

Abstract
The neurological effects of organophosphate pesticides, commonly used on foods and in households, are an important public health concern. Furthermore, subclinical exposure to combinations of organophosphates is implicated in Gulf War illness. Here we characterized the effects of the broadly-used insecticide chlorpyrifos on dopamine and glutamatergic neurotransmission effectors in corticostriatal motor/reward circuitry. Chlorpyrifos potentiated PKA-dependent phosphorylation of the striatal protein DARPP-32 and the GluR1 subunit of AMPA receptors in mouse brain slices. It also increased GluR1 phosphorylation by PKA when administered systemically. This correlated with enhanced glutamate release from cortical projections in rat striatum. Similar effects were induced by the sarin congener, diisopropyl fluorophosphate, alone or in combination with the putative neuroprotectant, pyridostigmine bromide and the pesticide DEET. This combination, meant to mimic the neurotoxicant exposure encountered by veterans of the 1991 Persian Gulf War, also induced hyperphosphorylation of the neurofibrillary tangle-associated protein tau. Diisopropyl fluorophosphate and pyrodostigmine bromide, alone or in combination, also increased the aberrant activity of the protein kinase, Cdk5, as indicated by conversion of its activating cofactor p35 to p25. Thus consistent with recent findings in humans and animals, organophosphate exposure causes dysregulation in the motor/reward circuitry and invokes mechanisms associated with neurological disorders and neurodegeneration.
Keywords: insecticide, dopamine, neurotoxicity, organophosphate, chlorpyrifos, Gulf War Illness
A wide variety of pesticides are known to have neurotoxic properties. For example, prenatal exposure to a class of compounds known as organophosphates (OP) reduces child IQ (Bouchard et al. 2011), impairs mental development (Engel et al. 2011), and causes cognitive deficits (Eaton et al. 2008; Bouchard et al. 2010; Rauh et al. 2011). Furthermore, exposure to cholinesterase-inhibiting compounds including OP, pesticides, pyridostigmine bromide (PB, an anti-nerve agent medication), and low-level sarin nerve gas is linked epidemiologically with a chronic multi-symptom illness in veterans from the Persian Gulf War (Staines 2005; Thomas et al. 2006; Haley et al. 2009). Symptoms include cognitive disturbances, muscle fatigue, fever, diarrhea, and insomnia.
OP nerve agents and insecticides disrupt cholinergic neurotransmission by inhibiting acetylcholinesterase (AChE) (Wiener and Hoffman 2004). Acetylcholine (ACh) is the major excitatory neurotransmitter at neuromuscular junctions of the peripheral nervous system and a widely distributed first messenger throughout the central nervous system. Termination of cholinergic transmission is completely dependent upon active AChE. Inactivation of AChE by OP increases nervous system levels of ACh, overstimulating both nicotinic (nAChR) and muscarinic (mAChR) receptors (Felder et al. 2000). Acute high level exposure to OP induces seizures, respiratory failure, coma, and death (Wiener and Hoffman 2004). Low level exposures to chlorpyrifos (CPF), the most well-known OP insecticide in agriculture and domestic use, alters the expression of genes involved in cell growth and differentiation, cAMP-related signaling, neurotransmitter synthesis and release, and receptors that target the actions of many neurotransmitters including ACh and dopamine (Auman et al. 2000; Slotkin and Seidler 2007; Eells and Brown 2009).
Dopaminergic pathways within the mesocorticolimbic circuitry mediate reward learning, motivation, and motor control (Philibin et al. 2011). At the hub of this circuitry, striatal medium spiny neurons (MSNs) receive cholinergic input via α4β2 nAChRs and α7 mAChRs at dopaminergic and glutamatergic terminals, respectively (Nomikos et al. 2000; Wonnacott et al. 2000). Furthermore, nicotine exhibits differential dose-dependent actions on pre-synaptic nAChRs, stimulating dopamine release and activation of D1- and D2-dependent signaling pathways in the neostriatum (Hamada et al. 2004). Interestingly, organophosphate exposure is linked to ADHD epidemiology, a disorder that targets corticostriatal circuitry (Bouchard et al. 2010; Drerup et al. 2010). Furthermore, single photon emission computed tomography (SPECT) revealed abnormal brain responses to cholinergic challenges in the basal ganglia of Gulf War veterans (Haley et al. 2009).
These findings suggest that the harmful effects of OP exposure are due, at least in part, to dysregulation of dopamine neurotransmission and neurotoxic effects on striatal neurons. Therefore, we assessed the effects of AChE-inhibiting agents on neostriatal dopamine signal transmission. The results revealed dysregulation of the D1 receptor/cAMP/PKA signaling pathway, potentiation of corticostriatal glutamatergic transmission, hyperphosphorylation of tau, and induction of aberrant activity of the neuronal protein kinase Cdk5. These data define novel molecular targets that may mediate OP neurotoxicity and provide an avenue for the development of therapeutic treatments following nerve agent exposure.
Experimental procedures
Materials
Adenosine deaminase, anti-phospho-tau (P-T205), CPF, diisopropyl fluorophosphate (DFP), N,N-Diethyl-meta-toluamide (DEET), N-Methyl-D-Aspartic Acid (NMDA), pyridostigmine bromide (PB), and R(+)-SCH-23390 hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phospho-DARPP-32 (P-T34), phospho-DARPP-32 (P-T75), and DARPP-32 antibodies were from Cell Signaling Technology, Inc. (Beverly, Massachusetts, USA). Chlorpyrifos oxon (CPO) was from Chem Service Inc. (West Chester, PA, USA), glutamate receptor 1 (GluR1) subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antibody from Abcam (Cambrigde, MA, USA), anti-phospho-GluR1 (P-S845) from PhosphoSolutions (Aurora, CO, USA), mouse (monoclonal) anti-tau from Biosource (Camarillo, CA, USA), and P35 (C-19) sc-820 antibody from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Goat anti-mouse IgG and goat anti-rabbit IgG peroxidase conjugated secondary antibodies were from Thermo Fisher Scientific (Rockford, IL). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animals
All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center or the CDC-NIOSH. For experiments involving systemic injections with nerve agents, drugs were administered via subcutaneous (s.c.) or intraperitoneal (i.p.) route to adult male FVB or adult male or female C57BL/6 mice at the indicated dose levels. For studies of the systemic effects of CPF, female C57BL/6 mice were dosed daily for 7 d (30 mg/kg, s.c.). On the seventh day, the mice were sacrificed 2 h following CPF treatment and striatal tissue was rapidly dissected for immunoblotting. Vehicle (Veh) treated mice received corn oil alone. Chlorpyrifos was dissolved in dimethylsulfoxide (DMSO) (for slice pharmacology experiments), and corn oil or peanut oil (for systemic injections). All experiments involving the use of DEET and DFP were performed at the CDC-NIOSH. In those experiments, for evaluation of phospho-Ser845 GluR1 and phospho-Thr205 tau, male FVB mice were dosed daily for 15 d with PB (2.5 mg/kg, s.c.) and DEET (5 mg/kg, s.c.). On the fifteenth day, a challenge dose of DFP (4 mg/kg, i.p.) was given and the mice were sacrificed 2 h following DFP for immunoblot analysis. The mice were sacrificed by focused microwave fixation. For evaluation of p25 levels, the dose of DEET was increased to 30 mg/kg (s.c.). Control mice received vehicle only. Mice that received DFP injections exhibited seizure-like behaviors and mortality was approximately 10% among all DFP treatment groups. For studies of the systemic effect of PB following a delay period from the time of exposure, male C57BL6 mice were dosed daily for 10 d (1 mg/kg, s.c.) and sacrificed 4 weeks after the last treatment for immunoblotting. Pyridostigmine bromide, diisopropyl fluorophosphates, and N,N-Diethyl-meta-toluamide were dissolved in 0.9% saline.
Acute slice pharmacology
Brains from male C57BL/6 mice (6-10 weeks old) were rapidly dissected and placed in ice-cold oxygenated Krebs buffer (124 mM NaCl, 4 mM KCl, 26 mM NaHCO3, 10 mM d-Glucose, 1.5 mM CaCl2, 1.5 mM MgSO4, 1.25 mM KH2PO4). Using a vibratome, coronal slices (350 μm) were prepared, and the striatum was microdissected and incubated at 30°C under 95% O2/5% CO2 for the indicated periods. Slices were separately treated with pharmacological reagents following a preincubation and recovery period in Kreb’s buffer containing adenosine deaminase (10 μg/mL) to allow homeostasis to be achieved. For experiments involving the D1 antagonist SCH-23390, a 10 min preincubation with the compound was performed before treatments with CPO as specified in the experiment. Following drug treatment, slices were snap frozen in dry ice and stored at −80°C.
Quantitative immunoblotting
Protein phosphorylation levels were evaluated by quantitative immunoblotting with phosphorylation state-specific antibodies. Samples were transferred to dry ice, then removed, immediately sonicated in boiling lysis buffer (10 mM NaF in 1% SDS), and boiled for 5 min. Protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Rockford, IL). Equal amounts of total protein (100 μg) were subjected to SDS-PAGE, followed by overnight transfer to nitrocellulose membranes. Membranes were blocked in 5% powdered skim milk dissolved in Tris-buffered saline-Tween, then probed with primary antibodies for P-T34 DARPP-32 (1:500), P-T75 DARPP-32 (1:1000), DARPP-32 (1:1000), P-S845 GluR1 (1:1000), GluR1 (1:1000), P-T205 tau (1:1000), tau (1:1000), and P35 (C-19) (1:500). Blots were washed several times and incubated with HRP-conjugated secondary antibodies (1:10,000). For detection of phosphorylation, blots were stripped and reprobed for total protein. Antibody was detected using enhanced chemiluminescence (GE Healthcare, Piscataway, NJ). Multiple exposure times were evaluated for each blot to ensure linearity.
Electrophysiological recordings
Following anesthetization of male or female p14-p18 Sprague-Dawley rats, brains were removed and immediately immersed in ice-cold, oxygenated, modified artificial cerebral spinal fluid (aCSF) containing the following (in mM): sucrose, 194; NaCl, 30; KCl, 4.5; MgCl2, 1; NaHCO3, 26; NaH2PO4, 1.2; D-glucose, 10. 300 μm slices were generated using a vibratome (Leica Microsystems, GmbH, Wetzlar, Germany) and subsequently placed in continuously oxygenated aCSF containing the following (in mM): NaCl, 124, KCl, 4.5; MgCl2, 1; CaCl, 2; NaHCO3, 26; NaH2PO4, 1.2; D-glucose, 10. The aCSF osmolarity was 320 mOsm and the pH was 7.3. Slices were equilibrated at 33°C for 1 h and subsequently incubated at room temperature until hemisected slices were transferred to the recording chamber.
Whole-cell electrophysiological recordings of dorsolateral striatal MSN miniature excitatory postsynaptic currents (mEPSCs) were performed in 29-31°C aCSF containing picrotoxin (50 μM), tetrodotoxin (TTX) (1 μM), and 0.1% DMSO. Intracellular glass recording electrodes were pulled from borosilicate glass on a Flaming/Brown micropipette puller (Sutter Instrument Co., Novato, CA) to a resistance of 2-5 mΩ. Recording electrodes were filled with a CsMeSO3–based solution of 295-300 mOsm containing (in mM); 120 CsMeSO3, 5 NaCl, 10 TEA-Cl, 10 HEPES, 5 QX-314, 1.1 EGTA, 0.3 Na-GTP, and 4 Mg-ATP. MSNs were voltage clamped at - 60 mV using a Multiclamp 700A amplifier (Axon Instruments) following rupture of the cell membrane and achievement of the whole-cell recording configuration. After a 3 min equilibration period, baseline mEPSC recordings were taken for 5 min, followed by a 10 min bath-application of CPO (100 μM, dissolved in DMSO) for a total of a 15 min recording session. The concentration of DMSO remained at 0.1% throughout the experiment. Recordings were filtered at 2 kHz and digitized at 10 kHz and were discarded if series resistance exceeded 25 MΩ or changed by more than 15%.
Data analysis
For quantitative immunoblotting analysis, immunoreactivity signals were captured by autoradiography and quantified with ImageJ software (NIH). The quantified effects were derived by normalization of the signals to total protein levels. Data are presented as mean ± standard error of the mean (SEM). Student’s t-test or analysis of variance (ANOVA) was performed to compare data sets using GraphPad Prism (GraphPad Software).
For electrophysiological analysis, baseline (0-5 min) and CPO (10-15 min) mEPSC data were analyzed using the Mini Analysis program version 6.0.7 (Synaptosoft, Decatur, GA, USA). Amplitude and area thresholds were manually set, and the accuracy of mEPSC detection was manually verified for all data sets. A paired t-test was used to compare data sets using GraphPad Prism (GraphPad Software). Data are expressed as mean ± SEM.
Results
Chlorpyrifos and pyridostigmine bromide enchance PKA-dependent phosphorylation of downstream effectors of dopamine neurotransmission in acute striatal slices
To explore the effect of nerve agents on dopamine neurotransmission we evaluated the influence of the organophosphate, CPF, on the PKA-dependent phosphorylation state of two important downstream effectors of striatal dopamine neurotransmission, DARPP-32 and the GluR1 subunit of the AMPA receptor. Treatment of acutely prepared mouse striatal slices with the CPF active metabolite, CPO caused a 1.9 ± 0.1-fold increase in phospho-Thr34 DARPP-32 (Fig. 1a). When PKA phosphorylates DARPP-32 at Thr34, it is converted into a potent inhibitor of protein phosphate 1 (PP1), thereby allowing dopamine to control the phosphorylation state of numerous phosphoproteins that regulate striatal neuron function (Greengard et al. 1999). CPO did not alter the phosphorylation state of DARPP-32 at Thr75, a Cdk5 site that converts it into a PKA inhibitor (Bibb et al. 1999) (Fig. 1b). GluR1 is another important downstream effector of the D1 receptor/cAMP/PKA cascade. Phosphorylation at Ser845 GluR1 is increased by D1 dopamine receptor agonists, and the phosphorylation of this site controls GluR1 trafficking, stability, and striatal neuron excitability (Song and Huganir 2002). Treatment of striatal slices with CPO enhanced PKA-dependent phosphorylation of Ser845 GluR1 by 2.1 ± 0.4-fold (Fig. 1c). Furthermore, treatment of striatal slices with the anti-nerve agent cholinesterase inhibitor PB revealed a 2.7 ± 0.5-fold enhancement in phospho-Ser845 GluR1 (Fig. 1d). Thus, CPO and PB regulate important downstream effectors of striatal dopamine neurotransmission.
Fig. 1.

Chlorpyrifos and pyridostigmine bromide activate PKA signaling in striatum. Effects of treatment of mouse striatal slices with CPO (100 μM, 60 min) on DARPP-32 (D32) phosphorylation by (a) PKA at Thr34 (P-T34) or (b) Cdk5 at Thr75 (P-T75) vs. vehicle (Veh) are shown as representative blots with quantification. The effects of treatment of CPO (c) or PB (d) (100 μM, 60 min) on GluR1 phosphorylation at Ser845 (P-S845) is shown. (e) The effect of systemic exposure to CPF on striatal P-S845 GluR1 is shown. (f) Effects of treatment of mouse striatal slices with CPO (100 μM, 60 min) on P-S845 GluR1 in the absence (−) and presence (+) of the D1 receptor antagonist SCH23390 (SCH, 1 μM). Data represent means ± SEM normalized for total levels. *p=0.0298, Student’s t-test, for a, n=3. *p=0.0305, Student’s t-test, for c, n=4. **p=0.0100, Student’s t-test, for d, n=4-5. **p=0.0085, Student’s t-test, for e, n=5. *p<0.05, **p<0.01, ***p<0.001, ANOVA with Newman-Keuls post-hoc test, for f, n=3-4.
Chlorpyrifos increases PKA signaling in vivo
To determine if OP exposure in vivo could activate striatal PKA, the effect of systemic administration CPO on PKA-dependent phosphorylation of GluR1 was assessed. Mice were treated with 8 daily injections of 1 mg/kg or 2.5 mg/kg of CPO. These doses were empirically determined to cause minimal lethality. However, no significant effect on phospho-Ser845 GluR1 was detected under these conditions. As an alternative, mice were injected subcutaneously with 30 mg/kg of the less lethal CPO parent compound, CPF, for 7 d while control mice received only vehicle. Striatal tissue was acutely dissected 2 h after the last dose of nerve agent. Interestingly, repeated treatment with CPF resulted in a 1.36 ± 0.04-fold increase in striatal phospho-Ser845 GluR1 levels (Fig. 1e). These in vivo data were consistent with the effects observed in striatal slices indicating that OP exposure activated striatal D1 receptor/cAMP/PKA effectors.
Chlorpyrifos modulation of PKA signaling may involve the activation of D1 dopamine receptors
To understand the mechanism by which nerve agents could dysregulate dopamine neurotransmission we evaluated the effects of CPO on phospho-Ser845 GluR1 levels in mouse striatal slices in the absence or presence of the D1 receptor antagonist, SCH 23390. SCH 23390 caused a reduction in the basal levels of phospho-Ser845 GluR1 to 50% (0.6 ± 0.1-fold) of control levels in untreated slices (1.2 ± 0.1-fold) (Fig. 1f). Furthermore, the increase in phospho-Ser845 GluR1 induced in the presence of the D1 receptor antagonist was 57.1% (1.2 ± 0.1-fold) of that induced by CPO treatment alone (2.1 ± 0.2-fold) (Fig. 1f). These findings suggest that at least a portion of the effect of the OP in phospho-Ser845 GluR1 was mediated by a pathway other than the activation of D1 receptors. These data also suggest that antagonists of D1 receptors may serve to counter some of the effects of nerve agents on dopamine signaling.
Chlorpyrifos augments corticostriatal glutamatergic transmission
To further define the pathophysiological effects of OP on striatal neuron function, the effects of CPO on corticostriatal glutamatergic neurotransmission was assessed by whole-cell patch clamp recording. For these neurophysiological analyses, TTX-insensitive mEPSCs from dorsolateral striatal MSNs were evaluated. Compared to baseline values, a 10 min bath-application of CPO did not affect mEPSC amplitude (baseline, 29.16 ± 1.32 pA vs. CPO, 28.81 ± 2.65 pA), but caused a significant decrease the inter-event interval (increase in frequency) of mEPSC events (baseline, 0.68 ± 0.16 s vs. CPO, 0.51 ± 0.15 s) (Fig. 2a-d). CPO application did not change the holding current (Fig. 2e). These results suggest that CPO alters striatal neurotransmission by enhancing glutamate release from corticostriatal terminals in an action potential-independent manner.
Fig. 2.

Chlorpyrifos enhances corticostriatal glutamatergic transmission. (a) Box plots demonstrating that bath application of CPO (100 μM) did not significantly change TTX-insensitive mEPSC amplitude. (b) A decrease in mEPSC inter-event interval during CPO application was observed in all recorded medium spiny neurons. (c) The cumulative distribution of mEPSC inter-event interval data during baseline (solid line) and CPO (dashed line) conditions is shown. (d) Example of mEPSC traces during baseline (top) and CPO application (bottom). (e) The holding current was unchanged by application of CPO. **p=0.0078, Student’s t-test, n=8.
DFP alone or in combination with other neurotoxicants alters PKA signaling in vivo
Studies using CPO and CPF revealed effects of this OP on striatal dopamine signaling and excitability. During the 1991 Gulf War, exposure to CPF used as an insecticide likely occurred in combination with use of the insect repellant, DEET, and, potentially, exposure to low levels of sarin gas (Golomb 2008). In addition, the reversible OP cholinesterase inhibitor, PB, was used prophylactically as a neuroprotectant in the event of nerve agent exposure. To mimic these conditions, mice were treated with different combinations of agents of potential relevance to Gulf War Illnesses. One cohort of subjects received a single subcutaneous injection of 4 mg/kg of DFP, a sarin surrogate compound. Another group was given 2.5 mg/kg PB and 5 mg/kg of DEET daily for 15 d. For still another group, to this regimen, a challenge dose of DFP was added on the final day. Quantitative immunoblot analysis was conducted on lysates from acutely dissected regions of the mesolimbic circuitry including striatum and hippocampus 2 h following the final treatment. Mice treated with DFP alone exhibited a trend toward enhanced phosphorylation of GluR1 at the Ser845 site (p=0.0586). However, PB/DEET treatment with or without the DFP challenge had no detectible effect at this phosphorylation site in the striatum (Fig. 3a).
Fig. 3.

Nerve agents exposure enhances PKA-dependent phosphorylation of GluR1 in vivo. Effects of systemic exposure to the different combinations of PB (15 d), DEET (15 d), and DFP (1 d, final day), indicated, and on P-S845 GluR1 levels in mouse striatum (a) and hippocampus (b) are shown. *p<0.05, **p<0.01, ANOVA with Newman-Keuls post-hoc test, n=4-5.
While the vast majority of dopamine neurotransmission occurs in the striatum, dopamine neurons also innervate the hippocampus. Nerve agents have been suggested to cause deficits in hippocampal-dependent tasks designed to measure cognition. Therefore, we also assessed the effects of systemic administration of PB, DEET, and DFP on phospho-Ser845 GluR1 levels in the hippocampus (Fig. 3b). Immunoblot analysis from hippocampal lysates of mice treated with the same dose described above showed a 1.5 ± 0.1-fold increase in phospho-Ser845 GluR1 following DFP treatment. Exposure to PB and DEET alone did not induce changes in phosho-Ser845 GluR1 compared to vehicle-treated mice. However, a 1.7 ± 0.1-fold increase in PKA-dependent phosphorylation was induced by the combination of these agents with DFP. Together, these data suggest that PKA signaling is enhanced in vivo by DFP alone or in combined exposure to other neurotoxicant acetylcholinesterase inhibitors.
Systemic exposure to nerve agents causes hyperphosphorylation of tau
It addition to the dysregulation of PKA-dependent phosphorylation of downstream effectors of dopamine neurotransmission, the effects of OP on neurological function suggest more severe neuronal injury may occur. The hyperphosphorylation of the neurofilament binding protein, tau, has been strongly implicated in an array of neurological and neurodegenerative diseases (Baumann et al. 1993; Lopes and Agostinho 2011). Moreover, phosphorylation of tau at Thr205 by the protein kinase Cdk5 has been associated with loss of neuronal function and death (Baumann et al. 1993; Lopes and Agostinho 2011). Interestingly, immunoblot analysis from mice exposed to PB, DEET, and DFP revealed a dramatic 17.0 ± 1.9-fold increase in the levels of phosphorylation of the aberrant Cdk5 substrate Thr205 tau in striatal lysates. DFP exposure alone caused a 15.5 ± 2.0-fold increase at that site. Furthermore, hippocampal lysates from mice exposed to all three neurotoxicants showed a 21.5 ± 1.3-fold increase in phospho-Thr205 tau levels, while DFP-treated mice exhibited a 17.1 ± 1.1-fold enhancement compared to mice that received vehicle treatment alone. These marked effects suggest that toxicity induced upon DFP exposure, alone or in the presence of PB and DEET, induces neuropathological effects in striatum and hippocampus.
Systemic exposure to nerve agents dysregulates Cdk5
Cdk5 regulates dopamine neurotransmission (Bibb et al. 1999) and contributes to many other neuronal functions (Cheung et al. 2006). However, neuronal injury and loss of homeostasis can cause activation of the Ca2+-dependent protease, calpain (Kusakawa et al. 2000; Lee et al. 2000). Cleavage of the Cdk5-activating neuronal cofactor p35 to p25 by calpain, results in hyper-activation and redirection of the kinase towards aberrant substrates including Thr205 tau. Cdk5 bound to p25 exhibits aberrant activity, contributes to neuronal death, and has been implicated in neurotoxicity and neurodegenerative diseases (Cruz et al. 2003; Lopes and Agostinho 2011). Furthermore, animal models of p25 overexpression exhibit behavioral deficits that correlate with dysregulation of dopamine signaling (Meyer et al. 2008).
To investigate the ability of nerve agents to induce aberrant Cdk5 activity, we examined p25 generation in different paradigms of AChE inhibitors exposure. While CPO alone as a systemic (1.0 or 2.5 mg/kg, s.c. 7 days) didn’t induce appreciable p25 generation, the effects of DFP alone or in combination with other OPs on tau hyperphosphorylation, suggested these, possibly more severe, treatments might have an effect on p25 generation. To investigate this possibility, mice were treated with the same dose regimen of PB, DEET, and DFP described above (i.e., Figs. 3 and 4) and lysates from acutely dissected regions of the striatum and hippocampus were analyzed 2 h following DFP exposure. Surprisingly, these conditions did not result in a detectable increase in p25 production. However, striatal lysates from a cohort of mice that received a higher dose regimen of the insecticide DEET (30 mg/kg, vs. 5 mg/kg in the earlier experiments) in combined exposure with PB and DFP reveal a 13.4 ± 3.3-fold increase in p25 generation 2 h following the last exposure (Fig. 5a). These mice displayed no changes in phosphorylation at Thr75 DARPP-32 (Fig. 5a). Interestingly, 6 h after the last exposure, mice exhibited a 10.0 ± 1.5-fold increase in p25 generation that was accompanied by a reduction in phospho-Thr75 DARPP-32 to 60% (0.6 ± 0.1) of control levels in vehicle-treated mice (1.0 ± 0.1) (Fig. 5b). Similar effects were also observed in hippocampus with 12.8 ± 3.9 and 17.4 ± 1.1-fold increases at 2 and 6 h, respectively, after the final treatment (Fig. 5c and d). These data indicate that nerve agents induce aberrant Cdk5 activity and shift its specificity away from physiological substrates, most likely, toward aberrant substrates such as tau.
Fig. 4.

Nerve agents exposure causes tau hyperphosphorylation in vivo. Effects of systemic exposure to different combinations of PB, DEET, and DFP, and on the phosphorylation state of Thr205 tau (P-T205 Tau) in mouse striatum (a) and hippocampus (b) are shown. ***p<0.001, ANOVA with Newman-Keuls post-hoc test, n=4-5.
Fig. 5.

Nerve agents exposure dysregulates Cdk5 activity in vivo. Effects of systemic exposure to PB, DEET, and DFP on p25 generation and Cdk5-dependent phosphorylation of P-T75 DARPP-32 in mouse striatum (a and b). Representative blots from tissue taken 2 or 6 h after treatments are shown above quantification. Effects of systemic exposure to PB, DEET, and DFP on p25 generation in hippocampus (c and d). *p=0.0108, **p=0.0045, ***p=0.0004, Student’s t-test, for a, n=3-4. *p=0.0102, #p<0.0001, Student’s t-test, for b, n=3-4.
Systemic exposure to pyridostigmine bromide dysregulates Cdk5 activity following a delay period from time of exposure
While neurotoxic exposure could be limited to self-administration of PB, the data indicated that PB in the presence of DEET had little initial neurotoxic effect. However, it has been suggested that the pathological effects to subclinical doses of OP such as PB may only manifest after a delay period of the last exposure (Kamel and Hoppin 2004). Therefore, we examined the effect of systemic administration of PB following a 4-week delay period after the last exposure. Mice were treated with 10 daily injections of 1 mg/kg PB and immunoblot analysis was conducted on lysates from acutely dissected regions following the delay period. Mice treated with PB exhibited a 1.6 ± 0.3-fold increase in p25 generation accompanied by a decrease in phospho-Thr75 DARPP-32 levels to 69% (0.69 ± 0.05) of control levels in vehicle-treated mice (1.0 ± 0.1) (Fig. 6a). Comparable effects were detected in hippocampus with 1.8 ± 0.2-fold increase following the delay treatment. The detection of high levels of p25 in these two brain regions and redirection of Cdk5 specificity support the hypothesis that this compound which was administered to Gulf War veterans during combat has the capacity to induce neuronal injury, even after considerable delay from the time of exposure to subclinical doses.
Fig. 6.
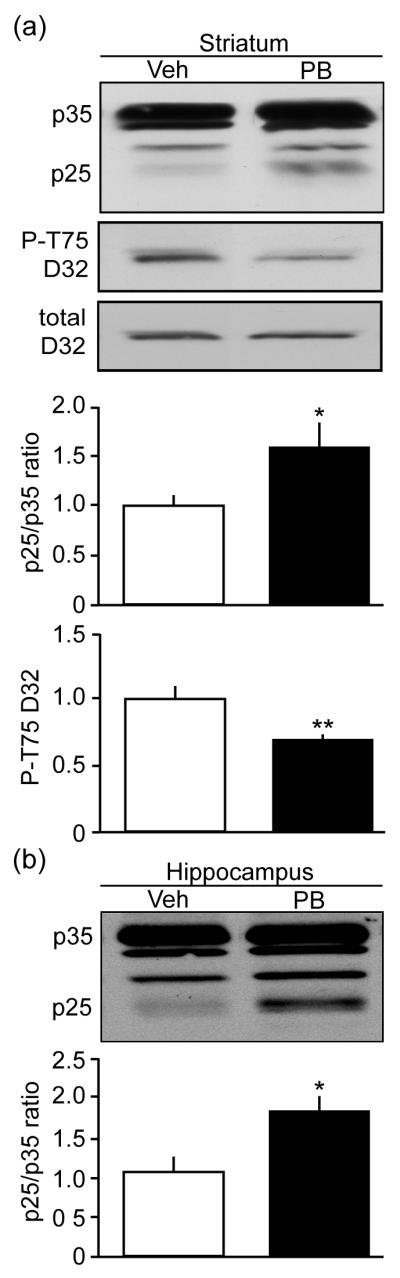
Exposure to pyridostigmine bromide dysregulates Cdk5 activity in vivo after a delay period. (a) Effects of systemic exposure to PB following a 4-week delay period after the last treatment on p25 generation and P-T75 DARPP32 in mouse striatum. (b) Delayed effects of systemic exposure to PB on p25 generation in mouse hippocampus. *p=0.0297, **p=0.0065, Student’s t-test, for a, n=6. *p=0.0106, Student’s t-test, for b, n=6.
Discussion
Nerve agents are thought to induce brain damage through hyperstimulation of cholinergic receptors (McDonough and Shih 1997). However, both glutamatergic and dopaminergic neurotransmitter systems mediate the biological and pathological effects of AChE inhibitors (McDonough and Shih 1997; Wonnacott 1997; Wonnacott et al. 2000). Furthermore, the neostriatum has been implicated as a target of the long-term neurotoxic effects of organophosphates in clinical studies (Haley et al. 2000; Haley et al. 2009). To better understand the deleterious and pathogenic effects of these compounds, we examined the consequences of nerve agent exposure on dopamine signaling pathways, glutamate neurotransmission, and neuronal injury mechanisms in the neostriatum in rodent brain slices and whole animals in vivo.
We found that CPF and PB increased PKA-dependent phosphorylation of downstream effectors of striatal dopamine neurotransmission including the GluR1 subunit of the AMPA receptor and/or DARPP-32 in slices and in vivo. A previous study found that, high concentrations of nicotine induced a D1 receptor-dependent activation of PKA, and increased phosphorylation of Thr34 DARPP-32 through activation of the striatonigral pathway (Hamada et al. 2004). However, given that a potent D1 receptor antagonist did not block the activation of striatal PKA, it is possible that at least a portion of the effects of CPF are mediated by pathways, such as those modulated by mAChR, which do not directly involve D1 receptor activation. Accordingly, acetylcholine released from striatal cholinergic interneurons interacts in a dynamic manner with dopamine signaling at multiple levels including presynaptic regulation of neurotransmitter release and postsynaptic effects (Threlfell and Cragg 2011). The degree to which CPF activates PKA through non-dopamine mediated mechanisms may warrant further study. Moreover, the deleterious effects of these agents may occur across a spectrum of biological and pathological mechanisms. At relatively low levels of exposure, effects may be occur via AChR activation and subsequent dopamine release, while higher doses may induce dysregulation of Cdk5, removal of tonic inhibition of PKA, or direct covalent modifications due to organophosphate reactivity.
The effects of CPF on the phosphorylation state of downstream effectors of the D1 receptor/cAMP/PKA pathway suggest that nerve agents may exert some of their pathological effects via alterations in dopamine neurotransmission. CPF has been shown to increase dopamine turnover and to alter dopamine and other monoamine metabolite levels (Moreno et al. 2008; Eells and Brown 2009). Furthermore, CPF exposure during the gestational days of developing rats evoked long-term increases in serotonin and dopamine turnover (Aldridge et al. 2005). Some of the positive effects of CPF on PKA activity in slices and whole animal subjects (see Fig. 1) were not replicated when combined organophosphate regimens, including higher DEET concentrations, were used (see Figs 3a, for example). However, these more toxic exposures may have invoke mechanisms of neuronal injury associated with Cdk5/p25 that possibly occluded effects observed at lower doses.
Excessive glutamate release may constitute a principle cause of neuronal injury and death. ACh can modulate glutamate release through presynaptic α7 nACh receptors in striatal synaptosomes (Marchi et al. 2002). Furthermore, nicotine has been shown to enhance hippocampal synaptic transmission on presynaptic terminals containing α7 nAChRs (Gray et al. 1996). Consistent with these observations, CPF attenuated the inter-interval period between excitatory mini-EPSC events, presumably as a result of enhanced striatal glutamate release. Surprisingly, there are few other reports of the potentiation of striatal glutamate neurotransmission by AChE inhibitors. On the other hand, a study on the effects of nerve agent soman-induced seizures has shown that the levels of excitatory amino acids are attenuated following intoxication (Shih and McDonough 1997). AChE inhibitors such as donepezil, rivastigmine, and galantamine are used as clinical treatments to delay or slow the onset of cognitive deficiencies accompanying Alzheimer’s disease pathology, and these compounds have been suggested to attenuate excitotoxicity in the hippocampus and cortex (Standridge 2004; Hansen et al. 2006). Furthermore, it has been suggested that the elicited neuroprotective effects of donepezil may involve down-regulation of NMDA receptors, thereby attenuating glutamate-induced Ca2+ increase following α7 nAChRs activation (Shen et al. 2010).
Nerve agents induce seizure-related brain damage through hyper-stimulation of cholinergic receptors. Subsequent excessive stimulation of the glutamatergic system triggers a prolonged intracellular increase in Ca2+, causing the activation of secondary pathways responsible for neuronal injury (McDonough and Shih 1997). NDMA receptor antagonists such as memantine, a drug used clinically to treat Alzheimer’s (Thomas and Grossberg 2009), have been recognized for their ability to reduce Ca2+ overload. However, controversy exists regarding their efficacy and resulting neurotoxic effects (Filbert et al. 2005). Current studies are focused on identifying secondary intracellular signaling pathways that could be targeted for the development of therapeutics that prevent or alleviate the symptoms following nerve agent exposure.
DFP alone or as part of a combination of neurotoxicants meant to mimic the exposure encountered by veterans of the 1991 Persian Gulf War caused hyper-phosphorylation of Thr205 tau. Tau functions to stabilize the cytoskeleton, and its hyper-phosphorylation can result in the formation of neurofibrillary tangles and neurodegeneration (Hanger et al. 2009). Phosphorylation at this site is Cdk5-dependent and it has been implicated as a biomarker for Alzheimer’s pathology (Cruz et al. 2003; Cruz and Tsai 2004). Cdk5 plays a critical role in corticogenesis, synaptic plasticity, drug addiction, and cognition (Bibb 2003; Angelo et al. 2006; Cheung et al. 2006). Dysregulation of its activity may depend on prolonged NMDA receptor activation. The resulting intracellular Ca2+ influx could then cause calpain-dependent conversion of p35 to p25 (Lee et al. 2000). Unnecessary NMDA receptor activation due to excessive cholinergic stimulation is one of the most prominent effects of nerve agent exposure that ultimately leads to neuropathology (McDonough and Shih 1997).
PB/DEET/DFP exposure also resulted in p25 generation and induction of aberrant Cdk5 activity in both striatum and hippocampus. Interestingly, the high levels of p25 in striatum correlated with a decrease in phospho-Thr75 DARPP-32, possibly disinhibiting PKA activity. Similar effects were induced by PB after an extensive delay from the time of exposure. High levels of nicotine also reduce Cdk5-dependent phosphorylation of DARPP-32 at Thr75, possibly through a dopamine-release dependent pathway (Hamada et al. 2005). Moreover, induction of transgenic p25 overexpression reduced Thr75 DARPP-32, altered dopamine signaling, and shifted Cdk5 specificity with regard to physiological and aberrant substrates (Meyer et al. 2008). Nonetheless, striatal signaling may be differentially dysregulated depending on the profile of exposure.
Our data indicate that dysregulation of Cdk5 is a component of the long-term effects that arise from exposure and is consistent with at least two reports suggesting organophosphates neurotoxicants dysregulate Cdk5 (Wang et al. 2006; Zhu et al. 2010). Thus, nerve agent-induced dysregulation of Cdk5 may dramatically affect striatal-dependent brain function, and may be relevant to subclinical neurotoxicity and disorders involving dopamine neurotransmission. For example, pregnant women exposed to pesticides such as chlorpyrifos and DEET have a higher risk to manifest deficits in fetal neurodevelopment and altered birth outcomes (Berkowitz et al. 2004; Barr et al. 2010). Prenatal insecticide exposure reduces IQ and delays mental and psychomotor development (Rauh et al. 2006; Bouchard et al. 2011; Engel et al. 2011; Rauh et al. 2011), and children with elevated urinary levels of OP metabolites had a higher ADHD prevalence (Bouchard et al. 2010). Furthermore, a recent study has shown that chlorpyrifos oxon alters the firing activity of the locus coeruleus noradrenergic neurons, a brain region associated with anxiety and stress (Cao et al. 2011). Taken together, the present study is among the first to examine the mechanisms by which organophosphates may alter brain function and cause long-term pathological effects. The secondary signaling targets identified here may contribute to the development of treatments to prevent or counter these neurotoxic effects.
Acknowledgments
We thank Suzanne Saldanha, Brenda Billig and Christopher Felton for technical assistance. This work was supported by U.S. National Institutes of Health Grants to J.A.B (MH79710, MH083711, and DA016672), Intramural funds from Centers for Disease Control and Prevention-NIOSH, and the Division of Intramural Clinical and Biological Research of NIAAA. Portions of this research were supported by IDIQ contract VA549-P-0027 (Robert W. Haley, M.D., PI), awarded and administered by the Department of Veterans Affairs Medical Center, Dallas, TX. The content does not necessarily reflect the position or the policy of the Federal government or the sponsoring agency, and no official endorsement should be inferred.
Abbreviations used
- ACh
acetylcholine
- AChE
acetylcholinesterase
- aCSF
artificial cerebral spinal fluid
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BCA
bicinchoninic acid
- Cdk5
cyclin-dependent kinase 5
- CPF
chlorpyrifos
- CPO
chlorpyrifos oxon
- DARPP-32 or D32
dopamine- and cAMP-regulated phosphoprotein of Mr 32 kDa
- DEET
N,N-Diethyl-meta-toluamide
- DFP
diisopropyl fluorophosphate
- DMSO
dimethylsulfoxide
- GluR1
glutamate receptor 1
- i.p.
intraperitoneal
- mAChR
muscarinic acetylcholine receptor
- mEPSC
miniature excitatory postsynaptic current
- MSN
medium spiny neuron
- nAChR
nicotinic acetylcholine receptor
- OP
organophosphates
- PB
pyridostigmine bromide
- PKA
protein kinase A
- PP1
protein phosphate 1
- s.c.
subcutaneous
References
- Aldridge JE, Meyer A, Seidler FJ, Slotkin TA. Alterations in central nervous system serotonergic and dopaminergic synaptic activity in adulthood after prenatal or neonatal chlorpyrifos exposure. Environ Health Perspect. 2005;113:1027–1031. doi: 10.1289/ehp.7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Plattner F, Giese KP. Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J Neurochem. 2006;99:353–370. doi: 10.1111/j.1471-4159.2006.04040.x. [DOI] [PubMed] [Google Scholar]
- Auman JT, Seidler FJ, Slotkin TA. Neonatal chlorpyrifos exposure targets multiple proteins governing the hepatic adenylyl cyclase signaling cascade: implications for neurotoxicity. Brain Res Dev Brain Res. 2000;121:19–27. doi: 10.1016/s0165-3806(00)00021-3. [DOI] [PubMed] [Google Scholar]
- Barr DB, Ananth CV, Yan X, Lashley S, Smulian JC, Ledoux TA, Hore P, Robson MG. Pesticide concentrations in maternal and umbilical cord sera and their relation to birth outcomes in a population of pregnant women and newborns in New Jersey. Science of The Total Environment. 2010;408:790–795. doi: 10.1016/j.scitotenv.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Letters. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- Berkowitz GS, Wetmur JG, Birman-Deych E, Obel J, Lapinski RH, Goldbold JH, Holzman IR, Wolff MS. In Utero pesticides esposure, maternal paraoxonase activity, and head circumference. Environmental Health Perspectives. 2004;112:388–391. doi: 10.1289/ehp.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA. Role of Cdk5 in neuronal signaling, plasticity, and drug abuse. Neurosignals. 2003;12:191–199. doi: 10.1159/000074620. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr., Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- Bouchard MF, Bellinger DC, Wright RO, Weisskopf MG. Attention-deficit/hyperactivity disorder and urinary metabolites of organophosphate pesticides. Pediatrics. 2010;125:e1270–1277. doi: 10.1542/peds.2009-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard MF, Chevrier J, Harley KG, Kogut K, Vedar M, Calderon N, Trujillo C, Johnson C, Bradman A, Barr DB, Eskenazi B. Prenatal Exposure to Organophosphate Pesticides and IQ in 7-Year Old Children. Environ Health Perspect. 2011;119:1189–1195. doi: 10.1289/ehp.1003185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JL, Varnell AL, Cooper DC. Gulf War Syndrome: A role for organophosphate induced plasticity of locus coeruleus neurons. Nature Precedings. 2011 [Google Scholar]
- Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends in molecular medicine. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 Activation by p25 Triggers Pathological Events Leading to Neurodegeneration and Neurofibrillary Tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Drerup JM, Hayashi K, Cui H, Mettlach GL, Long MA, Marvin M, Sun X, Goldberg MS, Lutter M, Bibb JA. Attention-deficit/hyperactivity phenotype in mice lacking the cyclin-dependent kinase 5 cofactor p35. Biol Psychiatry. 2010;68:1163–1171. doi: 10.1016/j.biopsych.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DL, Daroff RB, Autrup H, Bridges J, Buffler P, Costa LG, Coyle J, McKhann G, Mobley WC, Nadel L, Neubert D, Schulte-Hermann R, Spencer PS. Review of the toxicology of chlorpyrifos with an emphasis on human exposure and neurodevelopment. Crit Rev Toxicol. 2008;38(Suppl 2):1–125. doi: 10.1080/10408440802272158. [DOI] [PubMed] [Google Scholar]
- Eells JB, Brown T. Repeated developmental exposure to chlorpyrifos and methyl parathion causes persistent alterations in nicotinic acetylcholine subunit mRNA expression with chlorpyrifos altering dopamine metabolite levels. Neurotoxicol Teratol. 2009;31:98–103. doi: 10.1016/j.ntt.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Engel SM, Wetmur J, Chen J, Zhu C, Barr DB, Canfield RL, Wolff MS. Prenatal Exposure to Organophosphates, Paraoxonase 1, and Cognitive Development in Childhood. Environ Health Perspect. 2011;119:1182–1188. doi: 10.1289/ehp.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Bymaster FP, Ward J, DeLapp N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J Med Chem. 2000;43:4333–4353. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- Filbert M, Levine E, Ballough G. Neuroprotection for nerve agent-induced brain damage by blocking delayed calcium overload: A review. J Med CBR Def. 2005;3:1–21. [Google Scholar]
- Golomb BA. Acetylcholinesterase inhibitors and Gulf War illnesses. Proc Natl Acad Sci U S A. 2008;105:4295–4300. doi: 10.1073/pnas.0711986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the Dopamine Receptor: the DARPP-32/Protein Phosphatase-1 Cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Haley RW, Fleckenstein JL, Marshall WW, McDonald GG, Kramer GL, Petty F. Effect of Basal Ganglia Injury on Central Dopamine Activity in Gulf War Syndrome: Correlation of Proton Magnetic Resonance Spectroscopy and Plasma Homovanillic Acid Levels. Arch Neurol. 2000;57:1280–1285. doi: 10.1001/archneur.57.9.1280. [DOI] [PubMed] [Google Scholar]
- Haley RW, Spence JS, Carmack PS, Gunst RF, Schucany WR, Petty F, Devous MD, Sr., Bonte FJ, Trivedi MH. Abnormal brain response to cholinergic challenge in chronic encephalopathy from the 1991 Gulf War. Psychiatry Res. 2009;171:207–220. doi: 10.1016/j.pscychresns.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Hamada M, Higashi H, Nairn AC, Greengard P, Nishi A. Differential regulation of dopamine D1 and D2 signaling by nicotine in neostriatal neurons. Journal of Neurochemistry. 2004;90:1094–1103. doi: 10.1111/j.1471-4159.2004.02574.x. [DOI] [PubMed] [Google Scholar]
- Hamada M, Hendrick JP, Ryan GR, Kuroiwa M, Higashi H, Tanaka M, Nairn AC, Greengard P, Nishi A. Nicotine Regulates DARPP-32 (Dopamine- and cAMP-Regulated Phosphoprotein of 32 kDa) Phosphorylation at Multiple Sites in Neostriatal Neurons. Journal of Pharmacology and Experimental Therapeutics. 2005;315:872–878. doi: 10.1124/jpet.105.090852. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends in Molecular Medicine. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Hansen RA, Gartlehner G, Kaufer DJ, Lohr KN, Carey T. Drug Class Review on Alzheimer’s Drugs: Final Report. 2006. [PubMed]
- Kamel F, Hoppin JA. Association of pesticide exposure with neurologic dysfunction and disease. Environ Health Perspect. 2004;112:950–958. doi: 10.1289/ehp.7135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent Proteolytic Cleavage of the p35 Cyclin-dependent Kinase 5 Activator to p25. Journal of Biological Chemistry. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lopes JP, Agostinho P. Cdk5: Multitasking between physiological and pathological conditions. Progress in Neurobiology. 2011;94:49–63. doi: 10.1016/j.pneurobio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating α7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. Journal of Neurochemistry. 2002;80:1071–1078. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Shih TM. Neuropharmacological Mechanisms of Nerve Agent-induced Seizure and Neuropathology. Neuroscience & Biobehavioral Reviews. 1997;21:559–579. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- Meyer DA, Richer E, Benkovic SA, Hayashi K, Kansy JW, Hale CF, Moy LY, Kim Y, O’Callaghan JP, Tsai LH, Greengard P, Nairn AC, Cowan CW, Miller DB, Antich P, Bibb JA. Striatal dysregulation of Cdk5 alters locomotor responses to cocaine, motor learning, and dendritic morphology. Proc Natl Acad Sci U S A. 2008;105:18561–18566. doi: 10.1073/pnas.0806078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M, Cañadas F, Cardona D, Suñol C, Campa L, Sánchez-Amate MC, Flores P, Sanchez-Santed F. Long-term monoamine changes in the striatum and nucleus accumbens after acute chlorpyrifos exposure. Toxicology Letters. 2008;176:162–167. doi: 10.1016/j.toxlet.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Nomikos GG, Schilström B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH. Role of [alpha]7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behavioural Brain Research. 2000;113:97–103. doi: 10.1016/s0166-4328(00)00204-7. [DOI] [PubMed] [Google Scholar]
- Philibin SD, Hernandez A, Self DW, Bibb JA. Striatal signal transduction underlying drug addiction: behavior, neuroplasticity, and molecular perspectives. Frontiers in Neuroanatomy. 2011 doi: 10.3389/fnana.2011.00060. Manuscript in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh V, Arunajadai S, Horton M, Perera F, Hoepner L, Barr DB, Whyatt R. 7-Year Neurodevelopmental Scores and Prenatal Exposure to Chlorpyrifos, a Common Agricultural Pesticide. Environ Health Perspect. 2011;119:1196–1201. doi: 10.1289/ehp.1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh VA, Garfinkel R, Perera FP, Andrews HF, Hoepner L, Barr DB, Whitehead R, Tang D, Whyatt RW. Impact of Prenatal Chlorpyrifos Exposure on Neurodevelopment in the First 3 Years of Life Among Inner-City Children. Pediatrics. 2006;118:e1845–1859. doi: 10.1542/peds.2006-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Kihara T, Hongo H, Wu X, Kem WR, Shimohama S, Akaike A, Niidome T, Sugimoto H. Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of α7 nicotinic receptors and internalization of NMDA receptors. British Journal of Pharmacology. 2010;161:127–139. doi: 10.1111/j.1476-5381.2010.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih TM, McDonough JH. Neurochemical Mechanisms in Soman-induced Seizures. Journal of Applied Toxicology. 1997;17:255–264. doi: 10.1002/(sici)1099-1263(199707)17:4<255::aid-jat441>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Comparative developmental neurotoxicity of organophosphates in vivo: transcriptional responses of pathways for brain cell development, cell signaling, cytotoxicity and neurotransmitter systems. Brain Res Bull. 2007;72:232–274. doi: 10.1016/j.brainresbull.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends in Neurosciences. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Staines D. Do vasoactive neuropeptide autoimmune disorders explain pyridostigmine’s association with Gulf War syndrome? Medical Hypotheses. 2005;65:591–594. doi: 10.1016/j.mehy.2005.02.036. [DOI] [PubMed] [Google Scholar]
- Standridge JB. Pharmacotherapeutic approaches to the treatment of Alzheimer’s disease. Clinical Therapeutics. 2004;26:615–630. doi: 10.1016/s0149-2918(04)90064-1. [DOI] [PubMed] [Google Scholar]
- Thomas HV, Stimpson NJ, Weightman AL, Dunstan F, Lewis G. Systematic review of multi-symptom conditions in Gulf War veterans. Psychol Med. 2006;36:735–747. doi: 10.1017/S0033291705006975. [DOI] [PubMed] [Google Scholar]
- Thomas SJ, Grossberg GT. Memantine: a review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clin Interv Aging. 2009;4:367–377. doi: 10.2147/cia.s6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Cragg SJ. Dopamine signaling in dorsal versus ventral striatum: the dynamic role of cholinergic interneurons. Front Syst Neurosci. 2011;5:11. doi: 10.3389/fnsys.2011.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YP, Mou D. l., Song JF, Rao ZR, Li D, Ju G. Aberrant activation of CDK5 is involved in the pathogenesis of OPIDN. Journal of Neurochemistry. 2006;99:186–197. doi: 10.1111/j.1471-4159.2006.04027.x. [DOI] [PubMed] [Google Scholar]
- Wiener SW, Hoffman RS. Nerve agents: a comprehensive review. J Intensive Care Med. 2004;19:22–37. doi: 10.1177/0885066603258659. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends in Neurosciences. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Kaiser S, Mogg A, Soliakov L, Jones IW. Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. European Journal of Pharmacology. 2000;393:51–58. doi: 10.1016/s0014-2999(00)00005-4. [DOI] [PubMed] [Google Scholar]
- Zhu H, O’Brien JJ, O’Callaghan JP, Miller DB, Zhang Q, Rana M, Tsui T, Peng Y, Tomesch J, Hendrick JP, Wennogle LP, Snyder GL. Nerve agent exposure elicits site-specific changes in protein phosphorylation in mouse brain. Brain Res. 2010;1342:11–23. doi: 10.1016/j.brainres.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]