

Abstract
Chronic kidney disease constitutes an increasing medical burden affecting 26 million people in the United States alone. Diabetes, hypertension, ischemia, acute injury, and urological obstruction contribute to renal fibrosis, a common pathological hallmark of chronic kidney disease. Regardless of etiology, elevated TGF-β1 levels are causatively linked to the activation of profibrotic signaling pathways initiated by angiotensin, glucose, and oxidative stress. Unilateral ureteral obstruction (UUO) is a useful and accessible model to identify mechanisms underlying the progression of renal fibrosis. Plasminogen activator inhibitor-1 (PAI-1), a major effector and downstream target of TGF-β1 in the progression of several clinically important fibrotic disorders, is highly up-regulated in UUO and causatively linked to disease severity. SMAD and non-SMAD pathways (pp60c-src, epidermal growth factor receptor [EGFR], mitogen-activated protein kinase, p53) are required for PAI-1 induction by TGF-β1. SMAD2/3, pp60c-src, EGFR, and p53 activation are each increased in the obstructed kidney. This review summarizes the molecular basis and translational significance of TGF-β1-stimulated PAI-1 expression in the progression of kidney disease induced by ureteral obstruction. Mechanisms discussed here appear to be operative in other renal fibrotic disorders and are relevant to the global issue of tissue fibrosis, regardless of organ site.
Keywords: Fibrosis, PAI-1, TGF-β1, p53, Transcription
Cellular events in obstructive renal disease
Interstitial fibrosis is a common hallmark and major prognostic indicator of end-stage renal disease (Eddy 2000; Strutz and Neilson 2003; Eddy 2005; Bonventre 2010). The extent of associated tubulointerstitial pathology (i.e., inflammation, tubular atrophy, progressive fibrosis) has prognostic significance regardless of etiology (Grande et al. 2010; Truong et al. 2011). Congenital obstructive nephropathy, frequently involving the ureteropelvic junction, is the most significant contributor to chronic renal disease and eventual organ failure in children (Chevalier et al. 2010; Klein et al. 2010). Obstructive uropathy in adults generally reflects several age-associated disease processes, both benign and malignant. Whereas the pathophysiology of nephrofibrosis is complex regardless of origin, unilateral ureteral obstruction (UUO) in both neonatal and adult rodents is a well-established in vivo model of disease progression (Klahr and Morrissey 2002; Bascands and Schanstra 2005; Chevalier et al. 2009; Puri et al. 2010). Tubulointerstitial inflammatory and fibrotic responses following experimental UUO, orchestrated predominantly by dramatically elevated transforming growth factor-β1 (TGF-β1) levels in the injured kidney (Miyajima et al. 2000; Inazaki et al. 2004), closely mirror human obstructive uropathy (Hruska 2002; Moller et al. 1984) providing an accessible, translationally relevant, in vivo platform for the dissection of critical events and the identification of potential therapeutic targets. The extent of renal impairment or progression to eventual organ failure depends, to some degree, on the duration of obstruction but may be pronounced even upon short periods of blockage, and in many cases, injury progresses even upon release of the restriction (Truong et al. 2011). Persistence of elevated renal TGF-β1 expression, a potent profibrotic cytokine, even after relief of UUO (depending on the timing of release) leads to progressive tissue injury, impaired growth, and eventual loss of kidney function (Chevalier et al. 1999, 2009, 2010). Inflammation, interstitital fibrosis, tubular atrophy, and glomerular sclerosis all increase in the post-obstructed organ (Chevalier et al. 2009). Progressive fibrosis, moreover, is associated with capillary rarefaction and tissue hypoxia, exacerbating the fibrotic response (Boor 2010).
Within hours after experimental ureteral occlusion, the affected kidney exhibits changes in hydrostatic forces and increased oxidative stress (Schreiner et al. 1988; Klahr and Morrissey 2002; Dendoovan et al. 2010). Tubular stretch stimulates TGF-β1 expression (>20-fold), apoptosis, and inflammatory responses dependent upon nuclear factor kappa-B (Miyajima et al. 2000; Rohatgi and Flores 2010). Marked inflammation and excessive extracellular matrix (ECM) accumulation, the latter being attributable to the increased synthesis of collagen and fibronectin (predominantly by activated fibroblasts or myofibroblasts) coupled with reduced matrix degradation, accompanies tubular dilation, atrophy, progressive fibrosis, nephron loss, and scarring with eventual impairment of tissue function (Strutz and Neilson 2003; Eddy and Fogo 2006; Eddy 2000; Grande and Lopez-Novoa 2009; Higgins et al. 2003; Yabuki et al. 2005; Meng et al 2010; Manucha 2007; Yang et al. 2010; Truong et al. 2011). Myofibroblast density and/or persistence correlates with the severity of tubulointerstitial fibrosis and progression to renal failure (Qi et al. 2006b). Although their origin is uncertain, myofibroblasts are probably derived (1) from resident interstitial fibroblasts (e.g., Picard et al. 2008), (2) by Snai1- or Id1-dependent transcriptional reprogramming of renal pericytes or perivascular fibroblasts (Humphreys et al. 2010; Cook 2010; Roufosse et al. 2006; Lin et al. 2008), (3) as a consequence of tubular epithelial-to-mesenchymal transdifferentiation (EMT; Yamashita et al. 2005; Kalluri and Neilson 2003; Iwano et al. 2002; Eddy 2009; Iwano and Neilson 2004; Liu 2006, 2010; Grande and Lopez-Novoa 2009), or (4) by extra-renal recruitment from the circulation or bone marrow (Ricardo et al. 2008). Indeed, the mobilization of stem-like cells accompanies post-obstructive kidney regeneration suggesting that renal capsule-derived, hematopoietic, or mesenchymal progenitor cells participate in repair (Park et al. 2010a,Park et al. 2010b; Yamashita et al. 2005). Whether progenitor involvement has a positive or negative effect on outcome, however, is controversial, although renal-artery-delivered human mesenchymal stem cells appear to attenuate UUO-induced fibrosis (Asanuma et al. 2010).
TGF-β/SMAD signaling as a transducer of the fibrotic phenotype in obstructive nephropathy
TGF-β and downstream receptor-regulated SMAD transcriptional effectors are among the most significant inducers of fibrosis in multiple organ systems integrating, in some cases, signaling pathways initiated by other stimuli (Kalluri and Neilson 2003; Bottinger and Bitzer 2002; Stahl and Felsen 2001; Iwano and Neilson 2004; Ricardo et al. 2008; Bottinger 2007). Indeed, several factors that drive EMT in kidney cells and that promote myofibroblast differentiation and fibrosis in animal models of chronic renal disease utilize, at least in part, TGF-β/SMAD signaling pathways (Oldfield et al. 2001; Mezzano et al 2003; Fan et al. 2001; Liu 2010). Intraperitoneal injection of TGF-β alone, moreover, is sufficient to initiate a prominent renal fibrotic response (Ledbetter et al. 2000), and TGF-β-activated SMAD pathways are pivotal for the induction of EMT, myofibroblast differentiation, and disease progression (Derynck and Zhang 2003; Kalluri and Neilson 2003; Iwano and Neilson 2004). In the setting of UUO, tubulointerstitial fibrosis appears to be dependent on the interstitial upregulation of TGF-β1 expression (Miyajima et al. 2000). Prominent TGF-β1 reactivity in the tubular epithelium of senescence-prone SAMP1/Sku male mice following UUO is accompanied by an accelerated and severe onset of all the pathophysiologic hallmarks of mononuclear cell infiltration/fibrosis with rapid progression of EMT (Yabuki et al. 2005). Given the causative association between TGF-β1 and fibrotic disease, it is not surprising that SMAD3-deficient mice are protected from renal fibrosis by, at least in part, reduced EMT, inflammation, collagen deposition, tubular apoptosis, and the impaired expression of profibrotic TGF-β1 target genes (Sato et al. 2003; Arany et al. 2007; Zeisberg et al. 2003; Inazaki et al. 2004).
These findings provide a rationale for targeted intervention therapies directed to the TGF-β signaling network. Disease onset and progression is certainly attenuated by the blockade of TGF-β1 expression or function (Klahr 1991; Kaneto et al. 1993). The introduction of TGF-β1 antisense phosphorothioate oligodeoxynucleotides, by retrograde ureteral injection, or TGF-β1 small interfering RNA (siRNA) effectively inhibits collagen I mRNA expression and interstitial fibrosis in the obstructed kidney (Isaka et al. 2000; Hwang et al. 2006). The administration of TGF-β neutralizing antibodies significantly reduces UUO-initiated inflammation, tubular epithelial apoptosis, and fibrosis (Miyajima et al. 2000; Gagliardini and Benigni 2006). The overexpression of the latent form of TGF-β1, as one strategy to minimize active TGF-β1 in the tissue microenvironment, decreases the incidence of cells positive for α-smooth muscle actin (presumably myofibroblasts) in UUO and blocks SMAD2/3 activation (Huang et al. 2008). The peroxisome proliferator-activated receptor gamma agonist troglitazone, moreover, attenuates UUO-induced renal interstitial fibrosis and inflammation through suppression of TGF-β1 expression (Kawai et al. 2009). Collectively, these data are consistent with the current concept that TGF-β1 is, indeed, the key initiator of fibrosis in UUO (Inazaki et al. 2004), either directly or as a consequence of elevated angiotensin II expression (Shin et al. 2005; Pimentel et al. 1995; Gu et al. 2001; Fern et al. 1999; Satoh et al. 2001; Ishidoya et al. 1995). Moreover, angiotensin stimulates the expression of genes encoding both ECM structural elements, such as collagen, fibronectin, and laminin, and specific inhibitors of matrix degradation, including plasminogen activator inhibitor-1 (PAI-1; a clade E member of the serine protease inhibitor gene family, SERPINE1), via TGF-β1-dependent mechanisms, thus leading to ECM accumulation and tissue fibrogenesis (Kagami et al. 1994; Wolf et al. 1993; Wolf 2006). Angiotensin also increases TGF-β1 transcription in renal cells and its bioactivation via thrombospondin-1 (Naito et al. 2004). Obstructive nephropathies are additionally accompanied by an increased expression of TGF-β receptors (Sutaria et al. 1998) presenting, perhaps, another therapeutic opportunity. Oral administration of the TGF-β type I receptor (ALK5) kinase inhibitor IN-1130, indeed, suppresses UUO-induced SMAD activation, matrix accumulation, and interstitial fibrosis (Moon et al. 2006). Caution as to the overall usefulness of TGF-β1 network-neutralizing or signal-inhibitory approaches in the setting of obstructive renal disease, however, is highlighted by the finding that high-dose antibody administration reduces efficacy because of an exacerbated inflammatory reaction (Ma et al. 2004).
TGF-β1 signaling regulation in renal fibrosis
Once engaged, TGF-β1-mediated signaling mechanisms are highly-regulated, positively and negatively, at multiple levels, e.g., receptor activity, internalization, SMAD recruitment, activation, translocalization and promoter binding, nuclear retention/exit of SMADs, and SMAD partnering with co-factors and non-SMAD transcriptional factors (Moustakis and Heldin 2009; Massagué 1998). Mechanisms of SMAD activation and the canonical pathway of TGF-β signaling have been comprehensively reviewed previously (e.g., Bottinger and Bitzer 2002; Miyazono 2009; Moustakis and Heldin 2009; Itoh and ten Dijke 2007; Ikushima and Miyazono 2010a, 2010b). TGF-β1-stimulated receptor SMAD activation is negatively regulated by SMAD7 and by the SMAD co-repressors Ski and SnoN effectively suppressing transcriptional responses (Itoh and ten Dijke 2007). In obstructive nephropathies and chronic renal disease, endogenous levels of SMAD7, Ski, and SnoN are reduced (by ubiquitin-dependent degradation), probably leading to persistent TGF-β1 signaling (Fukasawa et al. 2004, 2006; Yang et al. 2003). Gene transfer of SMAD7 to the kidney dramatically reduces interstitial fibrosis and SMAD activation induced by UUO (Lan et al. 2003; Li et al. 2002). SnoN targeting might also have translational implications. Indeed, the inhibition of SnoN degradation effectively attenuates TGF-β-induced renal fibronectin and α-smooth muscle actin in response to UUO (Tan et al. 2006; Fukasawa et al. 2006). An additional level of control involves bone morphogenetic protein-7 (BMP-7), a member of the TGF-β superfamily of ligands (e.g., Wang and Hirschberg 2003; Tanaka et al. 2008). Expression of BMP-7 and its receptors are reduced during the progression of renal fibrosis (Wang and Hirschberg 2003). BMP-7 largely signals through SMAD1/5/8 to antagonize SMAD2/3-dependent transcription (Mitu and Hirschberg 2008; Patel and Dressler 2005). BMP-7 administration in animal models of progressive renal failure (e.g., UUO, diabetic nephropathies) significantly reverses interstitial fibrosis, EMT, and inflammation (Zeisberg et al. 2003) highlighting the potential therapeutic value of BMP-7 as an inhibitor of disease progression. Moreover, BMP-7 attenuates the TGF-β1 induction of matrix elements (e.g., collagen and fribonectin), certain matrix metalloproteinases, and the potent profibrotic factors CCN2 (previously known as connective tissue growth factor) and PAI-1 in both interstitial fibroblasts and mesengial cells. The mechanism appears to involve the suppression of TGF-β1-activated SMAD3 pathways via the upregulation of the inhibitory SMAD6 through SMAD5 signaling in response to BMP (Wang and Hirschberg 2003; Wang et al. 2005). BMP-7, moreover, promotes mesenchymal-to-epithelial conversion (MET) in renal fibroblasts (reminiscent of the early stages of nephron development) and reduces TGF-β1-mediated EMT in tubular epithelial cells, collectively contributing to kidney regeneration and inhibiting disease progression (Patel and Dressler 2005; Zeisberg et al. 2003; Zeisberg et al. 2005). BMP-7 action in the context of renal disease, however, is itself subject to complex controls. KCP (Kielin/chordin like protein), a secreted protein with cysteine-rich domains, positively regulates BMP-7 signaling by enhancing the binding of BMP-7 to its receptor (Lin et al. 2005). KCP stimulation of BMP-7 signaling to Smad1 transcriptional targets reduces renal fibrosis and EMT in response to UUO or acute ischemic tubular injury, and consistent with this putative protective role, KCP−/− mice are susceptible to the development of interstitial fibrosis (Lin et al. 2005). Conversely, ectodin (uterine sensitization-associated gene-1 [USAG-1] or sclerostin) is a natural suppressor of BMP-7 signaling expressed in the kidney. As predicted from this role, ectodin−/− mice have significantly less renal fibrosis and EMT attributable to reduced TGF-β1 activity and enhanced BMP-7 signaling compared with wild-type mice subjected to UUO or cisplatin-initiated renal damage (Yanagita et al. 2006). Thus, the magnitude of BMP-7 signaling, regulated by agonist (e.g., KCP, BMP-7) and antagonist (ectodin, TGF-β1) interplay, profoundly influences the progression of renal disease but also provides additional therapeutic candidates to address the pathophysiologic consequences of persistent TGF-β1 signaling in the kidney.
TGF-β target genes: causative roles in pathogenesis
TGF-β1 regulates the expression of subsets of genes involved in epithelial “plasticity”, morphologic transdifferentiation, and stromal remodeling (e.g., Zavadil et al. 2001; Higgins 2006; Freytag et al. 2009, 2010). Many of these same morphologic transitions reflect, in large part, changes in transcriptional outputs in response to increasing levels of TGF-β1 in the renal parenchyma and, therefore, are superimposed on the time course of tubulointersitial fibrosis. Several comprehensive databases, derived from microarray profiling, provide critical insights into the spectrum of differential gene expression patterns in renal disease (Murphy et al. 2008; Ju et al. 2009; Matsuo et al. 2005; Silverstein et al. 2003; Higgins et al. 2003; Eikmans et al 2003; Sadlier et al. 2004; Seseke et al. 2004) and in response to TGF-β1 (for reviews, see Freytag et al. 2009, 2010). PAI-1, in particular, is a prominent member of, if not the most highly upregulated gene in, the TGF-β1-induced gene set (Freytag et al. 2009; Zavadil et al. 2001; Akiyoshi et al. 2001) and is causatively involved in the development of the EMT phenotype (Freytag et al. 2010). Importantly, and consistent with in vitro data, TGF-β1 siRNA suppresses UUO-initiated interstitial pathology through the reduced expression of the profibrogenic TGF-β1 target genes PAI-1 and collagen I (Hwang et al. 2006). PAI-1 is a prominent downstream target of the TGF-β1/SMAD3 pathway (for reviews, see Samarakoon and Higgins 2008; Ha et al. 2009) and an established contributor to fibrogenesis in several organ systems (e.g., Eddy and Fogo 2006; Chuang-Tsai et al. 2003). A major inhibitor of plasmin generation, PAI-1 decreases ECM degradation and promotes its accumulation, contributing thereby to renal fibrotic disease (Oda et al. 2001; Eddy 2009). Indeed, the delivery of small PAI-1 decoy peptides that mimic uPA/tPA domains effectively reduces both UUO-initiated and established interstitial fibrosis (Gonzalez et al. 2009). Consistent with the activation of TGF-β1 signaling in UUO, a dramatic increase of SMAD2/3 phosphorylation and PAI-1 protein expression occurs in the obstructed kidney compared with contralateral controls (Fig. 1).
Fig. 1.
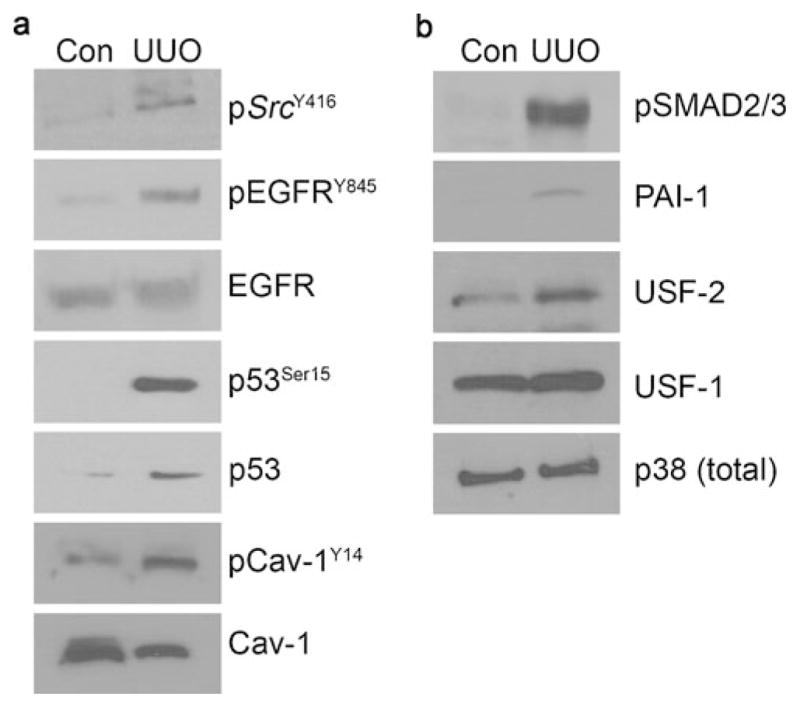
Activation of various profibrotic effector pathways in unilateral ureteral obstruction (UUO) in mice. UUO involved the ligation of the left ureter for 3 or 7 days; the contra-lateral (right) kidney that remained without surgical manipulation for the same period served as a control (Con). Obstructed and control kidneys were isolated, homogenized directly in SDS-containing buffer for Western analysis of tissue extracts. At 3 days after ureteral ligation, phosphorylation of c-SrcY416, epidermal growth factor receptorY845 (EGFRY845), and caveolin-1Y14 (Cav-1Y14; the two last-mentioned molecules being established targets of activated src family kinases) were increased, whereas total levels of EGFR and caveolin-1 largely remained unchanged providing relevant loading standards. p53Ser15 phosphorylation and total p53 levels were both markedly elevated in the obstructed kidney compared with the contra-lateral controls at day 3 (a). Consistent with increased TGF-β1 signaling in renal fibrotic disease, a robust induction of SMAD2/3 phosphorylation and an increase in plasminogen activator inhibitor-1 (PAI-1) expression were evident in the obstructed kidney at 7 days following UUO (b). A fivefold increase was noted in upstream stimulatory factor 2 (USF2) but not USF1 in the obstructed kidney. Total p38 levels remained constant providing a loading control
Increased PAI-1 expression occurs not only in UUO (Higgins et al. 2003), but also in other animal models of renal diseases including streptozotocin-initiated diabetic nephropathy (Nicholas et al. 2005) in which its induction is initiated by a number of profibrogenic and pro-inflammatory mediators including angiotensin, TGF-β, CCN2, interleukins, and tumor necrosis factor-α (Eddy and Fogo 2006). Although UUO is an important approach for identifying and assessing pathophysiologic events underlying induced renal fibrosis, some acknowledgement should be made that the underlying mechanisms might differ from those operative in diabetic animal models or human diabetic disease. In tubulointerstitial fibrosis induced by UUO, PAI-1 deficiency is renal-protective (Oda et al. 2001), whereas PAI-1 overexpression (in transgenic mice) promotes a fibrotic response with associated recruitment of macrophages and myofibroblasts (Matsuo et al. 2005). PAI-1−/− mice subjected to UUO, moreover, exhibit a significantly reduced inflammatory response compared with their wild-type counterparts suggesting that PAI-1 promotes the infiltration of macrophages and T-cells (Oda et al. 2001). PAI-1 also modulates TGF-β1 signaling, as PAI-1−/− animals have reduced TGF-β1 levels compared with wild-type mice similarly subjected to UUO (Krag et al. 2005). Indeed, recombinant PAI-1 stimulates TGF-β1 synthesis, perhaps through a urokinase plasminogen activator receptor (uPAR)-dependent mechanism involving downsteam mitogen-activated protein kinase (MAPK) signaling, to activate the TGF-β1 promoter suggesting the existence of a PAI-1/TGF-β1-positive feedback loop (Seo et al. 2009; Nicholas et al. 2005). However, significant examples exist in which PAI-1 can activate MAPK or Janus kinase(JAK)/signal transducer and activator of transcription (STAT) pathways via other co-receptors (e.g., low-density-lipoprotein receptor-related protein 1 [LRP1]; Degryse et al. 2004; Providence et al. 2008; Czekay et al. 2011). Modulation of PAI-1 expression levels may, therefore, have relevant effects on matrix accumulation through several proteolytic and non-proteolytic pathways, each of which may prove to have significant therapeutic value in retarding fibrosis (Huang et al. 2003).
Cooperative SMAD and non-SMAD factors mediate PAI-1 induction by TGF-β1
Although SMAD2/3 activation might be necessary, it is not sufficient for TGF-β1-stimulated PAI-1 expression in the absence of epidermal growth factor receptor (EGFR) signaling (Samarakoon et al. 2008; Samarakoon and Higgins 2008). These data are consistent with the realization that the progression of chronic renal disease involves the participation of several receptor and non-receptor tyrosine kinases (e.g., EGFR, platelet-derived growth factor, Abl, c-Src; see Wang et al. 2005; Floege et al. 2008; Eitner et al. 2008; Francois et al. 2004). Angiotensin-activated EGFR-mediated signaling (via the sheddase Adam 17), for example, is critically important in renal fibrosis induced by angiotensin infusion in mice (Lautrette et al. 2005). Transgenic mice expressing a dominant-negative EGFR (DN-EGFR) construct in the kidney, moreover, are also resistant to the development of renal fibrotic lesions following ischemic injury or subtotal nephrectomy (Terzi et al. 2000). Inhibition of EGFR by Gefitinib retards gromerular damage and collagen deposition in response to L-NAME-induced renal injury in rats (Francois et al. 2004). TGF-β1 also activates several tyrosine kinases including pp60c-src, c-Abl, and the EGFR (Wang et al. 2005; Samarakoon and Higgins 2008; Moustakis and Heldin 2009), and similar events occur in the setting of UUO (Fig. 1).
The available data suggest that SMADs and specific MAPK-targeted transcription factors occupy separate binding motifs at the TGF-β1-responsive PE2 region E-box in the human PAI-1 promoter (Dennier et al. 1998; Allen and Higgins 2004; Allen et al. 2005; Kutz et al. 2006; Samarakoon et al. 2005, 2008; Qi and Higgins 2003; Qi et al. 2006a). Complex formation at the PE2 site, moreover, requires cooperative signaling by the EGFR → extracellular signal-regulated kinase (ERK) (upstream stimulatory factor [USF]) and Rho/Rho-associated protein kinase (ROCK) (SMAD) pathways (Samarakoon and Higgins 2008). Extract immunodepletion and super-shift/complex-blocking experiments identified one PAI-1 E-box-binding protein to be USF2, a member of the basic helix-loop-helix-leucine zipper (bHLH-LZ) family of MYC-like proteins (Allen et al. 2005; Qi et al. 2006a). Replacement of USF1 homodimers with USF1/2 heterodimers or USF2 homodimers is associated with transcriptional activation of the PAI-1 gene (Qi et al. 2006a). Dominant-negative interference with USF DNA-binding ability significantly attenuates TGF-β1-mediated PAI-1 transcription (Qi and Higgins 2003; Qi et al. 2006a). Since MAPKs regulate the DNA-binding and transcriptional activities of USF (for a review, see Kutz et al. 2006), TGF-βR signaling through SMAD2/3 appears to require EGFR/mitogen-activated protein kinase kinase (MEK)-ERK-activated USF to attain high level PAI-1 expression (Samarakoon et al. 2005; Higgins 2006). SMADs might also interact with other E-box-binding HLH-LZ factors including TFE3 at the PE2 site, at least in one cell type (Hua et al. 1998). Such interacting complexes are likely to be important in PAI-1 gene control, since USF occupancy of the PAI-1 PE2 region E-box site modulates transcription in response to TGF-β1 or serum (Dennier et al. 1998; Qi and Higgins 2003), and USF2, but not USF1, levels are significantly increased in response to renal obstructive injury (Fig. 1).
The tumor suppressor p53 has also been implicated in the transcriptional regulation of profibrotic effectors (Vousden and Prives 2009). p53 family members are involved in a subset of TGF-β1 responses attributable to, in part, interactions between phosphorylated p53 and SMAD2, forming transcriptionally active multi-protein complexes (Piccolo 2008; Cordenonsi et al. 2003, 2007). DNase I footprinting/methylation interference and oligonucleotide mobility shift analyses have confirmed that p53 binds to motifs in the PAI-1 promoter, possibly the two p53 half-sites (AcACATGCCT, cAGCAAGTCC) at −224 bp to −204 bp relative to the transcription start site (Kunz et al. 1995; Riley et al. 2008). The DNA binding reflects both p53 sequence-driven reporter gene transcription and induced expression of the endogenous PAI-1 gene. TGF-β1-initiated PAI-1 expression is significantly attenuated in p53 knockdown cells (Cordenonsi et al. 2003). Consistent with these findings, p53−/− fibroblasts are not inducible for increased PAI-1 expression in response to TGF-β1 and pretreatment of Mv-1Lu mink lung cells (stably expressing a PAI-1 promoter-luciferase reporter construct) with the p53 inhibitor pifithrin-α effectively suppressed TGF-β1-dependent PAI-1 transcription (Fig. 2). Stimulation of PAI-1 gene activation in mouse fibroblasts by the nephrotoxin cisplatin, moreover, is p53-dependent (Kortlever et al. 2008). One hypothesis suggests that p53 interacts directly with SMAD2 (Dupont et al. 2004; Cordenonsi et al. 2003, 2007). In TGF-β1-stimulated cells, the binding of USF to the PE2 site might facilitate DNA bending. Phasing analysis has revealed that certain bHLH-LZ of the MYC family (including USF) orient the DNA bend toward the minor groove (Fisher et al. 1992); this could potentially promote interactions between p53, bound to its downstream half-site motif, with SMAD2 tethered to the upstream PE2 region SMAD site. Radiation-associated PAI-1 expression also requires TGF-β1-mediated p53/SMAD cooperative transcriptional interactions, further highlighting SMAD and non-SMAD interplay in TGF-β1-mediated signaling events (Milliat et al. 2008). Regardless of the precise mechanism, the available data suggest a central role for p53 in TGF-β1-initiated PAI-1 gene control (Figs. 1, 2).
Fig. 2.

Role of p53 in TGF-β1-induced PAI-1 expression. Confluent and serum-deprived p53−/− or wild-type (p53+/+) mouse embryonic fibroblasts (MEFs) were stimulated with TGF-β1 (0.1 ng/ml) for 24 h and disrupted in SDS-containing buffer; lysates were immunoblotted for the indicated proteins. Increases in PAI-1 (a, b) and p21 (a) protein 24 h after addition of TGF-β1 (T) were evident in wild-type but not in p53−/− MEFs (Con=control). ERK2 provided a loading control; p53 genetic status was confirmed by Western analysis (a). The histogram in c represents the mean±SD of triplicate studies highlighting the p53 requirement for transcriptional activation of the PAI-1 promoter in response to TGF-β1. Mink lung epithelial cells (Mv-1Lu) stably expressing a PAI-1 promoter-driven luciferase reporter (p800-Luc) were passaged and stimulated 8 h later with various doses of the p53 inhibitor pifithrin-α (10–20 μM; P-α(10), P-α(20)) for 24 hours prior to addition of TGF-β1 for an additional 18 h. Cellular lysates were used for luciferase measurements. TGF-β1-induced PAI-1 promoter activation was dose-dependently suppressed by pifithrin-α suggesting a requirement for p53 in PAI-1 transcription (c). TGF-β1 (0.1 ng/ml) and 200 μM H2O2 (serving as a positive control for oxidative stress) induced p53Ser15 phosphorylation, consistent with activation of this transcription factor by both stimuli in wild-type (p53+/+) but not p53 deficient MEFs (d). ERK2 provided a loading control
Increased expression and serine 15 phosphorylation (activation) of p53 is evident in the kidney following UUO (Fig. 1), ischemic-reperfusion- or nephrotoxin-induced (e.g., cisplatin, aristolochic acid) renal injury (Zhou et al. 2010; Wei et al. 2007). p53−/− mice largely retain their renal architecture and function following treatment with cisplatin or aristolochic acid, whereas their wild-type counterparts develop severe renal damage and compromised organ function consistent with a role for p53 in promoting tubular cell apoptosis and growth arrest (Wei et al. 2007; Zhou et al. 2010). siRNA-directed silencing of p53 in mice, moreover, abates the severity of cisplatin- and ischemic-induced kidney damage (Molitoris et al. 2009). Similarly, the p53 inhibitor pifithrin-α suppresses both the accumulation and phosphorylation of p53 and attenuates tubular apoptosis and renal damage (Wei et al. 2007). Recent studies, furthermore, link tubular epithelial growth arrest in response to both acute (e.g., ischemia-reperfusion, nephrotoxins) and more protracted (UUO) injury to the progression of renal fibrosis via p53 and JNK signaling with the retention of TGF-β signaling (Yang et al. 2010). Blockade of p53 activation by pifithrin-α effectively attenuates tubular cell G2/M arrest, decreases TGF-β1 and CCN2 levels, and reduces overall fibrosis (Yang et al. 2010).
Concluding remarks
MAPKs (e.g., p38, ERK1/2) phosphorylate and transcriptionally activate USF proteins (Higgins 2006), and molecular interference with the DNA-binding ability of USF (by dominant-negative approaches) significantly attenuates PAI-1 transcription in response to TGF-β1 in non-kidney cells (Qi et al. 2006a). Similarly, PAI-1 induction in renal and non-renal cells by TGF-β1 requires EGFR-ERK1/2 signaling (Cho et al. 2009; Samarakoon et al. 2005, 2008) and a USF1 → USF2 isoform switch at the PE2 E-box site in the PAI-1 promoter (Qi et al. 2006a). In this regard, the TGF-β1-initiated engagement of the MAPK (ERK, p38) pathways in UUO (Masaki et al. 2003) could result in USF2 mobilization and the induction of USF target genes, such as PAI-1, in cooperation with (TGF-β1-activated) SMADs and (MAPK-phosphorylated) p53 (summarized in Fig. 3). In human renal cells, moreover, autostimulation of TGF-β1 gene transcription by TGF-β1 also requires both SMAD3 and MAPK/ERK activity (Zhang et al. 2006).
Fig. 3.

TGFβ1/SMAD3 signaling as an integrator of fibrotic mechanisms in the kidney. The pathophysiology of obstructive and diabetic renal fibrosis involves the activation of multi-component receptor-initiated signaling events. Representation of the individual interacting systems discussed in this review. Whereas unique elements are found in each, TGF-β1/SMAD2/3 signaling appears to integrate downstream effectors of these collateral pathways, in certain circumstances, to promote a profibrotic microenvironment. Persistent TGF-β1 expression and SMAD2/3 phosphorylation consistently accompany progression of chronic renal disease in animal models and humans. Expression of negative regulatory TGF-β1/SMAD pathway elements (inhibitory SMAD-7, Ski, Sno) are reduced in the context of tissue fibrosis, thus, further amplifying TGF-β1-initiated fibrogenic processes (e.g., ECM synthesis, EMT, inflammation, CNN2 [previously designated connective tissue growth factor] and PAI-1 induction, and myofibroblast differentiation/persistence). Moreover, both the expression and signaling ability of negative effectors of TGF-β1 signaling (e.g., BMP-7, hepatocyte growth factor [HGF]) are also suppressed by a variety of mechanisms (detailed in the text) facilitating maintenance of TGF-β1 signaling in the injured tissue leading to excessive deposition of ECM components, disruption of renal parenchyma, and progressive loss of kidney function
Activated MEK/ERK, ataxia telangiectasia-mutated (ATM), and casein kinase sites 1 and 2 (CK1 and CK2) can phosphorylate p53 (at serine 15) promoting the acetylation of p53 (by CREB-binding protein/p300) necessary for subsequent transcriptional activation (Meek and Anderson 2009). p300/CREB-binding protein, a histone acetyltransferase, also interacts with and acetylates SMAD2/3 in response to TGF-β1 (Feng et al. 1998) facilitating the creation of a transcriptional complex (SMADs/p53/USF2) necessary for optimal PAI-1 induction (Tu and Luo 2007; Simonsson et al. 2006; Das et al. 2008). The importance of such interactions is underscored by the finding that RAP250, a protein that has no intrinsic enzymatic activity but that effectively recruits histone acetyltransferases and methylases to chromatin complexes, also interacts with SMAD2/3 and is essential for maximal TGF-β1-stimulated PAI-1 expression (Antonson et al. 2008). Selective targeting of p53/SMAD/USF2 interactions, therefore, might provide an attractive strategy to intervene in the progression of obstructive renal disease. Phosphorylation of p53, moreover, is critical for SMAD2/3 interactions, further demonstrating cross-talk between SMAD and non-SMAD elements in TGF-β signaling (Cordenonsi et al. 2007). USF2, which is upregulated in the obstructed kidney (Fig. 1), and the PE2 E-box motif with its adjoining SMAD-binding sequences probably constitute a platform for transcriptional complex formation with SMAD2/3 and p53. This hypothesis is supported by the finding that transgenic USF-2 mice overexpress TGF-β1 and components of the renin-angiotensin system, thereby modulating the progression of kidney disease in streptozotocin-induced diabetic nephropathies (Shi et al. 2009; Liu et al. 2007). Whether stimulation of the MAPK (ERK, p38) pathways in UUO (Masaki et al. 2003) by TGF-β1 with downstream USF activation subsequently induces the expression of USF-dependent genes, such as PAI-1, in cooperation with SMAD and p53 remains to be established. Clarification of this axis would have generalized implications with regard to current therapeutic options for the clinical management of renal fibrotic disease.
Acknowledgments
This work was supported by NIH grant R01 GM057242.
References
- Akiyoshi A, Ishii M, Nemoto N, Kawabata M, Aburatani H, Miyazono K. Targets of transcriptional regulation by transforming growth factor-β: expression profile analysis using oligonucleotide arrays. Jpn J Cancer Res. 2001;92:257–268. doi: 10.1111/j.1349-7006.2001.tb01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RR, Higgins PJ. Plasminogen activator inhibitor inhibitor type-1 expression and the pathophysiology of TGF-β1-induced epithelial-to-mesenchymal transition. Recent Res Dev Physiol. 2004;2:355–366. [Google Scholar]
- Allen RR, Qi L, Higgins PJ. Upstream stimulatory factor regulates E box-dependent PAI-1 transcription in human epidermal keratinocytes. J Cell Physiol. 2005;201:156–165. doi: 10.1002/jcp.20211. [DOI] [PubMed] [Google Scholar]
- Antonson P, Jakobsson T, Almlof T, Guldevall K, Steffensen KR, Gustafsson JA. RAP250 is a coactivator in the transforming growth factor β signaling pathway that interacts with Smad2 and Smad3. J Biol Chem. 2008;283:8995–9001. doi: 10.1074/jbc.M707203200. [DOI] [PubMed] [Google Scholar]
- Arany PR, Flanders KC, DeGraff W, Cook J, Mitchell JB, Roberts AB. Absence of Smad3 confers radioprotection through modulation of ERK-MAPK in primary dermal fibroblasts. J Dermatol Sci. 2007;48:35–42. doi: 10.1016/j.jdermsci.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma H, Vanderbrink BA, Campbell MT, Hile KL, Zhang H, Meldrum DR, Meldrum KK. Arterially delivered mesenchymal stem cells prevent obstruction-induced renal fibrosis. J Surg Res. 2010;168:e51–59. doi: 10.1016/j.jss.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bascands JL, Schanstra JP. Obstructive nephropathy: insights from genetically engineered animals. Kidney Int. 2005;68:925–937. doi: 10.1111/j.1523-1755.2005.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV. Pathophysiology of AKI: injury and normal and abnormal repair. Contrib Nephrol. 2010;165:9–17. doi: 10.1159/000313738. [DOI] [PubMed] [Google Scholar]
- Boor P. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6:643–656. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- Bottinger EP. TGF-β in renal injury and disease. Semin Nephrol. 2007;27:309–320. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Bottinger EP, Bitzer M. TGF-β signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- Chevalier RI, Kim A, Thornhill BA, Wolstenholme JT. Recovery following relief of unilateral ureteral obstruction in the neonatal rat. Kidney Int. 1999;55:793–807. doi: 10.1046/j.1523-1755.1999.055003793.x. [DOI] [PubMed] [Google Scholar]
- Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145–1152. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- Chevalier RL, Thornhill BA, Forbes MS, Kiley SC. Mechanisms of renal injury and progression of renal disease in congenital obstructive nephropathy. Pediatr Nephrol. 2010;25:687–697. doi: 10.1007/s00467-009-1316-5. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Kang JH, Kim T, Park KK, Kim CH, Lee IS, Min KS, Magae J, Nakajima H, Bae YS, Chang YC. Suppression of PAI-1 expression through inhibition of the EGFR-mediated signaling cascade in rat kidney fibroblast by ascofuranone. J Cell Biochem. 2009;107:335–344. doi: 10.1002/jcb.22130. [DOI] [PubMed] [Google Scholar]
- Chuang-Tsai S, Sisson TH, Hattori N, Tsai CG, Subbotina NM, Hanson KE, Simon RH. Reduction in fibrotic tissue formation in mice genetically deficient in plasminogen activator inhibitor-1. Am J Pathol. 2003;163:445–452. doi: 10.1016/S0002-9440(10)63674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook HT. The origin of renal fibroblasts and progression of kidney disease. Am J Pathol. 2010;176:22–24. doi: 10.2353/ajpath.2010.090898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S. Links between tumor suppressors: p53 is required for TGF-β gene responses by cooperating with SMADs. Cell. 2003;113:301–314. doi: 10.1016/s0092-8674(03)00308-8. [DOI] [PubMed] [Google Scholar]
- Cordenonsi M, Montagner M, Adorno M, Zacchigna L, Martello G, Mamidi A, Soligo S, Dupont S, Piccolo S. Integration of TGF-beta and Ras/MAPK signaling through p53 phosphorylation. Science. 2007;315:840–843. doi: 10.1126/science.1135961. [DOI] [PubMed] [Google Scholar]
- Czekay R-P, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet J, Klein RM, Higgins CE, Samarakoon R, Higgins PJ. PAI-1: an integrator of cell signaling and migration. Int J Cell Biol. 2011 doi: 10.1155/2011/562481. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das F, Ghosh-Choudhury N, Venkatesan B, Li X, Mahimainathan L, Choudhury GG. Akt kinase targets association of CBP with Smad 3 to regulate TGF-β-induced expression of plasminogen activator inhibitor-1. J Cell Physiol. 2008;214:513–527. doi: 10.1002/jcp.21236. [DOI] [PubMed] [Google Scholar]
- Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y, Loskutoff DJ. The low density lipoprotein receptor-related protein is a mitogenic receptor for plasminiogen activator inhibitor-1. J Biol Chem. 2004;279:22595–22604. doi: 10.1074/jbc.M313004200. [DOI] [PubMed] [Google Scholar]
- Dendooven A, Ishola DA, Jr, Nguyen TQ, Van der Giezen DM, Kok RJ, Goldschmeding R, Joles JA. Oxidative stress in obstructive nephropathy. Int J Exp Path. 2010 doi: 10.1111/j.1365-2613.2010.00730.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennier S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF-β1-inducible elements in the promoter of human plasminogen activator inhibotor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Dupont S, Zacchigna L, Adorno M, Soligo S, Volpin D, Piccolo S, Cordenonsi M. Convergence of p53 and TGF-β signaling networks. Cancer Lett. 2004;213:129–138. doi: 10.1016/j.canlet.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000;15:290–301. doi: 10.1007/s004670000461. [DOI] [PubMed] [Google Scholar]
- Eddy AA. Progression of chronic kidney disease. Adv Chronic Kidney Dis. 2005;12:353–365. doi: 10.1053/j.ackd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Eddy AA. Serine proteases, inhibitors and receptors in renal fibrosis. Thromb Haemost. 2009;101:656–664. [PMC free article] [PubMed] [Google Scholar]
- Eddy AA, Fogo AB. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol. 2006;17:2999–3012. doi: 10.1681/ASN.2006050503. [DOI] [PubMed] [Google Scholar]
- Eikmans M, Baelde JJ, de Heer E, Bruijn JA. ECM homeostasis in renal diseases: a genomic approach. J Pathol. 2003;200:526–536. doi: 10.1002/path.1417. [DOI] [PubMed] [Google Scholar]
- Eitner F, Bucher E, van Roeyen C, Kunter U, Rong S, Seikrit C, Villa L, Boor P, Fredriksson L, Backstrom G, Eriksson U, Ostman A, Floege J, Ostendorf T. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol. 2008;19:281–289. doi: 10.1681/ASN.2007030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JM, Huang XR, Ng YY, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am J Kidney Dis. 2001;37:820–831. doi: 10.1016/s0272-6386(01)80132-3. [DOI] [PubMed] [Google Scholar]
- Feng XH, Zhang Y, Wu RY, Derynck R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-b-induced transcriptional activation. Genes Dev. 1998;12:2153–2163. doi: 10.1101/gad.12.14.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern RJ, Yesko CM, Thornhill BA, Kim HS, Smithies O, Chevalier RL. Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest. 1999;103:39–46. doi: 10.1172/JCI4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher DE, Parent LA, Sharp PA. Myc/Max and other helix-loop-helix/leucine zipper proteins bend DNA toward the minor groove. Proc Natl Acad Sci USA. 1992;89:11779–11783. doi: 10.1073/pnas.89.24.11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- Francois H, Placier S, Flamant M, Tharaux PL, Chansel D, Dussaule JC, Chatziantoniou C. Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J. 2004;18:926–928. doi: 10.1096/fj.03-0702fje. [DOI] [PubMed] [Google Scholar]
- Freytag J, Wilkins-Port CE, Higgins CE, Carlson JA, Noel A, Foidart JM, Higgins SP, Samarakoon R, Higgins PJ. PAI-1 regulates the invasive phenotype in human cutaneous squamous cell carcinoma. J Oncol. 2009;2009:1–10. doi: 10.1155/2009/963209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag J, Wilkins-Port CE, Higgins CE, Higgins SP, Samarakoon R, Higgins PJ. PAI-1 mediates the TGF-β1+EGF-induced “scatter” response in transformed human keratinocytes. J Invest Dermatol. 2010;130:2179–2190. doi: 10.1038/jid.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, Uchida C, Kitagawa K, Hattori T, Suzuki S, Kitagawa M, Hishida A. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci USA. 2004;101:8687–8692. doi: 10.1073/pnas.0400035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, Uchida C, Kitagawa K, Hattori T, Suzuki S, Kitagawa M, Hishida A. Ubiquitin-dependent degradation of SnoN and Ski is increased in renal fibrosis induced by obstructive injury. Kidney Int. 2006;69:1733–1740. doi: 10.1038/sj.ki.5000261. [DOI] [PubMed] [Google Scholar]
- Gagliardini E, Benigni A. Role of anti-TGF-β antibodies in the treatment of renal injury. Cytokine Growth Factor Rev. 2006;17:89–96. doi: 10.1016/j.cytogfr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Gonzalez J, Klein J, Chauhan SD, Neau E, Calise D, Nevoit C, Chaaya R, Miravette M, Delage C, Bascands J-L, Schanstra JP, Buffin-Meyer B. Delayed treatment with plasminogen activator inhibitor-1 decoys reduces tubulointerstitial fibrosis. Exp Biol Med. 2009;234:1511–1518. doi: 10.3181/0903-RM-105. [DOI] [PubMed] [Google Scholar]
- Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol. 2009;5:319–328. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- Grande MT, Perez-Barriocanal F, Lopez-Novoa JM. Role of inflammation in tubulo-interstitial damage associated to obstructive nephropathy. J Inflamm (Lond) 2010;7:19. doi: 10.1186/1476-9255-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Morrissey J, McCracken R, Tolley T, Liapis H, Klahr S. Contributions of angiotensin II and tumor necrosis factor-α to the development of renal fibrosis. Am J Physiol Renal Physiol. 2001;280:F777–F785. doi: 10.1152/ajprenal.2001.280.5.F777. [DOI] [PubMed] [Google Scholar]
- Ha H, Oh EY, Lee HB. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat Rev Nephrol. 2009;5:203–211. doi: 10.1038/nrneph.2009.15. [DOI] [PubMed] [Google Scholar]
- Higgins PJ. TGF-β1-stimulated p21ras-ERK signaling regulates expression of the angiogenic SERPIN PAI-1. Recent Res Dev Biochem. 2006;7:31–45. [Google Scholar]
- Higgins DF, Lappin DW, Kieran NE, Anders HL, Watson RW, Strutz F, Schlondorff D, Haase VH, Fitzpatrick JM, Godson C, Brady HR. DNA oligonucleotide microarray technology identifies fisp-12 among other potential fibrogenic genes following murine unilateral ureteral obstruction (UUO): modulation during epithelial-mesenchymal transition. Kidney Int. 2003;64:2079–2091. doi: 10.1046/j.1523-1755.2003.00306.x. [DOI] [PubMed] [Google Scholar]
- Hruska KA. Treatment of chronic tubulointerstitital disease: a new concept. Kidney Int. 2002;61:1911–1922. doi: 10.1046/j.1523-1755.2002.00331.x. [DOI] [PubMed] [Google Scholar]
- Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and smad proteins in TGF-β-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 1998;12:3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Haraguchi M, Lawrence DA, Border WA, Yu L, Noble NA. A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J Clin Invest. 2003;112:379–388. doi: 10.1172/JCI18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XR, Chung AC, Wang XJ, Lai KN, Lan HY. Mice overexpressing latent TGF-β1 are protected against renal fibrosis in obstructive kidney disease. Am J Physiol Renal Physiol. 2008;295:F118–F127. doi: 10.1152/ajprenal.00021.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang M, Kim HJ, Noh HJ, Chang YC, Chae YM, Kim KH, Jeon JP, Lee TS, Oh HK, Lee YS, Park KK. TGF-β1 siRNA suppresses the tubulointerstitial fibrosis in the kidney of ureteral obstruction. Exp Mol Pathol. 2006;81:48–54. doi: 10.1016/j.yexmp.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K. Cellular context-dependent “colors” of transforming growth factor-β signaling. Cancer Sci. 2010a;101:306–312. doi: 10.1111/j.1349-7006.2009.01441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010b;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- Inazaki K, Kanamaru Y, Kojima Y, Sueyoshi N, Okumura K, Kaneko K, Yamashiro Y, Ogawa H, Nakao A. Smad3 deficiency attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66:597–604. doi: 10.1111/j.1523-1755.2004.00779.x. [DOI] [PubMed] [Google Scholar]
- Isaka Y, Tsujie M, Ando Y, Nakamura H, Kaneda Y, Imai E, Hori M. Transforming growth factor-β1 antisense oligodeoxynucleotides block interstitial fibrosis in unilateral ureteral obstruction. Kidney Int. 2000;58:1885–1892. doi: 10.1111/j.1523-1755.2000.00360.x. [DOI] [PubMed] [Google Scholar]
- Ishidoya S, Morrissey J, McCracken R, Reyes A, Klahr S. Angiotensin II receptor antagonist ameliorates renal tubulointerstitial fibrosis caused by unilateral ureteral obstruction. Kidney Int. 1995;47:1285–1294. doi: 10.1038/ki.1995.183. [DOI] [PubMed] [Google Scholar]
- Itoh S, ten Dijke P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19:176–184. doi: 10.1016/j.ceb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Iwano M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. Curr Opin Nephrol Hypertens. 2004;13:279–284. doi: 10.1097/00041552-200405000-00003. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Eichinger F, Bitzer M, Oh J, McWeeney S, Berthier CC, Shedden K, Cohen CD, Henger A, Krick S, Kopp JB, Stoeckert CJ, Jr, Dikman S, Schroppel B, Thomas DB, Schlondorff D, Kretzler M, Bottinger EP. Renal gene and protein expression signatures for prediction of kidney disease progression. Am J Pathol. 2009;174:2073–2075. doi: 10.2353/ajpath.2009.080888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J Clin Invest. 1994;93:2431–2437. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneto H, Morrissey J, Klahr S. Increased expression of TGF-β1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int. 1993;44:313–321. doi: 10.1038/ki.1993.246. [DOI] [PubMed] [Google Scholar]
- Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N. PPAR-gamma agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β. Lab Invest. 2009;89:47–58. doi: 10.1038/labinvest.2008.104. [DOI] [PubMed] [Google Scholar]
- Klahr S. New insights into the consequences and mechanisms of renal impairment in obstructive nephropathy. Am J Kidney Dis. 1991;18:689–699. doi: 10.1016/s0272-6386(12)80611-1. [DOI] [PubMed] [Google Scholar]
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002;283:F861–F875. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- Klein J, Gonzalez J, Miravete M, Caubet C, Chaaya R, Decramer S, Bandin F, Bascands J-L, Buffin-Meyer B, Joost P. Congenital ureteropelvic junction obstruction: human disease and animal models. Int J Exp Pathol. 2010 doi: 10.1111/j.1365-2613.2010.00727.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Brummelkamp TR, van Meeteren LA, Moolenaar WH, Bernards R. Suppression of the p53-dependent replicative senescence response by lysophosphatidic acid signaling. Mol Cancer Res. 2008;6:1452–1460. doi: 10.1158/1541-7786.MCR-08-0066. [DOI] [PubMed] [Google Scholar]
- Krag S, Danielsen CC, Carmeliet P, Nyengaard J, Wogensen L. Plasminogen activator inhibitor-1 gene deficiency attenuates TGF-β1-induced kidney disease. Kidney Int. 2005;68:2651–2666. doi: 10.1111/j.1523-1755.2005.00737.x. [DOI] [PubMed] [Google Scholar]
- Kunz C, Pebler S, Otte J, von der Ahe D. Differential regulation of plasminogen activator and inhibitor gene transcription by the tumor suppressor p53. Nucleic Acids Res. 1995;25:3710–3727. doi: 10.1093/nar/23.18.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutz SM, Higgins CE, Samarakoon R, Higgins SP, Allen RR, Qi L, Higgins PJ. TGF-β1-induced PAI-1 expression is E box/USF-dependent and requires EGFR signaling. Exp Cell Res. 2006;312:1093–1105. doi: 10.1016/j.yexcr.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu HJ, Morishita R, Johnson RJ. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol. 2003;14:1535–1548. doi: 10.1097/01.asn.0000067632.04658.b8. [DOI] [PubMed] [Google Scholar]
- Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med. 2005;11:867–874. doi: 10.1038/nm1275. [DOI] [PubMed] [Google Scholar]
- Ledbetter S, Kurtzberg L, Doyle S, Pratt BM. Renal fibrosis in mice treated with human recombinant transforming growth factor-β2. Kidney Int. 2000;58:2367–2376. doi: 10.1046/j.1523-1755.2000.00420.x. [DOI] [PubMed] [Google Scholar]
- Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY. Smad7 inhibits fibrotic effect of TGF-β on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol. 2002;13:1464–1472. doi: 10.1097/01.asn.0000014252.37680.e4. [DOI] [PubMed] [Google Scholar]
- Lin J, Patel SR, Cheng X, Cho EA, Levitan I, Ullenbruch M, Phan SH, Park JM, Dressler GR. Kielin/chordin-like protein, a novel enhancer of BMP signaling, attenuates renal fibrotic disease. Nat Med. 2005;11:387–393. doi: 10.1038/nm1217. [DOI] [PubMed] [Google Scholar]
- Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Renal fibrosis: new insights in to pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shi L, Wang S. Oversexpression of upstream stimulatory factor 2 accelerates diabetic kidney injury. Am J Physiol Renal Physiol. 2007;293:F1727–F1735. doi: 10.1152/ajprenal.00316.2007. [DOI] [PubMed] [Google Scholar]
- Ma LJ, Jha S, Ling H, Pozzi A, Ledbetter S, Fogo AB. Divergent effects of low versus high dose anti-TGF-beta antibody in puromycin aminonucleoside nephropathy in rats. Kidney Int. 2004;65:106–115. doi: 10.1111/j.1523-1755.2004.00381.x. [DOI] [PubMed] [Google Scholar]
- Manucha W. Biochemical-molecular markers in unilateral uretal obstruction. Biocell. 2007;31:1–12. [PubMed] [Google Scholar]
- Masaki T, Foti R, Hill PA, Ikezumi Y, Atkins RC, Nikolic-Paterson DJ. Activation of the ERK pathway preceeds tubular proliferation in the obstructed rat kidney. Kidney Int. 2003;63:1256–1264. doi: 10.1046/j.1523-1755.2003.00874.x. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Matsuo S, Lopez-Guisa JM, Cai X, Okamura DM, Alpers CE, Bumgarner RE, Peters MA, Zhang G, Eddy AA. Multi-functionality of PAI-1 in fibrogenesis: evidence from obstructive nephropathy in PAI-1-overexpressing mice. Kidney Int. 2005;67:2221–2238. doi: 10.1111/j.1523-1755.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, Ju W, Bottinger EP, Lan HY. Smad2 protects agains TGF-β/Smad3-mediated renal fibrosis. J Am Soc Nephrol. 2010;21:1477–1487. doi: 10.1681/ASN.2009121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzano SA, Aros CA, Droguett A, Burgos ME, Ardiles LG, Flores CA, Carpio D, Vio CP, Ruiz-Ortega M, Egido J. Renal angiotensin II up-regulation and myofibroblast activation in human membranous nephropathy. Kidney Int Suppl. 2003;86:S39–S45. doi: 10.1046/j.1523-1755.64.s86.8.x. [DOI] [PubMed] [Google Scholar]
- Milliat F, Sabourin JC, Tarlet G, Holler V, Deutsch E, Buard V, Tamarat R, Atfi A, Benderitter M, François A. Essential role of plasminogen activator inhibitor type-1 in radiation enteropathy. Am J Pathol. 2008;172:691–701. doi: 10.2353/ajpath.2008.070930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitu G, Hirschberg R. Bone morphogenetic protein-7 (BMP7) in chronic kidney disease. Front Biosci. 2008;13:4726–4739. doi: 10.2741/3035. [DOI] [PubMed] [Google Scholar]
- Miyajima A, Chen JC, Lawrence C, Ledbetter S, Soslow RA, Stern J, Jha S, Pigato J, Lemer ML, Poppas DP, Vaughan ED, Felsen D. Antibody to transforming growth factor-β ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000;58:2301–2313. doi: 10.1046/j.1523-1755.2000.00414.x. [DOI] [PubMed] [Google Scholar]
- Miyazono K. Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad B Phys Biol Sci. 2009;85:314–323. doi: 10.2183/pjab.85.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitoris BA, Dagher PC, Sandoval RM, Campos SB, Ashush H, Fridman E, Brafman A, Faerman A, Atkinson SJ, Thompson JD, Kalinski H, Skaliter R, Erlich S, Feinstein E. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol. 2009;20:1754–1764. doi: 10.1681/ASN.2008111204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller JC, Skiver E, Olsen S, Maunsbach AB. Ultrastructural analysis of human proximal tubules and cortical interstitium in chronic renal disease (hydronephrosis) Virchows Arch A Pathol Anat Histopathol. 1984;402:209–237. doi: 10.1007/BF00695077. [DOI] [PubMed] [Google Scholar]
- Moon JA, Kim HT, Cho IS, Sheen YY, Kim DK. IN-1130, a novel transforming growth factor-β type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 2006;70:1234–1243. doi: 10.1038/sj.ki.5001775. [DOI] [PubMed] [Google Scholar]
- Moustakis A, Heldin CH. The regulation of TGF-β signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- Murphy M, Crean J, Brazil DP, Sadlier D, Martin F, Godson C. Regulation and consequences of differential gene expression in diabetic kidney disease. Biochem Soc Trans. 2008;36:941–945. doi: 10.1042/BST0360941. [DOI] [PubMed] [Google Scholar]
- Naito T, Masaki T, Nikolic-Paterson DJ, Tanji C, Yorioka N, Kohno N. Angiotensin II induces thrombospondin-1 production in human mesangial cells via p38 MAPK and JNK: a mechanism for activation of latent TGF-β1. Am J Physiol Renal Physiol. 2004;286:F278–F287. doi: 10.1152/ajprenal.00139.2003. [DOI] [PubMed] [Google Scholar]
- Nicholas SB, Aguiniga E, Ren Y, Kim J, Wong J, Govindarajan N, Noda M, Wang W, Kawano Y, Collins A, Hsueh WA. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005;67:1297–1307. doi: 10.1111/j.1523-1755.2005.00207.x. [DOI] [PubMed] [Google Scholar]
- Oda T, Jung YO, Kim HS, Cai X, López-Guisa JM, Ikeda Y, Eddy AA. PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int. 2001;60:587–596. doi: 10.1046/j.1523-1755.2001.030002587.x. [DOI] [PubMed] [Google Scholar]
- Oldfield MD, Bach LA, Forbes JM, Nikolic-Paterson D, McRobert A, Thallas V, Atkins RC, Osicka T, Jerums G, Cooper ME. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J Clin Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HC, Yasuda K, Ratliff B, Stoessel A, Sharkovska Y, Yamamoto I, Jasmin JF, Bachmann S, Lisanti MP, Chander P, Goligorsky MS. Posobstructive regeneration of kidney is derailed when surge in renal stem cells during course of unilateral ureteral obstruction is halted. Am J Physiol Renal Physiol. 2010a;298:F357–F364. doi: 10.1152/ajprenal.00542.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HC, Yasuda K, Kuo MC, Ni J, Ratliff BB, Chander PN, Goligorsky MS. Renal capsule as stem cell niche. Am J Physiol Renal Physiol. 2010b doi: 10.1152/ajprenal.00406.2009. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SR, Dressler GR. BMP7 signaling in renal development and disease. Trends Mol Med. 2005;11:512–518. doi: 10.1016/j.molmed.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130:141–155. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S. p53 regulation orchestrates the TGF-β response. Cell. 2008;133:767–769. doi: 10.1016/j.cell.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Pimentel JL, Jr, Sundell CL, Wang S, Kopp JB, Montero A, Martinez-Maldonado M. Role of angiotensin II in the expression and regulation of transforming growth factor-β in obstructive nephropathy. Kidney Int. 1995;48:1233–1246. doi: 10.1038/ki.1995.407. [DOI] [PubMed] [Google Scholar]
- Providence KM, Higgins SP, Mullen A, Battista A, Samarakoon R, Higgins CE, Wilkins-Port CE, Higgins PJ. SERPINE1 (PAI-1) is deposited into keratinocyte migration “trails” and required for optimal monolayer wound repair. Arch Dermatol Res. 2008;300:303–310. doi: 10.1007/s00403-008-0845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri TS, Shakaib MI, Chang A, Mathew L, Olayinka O, Minto AW, Saray M, Hack BK, Quigg RJ. Chronic kidney disease induced in mice by reversible unilateral ureteral obstruction is dependent on genetic background. Am J Physiol Renal Physiol. 2010;298:F1024–F1032. doi: 10.1152/ajprenal.00384.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Higgins PJ. Use of chromatin immunoprecipitation to identify E box-binding transcription factors in the promoter of the growth state-regulated human PAI-1 gene. Recent Res Dev Mol Biol. 2003;1:1–12. [Google Scholar]
- Qi L, Allen RR, Lu Q, Higgins CE, Garone R, Staiano-Coico L, Higgins PJ. PAI-1 transcriptional regulation during the G0→G1 transition in human epidermal keratinocytes. J Cell Biochem. 2006a;99:495–507. doi: 10.1002/jcb.20885. [DOI] [PubMed] [Google Scholar]
- Qi W, Chen X, Poronnik P, Pollock CA. The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int J Biochem Cell Biol. 2006b;38:1–5. doi: 10.1016/j.biocel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Flores D. Intratubular hydrodynamic forces influence tubulointerstitial fibrosis in the kidney. Curr Opin Nephrol Hypertens. 2010;19:65–71. doi: 10.1097/MNH.0b013e32833327f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roufosse C, Bou-Gharios G, Prodromidi E, Alexakis C, Jeffrey R, Khan S, Otto WR, Alter J, Poulsom R, Cook HT. Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol. 2006;17:775–782. doi: 10.1681/ASN.2005080795. [DOI] [PubMed] [Google Scholar]
- Sadlier DM, Connolly SB, Kieran NE, Roxburgh S, Brazil DP, Kairaitis L, Wang Y, Harris DC, Doran P, Brady HR. Sequential extracellular matrix-focused and baited-global cluster analysis of serial transcriptomic profiles identifies candidate modulators of renal tubulointerstitial fibrosis in murine adriamycin-induced nephropathy. J Biol Chem. 2004;279:29670–29680. doi: 10.1074/jbc.M313408200. [DOI] [PubMed] [Google Scholar]
- Samarakoon R, Higgins PJ. Integration of non-SMAD and SMAD signaling in TGF-β1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Haemost. 2008;100:976–983. [PMC free article] [PubMed] [Google Scholar]
- Samarakoon R, Higgins CE, Higgins SP, Kutz SM, Higgins PJ. Plasminogen activator inhibitor type-1 gene expression and induced migration in TGF-β1-stimulated smooth muscle cells is pp 60c-src/MEK-dependent. J Cell Physiol. 2005;204:236–246. doi: 10.1002/jcp.20279. [DOI] [PubMed] [Google Scholar]
- Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. TGF-β1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscles requires pp 60c-src/EGFRY845 and Rho/ROCK signaling. J Mol Cell Cardiol. 2008;44:527–538. doi: 10.1016/j.yjmcc.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Kashihara N, Yamasaki Y, Maruyama K, Okamoto K, Maeshima Y, Sugiyama H, Sugaya T, Murakami K, Makino H. Renal interstitial fibrosis is reduced in angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2001;12:317–325. doi: 10.1681/ASN.V122317. [DOI] [PubMed] [Google Scholar]
- Schreiner GF, Harris KP, Purkerson ML, Klahr S. Immunological aspects of acute ureteral obstruction: immune cell infiltrate in the kidney. Kidney Int. 1988;34:487–493. doi: 10.1038/ki.1988.207. [DOI] [PubMed] [Google Scholar]
- Seo JY, Park J, Yu MR, Kim YS, Ha H, Lee HB. Positive feedback loop between plasminogen activator inhibitor-1 and transforming growth factor-β1 during renal fibrosis in diabetes. Am J Nephrol. 2009;30:481–490. doi: 10.1159/000242477. [DOI] [PubMed] [Google Scholar]
- Seseke F, Thelen P, Ringert RH. Characterization of an animal model of spontaneous congenital unilateral obstructive uropathy by cDNA microarray analysis. Eur Urol. 2004;45:374–381. doi: 10.1016/j.eururo.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Shi L, Nikolic D, Liu S, Lu H, Wang S. Activation of renal rennin-angiotensin system in upstream stimulatory factor 2 transgenic mice. Am J Physiol Renal Physiol. 2009;296:F257–F265. doi: 10.1152/ajprenal.90493.2008. [DOI] [PubMed] [Google Scholar]
- Shin GT, Kim WH, Yim H, Kim MS, Kim H. Effects of suppressing intrarenal angiotensinogen on renal transforming growth factor-β1 expression in acute ureteral obstruction. Kidney Int. 2005;67:897–908. doi: 10.1111/j.1523-1755.2005.00154.x. [DOI] [PubMed] [Google Scholar]
- Silverstein DM, Travis BR, Thornhill BA, Schurr JS, Kolls JK, Leung JC, Chevalier RL. Altered expression of immune modulator and structural genes in neonatal unilateral ureteral obstruction. Kidney Int. 2003;64:25–35. doi: 10.1046/j.1523-1755.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- Simonsson M, Kanduri M, Gronroos E, Heldin CH, Ericsson J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J Biol Chem. 2006;281:39870–39880. doi: 10.1074/jbc.M607868200. [DOI] [PubMed] [Google Scholar]
- Stahl PJ, Felsen D. Transforming growth factor-β, basement membrane, and epithelial-mesenchymal transdifferentiation: implications for fibrosis in kidney disease. Am J Pathol. 2001;159:1187–1192. doi: 10.1016/s0002-9440(10)62503-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutz F, Neilson EG. New insights into mechanisms of fibrosis in immune renal injury. Springer Semin Immunopathol. 2003;24:459–476. doi: 10.1007/s00281-003-0123-5. [DOI] [PubMed] [Google Scholar]
- Sutaria PM, Ohebshalom M, McCaffrey TA, Vaughan ED, Jr, Felsen D. Transforming growth factor-β receptor types I and II are expressed in renal tubules and are increased after chronic unilateral ureteral obstruction. Life Sci. 1998;62:1965–1972. doi: 10.1016/s0024-3205(98)00166-0. [DOI] [PubMed] [Google Scholar]
- Tan R, Zhang J, Tan X, Zhang X, Yang J, Liu Y. Down-regulation of SnoN expression in obstructive nephropathy is mediated by an enhanced ubiquitin-dependent degradation. J Am Soc Nephrol. 2006;17:2781–2791. doi: 10.1681/ASN.2005101055. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Endo S, Okuda T, Economides AN, Valenzuela DM, Murphy AJ, Robertson E, Sakurai T, Fukatsu A, Yancopoulos GD, Kita T, Yanagita M. Expression of BMP-7 and USAG-1 (a BMP antagonist) in kidney development and injury. Kidney Int. 2008;73:181–191. doi: 10.1038/sj.ki.5002626. [DOI] [PubMed] [Google Scholar]
- Terzi F, Burtin M, Hekmati M, Federici P, Grimber G, Briand P, Friedlander G. Targeted expression of a dominant-negative EGF-R in the kidney reduces tubulo-interstitial lesions after renal injury. J Clin Invest. 2000;106:225–234. doi: 10.1172/JCI8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong LD, Gaber L, Eknoyan G. Obstructive uropathy. Contrib Nephrol. 2011;169:311–326. doi: 10.1159/000314578. [DOI] [PubMed] [Google Scholar]
- Tu AW, Luo K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor β response. J Biol Chem. 2007;282:21187–21196. doi: 10.1074/jbc.M700085200. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang S, Hirschberg R. BMP7 antagonizes TGF-beta-dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol. 2003;284:F1006–F1013. doi: 10.1152/ajprenal.00382.2002. [DOI] [PubMed] [Google Scholar]
- Wang S, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non-Smad TGF-β pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005;19:1–11. doi: 10.1096/fj.04-2370com. [DOI] [PubMed] [Google Scholar]
- Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2007;293:F1282–F1291. doi: 10.1152/ajprenal.00230.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-β pathway. Kidney Int. 2006;70:1914–1919. doi: 10.1038/sj.ki.5001846. [DOI] [PubMed] [Google Scholar]
- Wolf G, Mueller E, Stahl RA, Ziyadeh FN. Angiotensin II-induced hypertrophy of cultured murine proximal tubular cells is mediated by endogenous transforming growth factor-β. J Clin Invest. 1993;l92:1366–1372. doi: 10.1172/JCI116710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki A, Maeda M, Matsumoto M, Kamimura R, Masuyama T, Suzuki S. SAMP1/Sku as a murine model for tubulointerstitial nephritis: a study using unilateral ureteral obstruction. Exp Anim. 2005;54:53–60. doi: 10.1538/expanim.54.53. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Maeshima A, Nojima Y. Involvement of renal progenitor tubular cells in epithelial-to-mesenchymal transition in fibrotic rat kidneys. J Am Soc Nephrol. 2005;16:2044–2051. doi: 10.1681/ASN.2004080681. [DOI] [PubMed] [Google Scholar]
- Yanagita M, Okuda T, Endo S, Tanaka M, Takahashi K, Sugiyama F, Kunita S, Takahashi S, Fukatsu A, Yanagisawa M, Kita T, Sakurai T. Uterine sensitization-associated gene-1 (USAG-1), a novel BMP antagonist expressed in the kidney, accelerates tubular injury. J Clin Invest. 2006;116:70–79. doi: 10.1172/JCI25445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang X, Li Y, Liu Y. Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-β1 signaling. J Am Soc Nephrol. 2003;14:3167–3177. doi: 10.1097/01.asn.0000099373.33259.b2. [DOI] [PubMed] [Google Scholar]
- Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci USA. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Shah AA, Kalluri R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J Biol Chem. 2005;280:8094–8100. doi: 10.1074/jbc.M413102200. [DOI] [PubMed] [Google Scholar]
- Zhang M, Fraser D, Phillips A. ERK, p38, and Smad signaling pathways differentially regulate transforming growth factor-β1 autoinduction in proximal tubular epithelial cells. Am J Pathol. 2006;169:1282–1293. doi: 10.2353/ajpath.2006.050921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Fu P, Huang XR, Liu F, Lai KN, Lan HY. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol. 2010;21:31–41. doi: 10.1681/ASN.2008111133. [DOI] [PMC free article] [PubMed] [Google Scholar]