566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott J E, Dyne K M, Thomlinson A M, Ritchie M, Bateman J, Cetta G, Valli M. Exp Cell Res. 1998;243:59–66. doi: 10.1006/excr.1998.4089. [DOI] [PubMed] [Google Scholar]
- 33.Hahn R A, Birk D E. Development (Cambridge, UK) 1992;115:383–393. doi: 10.1242/dev.115.2.383. [DOI] [PubMed] [Google Scholar]
- 34.Danielson K G, Baribault H, Holmes D F, Graham H, Kadler K E, Iozzo R V. J Cell Biol. 1997;136:729–743. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chakravarti S, Magnuson T, Lass J H, Jepsen K J, LaMantia C, Carroll H. J Cell Biol. 1998;141:1277–1286. doi: 10.1083/jcb.141.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leygue E, Snell L, Dotzlaw H, Troup S, Hiller-Hitchcock T, Murphy L C, Roughley P J, Watson P H. J Pathol. 2000;192:313–320. doi: 10.1002/1096-9896(200011)192:3<313::AID-PATH694>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 37.Bert J L, Pearce R H. In: Handbook of Physiology: The Cardiovascular System. Berne R M, Sperelakis N, editors. Vol. 4. Bethesda, MD: Am. Physiol. Soc.; 1990. pp. 521–547. [Google Scholar]
- 38.Parameswaran S, Brown L V, Ibbott G S, Lai-Fook S J. Microvasc Res. 1999;58:114–127. doi: 10.1006/mvre.1999.2168. [DOI] [PubMed] [Google Scholar]
- 39.Qiu X L, Brown L V, Parameswaran S, Marek V W, Ibbott G S, Lai-Fook S J. Lung. 1999;177:273–288. doi: 10.1007/pl00007647. [DOI] [PubMed] [Google Scholar]
- 40.Iozzo R V, Cohen I. Exs. 1994;70:199–214. doi: 10.1007/978-3-0348-7545-5_12. [DOI] [PubMed] [Google Scholar]
- 41.Kimata K, Takeda M, Suzuki S, Pennypacker J P, Barrach H J, Brown K S. Cancer Res. 1983;43:1347–1354. [Google Scholar]
- 42.Knudson W. Am J Pathol. 1996;148:1721–1726. [PMC free article] [PubMed] [Google Scholar]
- 43.Noel A, Munaut C, Nusgens B, Foidart J M, Lapiere C M. Matrix. 1992;12:213–220. doi: 10.1016/s0934-8832(11)80064-8. [DOI] [PubMed] [Google Scholar]