

Abstract
The discovery, syntheses, and structure-activity relationships (SAR) of a new family of heterocyclic antibacterial compounds based on N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffolds are described. A structurally diverse library of ~100 heterocyclic molecules generated from Lewis acid-mediated nucleophilic ring opening reactions with nitroso Diels-Alder cycloadducts and nitroso ene reactions with substituted alkenes was evaluated in whole cell antibacterial assays. Compounds containing the N-alkyl-N-(pyridin-2-yl)hydroxylamine structure demonstrated selective and potent antibacterial activity against the Gram-positive bacterium Micrococcus luteus ATCC 10240 (MIC90 = 2.0 μM or 0.41 μg/mL) and moderate activity against other Gram-positive strains including antibiotic resistant strains of Staphylococcus aureus (MRSA) and Enterococcus faecalis (VRE). A new synthetic route to the active core was developed using palladium-catalyzed Buchwald-Hartwig amination reactions of N-alkyl-O-(4-methoxybenzyl)hydroxylamines with 2-halo-pyridines that facilitated SAR studies and revealed the simplest active structural fragment. This work shows the value of using a combination of diversity-oriented synthesis (DOS) and parallel synthesis for identifying new antibacterial scaffolds.
Introduction
The development of bacterial resistance to current antibacterial chemotherapies is one of modern medicine’s most important problems.1–3 The situation is confounded by a complex balance of medical, ethical, social, political, and economic factors all resulting in a decline of antibiotic development and rise in antibiotic resistance.4 With only a few last resort drugs available, such as vancomycin and linezolid, for treating highly resistant Gram-positive bacterial infections, there is a desperate need for new antibacterial agents based on new molecular scaffolds with novel modes of action.5 Historically, the antibiotic arms race has been maintained through the diligent work of medicinal chemists synthetically tailoring existing antibacterial scaffolds to keep one step ahead of developing microbial resistance. The evolution of β-lactam antibiotics into many useful generations is the best example of this method.5,6 Modification of existing scaffolds is still the primary strategy of antibiotic resistance management employed today,7 but time is running out, the antibiotic pipeline is depleted, and a more long-term approach to managing antibiotic resistance is essential to regaining our medicinal advantage.
One promising approach to managing resistance involves identifying and developing new small molecules8 that hit new antibacterial targets and do not develop cross resistance with antibiotics acting on existing antibacterial targets.9 This approach is challenging since accessing such novel targets will likely require screening new molecular scaffolds that have not been isolated from nature or chemically synthesized. Existing compound libraries suffer from low structural novelty5,10,11 and often favor compounds with physical properties suitable for accessing eukaryotic cellular targets,5 while prokaryotes require different physiochemical properties for cell penetration.12,13 Modern medicinal chemistry techniques including fragment-based drug discovery (FBDDa),14,15 computational structure-based design methods,16 high-throughput screening (HTS),17 and other target-based genomic approaches have been validated for identifying antibacterial compounds with new biological targets by the so called reverse chemical genetic approach.18 Overall, these methods alone have proven difficult to translate into well established whole cell antibiotic susceptibility assays due primarily to cell permeability issues. Additionally, most compound libraries used for virtual and in vitro screening lack the structural diversity for exploring unique chemical space.19
Traditional combinatorial library synthesis and HTS continue to play an important role in drug discovery, but have yielded limited success in bringing new chemical entities, including antibacterials, to market.20 This lack of success is often attributed to the lack of structural diversity found in combinatorial compound libraries.19,20 Diversity-oriented synthesis (DOS) has emerged as an important strategy for producing structurally diverse and unbiased collections of small molecules covering unique chemical space.21,22 The use of DOS has already contributed to the discovery of new antibacterial scaffolds using whole cell HTS assays by the so called forward chemical genetic approach.18,23 Inclusion of DOS into antibacterial discovery programs provides a valuable source of structurally diverse molecular scaffolds for target-blind, unbiased screening.23
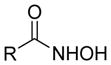
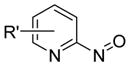
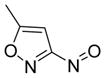
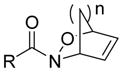
Our group recently published new synthetic methodologies for the syntheses of highly functionalized hydroxylamine-containing compounds using Lewis acid-mediated nucleophilic ring opening reactions of nitroso Diels-Alder cycloadducts with alcohols24,25 and nitroso ene reactions with substituted alkenes.26 These methodologies were an extension of our Modular Enhancement of Nature’s Diversity (MEND) natural product derivatization program into a DOS strategy for rapidly producing structurally complex small molecules from simple starting materials.27–31 Biological screening of the hydroxylamine-containing compounds revealed selective and potent antibacterial activity against Micrococcus luteus ATCC 10240 for compounds with pyridyl or isoxazole heterocycles. Discovery of the antibacterial activity coincided with a report from our group describing similar activity of pyridyl-hydroxylamines derived from the ene reaction of 2-nitrosopyridines with the sesquiterpene natural product caryophyllene.32 Further literature searching revealed only one other report of N-alkyl-N-(pyridin-2-yl)hydroxylamines possessing moderate antibacterial activity against Staphylococcus aureus SG511, which also came from our group.33 While other structurally unique hydroxylamine-containing antibacterial scaffolds are known (Figure 1),34–38 the novelty of the structures and obscurity of documented antibacterial activity inspired us to pursue this new antibacterial lead further.
Figure 1.

Examples of known antibacterial agents containing an N-O bond and the generic N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffold from this work.34–38
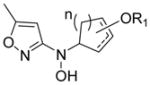
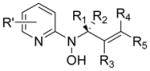
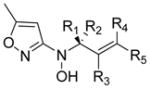
Herein is described the discovery and elucidation of the structure-activity relationships (SAR) of a new class of Gram-positive selective antibacterial agents based on a N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffold (Figure 1). Application of a DOS strategy using simple starting materials and the rich chemistry of nitroso Diels-Alder cycloaddition and nitroso ene reactions generated a structurally diverse library of oxyamino-containing compounds including hydroxamic acids, bicyclic oxazines, and N-alkyl-N-aryl-hydroxylamines (compounds 1–5, Figure 2). The compounds were screened for biological activity in an unbiased, target-blind approach using whole cell antibacterial susceptibility assays. Nitroso ene reactions were used in a parallel synthesis strategy to explore SAR on the pyridine ring and the hydroxylamine side chain. A new convergent synthetic route was developed using palladium-catalyzed Buchwald-Hartwig cross-coupling amination reactions of N-alkyl-O-(4-methoxybenzyl)hydroxylamines with 2-halo-pyridines to achieve further SAR exploration. The simplest active fragment molecule was identified by synthesizing and testing all major molecular fragments. The discovery process outlined here highlights the combination of DOS and parallel synthesis as a useful strategy for identifying new antibacterial scaffolds (Figure 2).
Figure 2.

(a) Generalized DOS and combinatorial chemistry strategy for identifying new biologically active lead scaffolds. (b) Combination strategy of DOS and combinatorial chemistry used for discovering the new antibacterial N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffold in this work.
Results and Discussion
Chemistry
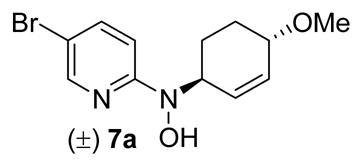
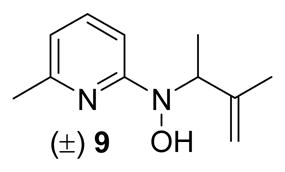
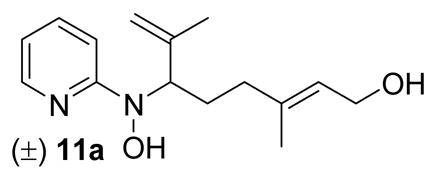
The majority of molecules from the structurally diverse DOS library (compounds 1–5 generalized in Figure 2 and Table 4; exact structures as shown in the supporting information Table S1) were synthesized and characterized as described previously by our group using Lewis acid-mediated nucleophilic ring-opening reactions of nitroso Diels-Alder cycloadducts and nitroso ene reactions.24–26 Several important and highly biologically active compounds (7, 9, and 11) from this library were resynthesized according to the previously described methods (Scheme 1). Separable isomers 7a and 7b were prepared using an indium(III) triflate-mediated nucleophilic ring opening of 5-bromo-2-nitrosopyridine Diels-Alder cycloadduct 6 with methanol.24 Compounds 9 and 11a,b were synthesized using nitroso ene reactions with 2-methyl-2-butene and geraniol, respectively.26 The major isomer from the ene reaction with geraniol (11a) was readily obtained in pure form by standard chromatographic methods, while the minor isomer (11b) was always isolated as a mixture with 11a.
Table 4.
Structure classes and antibiotic activity against M. luteus ATCC 10240 in the agar diffusion assay.a,b,c
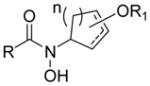
| Structure Type | Class A | Active? | Class B | Active? | Class C | Active? |
|---|---|---|---|---|---|---|
| Nitrosos and Hydroxamic Acids (1) |
|
Nod |
|
No |
|
No |
| Diels-Alder Cyloadducts (2) |
|
No |
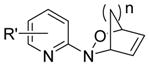
|
No |
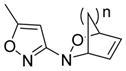
|
No |
| Ring Opened Compounds (3, 4) |
|
No |
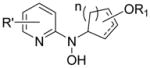
|
Yese |
|
Yes |
| Ene Products (5) |

|
No |
|
Yes |
|
Yes |
See supporting information for a complete list of structures and antibacterial data (Tables S1 and S2).
All compounds were racemic. R = alkyl, O-alkyl; R′ = alkyl, halo, carboxyl; R1, R2, R3, R4, R5 = alkyl, H; n = 1,2.
Exactly 50 μL of each compound solution (2.0 mM in 10:1 MeOH:DMSO) were added to 9 mm wells in agar media (MHII) inoculated with ~5×103 CFU/mL of M. luteus ATCC 10240. Diameters of growth inhibition zones were measured (mm) after incubation at 37 °C for 24 h.50
No: indicates a growth inhibition zone ≤14 mm.
Yes: indicates a growth inhibition zone >14 mm.
Scheme 1.

Syntheses of N-alkyl-N-(pyridin-2-yl)hydroxylamines 7a, 7b, 9, and 11a,and 11b.a
aReagents and conditions: (a) In(OTf)3 (1.0 equiv), MeOH, 70 °C, 4 h, 95% (1:1 ratio of 7a:7b); (b) 2-methyl-2-butene (2.0 equiv), CH2Cl2, 0 °C–rt, 20 min, 38%; (c) geraniol (2.0 equiv), CH2Cl2, 0 °C–rt, 25 min, 71% (72:28 ratio of 11a:11b).
As described later, preliminary antibacterial data revealed that hydroxylamine compounds containing pyridine or isoxazole heterocycles showed strong antibacterial activity against M. luteus ATCC 10240, with pyridine being the preferred heterocycle. To confirm the preference for pyridine, compounds 12, 14, and 16 featuring pyridine, quinoline, and isoxazole heterocycles, respectively, with identical isoprenyl side chains were synthesized via ene reactions of the nitroso compounds with 2-methyl-2-butene (Scheme 2).
Scheme 2.
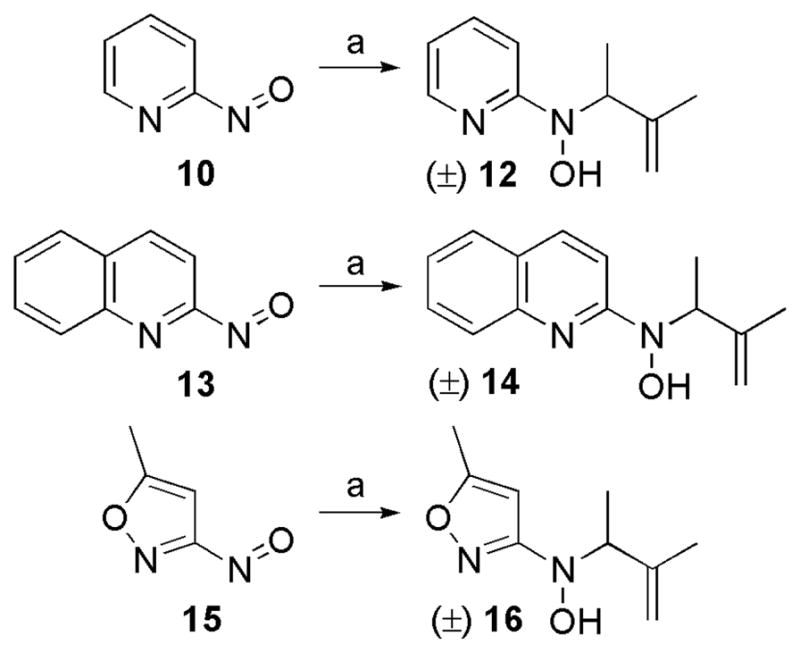
Syntheses of pyridine (12), quinoline (14), and isoxazole (16) hydroxylamine analogs with an isoprenyl side chain.a
aReagents and conditions: (a) 2-methyl-2-butene (2.0 equiv), CH2Cl2, 0 °C–rt; 15 min, 48% for 12; 1 h, 53% for 14; 10 min, 94% for 16.
Nitroso ene chemistry was also used to rapidly synthesize a panel of substituted pyridine analogs with an isoprenyl side chain. Parallel reaction of 2-nitrosopyridine enophiles (17a–i) with 2-methyl-2-butene gave the corresponding racemic N-isoprenyl-N-(pyridin-2-yl)hydroxylamines (18a–i; Table 1). These analogs were designed to probe SAR of the pyridine ring using substituents with differing electronics, sterics, and substitution patterns.
Table 1.
Syntheses of substituted pyridine analogs (18a–i) with an isoprenyl side chain.
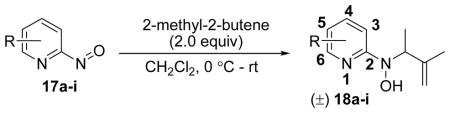 | |||
|---|---|---|---|
| Comp. | R | Time | % Yield |
| 18a | 6-ethyl | 30 min | 46 |
| 18b | 4-methyl | 30 min | 47 |
| 18c | 3-methyl | 9 h | 8 |
| 18d | 5-chloro | 30 min | 59 |
| 18e | 5-bromo | 10 min | 64 |
| 18f | 5-iodo | 1 h | 68 |
| 18g | 3,5-dichloro | 1 h | 67 |
| 18h | 5-bromo-6-methyl | 30 min | 62 |
| 18i | 3-chloro-5-CF3 | 45 min | 75 |
Nitroso ene chemistry was also used to generate a small panel of analogs that probed SAR of the hydroxylamine side chain. Reaction of 2-nitrosopyridine 10 with a variety of substituted alkenes gave the corresponding ene products (19a–c; Table 2). Compound 19a has an α-methyl substituted isoprenyl side chain and is achiral. Compound 19b lacks substitution and chirality at the carbon alpha to the hydroxylamine nitrogen and introduces a styrene unit. Compound 19c retains the isoprenyl stereocenter and introduces a ketone into the side chain.
Table 2.
Syntheses of pyridine analogs (19a–c) with various hydroxylamine side chains.
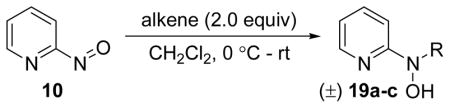 | |||
|---|---|---|---|
| Comp. | R | Time | % Yield |
| 19a |
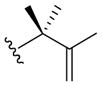
|
30 min | 71 |
| 19b |
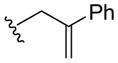
|
3 h | 6 |
| 19c |
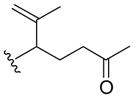
|
10 min | 94 |
To probe deeper into the SAR and identify the simplest active molecular fragment, a new convergent synthesis of the general N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffold was designed. The simplified core structure 20 was viewed retrosynthetically (Figure 3) by disconnecting the hydroxylamine bond at the 2-position of the pyridine ring. The C-N bond could be formed using Pd(0)-catalyzed Buchwald-Hartwig39,40 cross-coupling amination reactions of commercially available 2-halopyridines 21 with suitable O-protected hydroxylamines 22. The N-substituted hydroxylamines 22 were available via N-alkylation of N-Boc-O-protected hydroxylamine 23 with alkyl halides 24 under SN2 conditions followed by removal of the N-Boc protecting group with anhydrous acid.41,42 This synthetic route provided convergent access to the active core 20 where advanced functionality could be incorporated into both major fragments (21 and 22) separately.
Figure 3.

Retrosynthetic analysis of N-alkyl-N-(pyridin-2-yl)hydroxylamines (20).
An O-(4-methoxybenzyl)ether (PMB) protecting group was chosen for the hydroxylamine since PMB-ethers are easily removed by exposure to anhydrous acid.43 The PMB-protected hydroxylamine derivative, N-Boc-O-(4-methoxybenzyl)hydroxylamine (BocNHOPMB, 27) was synthesized on multigram scale (10.5 g) starting from N-hydroxyphthalimide (25) using a modified literature protocol (Scheme 3).44–46
Scheme 3.
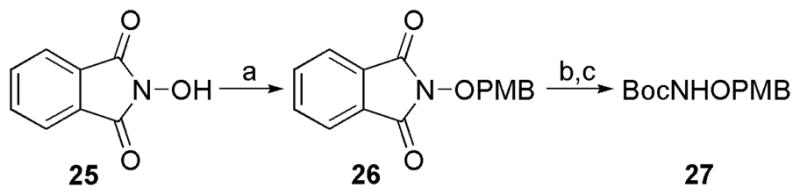
Synthesis of N-Boc-O-(4-methoxybenzyl)hydroxylamine (BocNHOPMB, 27).a
aReagents and conditions: (a) 4-(methoxybenzyl)chloride, Et3N, DMF, 90 °C, 1 h, 71%. (b) NH2NH2, DMF:MeOH, 60 °C, 10 min. (c) Boc2O, Et3N, THF:H2O, 2 h, 90% (steps b,c).
The target N-alkyl-N-(pyridin-2-yl)hydroxylamines were synthesized using a four step sequence starting from BocNHOPMB (27). Deprotonation of BocNHOPMB (27) with sodium hydride in DMF followed by SN2 reaction with a variety of alkyl halides gave N-alkylated hydroxylamines 28a–i in excellent yields. Brief exposure of compounds 28a–i to anhydrous trifluoroacetic acid (TFA) selectively removed the N-Boc protecting groups and an aqueous NaHCO3 wash gave the N-alkyl-O-PMB-hydroxylamines 29a–i in good yields. The original aryl amination conditions reported by Buchwald and coworkers (Pd2(dba)3, (±}) BINAP, NaOtBu, toluene, 70 °C)47 gave successful cross couplings of hydroxylamines 29a–i with 2-bromopyridine yielding N-alkyl-N-(pyridin-2-yl)-O-PMB-hydroxylamines 30a–i. Use of hydroxylamines 29a–i in crude form gave the desired products, but chromatographic purification of the hydroxylamines prior to coupling improved yields. Increasing the size of the hydroxylamine alkyl substituent resulted in decreased cross-coupling yields following the trend hexyl (39%) < propyl (51%) < methyl (77%). The final deprotection of the PMB group under acidic conditions (TFA) in the presence of a cation scavenger (Et3SiH) proceeded smoothly giving moderate to good yields (55–84%) of the desired N-alkyl-N-(pyridin-2-yl)hydroxylamine products (31a–i) after freebasing with aqueous NaHCO3. The PMB-deprotection reactions were monitored closely since overexposure to TFA and Et3SiH caused reduction of the N-O bond. The choice of Et3SiH as a cation scavenger was important since other cation scavengers such as anisole led to decomposition of the products. The results from the four step synthetic sequence are summarized in Table 3.
Table 3.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp | Step a | Step b | Step c | Step d | |||||
| 28–31 | R | Time (h) | % Yield | Time (min) | % Yield | Time (h) | % Yield | Time (h) | % Yield |
|
(min)
| |||||||||
| a | Me | 26 | 91 | 15 | 99c | 96 | 77 | 7 | 64e |
| b | Pr | 5 | 98d | 15 | 99c | 72 | 51 | 5 | 75 |
| c | n-hexyl | 14 | 97 | 15 | 77 | 72 | 39d | 12 | 62 |
| d | Bn | 6 | 98d | 15 | 75d | 64 | 63d | 5.5 | 84d |
| e | (CH2)2Ph | 10 | 88d | 15 | 73 | 60 | 49 | 4.5 | 64d |
| f | allyl | 10 | 87 | 15 | 58 | 66 | 54 | 5 | 55 |
| g | 3-(OPh)Bn | 24 | 50 | 15 | 70 | 70 | 59 | 7 | 60 |
| h | 4-(CF3)Bn | 22 | 98 | 15 | 62 | 96 | 56 | 7 | 81 |
| i | 4-(OCF3)Bn | 22 | 86 | 15 | 72d | 96 | 53d | 19 | 70 |
|
| |||||||||
| Average | 15 | 88 | 15 | 70f | 77 | 56 | 8 | 68 | |
| Range | 5–26 | 50–98 | 15 | 58–77f | 60–96 | 39–77 | 4.5–19 | 55–84 | |
Reagents and conditions: (a) NaH, RX (alkyl halide), DMF, 0 °C–rt. (b) TFA, CH2Cl2, rt; then aq. NaHCO3. (c) 2-bromopyridine, Pd2(dba)3, (±) BINAP, NaOtBu, toluene, 70 °C. (d) TFA, Et3SiH, CH2Cl2, rt; then aq. NaHCO3.
Yields are based on isolated, purified, and characterized material unless otherwise stated and optimal reaction time listed if reactions were replicated.
Crude yield.
Average of two yields.
Average of three yields.
Calculated for purified products only.
The analogs generated from the Buchwald-Hartwig amination protocol were designed to investigate some key SAR points including the presence of a chiral center and allylic unsaturation. A number of analogs with more drastic structural changes were also synthesized to address several other important SAR questions and identify the simplest active molecular fragment. To test the importance of the hydroxyl group an analog with the N-O bond reduced, N-benzylpyridin-2-amine (32), was prepared by subjecting compound 31d to Pd-catalyzed hydrogenolysis. The O-acetyl analog (33) was prepared by treatment of 31d with acetic anhydride (Scheme 4). Analogous N-O reduced (34) and O-acetyl (35) compounds were prepared from the more structurally complex compound 7b (Scheme 4). Selective reduction of the N-O bond of compound 7b was achieved using a titanium-mediated reduction procedure which gave analog 34 in good yield.48 Acetylation of compound 7b was achieved using acetic anhydride which provided analog 35 in excellent yield.
Scheme 4.
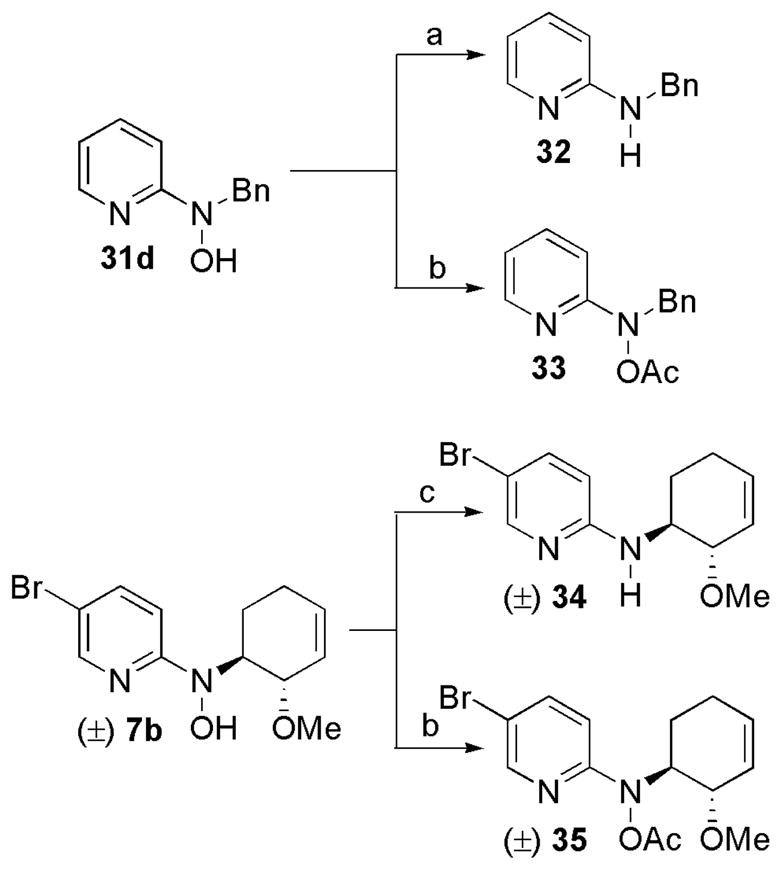
Syntheses of N-O reduced analogs (32 and 34) and O-acetylated analogs (33 and 35).a
aReagents and conditions: (a) 10% Pd-C, H2 (1 atm), MeOH, rt, 30 min, 97%. (b) Ac2O, iPr2EtN, DMAP, CH2Cl2, rt; 12 h, 72% for 33; 9 h, 94% for 35 (c) Cp2TiCl2, Zn, THF/MeOH, −30 °C, 1 h, 73%.
All the analogs presented thus far featured placement of the hydroxylamine at the 2-position of pyridine. The same Buchwald-Hartwig amination sequence discussed earlier was used to synthesize an analog with the hydroxylamine placed at the 3-position of the pyridine ring (compound 37; Scheme 5). The Pd-catalyzed cross coupling of hydroxylamine 29d was less effective at the 3-position of pyridine (compound 36, 36% yield) relative to coupling at the 2-position (compound 30d, 63% yield; Table 3), but still afforded the desired product (36). Deprotection of the PMB group with TFA/Et3SiH and quenching with aqueous NaHCO3 provided N-benzyl-N-(pyridin-3-yl)hydroxylamine (37).
Scheme 5.
Synthesis of 3-pyridyl analog 37.a
aReagents and conditions: (a) 3-bromopyridine, Pd2(dba)3, (±}) BINAP, NaOtBu, toluene, 70 °C, 67 h, 36%. (b) TFA, Et3SiH, CH2Cl2, rt, 30 h; then aq. NaHCO3, 45%.
The homologated analog 39 was designed to determine if direct attachment of the hydroxylamine to the pyridine ring was essential for activity. Alkylation of hydroxylamine 29d with 2-(bromomethyl)pyridine gave O-PMB-protected hydroxylamine 38. Removal of the PMB group with TFA/Et3SiH gave the desired homologated analog 39 (Scheme 6).
Scheme 6.
Synthesis of homologated analog 39.a
aReagents and conditions: (a) 2-(bromomethyl)pyridine hydrobromide, iPr2EtN, CH3CN, 50 °C, 17 h, 44%. (b) TFA, Et3SiH, CH2Cl2, rt; then aq. NaHCO3, 5 h, 73%.
As described later, preliminary testing indicated that nitroso ene reactions with geraniol generated highly antibacterially active compounds and that 6-alkyl substitution was favorable. Therefore, a 6-ethyl geraniol analog (40a) was synthesized via ene reaction of 6-ethyl-2-nitrosopyridine with geraniol (Scheme 7). The major isomer (40a) was isolated in pure form after chromatographic purification. A crystal of 40a was obtained from CH2Cl2/hexanes and X-ray diffraction confirmed the structure of the major isomer (Figure 4; see supporting information for experimental details).
Scheme 7.
Synthesis of geranyl analogs 40a and 40b.a
aReagents and conditions: (a) geraniol (2.0 equiv), CH2Cl2, 0 °C–rt, 30 min, 75% (70:30 ratio of 40a:40b).
Figure 4.
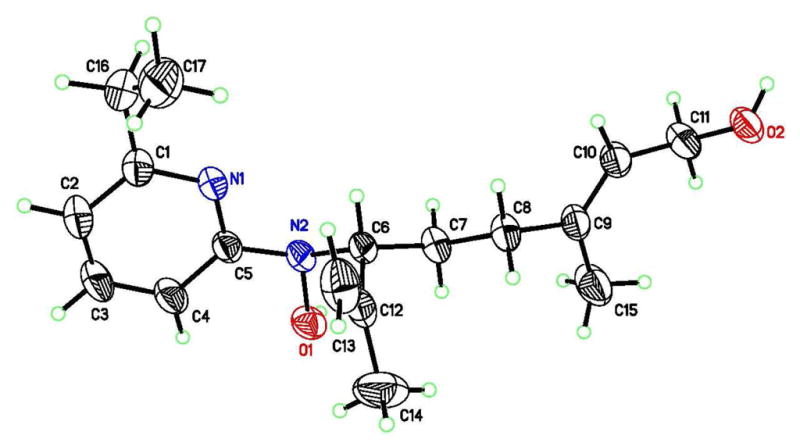
X-ray diffraction structure of analog 40a displayed with 50% probability ellipsoids.
Biology
Selected compounds from the nitroso chemistry (compounds 1–5, Figure 2)24–26 were compiled into a DOS library of ~100 small molecules with a high degree of appendage, functional group, stereochemical, and skeletal diversity.23 The representative structural classes are given in Table 4. A complete list of exact structures is given in the supporting information (Table S1). The entire DOS compound library was screened against Micrococcus luteus ATCC 10240 using an in-house Kirby-Bauer agar diffusion antibiotic susceptibility whole cell assay (Table 4).49,50 The agar diffusion assay facilitated rapid identification of active compounds and comparison of relative potencies (increased size of the growth inhibition zone corresponds to an increase in potency; i.e., a lower minimum inhibitory concentration value).
The nitroso compounds and hydroxamic acids (1; Class A–C), bicyclic Diels-Alder cycloadducts (2; Class A–C), monocyclic hydroxamate-derived ring opened compounds (3,4; Class A), and acyclic hydroxamate derived ene products (5; Class A) were not active against M. luteus. The pyridyl and isoxazole ring opened compounds (3,4; Class B,C) and ene products (5, Class B,C) all showed strong antibacterial activity against M. luteus (Table 4). A complete list of biological data for all compounds is provided in the supporting information (Table S2).
In general, the N-alkyl-N-(pyridin-2-yl)hydroxylamines (3, 4, and 5; Class B) gave larger zones of growth inhibition in the agar diffusion assay than the N-alkyl-N-(5-methylisoxazol-3-yl)hydroxylamines (3, 4, and 5; Class C). The most active pyridyl compounds (7a, 9, and 11a) from the Class B N-alkyl-N-(pyridin-2-yl)hydroxylamines were selected for broader antibacterial testing against a panel of Gram-negative, Gram-positive, and yeast organisms, including antibiotic resistant strains of S. aureus (MRSA) and E. faecalis (VRE). The results of this broad antibacterial screening are summarized in Table 5 using a generalized scale to clearly show the narrow spectrum of antibacterial activity of compounds 7a, 9, and 11a. Ciprofloxacin was included as a control antibiotic with known broad-spectrum activity.51
Table 5.
| Test Organism | Test Compound
|
|||
|---|---|---|---|---|
 |
 |
 |
ciprofloxacind | |
| M. luteus ATCC 10240 | +++ | +++ | +++ | − |
| S. aureus SG511 | + | + | ++ | +++ |
| S. aureus 134/94 (MRSA) | + | + | ++ | + |
| E. faecalis ATCC 49532 | ++* | ++* | ++* | ++ |
| E. faecalis 1528 (VRE) | ++* | ++* | ++* | ++ |
| B. subtilis ATCC 6633 | − | − | + | +++ |
| M. vaccae IMET 10670 | + | ++ | ++ | +++ |
| E. coli DC0 | − | − | − | +++ |
| E. coli DC2 | − | − | − | +++ |
| P. aeruginosa 799/WT | − | − | − | +++ |
| P. aeruginosa 799/61 | − | − | − | +++ |
| S. salmonicolor 549 (yeast) | − | − | − | ++e |
Exactly 50 μL of each compound solution (2.0 mM in 10:1 MeOH:DMSO) were added to 9 mm wells in agar media (MHII) inoculated with ~5×103 CFU/mL. Diameters of growth inhibition zones were measured (mm) after incubation at 37 °C for 24 h.50
See supporting information for exact diameters (mm) of inhibition zones (Table S2).
Diameters of inhibition zones are generalized as follows: +: inhibition zone of 11 – 15 mm; ++: inhibition zone of 16 – 20 mm; +++: inhibition zone of ≥ 21 mm; −: inhibition zone of ≤ 10 mm.
Ciprofloxacin was used as a control at 5 μg/mL in H2O.
Amphotericin B was used as the control (10 μg/mL in 10:1 DMSO:MeOH) instead of ciprofloxacin.
Indicates partially unclear inhibition zone.
Compounds 7a, 9, and 11a all displayed a similar narrow spectrum of antibacterial activity and the ciprofloxacin control (5 μg/mL in H2O) showed broad spectrum activity against all strains except M. luteus. Compounds 7a, 9, and 11a exclusively inhibited the growth of Gram-positive bacteria with particularly strong and selective potency against M. luteus ATCC 10240. The compounds showed comparable, although moderate, activity against both antibiotic susceptible (S. aureus SG511; E. faecalis ATCC 49532) and antibiotic resistant (S. aureus 134/94 MRSA; E. faecalis 1528 VRE) Gram-positive strains. Due to the highly selective antibacterial activity against Gram-positive organisms, M. luteus ATCC 10240, S. aureus SG511, and E. faecalis ATCC 49532 were used as model test organisms to evaluate SAR of the N-alkyl-N-(pyridin-2-yl)hydroxylamine analogs described from this point forward.
Two antibacterial susceptibility assays were used to evaluate the antibacterial activity of all the synthesized analogs, an agar diffusion assay49,50 and a visual end point broth microdilution assay.52 Data from the agar diffusion assay are reported as diameters of growth inhibition (mm) and data from the broth microdilution assay are reported as minimum inhibitory concentrations (MIC90; μM). The fluoroquinolone antibiotic ciprofloxacin was included as a control.51 The results from the two assays are provided in Table 6.
Table 6.
Antibacterial activity of test compounds in the agar diffusion assaya (diameter of growth inhibition zones given in mm) and the broth microdilution assayb,c (MIC90 values given in μM).
| Comp. | Test Organism
|
|||||
|---|---|---|---|---|---|---|
| M. luteus ATCC 10240 | S. aureus SG511 | E. faecalis ATCC 49532 | ||||
| Zone (mm) | MIC90 (μM) | Zone (mm) | MIC90 (μM) | Zone (mm) | MIC90 (μM) | |
| 7a | 34 | 4.0 | 12 | >128 | 16* | >128 |
| 7b | 34 | 4.0 | 18* | ≥128 | 20* | >128 |
| 9 | 48 | 4.0 | 12 | >128 | 15* | >128 |
| 11a | 35 | 8.0 | 19 | 64 | 20* | >128 |
| 12 | 31 | 8.0 | 18 | 128 | 22* | >128 |
| 14 | 31 | 8.0 | 13 | 128 | 13* | >128 |
| 16 | 21 | 64 | 0 | >128 | 13* | >128 |
| 18a | 52 | 2.0 | 11 | >128 | 22* | >128 |
| 18b | 29 | 4.0 | 20 | 128 | 22* | >128 |
| 18c | 25 | 16 | 10 | >128 | 19* | >128 |
| 18d | 40 | 2.0 | 18 | 128 | 21* | ≥128 |
| 18e | 35 | 4.0 | 17* | >128 | 19* | >128 |
| 18f | 38 | 2.0 | 12 | >128 | 19* | >128 |
| 18g | 27 | 16.0 | 0 | >128 | 14* | >128 |
| 18h | 35 | 4.0 | 13 | >128 | 12* | >128 |
| 18i | 15 | 128 | 0 | >128 | 13* | >128 |
| 19a | 33 | 4.0 | 15 | 128 | 18* | >128 |
| 19b | 20 | 64 | 19 | 32 | 18* | >128 |
| 19c | 30 | 16 | 15* | 128 | 16* | >128 |
| 30d | 0 | >128 | 0 | >128 | 0 | >128 |
| 31a | 18* | ≥128 | 0 | >128 | 0 | >128 |
| 31b | 13* | >128 | 0 | >128 | 0 | >128 |
| 31c | 15 | 128 | 17* | >128 | 16* | >128 |
| 31d | 24 | 64 | 18 | 128 | 20* | >128 |
| 31e | 16 | 128 | 0 | >128 | 15* | >128 |
| 31f | 14 | >128 | 0 | >128 | 12* | >128 |
| 31g | 12* | >128 | 13 | >128 | 0 | >128 |
| 31h | 16 | 128 | 15* | >128 | 15* | >128 |
| 31i | 17 | 128 | 14* | >128 | 15* | >128 |
| 32 | 0 | >128 | 0 | >128 | 0 | >128 |
| 33 | 20 | 128 | 12 | >128 | 11* | >128 |
| 34 | 0 | >128 | 0 | >128 | 0 | >128 |
| 35 | 22 | 32 | 0 | >128 | 0 | >128 |
| 37 | 15* | ≥128 | 16* | >128 | 0 | >128 |
| 39 | 0 | >128 | 0 | >128 | 0 | >128 |
| 40a | 31 | 2.0 | 18* | >128 | 19* | >128 |
| ciprod | 0 | 8.0 | 26 | 0.5 | 17 | 2.0 |
Exactly 50 μL of each compound solution (2.0 mM in 10:1 MeOH:DMSO) were added to 9 mm wells in agar media (MHII) inoculated with ~5×103 CFU/mL. Diameters of growth inhibition zones were measured (mm) after incubation at 37 °C for 24 h.50
MIC90 values were determined by the visual end point broth microdilution method following CLSI guidelines.52
Each compound was tested in triplicate.
Ciprofloxacin was used as a standard at 5 μg/mL in H2O in the agar diffusion assay.
Indicates partially unclear inhibition zone.
The MIC90 data for compounds 7a, 9, and 11a mirrored the agar diffusion data and confirmed the selective potency against M. luteus (MIC90 values of 4, 4, and 8 μM, respectively). Geranyl derivative 11a also had a moderate MIC90 of 64 μM against S. aureus. Similarly, analogs 12, 14, and 16 featuring pyridine, quinoline, and isoxazole heterocycles, respectively, all displayed selective antibacterial activity against M. luteus. The anti-M. luteus activity, based on MIC90 values, of the heterocylic cores increased in the order of isoxazole (64 μM) < quinoline (8 μM) = pyridine (8 μM), which confirmed the initial observation of pyridine as a favorable core.
The substituted pyridine analogs (18a–i) with identical isoprenyl side chains also preferentially inhibited the growth of M. luteus relative to S. aureus and E. faecalis. Methyl substitution at the 4- and 6-positions (compounds 9 and 18b, respectively) enhanced anti-M. luteus activity (MIC90 values of 4 μM) compared to the 3-position (compound 18c) of the pyridine ring (MIC90 value of 16 μM). There was a noticeable increase in potency in both assays for the 6-ethyl substituted compound (18a, 2 μM) compared to the unsubstituted (12, 8 μM) and 6-methyl substituted analogs (9, 4 μM). Halogen substitution (5-Cl, 5-Br, and 5-I) at the 5-position (compounds 18d–f) of the pyridine ring also increased anti-M. luteus activity (MIC90 values from 2–4 μM). 3,5-Dichloro substitution (compound 18g) gave decreased anti-M. luteus activity (16 μM) relative to 5-chloro substitution (compound 18d, 2 μM). Anti-M. luteus activity was not improved by the use of mixed alkyl-halogen substitution (5-Br-6-Me, 18h; MIC90 value of 4 μM) relative to monohalogen (5-Br, 18e; MIC90 value of 4 μM) or alkyl substitution alone (6-Me, 9; MIC90 value of 4 μM). Substitution of the pyridine using 3-Cl-5-CF3 substituents (compound 18i) was detrimental (MIC90 value of 128 μM against M. luteus). As supported by compounds 18c, 18g, and 18i, any substitution at the 3-position of the pyridine ring resulted in decreased anti-M. luteus activity. Most of the analogs had no significant activity against E. faecalis or S. aureus, but the 4-methyl (18b) and 5-bromo (18e) derivatives showed a noticeable increase in activity against S. aureus relative to the other analogs. In general for the substituted pyridine analogs (18a–i), 5-halo and 6-alkyl substitution was favored for increased anti-M. luteus activity and the 6-ethyl analog (18a) emerged as the most potent compound in both assays (inhibition zone of 52 mm; MIC90 value of 2 μM). The preference for 6-ethyl substitution was confirmed by the 6-ethyl substituted geranyl derivative 40a which showed enhanced anti-M. luteus activity (MIC90 value of 2 μM) relative to the unsubstituted geranyl derivative 11a (MIC90 value of 8 μM).
Evaluation of the pyridine analogs with differing hydroxylamine alkyl side chains (19a–c) showed that substitution of the hydroxylamine nitrogen is tolerated, but does noticeably influence the potency and selectivity. Compound 19a had a gem-dimethyl isoprene unit and maintained strong potency against M. luteus (MIC90 value of 4 μM) and weak anti-S. aureus activity (MIC90 value of 128 μM). Compound 19b featured a styrene side chain and showed decreased anti-M. luteus activity (MIC90 value of 64 μM) and increased anti-S. aureus activity (MIC90 value of 32 μM). Compound 19c gave MIC90 values of 16 μM against M. luteus and 128 μM against S. aureus which showed that an alkyl ketone was tolerated as a side chain. Antibacterial data from compounds 19a and 19c and the other unsubstituted pyridyl nitroso ene products 11a and 12 showed that high substitution is tolerated on the alpha carbon of the isoprenyl hydroxylamine side chain. Data from compound 19b indicated that alkyl substitution of the alpha carbon is important for enhancing the potency against M. luteus and that modification of the hydroxylamine side chain can enhance potency against S. aureus.
The N-alkyl-N-(pyridin-2-yl)hydroxylamine products 31a–i from the Buchwald-Hartwig chemistry revealed some interesting SAR trends. Compound 31a with a simple N-methyl side chain showed weak, but noticeable activity against M. luteus. This observation was important because compound 31a represents the simplest active molecular fragment with all the critical structural components (pyridine heterocycle, N-OH at 2-position, and alkyl substitution of the hydroxylamine nitrogen). There was no drastic change in anti-M. luteus activity observed when the size of the alkyl side chain was increased from Me to Pr to n-hexyl (compounds 31a–c). However, an N-benzyl group was found to be a suitable side chain and compound 31d was the most active analog generated from the Buchwald-Hartwig chemistry against both M. luteus (MIC90 value of 64 μM) and S. aureus (MIC90 value of 128 μM). The benzyl derivative 31d showed increased activity relative to the phenethyl derivative (31e) where the unsaturated carbon system was homologated. Compound 31f had a simple N-allyl side chain and showed only weak anti-M. luteus activity in the agar diffusion assay.
The presence of an allylic unsaturated system appeared to be important for antibacterial activity and a benzyl side chain seemed to be a good choice especially since the phenyl ring offered a versatile point of derivatization. Preliminary SAR studies around the phenyl ring of benzyl derivative 31d were performed using 3-phenoxy (31g), 4-trifluoromethyl (31h), and 4-trifluoromethoxy (31i) substituted compounds generated from the Buchwald-Hartwig cross-coupling chemistry. The phenyl substituted analogs were less active than the parent benzyl compound and the anti-M. luteus activity increased in the order of 3-(PhO)benzyl (31g) < 4-(CF3)benzyl (31h) ≈ 4-(CF3O)benzyl (31i) < benzyl (31d). Electron withdrawing groups appeared to be tolerated better than electron donating groups, but more data is needed to confirm this trend.
Comparison of the anti-M. luteus activity of allyl derivative 31f (inhibition zone of 14 mm; MIC90 value of >128 μM) to isoprenyl derivative 12 (inhibition zone of 31 mm; MIC90 value of 8 μM) revealed a drastic difference in activity for only subtle differences in structure and molecular weight. The same trend was apparent when comparing all the simplified N-alkyl-N-(pyridin-2-yl)hydroxylamines 31a–i to the unsubstituted pyridyl compounds with isoprenyl or substituted isoprenyl side chains generated from nitroso ene chemistry (11a, 12, 19a, and 19c). The increased activity associated with the isoprenyl side chains must be attributed to substitution at the carbon alpha to the hydroxylamine or alkyl branching from the double bond, which are the only clear structural differences.
The scaffold SAR was expanded by evaluating compounds with more drastic structural modifications (32–35, 37, and 39). The N-O reduced analogs 32 and 34 had no antibacterial activity in both susceptibility assays. Additionally, testing of O-PMB protected compound 30d revealed no antibacterial activity. These observations confirmed that the presence of the N-OH group is critical to activity. The O-acetyl analogs 33 and 35 had somewhat reduced activity against M. luteus (MIC90 values of 128 μM and 32 μM, respectively) compared to the parent compounds 31d and 7b (MIC90 values of 64 μM and 4 μM, respectively). The acetylated derivatives likely acted as prodrugs for the parent hydroxylamine compounds triggered by hydrolysis in the testing media. The 3-substituted pyridine analog 37 had reduced activity against M. luteus relative to 2-substituted pyridine analog 31d. The homologated analog 39 showed no antibacterial activity. The anti-M. luteus data for compounds 37 and 39 suggested that N-arylation of the hydroxylamine is essential for activity and that N-arylation at the 2-position of pyridine is optimal.
Overall the agar diffusion data and the MIC90 data in Table 6 correlated nicely. Compounds with the largest diameter growth inhibition zones (mm) in the agar diffusion assay gave the lowest MIC90 values (μM) in the broth microdilution assay. Compounds with the N-alkyl-N-(pyridin-2-yl)hydroxylamine core structure had selectively potent activity against M. luteus (MIC90 values as low as 2 μM) and weak to moderate activity against S. aureus (MIC90 values as low as 32 μM). Although many compounds gave larger diameter zones of inhibition against E. faecalis compared to S. aureus in the agar diffusion assay, these zones were hazy and correlated to high MIC90 values of >128 μM. Although the N-alkyl-N-(pyridin-2-yl)hydroxylamines only have weak to moderate activity against S. aureus and E. faecalis, the agar diffusion antibacterial data indicated that the compounds had comparable activity against antibiotic resistant strains (MRSA and VRE, Table 5). The SAR studies showed that the structure can be optimized to enhance activity against these clinically relevant Gram-positive pathogens. The SAR trends for anti-M. luteus activity of the N-alkyl-N-(pyridin-2-yl)hydroxylamines revealed in this study are summarized in Figure 5.
Figure 5.

General antibacterial SAR trends for N-alkyl-N-(pyridin-2-yl)hydroxylamines.
Representative compounds were also evaluated for growth inhibition of cancer cell lines (MCF-7, PC-3, and HeLa). None of the compounds showed significant toxicity towards the mammalian cell lines which indicated a potentially favorable therapeutic index. Experimental details and results from the anticancer assay are given in the supporting information (Table S2).
Further Discussion and Perspective
The selective antibacterial activity observed for the N-alkyl-N-(pyridin-2-yl)hydroxylamines against M. luteus was intriguing. While M. luteus is a commonly encountered Gram-positive bacterium that is part of normal human flora, it has a unique biochemistry53–55 and pattern of antibiotic susceptibility (nitrofurantoin and ciprofloxacin resistant).51,56 Consequently, M. luteus is commonly used in antibacterial screening programs to identify compounds with potential activity against resistant Gram-positive strains.57 Beyond its use as a model laboratory organism, M. luteus has clinical relevance as an infectious pathogen. While this bacterium was long considered nonpathogenic, recent literature recognizes M. luteus and other Microccoci as commonly misdiagnosed opportunistic pathogens that cause a variety of infections in immunocompromised patients that have been documented to be lethal in some cases.58,59 Clearly, Microccoci are clinically relevant pathogens that merit the development of new effective antibiotics especially since there is renewed interest in finding markets for species selective antimicrobial agents as a means of managing the development of resistance.5,60
Initially it was suspected that the N-alkyl-N-(pyridin-2-yl)hydroxylamines might be acting as prodrugs for releasing a toxic nitroso compound inside of the bacterial cells61 or possibly generating reactive oxygen species from the aryl hydroxylamine.62,63 However, the parent nitroso compounds (1, Class B,C; Table 4) and Diels-Alder cycloadducts (2, Class A–C; Table 4), which can undergo retro Diels-Alder reactions to generate a nitroso species,64 had no significant antibacterial activity. Additionally, establishment of clear SAR trends suggested the existence of a discrete, conserved cellular target. This hypothesis was also supported by the selective biological activity against Gram-positive bacteria, since a generally toxic molecule should show a broader spectrum of activity.
While any discussion of potential modes of action for these compounds is speculation at this point, some key structural features of the N-alkyl-N-(pyridin-2-yl)hydroxylamines showed striking similarities to known antibacterial compounds in the literature. The presence of a metal chelating group, the 2-pyridylhydroxylamine functionality,65,66 was similar to other known antibiotics containing N-OH bonds (Figure 1). For these known antibacterial agents, the N-OH bond played a key role by aiding in chelation of a metal in the active site of the target enzyme.34–37,67 Recent literature reports of fosmidomycin analogs68–71 revealed many structural similarities of N-alkyl-N-(pyridin-2-yl)hydroxylamines to known inhibitors (41–44) of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) in the methylerythritol phosphate (MEP) isoprene biosynthesis pathway,72 a validated target for antibacterial agents (Figure 6).73
Figure 6.

Structural similarities of N-benzyl-N-(pyridin-2-yl)hydroxylamine (31d) to known DXR inhibitors (fosmidomycin and compounds 41–44).
The preference for isoprenyl and geranyl hydroxylamine side chains of the most active N-alkyl- N-(pyridin-2-yl)hydroxylamine analogs found in this work might aid in binding to enzymes along terpenoid biosynthetic pathways. This might explain the selective antibacterial activity of these compounds against M. luteus which is known to express high levels of terpenoid biosynthesis related enzymes in its cell membrane. Furthermore, M. luteus is known to possess a more promiscuous set of magnesium-dependent prenylation enzymes, relative to Gram-negative (E. coli) and other Gram-positive (S. aureus) strains.55 Therefore, it is important to investigate these compounds for inhibition of isoprenoid and lipid biosynthetic pathways.73,74
Summary and Conclusions
A DOS library of compounds was generated using nitroso Diels-Alder and nitroso ene chemistry which led to the discovery of N-alkyl-N-(pyridin-2-yl)hydroxylamine scaffolds as selective antibacterial agents against Micrococcus luteus ATCC 10240. The SAR was investigated using parallel synthesis with nitroso ene chemistry and a new Buchwald-Hartwig amination cross-coupling reaction of O-PMB-protected hydroxylamines with 2-bromopyridine. From these SAR studies the simplest active fragment molecule, N-methyl-N-(pyridin-2-yl)hydroxylamine (31a) was determined. New lead structures (11a, 18a, 18d, 31d, and 40a) were identified with potent activity against M. luteus (MIC90 values as low as 2.0 μM) that will direct the production of future analogs. Structural similarities of N-alkyl-N-(pyridin-2-yl)hydroxylamines to known inhibitors of the MEP isoprenoid biosynthesis pathway (41–44) have inspired the direction for future mode of action studies and the design of new analogs. This work demonstrated that an interdisciplinary combination of DOS, parallel synthesis, and whole-cell antibacterial susceptibility assays is an effective strategy for the discovery of new, structurally novel antibacterial scaffolds.
Experimental
Chemistry
General materials and methods
All reactions were performed under a dry argon atmosphere, unless otherwise stated. All solvents and reagents were obtained from commercial sources and used without further purification unless otherwise stated. Dichloromethane (CH2Cl2) was distilled from calcium hydride. Tetrahydrofuran (THF) was distilled from Na/benzophenone. Dimethylformamide (DMF), diisopropylethylamine (iPr2EtN), acetonitrile (CH3CN), and triethylsilane (Et3SiH) were used from Acros Seal® anhydrous bottles. All nitroso compounds were prepared from aminoheterocyclic precursors by a literature protocol75 and were stored at −20 °C. Cycloadduct 6 was obtained from previous work published by our group.24 Silica gel column chromatography was performed using Sorbent Technologies silica gel 60 (32–63 Zm). 1H-NMR and 13C-NMR spectra were obtained on a 300 MHz, 500 MHz, or 600 MHz Varian DirectDrive spectrometer and FIDs were processed using ACD/ChemSketch version 10.04. Chemical shifts (δ) are given in parts per million (ppm) and are referenced to residual non-deuterated solvent. Coupling constants (J) are reported in hertz (Hz). High resolution, accurate mass measurements were obtained with a Bruker micrOTOF II electrospray ionization time-of-flight mass spectrometer in positive ion mode. Sample was introduced via flow injection at a rate of 4 μL/min, and mass spectra were accumulated from 50–3000 m/z for two minutes. X-ray diffraction analysis was performed on a Bruker APEX-II diffractometer (full experimental details and data processing are given in the supporting information). High-performance liquid chromatography (HPLC) was performed on a Waters 1525 Binary HPLC Pump instrument with a Waters 2487 Dual λ Absorbance Detector set at 254 nm operated by Breeze version 3.30 software. A YMC Pro C18 reverse phase column (3.0 × 50 mm) fit with precolumn frit (0.5 μm) and YMC Pro C18 guard column (2.0 × 10 mm) was used for all analyses. Mobile phases used were 10 mM ammonium acetate in HPLC grade water (A) and HPLC grade acetonitrile (B). A gradient was formed from 5%–80% of B in 10 min, then 80%–95% of B in 2 min, and then 95%–5% of B in 3 min at a flow rate of 0.7 mL/min (total run time of 15 min). Thin layer chromatography (TLC) was performed with aluminum-backed Merck 60-F254 silica gel plates using a 254 nm lamp, aq. FeCl3, aq. KMnO4, or iodine vapor for visualization. Infrared (IR) spectra were recorded on a Bruker Tensor series FT-IR spectrometer using a diamond ATR accessory and signals are reported as wavenumbers in reciprocal centimeters (cm−1). Melting points were determined in capillary tubes using a Thomas Hoover melting point apparatus and are uncorrected. The purity of compounds tested in biological assays was evaluated by analytical HPLC and verified to be ≥95%, unless otherwise noted.
(±) N-(5-Bromopyridin-2-yl)-N-((1S/R,4S/R)-4-methoxycyclohex-2-enyl)hydroxylamine (7a) and N-(5-Bromopyridin-2-yl)-N-((1S/R,2S/R)-2-methoxycyclohex-3-enyl)hydroxylamine (7b)
The synthesis of compounds 7a and 7b was replicated from work reported by Yang and Miller.24 A solution of nitroso Diels-Alder cycloadduct 6 (103.0 mg, 0.39 mmol) in 3 mL of MeOH was treated with In(OTf)3 (216.7 mg, 0.39 mmol) and heated to 70 °C (oil bath temperature). After 4 h, TLC (20% EtOAc in hexanes) showed complete consumption of starting material. The solution was cooled to rt and concentrated under reduced pressure. The resulting oil was partitioned between EtOAc (20 mL) and water (10 mL). The layers were separated and the water was extracted with EtOAc (1 × 10 mL). The EtOAc layers were combined, dried over anhydrous MgSO4, filtered, and concentrated. The crude product (~1:1 mixture of 7a:7b) was purified by column chromatography (0.75 × 8 in silica gel; 10%–25% EtOAc in hexanes) to give isomer 7a as an off-white solid (49.2 mg, 0.16 mmol), isomer 7b as a clear, viscous oil (40.0 mg, 0.13 mmol), and a mixed fraction of 7a/7b (20.0 mg, 0.07 mmol) as a clear, viscous oil (95% overall yield). (7a) HPLC retention time 6.86 min. (7b) HPLC retention time 7.32 min. All characterization data matched the previously reported data.24
Typical procedure for nitroso ene reactions
(±) N-(6-Ethylpyridin-2-yl)-N-(3-methylbut-3- en-2-yl)hydroxylamine (18a)
This procedure followed a general method reported previously.26 6-Ethyl-2-nitrosopyridine (17a; 271.0 mg, 1.99 mmol) was added as a solid to a solution of 2-methyl-2-butene (0.42 mL, 3.97 mmol) in 4 mL of anhydrous CH2Cl2 at 0 °C (ice bath temperature) and the solution turned orange. After 30 min, no starting nitroso compound (17a) was detected by TLC (20% EtoAc in hexanes). The solution was warmed to rt and the CH2Cl2 was evaporated under reduced pressure. The crude product was purified by column chromatography (1.25 × 7.5 in silica gel; 5%–30% EtOAc in hexanes) to give the desired ene adduct (18a) in 46% yield as a tan solid (190.0 mg, 0.92 mmol). Mp 64–65 °C; 1H-NMR (600 MHz, DMSO-d6) δ 8.68 (br s, N-OH, 1 H), 7.48 (dd, J = 8.2, 7.3 Hz, 1 H), 6.85 (d, J = 8.2 Hz, 1 H), 6.57 (d, J = 7.0 Hz, 1 H), 5.12 (q, J = 6.5 Hz, 1 H), 4.88–4.86 (m, 1 H), 4.84–4.82 (m, 1 H), 2.59 (q, J = 7.6 Hz, 3 H), 1.71 (s, 3 H), 1.18–1.15 (m, 6 H); 13C-NMR (150 MHz, DMSO-d6) δ 162.5, 160.2, 146.1, 137.8, 112.2, 111.6, 105.8, 58.2, 30.5, 21.5, 13.7, 13.6; HRMS-ESI (m/z): [M+H]+ calcd. for C12H19N2O: 207.1492, found 207.1488; IR (neat): 3139.7 (br), 3092.3, 1589.6, 1572.1, 1441.2, 1108.9, 968.9 cm−1; HPLC retention time 8.34 min.
N-Boc-O-(4-Methoxybenzyl)hydroxylamine (BocNHOPMB, 27)
This compound was prepared in 64% overall yield following a modified literature procedure.44 N-Hydroxyphthalimide (10.53 g, 64.5 mmol), 25, was treated with 4-methoxybenzyl chloride (8.9 mL, 65.4 mmol) and triethylamine (22.0 mL, 158.0 mmol), respectively, in 200 mL of DMF. The dark red solution was heated to 90 °C (oil bath temperature) for 1 h. The mixture was cooled to ~45 °C and poured over ice water. Product 26 precipitated as a light brown solid and was isolated via vacuum filtration and dried overnight under vacuum (12.99 g, 71%). 1H-NMR (500 MHz, CDCl3) δ 7.80–7.75 (m, 2 H), 7.73–7.68 (m, 2 H), 7.43 (d, J = 8.4 Hz, 2 H), 6.86 (d, J = 8.4 Hz, 2 H), 5.13 (s, 2 H), 3.78 (s, 3 H); 13C-NMR (125 MHz, CDCl3) δ 163.4, 160.3, 134.3, 131.5, 128.7, 125.7, 123.3, 113.8, 79.4, 55.1. The solid was dissolved in 188 mL of DMF:MeOH (12:25) while at 60 °C (oil bath temperature) and treated with hydrazine hydrate (65% v/v; 7.0 mL, 146.5 mmol). After 10 min, the mixture was cooled to rt and diluted with 100 mL of H2O. The MeOH was removed under reduced pressure and the DMF/H2O mixture was extracted with EtOAc (4 × 100 mL). The combined EtOAc layers were dried over anhydrous MgSO4, filtered, and concentrated to give 11.0 g of a faint, orange liquid. This liquid was immediately dissolved in 100 mL of THF:H2O (1:1) and treated with triethylamine (6.5 mL, 46.6 mmol) and Boc anhydride (12.0 g, 55.0 mmol), respectively. After 2 h, the THF was removed under reduced pressure and the H2O was extracted with EtOAc (2 × 50 mL). The combined EtOAc layers were washed with 5% aq. citric acid (1 × 25 mL) and water (1 × 25 mL), dried over anhydrous MgSO4, filtered, and concentrated to give 14.7 g of a clear, orange liquid. Purification by column chromatography (3 × 6 in silica gel; 20% EtOAc in hexanes) provided the desired product (27) in 90% yield (2 steps) as a clear, colorless liquid (10.49 g, 41.4 mmol). 1H-NMR (600 MHz, CDCl3) δ 7.35–7.32 (m, 2 H), 7.07 (s, 1 H), 6.91–6.88 (m, 2 H), 4.80 (s, 2 H), 3.82 (s, 3 H), 1.49 (s, 9 H); 13C-NMR (150 MHz, CDCl3) δ 159.8, 156.7, 130.8, 127.8, 113.8, 81.6, 78.0, 55.3, 28.2; HRMS-ESI (m/z): [M+Na]+ calcd. for C13H19NNaO4: 276.1206, found 276.1202; IR (neat): 3285.8 (br), 1715.8, 1245.3, 1162.8, 1099.8, 1032.4 cm−1.
Typical procedure for the alkylation of BocNHOPMB
N-Boc-N-Benzyl-O-(4-methoxybenzyl)hydroxylamine (28d)
Sodium hydride (60% in mineral oil; 400 mg, 10.00 mmol) solid was added to a solution of compound 27 (1.955 g, 7.72 mmol) in 20 mL of anhydrous DMF at 0 °C (ice bath temperature). The solution bubbled and turned faint yellow. After stirring for 30 min, benzylbromide (1.1 mL, 9.26 mmol) was added dropwise over 5 min and the mixture was warmed to rt. After 6 h, no starting material (27) was detected by TLC (10% EtOAc in hexanes). The off-white, opaque mixture was cooled to 0 °C and 10% aq. NaHCO3 (10 mL) was added slowly. The DMF and H2O were removed by high vacuum rotary evaporation (~1 mm Hg). The resulting slurry was partitioned between EtOAc (30 mL) and H2O (30 mL). The layers were separated and the H2O was extracted with EtOAc (1 × 30 mL). The EtOAc layers were combined, washed with brine (1 × 30 mL), dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (1 × 6 in silica gel; 10%–20% EtOAc in hexanes) to give the desired product (28d) in 99% yield as a clear, colorless liquid (2.64 g, 7.69 mmol). 1H-NMR (600 MHz, CDCl3) δ 7.38–7.28 (m, 5 H), 7.21–7.18 (m, 2 H), 6.87–6.84 (m, 2 H), 4.63 (s, 2 H), 4.55 (s, 2 H), 3.81 (s, 3 H), 1.52 (s, 9 H); 13C-NMR (150 MHz, CDCl3) δ 159.7, 156.6, 136.9, 131.0, 128.7, 128.3, 127.7, 127.4, 113.7, 81.4, 76.8, 55.2, 53.9, 28.3; HRMS-ESI (m/z): [M+H]+ calcd. for C20H26NO4: 344.1856, found 344.1851; IR (neat): 3064.4, 3033.1, 3003.0, 1697.9, 1247.4, 1157.1, 1089.4, 1032.2 cm−1.
Typical procedure for the N-Boc-deprotections of N-Boc-N-alkyl-O-(4-methoxybenzyl)hydroxylamines
N-Benzyl-O-(4-methoxybenzyl)hydroxylamine (29d)
Anhydrous TFA (2 mL) was added slowly to a solution of compound 28d (1.14 g, 3.32 mmol) in 8 mL of anhydrous CH2Cl2. The clear, colorless solution bubbled upon addition of TFA. After 15 min, the TFA and CH2Cl2 were evaporated under reduced pressure giving a clear, colorless liquid. This material was dissolved in CHCl3 (25 mL) and washed with satd. aq. NaHCO3 (1 × 50 mL). The CHCl3 was separated, dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (1.25 × 6 in silica gel; 10%–20% EtOAc in hexanes) to give the desired product (29d) in 77% yield as a clear, colorless liquid (623.8 mg, 2.56 mmol). 1H-NMR (600 MHz, CDCl3) δ 7.38–7.28 (m, 5 H), 7.27–7.24 (m, 2 H), 6.89–6.86 (m, 2 H), 5.69 (br s, NH, 1 H), 4.60 (s, 2 H), 4.05 (s, 2 H), 3.81 (s, 3 H); 13C-NMR (150 MHz, CDCl3) δ 159.3, 137.6, 130.1, 129.9, 129.0, 128.4, 127.4, 113.7, 75.9, 56.5, 55.2; HRMS-ESI (m/z): [M+H]+ calcd. for C15H18NO2: 244.1332, found 244.1320; IR (neat): 3260.3 (br), 3062.0, 3030.5, 3001.4, 1611.0, 1511.7, 1244.7, 1031.9 cm−1.
Typical procedure for the Buchwald-Hartwig amination cross-coupling reactions
N-Benzyl-O-(4-methoxybenzyl)-N-(pyridin-2-yl)hydroxylamine (30d)
This procedure followed a general method reported previously by Buchwald and coworkers.47 Hydroxylamine 29d (300.0 mg, 1.23 mmol), 2-bromopyridine (0.10 mL, 1.00 mmol), Pd2(dba)3 (18.0 mg, 0.02 mmol), (±}) BINAP (25.0 mg, 0.04 mmol), and NaOtBu (134.0 mg, 1.40 mmol) were dissolved in 9 mL of anhydrous toluene, respectively, in an oven-dried, argon-flushed Schlenk tube. The brown mixture was heated to 70 °C (oil bath temperature) overnight. The mixture turned darker brown over the course of the reaction. After 64 h, the mixture was cooled to rt, diluted with Et2O (20 mL), and filtered through celite. The clear, orange solution was washed with brine (1 × 20 mL), dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by column chromatography (1 × 6 in silica gel; CH2Cl2 as eluent) to give the desired product (30d) in 63% yield as a clear, colorless liquid (201.2 mg, 0.63 mmol). 1H-NMR (600 MHz, CDCl3) δ 8.31 (ddd, J = 4.8, 1.9, 0.9 Hz, 1 H), 7.60–7.57 (m, 1 H), 7.46–7.44 (m, 2 H), 7.36–7.32 (m, 2 H), 7.32–7.28 (m, 1 H), 7.19–7.16 (m, 2 H), 7.10 (dt, J = 8.2, 0.9 Hz, 1 H), 6.87–6.85 (m, 2 H), 6.82 (ddd, J = 7.3, 4.8, 0.9 Hz, 1 H), 4.87 (s, 2 H), 4.56 (s, 2 H), 3.81 (s, 3 H); 13C-NMR (150 MHz, CDCl3) δ 162.4, 159.6, 147.8, 137.9, 137.6, 130.8, 129.7, 128.1, 128.0, 127.2, 116.3, 113.8, 109.2, 76.2, 57.4, 55.2; HRMS-ESI (m/z): [M+H]+ calcd. for C20H21N2O2: 321.1598, found 321.1599; IR (neat): 3061.7, 3031.0, 3004.1, 1587.0, 1432.8, 1247.3, 1030.9 cm−1; HPLC retention time 9.78 min.
Typical procedure for the PMB-deprotection reactions
N-Benzyl-N-(pyridin-2- yl)hydroxylamine (31d)
Anhydrous TFA (3 mL) was added slowly to a solution of compound 30d (100.0 mg, 0.31 mmol) and triethylsilane (0.10 mL, 0.63 mmol) in 7 mL of anhydrous CH2Cl2. After 11 h, a small aliquot of the solution was quenched with satd. aq. NaHCO3 and no starting material (30d) was detected by TLC (20% EtOAc in hexanes). The solution was diluted with CH2Cl2 (20 mL) and washed with satd. aq. NaHCO3 (1 × 30 mL). The CH2Cl2 was separated, dried over anhydrous MgSO4, filtered, and concentrated to give a faint green liquid. The desired product (31d) was precipitated by trituration with cold pentanes and isolated in 82% yield as white crystals (51.0 mg, 0.26 mmol). Mp 116–118 °C; 1H-NMR (600 MHz, CDCl3) δ 8.25 (d, J = 4.7 Hz, 1 H), 7.64–7.58 (m, 1 H), 7.41 (d, J = 7.0 Hz, 2 H), 7.35 (t, J = 7.3 Hz, 2 H), 7.32–7.28 (m, 1 H), 7.13 (d, J = 8.5 Hz, 1 H), 6.83 (dd, J = 7.0, 5.3 Hz, 1 H), 4.84 (s, 2 H); 13C-NMR (150 MHz, CDCl3) δ 162.1, 146.9, 137.8, 137.0, 129.0, 128.4, 127.5, 116.3, 109.7, 58.7; HRMS-ESI (m/z): [M+H]+ calcd. for C12H13N2O: 201.1022, found 201.1019; IR (neat): 3120.2 (br), 3068.3, 3029.9, 1591.2, 1432.2, 1213.4, 986.2 cm−1; HPLC retention time 6.30 min.
N-Benzylpyridin-2-amine (32)
Compound 31d (2.7 mg, 0.013 mmol) was dissolved in 3 mL of MeOH in a round bottom flask. The flask was charged with 10% Pd-C (0.2 mg) and sealed under argon. The flask was flushed several times with hydrogen gas using intermediate vacuum evacuations. The mixture was then left to stir at rt under a hydrogen atmosphere (1 atm). After 30 min, no starting material (31d) was detected by TLC (20% EtOAc in hexanes). The flask was flushed with argon and the mixture was vacuum filtered through celite. Evaporation of the MeOH gave pure product (32) in 97% yield as a white solid (2.4 mg, 0.013 mmol). 1H-NMR (300 MHz, CDCl3) δ ppm 8.04 (ddd, J = 5.0, 2.0, 1.0 Hz, 1 H), 7.40–7.16 (m, 5 H), 6.53 (ddd, J = 7.1, 5.0, 0.8 Hz, 1 H), 6.31 (dt, J = 8.4, 1.1 Hz, 1 H), 4.87 (br s, NH, 1 H), 4.44 (d, J = 5.8 Hz, 2 H); HRMS-ESI (m/z): [M+H]+ calcd. for C12H13N2: 185.1073, found 185.1066; HPLC retention time 6.42 min. Data was consistent with material from commercial sources (Sigma- Aldrich Co.).
Typical procedure for the O-acetylation of N-alkyl-N-(pyridin-2-yl)hydroxylamines
O-Acetyl- N-benzyl-N-(pyridin-2-yl)hydroxylamine (33)
Compound 31d (8.0 mg, 0.04 mmol), Ac2O (4.9 mg, 0.05 mmol), iPr2EtN (7.8 mg, 0.06 mmol), and catalytic DMAP (1.0 mg, 0.01 mmol) were dissolved in 4 mL of anhydrous CH2Cl2, respectively. After 12 h, no starting material (31d) was detected by TLC (20% EtOAc in hexanes). The CH2Cl2 was evaporated and the crude product was purified by column chromatography (0.25 × 5 in silica gel; 10%–20% EtOAc in hexanes) to give the desired product (33) in 72% yield as a clear, faint yellow oil (7.0 mg, 0.03 mmol). 1H-NMR (300 MHz, CDCl3) δ 8.36 (ddd, J = 4.9, 1.7, 0.8 Hz, 1 H), 7.65–7.57 (m, 1 H), 7.42–7.25 (m, 5 H), 6.94 (ddd, J = 7.2, 4.9, 0.8 Hz, 1 H), 6.80 (dt, J = 8.5, 0.9 Hz, 1 H), 5.01 (s, 2 H), 2.04 (s, 3 H); 13C-NMR (75 MHz, CDCl3) δ 176.3, 160.4, 147.7, 137.9, 135.9, 129.3, 128.3, 127.6, 117.8, 109.9, 57.4, 18.8; HRMS-ESI (m/z): [M+H]+ calcd. for C14H15N2O2: 243.1128, found 243.1137; IR (neat): 3063.1, 3031.2, 3008.4, 1780.6, 1590.0, 1467.2, 1434.7, 1184.1 cm−1; HPLC retention time 7.19 min.
(±) 5-Bromo-N-((1S/R,4S/R)-4-methoxycyclohex-2-enyl)pyridin-2-amine (34)
This compound was prepared following a general method reported previously.48 Activated zinc (65 mg, 1.00 mmol) was added to a solution of Cp2TiCl2 (124.0 mg, 0.50 mmol) in 3 mL of anhydrous THF in a flame-dried round bottom flask purged with argon. This mixture was stirred at rt for 45 min. The reaction mixture changed color from dark red to olive green. The mixture was cooled to −30 °C and a solution of 7b (60.0 mg, 0.20 mmol) in 2 mL of MeOH was added dropwise over 3 min. The mixture was stirred for 1 h while the bath temperature was maintained between −10 and −30 °C. The mixture was warmed to rt and partitioned between satd. aq. K2CO3 (5 mL) and EtOAc (15 mL). The organic layer was separated and filtered through a Whatman glass microfiber filter (type GF/F). The aq. layer was extracted with EtOAc (2 × 10 mL) and the separated organic layer was filtered through a Whatman glass microfiber filter (type GF/F) each time. The combined, filtered organics were dried over Na2SO4, filtered through a Whatman glass microfiber filter (type GF/F), and concentrated. The crude product was purified by column chromatography (0.5 × 6 in silica gel; 25% EtOAc in hexanes) to give the desired product (34) in 73% yield as a white solid (41.6 mg, 0.15 mmol). Mp 50–52 °C; 1H-NMR (600 MHz, DMSO-d6) δ 8.01 (dd, J = 2.5, 0.4 Hz, 1 H), 7.48 (dd, J = 8.9, 2.5 Hz, 1 H), 6.74 (d, J = 6.5 Hz, 1 H), 6.51 (dd, J = 8.9, 0.7, 1 H), 5.86–5.81 (m, 1 H), 5.74–5.70 (m, 1 H), 3.99–3.93 (m, 1 H), 3.71 (td, J = 4.4, 2.0 Hz, 1 H), 3.28 (s, 3 H), 2.12–2.05 (m, 2 H), 1.89–1.81 (m, 1 H), 1.55–1.46 (m, 1 H); 13C-NMR (150 MHz, DMSO-d6) δ 157.1, 147.4, 138.8, 129.9, 126.3, 110.7, 104.7, 77.2, 55.4, 49.1, 25.7, 23.2; HRMS-ESI (m/z): [M+H]+ calcd. for C12H16BrN2O: 283.0441, found 283.0455; IR (neat): 3301.6 (br), 3161.9, 3131.0, 3071.3, 3032.0, 3001.8, 1593.8, 1498.8, 1099.3, 1085.8 cm−1; HPLC retention time 7.48 min.
N-(4-Methoxybenzyloxy)-N-benzyl(pyridin-2-yl)methanamine (38)
Compound 29d (36.0 mg, 0.15 mmol), 2-(bromomethyl)pyridine hydrobromide (56.2 mg, 0.22 mmol), and iPr2EtN (0.10 mL, 0.57 mmol) were dissolved in 6 mL of anhydrous CH3CN, respectively. This clear, red solution was heated to 50 °C (oil bath temperature) overnight. After 17 h, only trace starting material (29d) was detected by TLC (33% EtOAc in hexanes). The reaction mixture was cooled to rt and the CH3CN was evaporated under reduced pressure. The resulting material was purified by silica gel column chromatography (0.75 × 4 in silica gel; 20%–40% EtOAc in hexanes) to give the desired product (38) in 44% yield as a clear, faint yellow oil (22.0 mg, 0.07 mmol). 1H-NMR (600 MHz, CDCl3) δ 8.61 (ddd, J = 4.9, 1.8, 0.9 Hz, 1 H), 7.67 (td, J = 7.6, 1.8 Hz, 1 H), 7.47–7.43 (m, 3 H), 7.37–7.33 (m, 2 H), 7.33–7.28 (m, 1 H), 7.21 (ddd, J = 7.6, 4.9, 1.3 Hz, 1 H), 6.88–6.84 (m, 2 H), 6.76–6.73 (m, 2 H), 4.15 (s, 2 H), 4.07 (s, 2 H), 3.96 (s, 2 H), 3.76 (s, 3 H); 13C-NMR (150 MHz, CDCl3) δ 159.2, 157.8, 149.2, 137.4, 136.1, 130.5, 130.0, 129.1, 128.1, 127.3, 124.2, 122.2, 113.5, 75.4, 64.7, 63.2, 55.2; HRMS-ESI (m/z): [M+H]+ calcd. for C21H23N2O2: 335.1754, found 335.1738; IR (neat): 3062.3, 3031.4, 3007.4, 1611.97, 1588.8, 1513.3, 1247.4, 1032.5 cm−1; HPLC retention time 8.81 min.
Microbiology
General materials and methods
All liquids and media were sterilized by autoclaving (121 °C, 15 min) before use. All aq. solutions and media were prepared using distilled, deionized, and filtered water (Millipore Milli-Q Advantage A10 Water Purification System). Luria broth (LB) was purchased from VWR. Mueller-Hinton No. 2 broth (MHII broth; cation adjusted) was purchased from Sigma-Aldrich (St. Louis, MO). Mueller-Hinton No. 2 agar (MHII agar; HiMedia Laboratories) was purchased from VWR. McFarland BaSO4 turbidity standards were purchased from bioMerieux, Inc. Sterile plastic petri dishes (145 mm × 20 mm; Greiner Bio-One) were purchased from VWR. Ciprofloxacin was purchased from Sigma-Aldrich (St. Louis, MO). Amphotericin B was purchased from Amresco (Solon, OH). All test organisms used in this work were from sources given in the supporting information (Table S3).
Antibiotic susceptibility testing by the agar diffusion method
Antibacterial activity of the compounds was determined by a modified Kirby-Bauer agar diffusion assay.50 Overnight cultures of test organisms were grown in LB broth for 18–24 h and standard suspensions of 1.5 × 106 CFU/mL were prepared in saline solution (0.9% NaCl) according to a 0.5 BaSO4 McFarland Standard.76 Each standardized suspension (0.1 mL) was added to 34 mL of sterile, melted MHII agar tempered to 47–50 °C. After gentle mixing, the inoculated agar media was poured into a sterile plastic petri dish (145 mm × 20 mm) and allowed to solidify near a flame with the lid cracked for ~30 min. Wells of 9.0 mm diameter were cut from the petri dish agar and filled with exactly 50 μL of the test sample solution. The petri dish was incubated at 37 °C for 18–24 h and the inhibition zone diameters were measured (mm) with an electronic caliper after 24–48 h.
Antibiotic susceptibility testing by the broth microdilution method
Antibacterial activity of the compounds was determined by measuring their minimum inhibitory concentrations (MIC90’s) using the broth microdilution method according to the Clinical and Laboratory Standards Institute (CLSI, formerly the NCCLS) guidelines.52 Each well of a 96-well microtiter plate was filled with 50 μL of sterile MHII broth. Each test compound was dissolved in DMSO making a 20 mM solution, then diluted with sterile MHII broth to 512 μM. Exactly 50 μL of the compound solution was added to the first well of the microtiter plate and 2-fold serial dilutions were made down each row of the plate. Exactly 50 μL of bacterial inoculum (5 × 105 CFU/mL in MHII broth) was then added to each well giving a total volume of 100 μL/well and a final compound concentration gradient of 128 μM–0.0625 μM. The plate was incubated at 37 °C for 18 h and then each well was examined for bacterial growth. The MIC90 was recorded as the lowest compound concentration (μM) required to inhibit 90% of bacterial growth as judged by turbidity of the culture media relative to a row of wells filled with a DMSO standard. Ciprofloxacin was included in a control row at a concentration gradient of 32 μM–0.0156 μM.
Supplementary Material
Acknowledgments
We gratefully acknowledge the use of NMR facilities provided by the Lizzadro Magnetic Resonance Research Center at The University of Notre Dame (UND) and the mass spectrometry services provided by The UND Mass Spectrometry & Proteomics Facility (Mrs. N. Sevova, Dr. W. Boggess, and Dr. M. V. Joyce; supported by the National Science Foundation under CHE-0741793). We thank Mrs. Patricia A. Miller (UND) for anticancer testing and assistance with antibacterial susceptibility testing. We thank Dr. Ute Mollmann (HKI) for antibacterial testing against the resistant strains S. aureus 134/94 (MRSA) and E. faecalis 1528 (VRE) and for many helpful discussions. We thank Dr. Viktor Krchňak (UND) for assistance with analytical and preparative HPLC and for helpful discussions. TAW gratefully acknowledges The UND Chemistry-Biochemistry-Biology Interface (CBBI) Program funded by NIH (T32GM075762) for three years of fellowship support. TAW also thanks The UND Department of Chemistry and Biochemistry (Grace Fellowship), the Center for Environmental Science and Technology (CEST), and Bayer for additional fellowship support. BY acknowledges a Reilly Graduate Fellowship and JRR acknowledges a College of Science Summer Undergraduate Research Fellowship (COS-SURF), both sponsored by UND. We acknowledge The University of Notre Dame and the NIH (GM 075855) for supporting this work.
Footnotes
Abbreviations: Ac, acetyl; ATCC, American Type Culture Collection; BINAP, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; Bn, benzyl; Boc, tert-butoxycarbonyl; CFU, colony forming unit; Cp, cyclopentadienyl; dba, dibenzylideneacetone; DMAP, 4-dimethylaminopyridine; DMF, dimethylformamide; DMSO, dimethylsulfoxide; DOS, diversity-oriented synthesis; DXR, 1-deoxy-D-xylulose-5-phosphate reductoisomerase; ESI, electrospray ionization; FBDD, fragment-based drug design; HPLC, high-performance liquid chromatography; HRMS, high-resolution mass spectrometry; HTS, high-throughput screening; IR, infrared; MEND, modular enhancement of nature’s diversity; MEP, methylerythritol phosphate; MHII, Mueller-Hinton Media No. II; MIC, minimum inhibitory concentration; MRSA, methicillin-resistant Staphylococcus aureus; NMR, nuclear magnetic resonance; PG, protecting group; PMB, 4-methoxybenzyl; SAR, structure-activity relationship; TFA, trifulouroacetic acid; THF, tetrahydrofuran; TLC, thin layer chromatography; VRE, vancomycin-resistant Enterococci;
Supporting Information Available
Complete list of compounds subjected to biological testing (Table S1). Complete list of biological data collected during this study (Table S2). Complete list of strains, markers, and origins for microorganisms used in this work (Table S3). Experimental procedures for the preparation of unpublished compounds not given in main article text. Copies of 1H-NMR and 13C-NMR spectra for all unpublished compounds (9,11a, 12, 14, 16, 18a–i, 19a–c, 27, 28c–i, 29a–i, 30a–i, 31a–i, 33–40a). Copies of HPLC chromatograms for compounds subjected to biological testing (7a, 7b, 9, 11a, 12, 14, 16, 18a–i, 19a–c, 30a–i, 31a–i, 32–40a). Crystal data, images of unit cell, and tables of atom coordinates and thermal parameters for the X-ray diffraction analysis of compound 40a. This material is available free of charge via the Internet at http://pubs.acs.org. CCDC 824255 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Jr, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clinical Infectious Diseases. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J., Jr The Epidemic of Antibiotic-Resistant Infections: A Call to Action for the Medical Community from the Infectious Diseases Society of America. Clinical Infectious Diseases. 2008;46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 3.Bad Bugs, No Drugs As Antibiotic Discovery Stagnates, A Public Health Crisis Brews. [Accessed April 4, 2011.];Infectious Disease Society of America (ISDA) Report. 2004 July; Available at: < www.idsociety.org>.
- 4.Laxminarayan R, Malani A. Resources for the Future. Washington, DC: 2007. Extending the Cure: Policy Responses to the Growing Threat of Antibiotic Resistance; pp. 1–175. [Google Scholar]
- 5.Fischback MA, Walsh CT. Antibiotics for Emerging Pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Testero SA, Fisher JF, Mobashery S. β-Lactam Antibiotics. Burger’s Medicinal Chemistry. In: Abraham DJ, Rotella DP, editors. Drug Discovery and Development. 7. Wiley; Hoboken, NJ: 2010. pp. 259–404. [Google Scholar]
- 7.Silver LL. Multi-Targeting by Monotherapeutic Antibacterials. Nat Rev Drug Discovery. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber SL. Small Molecules: The Missing Link in the Central Dogma. Nat Chem Biol. 2005;1:64–66. doi: 10.1038/nchembio0705-64. [DOI] [PubMed] [Google Scholar]
- 9.Amer FA, El-Behedy EM, Mohtady HA. New Targets for Antibacterial Agents. Biotechnol Mol Biol Rev. 2008;3:46–57. [Google Scholar]
- 10.Miller JR, Dunham S, Mochalkin I, Banotai C, Bowman M, Buist S, Dunkle B, Hanna D, Harwood HJ, Huband MD, Kamovsky A, Kuhn M, Limberakis C, Liu JY, Mehrens S, Mueller WT, Narasimhan L, Ogden A, Ohren J, Prasad JVNV, Shelly JA, Skerlos L, Sulavik M, Thomas VH, VanderRoest S, Wang L, Wang Z, Whitton A, Zhu T, Stover CK. A Class of Selective Antibacterials Derived from a Protein Kinase Inhibitor Pharmacophore. Proc Nat Acad Sci US A. 2009;106:1737–1742. doi: 10.1073/pnas.0811275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh CT, Fischbach MA. Repurposing Libraries of Eukaryotic Protein Kinase Inhibitors for Antibiotic Discovery. 2009;106:1689–1690. doi: 10.1073/pnas.0813405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silver LL. Are Natural Products Still the Best Source for Antibacterial Discovery? The Bacterial Entry Factor. Expert Opin Drug Discovery. 2008;3:487–500. doi: 10.1517/17460441.3.5.487. [DOI] [PubMed] [Google Scholar]
- 13.O’Shea R, Moser HE. Physiochemical Properties of Antibacterial Compounds: Implications in Drug Discovery. J Med Chem. 2008;51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 14.For reviews of FBDD see: Murray CW, Rees DC. The Rise of Fragment-Based Drug Discovery. Nat Chem. 2009;1:187–192. doi: 10.1038/nchem.217.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent Developments in Fragment-Based Drug Discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373.Erlanson DA, McDowell RS, O’Brien T. Fragment-Based Drug Discovery. J Med Chem. 2004;47:3463–3482. doi: 10.1021/jm040031v.
- 15.For successful application of FBDD for antibacterial discovery see: Mochalkin I, Miller JR, Narasimhan L, Thanabal V, Erdman P, Cox PB, Prasad JVNV, Lightle S, Huband MD, Stover CK. Discovery of Antibacterial Biotin Carboxylase Inhibitors by Virtual Screening and Fragment-Based Approaches. ACS Chem Biol. 2009;4:473–483. doi: 10.1021/cb9000102.Waldrop GL. Smaller is Better for Antibiotic Discovery. ACS Chem Biol. 2009;4:397–399. doi: 10.1021/cb900122j.
- 16.For successful application of computational structure-based design methods for antibacterial discovery see: Shen Y, Liu J, Estiu G, Isin B, Ahn YY, Lee DS, Barabasi AL, Kapatral V, Wiest O, Oltvai ZN. Blueprint for Antimicrobial Hit Discovery Targeting Metabolic Networks. Proc Nat Acad Sci US A. 2010;107:1082–1087. doi: 10.1073/pnas.0909181107.
- 17.For successful applications of target-based HTS for antibacterial discovery see: Jarvest RL, Berge JM, Berry V, Boyd HF, Brown MJ, Elder JS, Forrest AK, Fosberry AP, Gentry DR, Hibbs MJ, Jaworski DD, O’Hanlon PJ, Pope AJ, Rittenhouse S, Sheppard RJ, Slater-Radosti C, Worby A. Nanomolar Inhibitors of Staphylococcus aureus Methionyl tRNA Synthetase with Potent Antibacterial Activity Against Gram-Positive Pathogens. J Med Chem. 2002;45:1959–1962. doi: 10.1021/jm025502x.Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, DeWolf WE, Jr, Fosberry AP, Greenwood R, Head MS, Heerding DA, Janson CA, Jaworski DD, Keller PM, Manley PJ, Moore TD, Newlander KA, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Slater-Radosti C, Salyers KL, Seefeld MA, Smyth MG, Takata DT, Uzinskas IN, Vaidya K, Wallis NG, Winram SB, Yuan CCK, Huffman WF. Discovery of a Novel and Potent Class of FabI-Directed Antibacterial Agents. Antimicrob Agents Chemother. 2002;46:3118–3124. doi: 10.1128/AAC.46.10.3118-3124.2002.Fan F, Yan K, Wallis NG, Reed S, Moore TD, Rittenhouse SF, DeWolf WE, Jr, Huang J, McDevitt D, Miller WH, Seefeld MA, Newlander KA, Jakas DR, Head MS, Payne DJ. Defining and Combating the Mechanisms of Triclosan Resistance in Clinical Isolates of Staphylococcus aureus. Antimicrob Agents Chemother. 2002;46:3343–3347. doi: 10.1128/AAC.46.11.3343-3347.2002.
- 18.Walsh DP, Chang YT. Chemical Genetics. Chem Rev. 2006;106:2476–2530. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]
- 19.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat Rev Drug Discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 20.As of 2007, only one combinatorial chemistry-derived compound has been approved by the FDA: Newman DJ, Cragg GM. Natural Products as Sources of New Drugs over the Last 25 Years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v.
- 21.Schreiber SL. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 22.Burke MD, Schreiber SL. A Planning Strategy for Diversity-Oriented Synthesis. Ang Chem, Int Ed. 2004;43:46–58. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 23.For application of DOS for antibacterial discovery and definitions of structural diversity see: Galloway WRJD, Bender A, Welch M, Spring DR. The Discovery of Antibacterial Agents Using Diversity-Oriented Synthesis. Chem Comm. 2009:2446–2462. doi: 10.1039/b816852k.
- 24.Yang B, Miller MJ. Regio- and Stereoselective Indium Triflate-Mediated Nucleophilic Ring-Opening Reactions of 3-Aza-2-oxabicyclo[2.2.1]hept-5-ene and –[2.2.2]oct-5-ene Systems. J Org Chem. 2009;74:7990–7993. doi: 10.1021/jo9016343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang B, Miller MJ. Regio- and Stereochemically Controlled Formation of Hydroxamic Acids From Indium Triflate-Mediated Nucleophilic Ring-Opening Reactions with Acylnitroso-Diels-Alder Adducts. Tetrahedron Lett. 2010;51:889–891. doi: 10.1016/j.tetlet.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang B, Miller MJ. Iminonitroso Ene Reactions: Experimental Studies on Reactivity, Regioselectivity, and Enantioselectivity. Tetrahedron Lett. 2010;51:328–331. doi: 10.1016/j.tetlet.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogt PF, Miller MJ. Development and Applications of Amino Acid-Derived Chiral Acylnitroso Hetero Diels-Alder Reactions. Tetrahedron. 1998;54:1317–1348. [Google Scholar]
- 28.Li F, Yang B, Miller MJ, Zajicek J, Noll BC, Mollmann U, Dahse HM, Miller PA. Iminonitroso Diels-Alder Reactions for Efficient Derivatization and Functionalization of Complex Diene-Containing Natural Products. Org Lett. 2007;9:2923–2926. doi: 10.1021/ol071322b. [DOI] [PubMed] [Google Scholar]
- 29.Krchňak V, Waring KR, Noll BC, Moellmann U, Dahse HM, Miller MJ. Evolution of Natural Product Scaffolds by Acyl- and Arylnitroso Hetero-Diels-Alder Reactions: New Chemistry on Piperine. J Org Chem. 2008;73:4559–4567. doi: 10.1021/jo8004827. [DOI] [PubMed] [Google Scholar]
- 30.Yang B, Miller PA, Mollmann U, Miller MJ. Syntheses and Biological Activity Studies of Novel Sterol Analogs from Nitroso Diels-Alder Reactions of Ergosterol. Org Lett. 2009;11:2828–2831. doi: 10.1021/ol900997t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bodnar B, Miller MJ. The Nitrosocarbonyl Hetero-Diels-Alder Reaction as a Useful Tool for Organic Syntheses. Ang Chem Int Ed. 2011;50:5630–5647. doi: 10.1002/anie.201005764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krchňak V, Zajiček J, Miller PA, Miller MJ. Selective Molecular Sequestration with Concurrent Natural Product Functionalization and Derivatization: From Crude Natural Product Extracts to a Single Natural Product Derivative in One Step. 2011 doi: 10.1021/jo201361s. To be submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krchňak V, Moellmann U, Dahse HM, Miller MJ. Solid-Supported Nitroso Hetero-Diels-Alder Reactions. 3. Acid-Mediated Transformation of Cycloadducts by Scission of the Oxazine C-O Bonds. J Comb Chem. 2008;10:112–117. doi: 10.1021/cc700142d. [DOI] [PubMed] [Google Scholar]
- 34.Credito K, Lin G, Ednie LM, Appelbaum PC. Antistaphylococcal Activity of LBM415, a New Peptide Deformylase Inhibitor, Compared with Those of Other Agents. Antimicrob Agents Chemother. 2004;48:4033–4036. doi: 10.1128/AAC.48.10.4033-4036.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, Margolis PS, Wu C, Ni ZJ, Trias J, White RJ, Yuan Z. Actinonin, a Naturally Occurring Antibacterial Agent, Is a Potent Deformylase Inhibitor. Biochemistry. 2000;39:1256–1262. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- 36.Mine Y, Kamimura T, Nonoyama S, Nishida M, Goto S, Kuwahara S. In Vitro and In Vivo Antibacterial Activities of FR-31564, a New Phosphonic Acid Antibiotic. J Antibiot. 1980;33:36–43. doi: 10.7164/antibiotics.33.36. [DOI] [PubMed] [Google Scholar]
- 37.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial Agents That Inhibit Lipid A Biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 38.Woulfe SR, Miller MJ. Heteroatom Activated β-Lactam Antibiotics: Synthesis of Biologically Active Substituted N-Oxy-3-amino-2-azetidinones (Oxamazins) Tetrahedron Lett. 1984;25:3293–3296. [Google Scholar]
- 39.Muci AR, Buchwald SL. Practical Palladium Catalysts for C-N and C-O Bond Formation. Top Curr Chem. 2002;219:131–209. [Google Scholar]
- 40.Hartwig JF. Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides. Acc Chem Res. 2008;41:1534–1544. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roosenberg JM, Miller MJ. Total Synthesis of the Siderophore Danoxamine. J Org Chem. 2000;65:4833–4838. doi: 10.1021/jo000050m. [DOI] [PubMed] [Google Scholar]
- 42.Gebhardt P, Crumbliss AL, Miller MJ, Mollmann U. Synthesis and Biological Activity of Saccharide Based Lipophilic Siderophore Mimetics as Potential Growth Promoters for Mycobacteria. Biometals. 2008;21:41–51. doi: 10.1007/s10534-007-9091-x. [DOI] [PubMed] [Google Scholar]
- 43.Yan L, Kahne D. p-Methoxybenzyl Ethers as Acid-Labile Protecting Groups in Oligosaccharide Synthesis. Synlett. 1995:523–524. [Google Scholar]
- 44.Zhu G, Yang F, Balachandran R, Hook P, Vallee RB, Curran DP, Day BW. Synthesis and Biological Evaluation of Purealin and Analogues as Cytoplasmic Dynein Heavy Chain Inhibitors. J Med Chem. 2006;49:2063–2076. doi: 10.1021/jm051030l. [DOI] [PubMed] [Google Scholar]
- 45.Schumann EL, Heinzelman RV, Greig ME, Veldkamp W. Hydroxylamine Chemistry. IV. O-Aralkylhydroxylamines. J Med Chem. 1964;7:329–334. doi: 10.1021/jm00333a017. [DOI] [PubMed] [Google Scholar]
- 46.McKay AF, Garmaise DL, Paris GY, Gelblum S. Bacteriostats. III. Oxyamines and Their Derivatives. Can J Chem. 1960;38:343–358. [Google Scholar]
- 47.Wagaw S, Buchwald SL. The Synthesis of Aminopyridines: A Method Employing Palladium-Catalyzed Carbon-Nitrogen Bond Formation. J Org Chem. 1996;61:7240–7241. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]
- 48.Cesario C, Tardibono LP, Miller MJ. Titanocene(III) Chloride-Mediated Reductions of Oxazines, Hydroxamic Acids, and N-Hydroxy Carbamates. J Org Chem. 2009;74:448–451. doi: 10.1021/jo802184y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.For the original descriptions of the Kirby-Bauer agar diffusion assay see: Bauer AW, Perry DM, Kirby WMM. Single Disc Antibiotic Sensitivity Testing of Staphylococci: An Analysis of Technique and Results. Arch Int Med. 1959;104:208–216. doi: 10.1001/archinte.1959.00270080034004.Bauer AW, Kirby WMM, Sherris JC, Turck M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am J Clin Pathol. 1966;45:493–496.
- 50.For a specific example general description of the modified Kirby-Bauer agar diffusion assay used in this study see: Afonin S, Glaser RW, Berditchevskaia M, Wadhwani P, Guhrs K-H, Mollmann U, Perner A, Ulrich AS. 4-Fluoro-phenylglycine as a Label for 19F-NMR Structure Analysis of Membrane Associated Peptides. ChemBioChem. 2003;4:1151–1163. doi: 10.1002/cbic.200300568.
- 51.Ciprofloxacin [package insert] Bayer HealthCare Pharmaceuticals, Inc; Wayne, NJ: Apr, 2009. [Accessed March 8, 2011.]. http://www.univgraph.com/bayer/inserts/ciprotab.pdf. [Google Scholar]
- 52.Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. 8. Villanova, PA, USA: Clinical and Laboratory Standards Institute (CLSI); 2009. approved standard document M07–A7. [Google Scholar]
- 53.Eaton RW, Ribbons DW. Metabolism of Dibutylphthalate and Phthalate by Micrococcus sp. Strain 12B. J Bacteriol. 1982;151:48–57. doi: 10.1128/jb.151.1.48-57.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukamolova GV, Murzin AG, Salina EG, Demina GR, Kell DB, Kaprelyants AS, Young M. Muralytic Activity of Micrococcus luteus Rpf and its Relationship to Physiological Activity in Promoting Bacterial Growth and Resuscitation. Mol Microbiol. 2006;59:84–98. doi: 10.1111/j.1365-2958.2005.04930.x. [DOI] [PubMed] [Google Scholar]
- 55.Saito Y, Ogura K. Biosynthesis of Menaquinones. Enzymatic Prenylation of 1,4-Dihydroxy-2-Naphthoate by Micrococcus luteus Membrane Fractions. J Biochem. 1981;89:1445–1452. doi: 10.1093/oxfordjournals.jbchem.a133337. [DOI] [PubMed] [Google Scholar]
- 56.Garcia-Lopez M–L, Santos J–A, Otero A. Micrococcus. In: Robinson RK, Batt CA, Patel PD, editors. Encyclopedia of Food Microbiology. Vol. 2. Academic Press; San Diego, CA: 2000. pp. 1344–1350. [Google Scholar]
- 57.Friedman M, Buick R, Elliott CT. Antimicrobial Activities of Plant Compounds Against Antibiotic-Resistant Micrococcus luteus. Int J Antimicrob Agents. 2006;28:151–158. doi: 10.1016/j.ijantimicag.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 58.Smith KJ, Neafie R, Yeager J, Skelton HG. Micrococcus folliculitis in HIV-1 Disease. Br J Dermatol. 1999;141:558–561. doi: 10.1046/j.1365-2133.1999.03060.x. and references therein. [DOI] [PubMed] [Google Scholar]
- 59.Yang S, Sugawara S, Monodane T, Nishijima M, Adachi Y, Akashi S, Miyake K, Hase S, Takada H. Micrococcus luteus Teichuronic Acids Activate Human and Murine Monocyte Cells in a CD14− and Toll-Like Receptor 4-Dependent Manner. Infect Immun. 2001;69:2025–2030. doi: 10.1128/IAI.69.4.2025-2030.2001. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.For recent examples of selective antibacterial agents see: Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AHJ, Oldfield E. A Cholesterol Biosynthesis Inhibitor Blocks Staphylococcus aureus Virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018.Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. An Inhibitor of FtsZ with Potent and Selective Anti-Staphylococcal Activity. Science. 2008;321:1673–1675. doi: 10.1126/science.1159961.
- 61.Adam W, Krebs O. The Nitroso Ene Reaction: A Regioselective and Stereoselective Allylic Nitrogen Functionalization of Mechanistic Delight and Synthetic Potential. Chem Rev. 2003;103:4131–4146. doi: 10.1021/cr030004x. [DOI] [PubMed] [Google Scholar]
- 62.Hwu JR, Tsay S–C, Chen B–L, Patel HV, Chou C–T. N-Arylalkyl-N-phenylhydroxylamines as Novel Photo-Induced DNA-Cleaving Agents. J Chem Soc, Chem Commun. 1994:1427–1428. [Google Scholar]
- 63.Kovacic P, Somanathan R. Novel, Unifying Mechanism for Aromatic Primary-Amines (Therapeutics, Carcinogens and Toxins): Electron Transfer, Reactive Oxygen Species, Oxidative Stress and Metabolites. Med Chem Comm. 2011;2:106–112. [Google Scholar]
- 64.Yang B, Lin W, Krchňak V, Miller MJ. Retro Iminonitroso Diels-Alder Reactions: Interconversion of Nitroso Cycloadducts. Tetrahedron Lett. 2009;50:5879–5883. doi: 10.1016/j.tetlet.2009.07.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.For examples of N-2-pyridylhydroxylamine metal chelation see: Okazawa A, Hashizume D, Ishida T. Ferro- and Antiferromagnetic Coupling Switch Accompanied by Twist Deformation Around the Copper(II) and Nitroxide Coordination Bond. J Am Chem Soc. 2010;132:11516–11524. doi: 10.1021/ja102163d.Okazawa A, Ishida T. Spin-Transition-Like Behavior on One Side in a Nitroxide-Copper(II)-Nitroxide Triad System. Inorg Chem. 2010;49:10144–10147. doi: 10.1021/ic101536q.Osanai K, Okazawa A, Nogami T, Ishida T. Strong Ferromagnetic Exchange Couplings in Copper(II) and Nickel(II) Complexes with a Paramagnetic Tridentate Chelate Ligand, 2,2′-Bipyridin-6-yl tert-Butyl Nitroxide. J Am Chem Soc. 2006;128:14008–14009. doi: 10.1021/ja065623c.Okazawa A, Nagaichi Y, Nogami T, Ishida T. Magneto-Structure Relationship in Copper(II) and Nickel(II) Complexes Chelated with Stable tert-Butyl 5-Phenyl-2-pyridyl Nitroxide and Related Radicals. Inorg Chem. 2008;47:8859–8868. doi: 10.1021/ic800937n.
- 66.For a review on the metal chelation properties of pyridyl oximes see: Milios CJ, Stamatatos TC, Perlepes SP. The Coordination Chemistry of Pyridyl Oximes. Polyhedron. 2006;25:134–194.
- 67.Kuzuyama T, Shimizu T, Takahashi S, Seto H. Fosmidomycin, a Specific Inhibitor of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase in the Nonmevalonate Pathway for Terpenoid Biosynthesis. Tetrahedron Lett. 1998;39:7913–7916. [Google Scholar]
- 68.Haemers T, Wiesner J, Van Poecke S, Goeman J, Henschker D, Beck E, Jomaa H, Van Calenbergh S. Synthesis of α-Substituted Fosmidomycin Analogues as Highly Potent Plasmodium falciparum Growth Inhibitors. Bioorg Med Chem Lett. 2006;16:1888–1891. doi: 10.1016/j.bmcl.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 69.Deng L, Endo K, Kato M, Cheng G, Yajima S, Song Y. Structures of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase/Lipophilic Phosphonate Complexes. ACS Med Chem Lett. 2011;2:165–170. doi: 10.1021/ml100243r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yajima S, Hara K, Sanders JM, Yin F, Ohsawa K, Wiesner J, Jomaa H, Oldfield E. Crystallographic Structures of Two Bisphosphonate: 1-Deoxyxylulose-5-Phosphate Reductoisomerase Complexes. J Am Chem Soc. 2004;126:10824–10825. doi: 10.1021/ja040126m. [DOI] [PubMed] [Google Scholar]
- 71.Deng L, Sundriyal S, Rubio V, Shi ZZ, Song Y. Coordination Chemistry Based Approach to Lipophilic Inhibitors of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase. J Med Chem. 2009;52:6539–6542. doi: 10.1021/jm9012592. [DOI] [PubMed] [Google Scholar]
- 72.Hunter WN. The Non-Mevalonate Pathway of Isoprenoid Precursor Biosynthesis. J Biol Chem. 2007;282:21573–21577. doi: 10.1074/jbc.R700005200. [DOI] [PubMed] [Google Scholar]
- 73.Testa CA, Brown MJ. The Methylerythritol Phosphate Pathway and its Significance as a Novel Drug Target. Curr Pharm Biotechnol. 2003;4:248–259. doi: 10.2174/1389201033489784. [DOI] [PubMed] [Google Scholar]
- 74.Heath RJ, White SW, Rock CO. Lipid Biosynthesis as a Target for Antibacterial Agents. Prog Lipid Res. 2001;40:467–497. doi: 10.1016/s0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 75.Taylor EC, Tseng CP, Rampal JB Conversion of a Primary Amino Group into a Nitroso Group. Synthesis of Nitroso-Substituted Heterocycles. J Org Chem. 1982;4:552–555. [Google Scholar]
- 76.Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH. Manual of Clinical Microbiology. 7. American Society for Microbiology; Washington, DC: 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.