

Abstract
The unique pharmacological profile of buprenorphine has led to its considerable success as an analgesic and as a treatment agent for drug abuse. Activation of nociceptin/orphanin FQ peptide (NOP) receptors has been postulated to account for certain aspects of buprenorphine’s behavioural profile. In order to investigate the role of NOP activation further, a series of buprenorphine analogues has been synthesised with the aim of increasing affinity for the NOP receptor. Binding and functional assay data on these new compounds indicate that the area around C20 in the orvinols is key to NOP receptor activity, with several compounds displaying higher affinity than buprenorphine. One compound, 1b, was found to be a mu opioid receptor partial agonist of comparable efficacy to buprenorphine, but with higher efficacy at NOP receptors.
Introduction
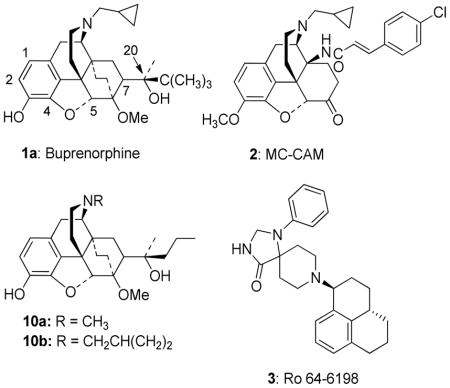
The treatment of opiate abuse and dependence by detoxification, substitution and maintenance with methadone, LAAM and more recently buprenorphine (1a), is the most successful pharmacotherapeutic programme of substance abuse treatment.1 The pharmacological profile of buprenorphine has long been recognized as having some unique features.2,3 Some of these effects are explicable by the slow onset and even slower offset of its interaction with mu opioid (MOP) receptors. These effects include its long duration of action and the mildness of the abstinence syndrome following its chronic administration and the delay in the appearance of these effects.4 Of particular significance to its use as a pharmacotherapy for heroin addiction is its acute safety in overdosage which clearly differentiates buprenorphine from the abused opiates and the therapeutic substitute, methadone. Though, as a MOP receptor partial agonist, buprenorphine would be expected to show a ceiling to its opiate effects, including respiratory depression, in fact the level of opiate effects from very high doses are lower than from intermediate doses, i.e. buprenorphine has bell-shaped dose-response curves for its opiate effects including respiratory depression, thus explaining its exceptional acute safety.5,6
The mechanism of the bell-shaped dose-response curves for buprenorphine’s MOP receptor effects in vivo has been the subject of considerable speculation. This has been addressed in a review of buprenorphine’s basic pharmacology and the two most favoured explanations recognized as being (a) interaction with an opposing pronociceptive system and (b) induction of ‘autoinhibition’ at higher doses which can be attributed to buprenorphine’s slow off-set kinetics.6 The pronociceptive system in question is the NOP receptor (nociceptin/orphanin FQ receptor); a receptor to which most standard opioid ligands do not bind with any significant affinity.7 Buprenorphine is therefore somewhat unusual among opioids in having moderate affinity at the NOP site.7 Although the affinity is two orders of magnitude lower than the affinity for the MOP and KOP receptors, buprenorphine seems to have some agonist activity at NOP.8,9,10 Lutfy and colleagues discovered that the bell-shaped antinociception curve lost its downward component in the presence of an NOP antagonist and in NOP knock-out mice in the absence of an antagonist.11 This finding led to the conclusion that NOP activation at high buprenorphine concentrations was the cause of the downward part of the dose-response curve. Further support for the influence of buprenorphine’s NOP activity was supplied by the findings of Ciccocioppo and colleagues that low doses of buprenorphine enhanced alcohol consumption in rats but high doses of buprenorphine reduced consumption of alcohol.12 The reduced consumption produced by high buprenorphine dosage was additive with the opiate antagonist naltrexone’s similar effect but inhibited by a selective NOP antagonist.
Though the case for NOP involvement to explain buprenorphine’s bell-shaped dose-response curves in MOP receptor mediated behavioural assays is compelling, it seems unlikely that this is the whole story. This follows from the effect of the competitive MOP receptor antagonist naloxone on buprenorphine’s antinociception bell-shaped dose-response curve.13 Naloxone induced a parallel displacement of the dose-response curve to a higher dose range, without changing the bell-shape. This is only possible when all components of the curve are susceptible to the competitive interaction with naloxone. The antagonist has no measurable affinity for NOP so could not affect the NOP component.14,15
The slow dissociation of buprenorphine from the MOP receptor, though not involving any covalent interactions with the receptor, corresponds to a pseudo-irreversible binding. It can be hypothesized that pseudo-irreversible binding is preceded by reversible binding during which time the ligand displays its agonist effects.16 The prolonged occupation of the receptor produces receptor blockade which is manifested as an antagonist effect of the ligand.6,17 The lower MOP receptor agonist effect of higher doses of buprenorphine may be related to the observation by Bidlack and coworkers that onset of MOP receptor antagonist effect in ligands related to methoclocinnamox (MC-CAM, 2: a ligand with very similar profile to buprenorphine) is dose-dependent with higher doses having a very much faster onset.18,19
It is therefore unclear whether direct activation of NOP receptors is the mechanism by which buprenorphine produces a bell-shaped dose-response curve. This issue can be investigated by identifying and characterising novel ligands with differing affinity and activity profiles at MOP and NOP receptors. Such ligands might also allow development of substitution therapies for opiate dependence with duration of effect and acute safety comparable to buprenorphine but with, for example, a higher level of morphine-like effects to appeal to addicts for whom methadone is presently prescribed. Alternatively it may allow the identification of novel analgesics with low or no abuse potential and low tolerance development.
Design and Synthesis
The majority of NOP receptor ligands can be described by a two-dimensional (2-D) pharmacophore as previously reported.20 A major feature of the pharmacophore is the presence of a basic nitrogen to which is attached a large, lipophilic moiety. We have previously demonstrated that replacement of the N-cyclopropylmethyl group of buprenorphine with the lipophilic cyclooctylmethyl group found in selective NOP receptor antagonists such as J-11339715 resulted in a significant decrease in affinity for the NOP receptor7 suggesting that the N-17 substituent of buprenorphine does not play the same role as the N-substituent in high affinity NOP receptor ligands and that an alternative group in the buprenorphine structure interacts with the lipophilic site. Alignment (using the “flexible alignment” feature of MOE, v.2005.06) of buprenorphine with known NOP receptor ligands (Figures 1 and 2 show alignments with Ro 64-6198 (3)21) indicates that either the t-butyl group (Fig. 1) or the aromatic A-ring (Fig. 2) of buprenorphine could be acting as the lipophilic group while still allowing the basic nitrogen to occupy a position similar to that in 3. These alignments suggest that either increasing the steric bulk of the t-butyl substituent or adding a substituent to the aromatic ring of buprenorphine could provide compounds with increased affinity for NOP receptors.
Figure 1.
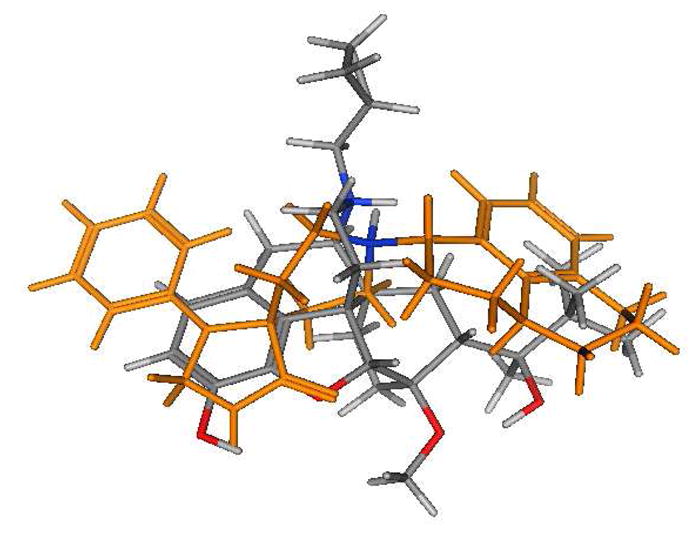
Overlay of buprenorphine (colored by atom type) with Ro 64-6198 (gold, except for the NH, which is colored by atom type).
Figure 2.
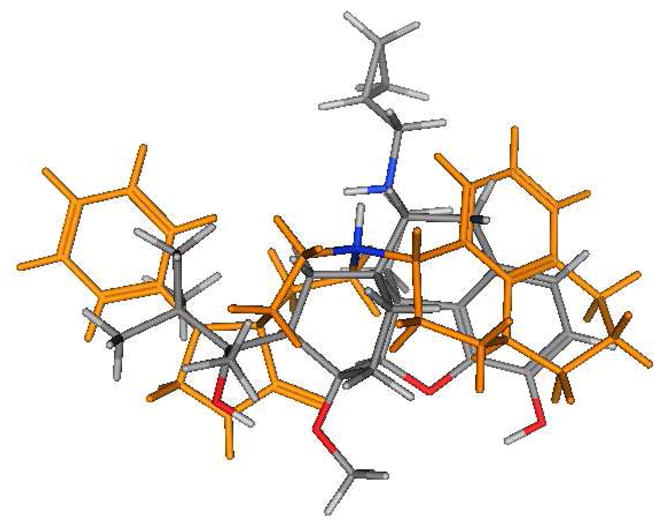
Overlay of buprenorphine (colored by atom type) and Ro 64-6198 (gold except for the NH, which is colored by atom type).
To this end, 1-chloro- and 1-bromo-buprenorphine were prepared using methods previously reported for the halogenation of hydrocodone and oxycodone.22 Thus, buprenorphine 3-O-methyl ether (4) was halogenated using the appropriate N-halosuccinimide to give the 1-substituted thevinol (5) before 3-O-demethylation with propane thiolate (Scheme 1), while bromination of buprenorphine itself with N-bromosuccinimide gave 2-bromobuprenorphine (7) directly, analogous to a previous iodination of buprenorphine.23 Attempts to synthesize 2-chlorobuprenorphine using a identical procedure with N-chlorosuccinimide failed to give the desired product due to, amongst other things, competing 6-O-demethylation.
Scheme 1.

(i) N-bromosuccinimide, 0.1N H2SO4, 38% or N-chlorosuccinimide, 0.1N HCl, 45% (ii) PrSNa, HMPA, 120°C, 56–89%
Access to analogues of buprenorphine in which the t-butyl group is replaced by alternative bulky groups was by the standard procedure of Grignard addition to ketone (8). This is a reaction that is low yielding (30 – 40%) for buprenorphine itself, due to the size of the Grignard reagent. Unsurprisingly it proved difficult to increase bulk without adversely affecting this addition step. p-(t-Butyl)phenylmagnesium bromide, phenanthren-9-ylmagnesium bromide, t-pentylmagnesium chloride were prepared from their corresponding halides and each gave the expected addition product (9b,e, 9g) on reaction with (8). Attempted addition of (3-methylbut-2-en-2-yl)magnesium bromide or 1-methylcyclopentylmagnesium bromide failed to yield any of the desired tertiary alcohol. In the case of the alkyl and arylalkyl Grignard reagents, the addition of a methylene group to separate the reacting and bulky centres resulted, as expected, in more facile Grignard addition (9c,d,f). The generally low yields (15 – 45%) were the result of rearrangements leading to C4-phenols and, in the reaction with t-pentylmagnesium chloride, the secondary alcohol product of Grignard reduction.24 In each case the thevinol (9) was then 3-O-demethylated to the corresponding orvinol (1) using propane thiolate. Despite numerous attempts this step failed for 9g (no reaction took place) and the corresponding orvinol could not be obtained.
Results and Discussion
Binding affinities of the new compounds, plus various standards, were determined in CHO cells transfected with human receptor cDNA, as previously described;25 the displaced selective radioligands were [3H]DAMGO, [3H]Cl-DPDPE, [3H]U69593 and [3H]N/OFQ for binding to MOP, delta opioid (DOP), kappa opioid (KOP) and NOP receptors respectively. Buprenorphine (1a) was found to bind with nM affinity to MOP, KOP and DOP receptors with around 50-fold lower affinity for NOP receptors compared to MOP receptors (Table 1). Halogenation at C-1 or C-2 (6a, 6b, 7) had little effect on NOP and DOP receptor affinity but resulted in somewhat lower affinity for MOP and KOP receptors. This might imply that the aryl ring of buprenorphine does not occupy the lipophilic site described within the NOP receptor pharmacophore. Due to this result no further aryl ring substituted analogues were synthesised and evaluated and attention focussed on the C20 group.
Table 1.
Binding affinities to opioid and NOP receptors.
| Ki (nM) ± SEMa | ||||
|---|---|---|---|---|
| NOP | MU | DELTA | KAPPA | |
| Nociceptin | 0.08 ± 0.03 | 437 ± 13 | 2846 ± 512 | 147 ± 3.4 |
| DAMGO | > 10,000 | 1.59 ± 0.17 | 300 ± 59 | 306 ± 46 |
| DPDPE | > 10,000 | 504 ± 10 | 1.24 ± 0.09 | > 10,000 |
| U69,593 | > 10,000 | > 10,000 | > 10,000 | 1.6 ± 0.26 |
| 1a | 77.4 ± 16 | 1.5 ± 0.8 | 6.1 ± 0.4 | 2.5 ± 1.2 |
| 1b | 8.46 ± 1.3 | 2.14 ± 0.79 | 1.59 ± 0.28 | 5.63 ± 1.3 |
| 1c | 18.5 ± 0.68 | 2.95 ± 0.65 | 1.27 ± 0.21 | 1.59 ± 0.25 |
| 1d | 22.0 ± 0.55 | 2.19 ± 0.65 | 3.66 ± 0.92 | 4.15 ± 1.3 |
| 1e | 133 ± 8.2 | 5.56 ± 1.3 | 3.03 ± 0.61 | 8.92 ± 0.34 |
| 1f | 16.0 ± 1.5 | 11.4 ± 0.17 | 7.13 ± 1.3 | 9.87 ± 1.3 |
| 6a | 237 ± 64 | 14.3 ± 0.88 | 6.79 ± 0.99 | 9.93 ± 3.7 |
| 6b | 67.8 ± 20 | 13.2 ± 0.97 | 17.2 ± 0.26 | 45.6 ± 3.4 |
| 7 | 159 ± 1.4 | 10.5 ± 3.86 | 8.67 ± 0.81 | 35.5 ± 3.9 |
Of the analogues of 1a with variation in the C20 substituent, 1b and 1c, having t-pentyl and neopentyl groups, are the closest in structure to 1a, being homologues with one extra methylene group. Compared to 1a, 1b retained affinity at opioid receptors (all 1.6 – 5.6 nM) and was one order of magnitude higher affinity at NOP receptors (8.5 nM versus 77 nM). Therefore 1b has essentially equivalent affinities for each of the opioid and NOP receptors. 1c behaves similarly in binding, though the effect at NOP receptors is not as profound, being 4–5 – fold higher affinity than 1a. Once again, 1c has high, non-selective affinity for the three opioid receptors comparable with 1a. 1d, which is structurally related to 1c, having a phenyl group in place of one of the methyls of the neopentyl group, has similar affinity to 1c for all four receptors and is 10-fold selective for MOP over NOP receptors. 1f, in which the C20 substituent is a bulky tricyclic group attached through a methylene spacer to C20, also shows increased affinity for NOP receptors compared to 1a, but couples this with reduced affinity for opioid receptors such that it has near 10 nM affinity for each of the four receptors. The final ligand, 1e, has the t-butyl group separated from C20 by a phenyl ring; it displays no increase in affinity for NOP receptors over 1a, while having similar or slightly lower opioid receptor affinity.
Overall, the binding data suggest that it is the C20 substituent of 1a and related orvinols that occupies the putative lipophilic site in the NOP receptor, as required for high affinity binding.
The ligands were assessed for functional activity in the [35S]GTPγS-binding assay in human receptor transfected CHO cells as described previously.25 The results from these assays are shown in Table 2. Buprenorphine (1a) was confirmed as a low efficacy partial agonist at the MOP receptor, with 29% stimulation relative to the standard MOP receptor agonist DAMGO and as a low potency (116 nM) and low efficacy (21% relative to nociceptin) partial agonist at NOP receptors. The data for 1a is consistent with the suggestion that its in vivo profile is due to it having MOP receptor mediated effects at low doses with NOP receptor activation being noticeable at higher doses.12,26
Table 2.
Stimulation (EC50/nM and % stimulation)of [35S]GTPγS binding to opioid and NOP receptors
| MOR | NOP | DOR | KOR | |||||
|---|---|---|---|---|---|---|---|---|
| EC50 | %stima | EC50 | % stima | EC50 | % stima | EC50 | % stima | |
| DAMGO | 35.3 ± 0.53 | 100 | ----- | ----- | 6.86 ± 0.4 | 100 | ----- | ----- |
| Nociceptin | ----- | ----- | 8.1 ± 1.4 | 100 | ----- | ----- | 78.5 ± 8.80 | 100 |
| 1a | 10.2 ± 2.2 | 28.7 ± 1.1 | 116 ± 88 | 21.0 ± 8.4 | >10,000 | >10,000 | ||
| 1b | 6.0 ± 2.1 | 20.1 ± 8.7 | 78.6 ± 49 | 48 ± 13 | * | 10.8 ± 6.8 | * | 0 |
| 1c | 2.6 ± 1.2 | 26.5 ± 8.0 | * | 5.4 ± 3.4 | 50.1 ± 7.1 | 20.6 ± 0.6 | * | 16.9 ± |
| 1d | * | 16.8 ± 1.3 | 2380 ± 156 | 42.7 ± 21 | 3411 ± 819 | 42.7 ± 6.7 | 0.88 ± 0.58 | 33.7 ± 3.4 |
| 1e | * | 0 | * | 9.3 | * | 0 | * | 0 |
| 1f | 6.1 ± 2.7 | 67.7 ± 7.0 | 111 ± 10 | 58.2 ± 20 | 49.3 ± 4.6 | 61.3 ± 21 | 5.33 ± 1.8 | 52.4 ± 7.9 |
| 6a | * | 18.6 ± 19.0 | * | 0 | * | 17.0 ± 4.8 | * | 0 |
| 6b | * | 13.8 ± 4.8 | * | 0 | * | 0 | * | 12.4 |
| 7 | * | 17.1 ± 1.1 | * | 17.0 ± 1.9 | * | 12.9 ± 4.9 | * | 6.05 ± 3.6 |
Percent maximal stimulation with respect to the standard agonists DAMGO (MOP), U69593 (KOP), DPDPE (DOP) and nociceptin (NOP). Values are the means of 5 or 6 experiments.
The halogenated analogues (6a, 6b, 7) had little efficacy at any receptor studied. Each had around half the efficacy of buprenorphine at MOP receptors with only the 2-bromo analogue (7) demonstrating any efficacy at NOP receptors. 1-Chlorobuprenorphine (6b) is interesting as a highly lipophilic, universal opioid and NOP receptor antagonist. In vitro, 6b inhibited agonist stimulated [35S]GTPγS activity at MOP, DOP, KOP, and NOP receptors (see Supporting Information Figure S1). Its MOP receptor antagonist activity has been confirmed in vivo in mice, where it was found to have little to no efficacy in the tail flick assay up to a dose of 30mg/kg (peak effect of 15% at 30 mg/kg) and only reached a peak of 20% in the much lower stimulus intensity PPQ-induced antiwrithing assay, in which even low efficacy partial agonists are active.27 6b did act as an antagonist versus morphine in the tail flick assay, AD50 4.7 mg/kg, which is comparable to that obtained with 1a (AD50 1 mg/kg). However, 1a was an agonist in both the tail flick (ED50 0.14 mg/kg) and PPQ-induced writhing assays (ED50 0.016 mg/kg) confirming it as a partial agonist in vivo.
Of the C20 analogues of buprenorphine (1a), the one carbon homologues 1b and 1c displayed similar efficacy to 1a at MOP receptors (all 20–29% relative to DAMGO), and were of very similar potency (2.6 – 10.2 nM). Quite substantial differences in efficacy were found between these three ligands at the NOP receptor (5% – 48% relative to nociceptin). 1a was of similar efficacy at the NOP receptor as at the MOP receptor, but an order of magnitude less potent at the former, while the homologue having a methylene between C20 and the t-butyl group (1c) had little or no efficacy at NOP receptors. In contrast 1b was more efficacious, and of similar or higher potency, at NOP receptors than 1a. As found with 1a, 1b was around 10-fold more potent at the MOP receptor compared to the NOP receptor. The low efficacy at MOP and KOP receptors displayed by 1b and 1c is notable due to their structural relationship to the n-propyl orvinols 10a and 10b. In the original orvinol and N-CPM nororvinol series, a C20 R-group three carbon atoms in length was associated with a peak in analgesic activity, whether through activation of MOP or KOP receptors.28 Both 1b and 1c have a substituted three carbon chain and yet have low efficacy at both MOP and KOP receptors indicating that the effect of branching in lowering efficacy at opioid receptors outweighs the higher efficacy associated with a three carbon-long chain. 1d, in which a phenyl ring replaces a methyl group in 1c, had low efficacy at MOP receptors and only very low potency at NOP receptors. The most noticeable feature about 1d’s profile in this assay was the increase in efficacy at KOP receptors, such that it was a potent low efficacy partial agonist. This compares with the equivalent phenethyl compound (1, R = (CH2)2Ph; unpublished) that demonstrates negligible efficacy at any of the opioid receptors under these conditions, but was a partial agonist at NOP receptors (35% stimulation). Similarly, 1f displayed KOP receptor partial agonist activity, suggesting that the introduction of the methylene group between C20 and the bulky substituent leads to a rise in KOP receptor activation, as would be predicted from earlier studies.29,30 1f profiled as a partial agonist at each of the four receptors, with highest potency at MOP and KOP receptors. 1e had little to no efficacy at any receptor, and like the 1-chloro analogue (6b) may prove to be a universal opioid and NOP receptor antagonist.
Since 1b has a similar in vitro profile to 1a, but with higher affinity and efficacy for the NOP receptor, it was evaluated in an antinociceptive assay (tail flick) in the mouse and has been found to be an agonist of higher efficacy than buprenorphine and equivalent in efficacy to morphine.31 This may indicate that the MOP receptor mediated effects of BU08028 predominate in vivo.
Conclusions
The notable rise in affinity at NOP receptors for compounds 1b, 1c, 1d and 1f, but not with 6a, 6b and 7, indicates that it is the area around the C20 carbon of buprenorphine (1a) and analogues that fulfils the role of the large lipophilic group found in NOP receptor ligands. A methylene group inserted between the bulky lipophilic group and C20 (1c, 1d, 1f) led to an increase in efficacy at KOP receptors compared to 1a with less consistent effects on MOP receptor activity. One compound, 1b, profiled as a MOP receptor partial agonist of comparable efficacy to buprenorphine, but with higher efficacy at NOP receptors, is the subject of more extensive pharmacological evaluation in vivo.31
Experimental Part
Full details are available in the Supporting Information. Reagents and solvents were purchased from Aldrich or Lancaster and used as received. M.p.: Gallenkamp MFB-595 melting point apparatus; uncorrected. NMR Spectra: Jeol Delta-270-MHz instrument: 1H at 270 MHz, and Varian Mercury-400-MHz instrument: 1H at 400 MHz, 13C at 100 MHz; δ in ppm, J in Hz with TMS as an internal standard. ESIMS: micrOTOF (BRUKER). Microanalysis: Perkin-Elmer 240C analyser. Ligands were tested as their hydrochloride salts, prepared by adding one equivalent of (5N hydrochloric acid in isopropanol) to an ether solution of the compound. All compounds were > 95% pure by microanalysis.
General Procedure A, halogenation
Buprenorphine or buprenorphine 3-O-methyl ether (0.31 mmol), was dissolved in H2SO4 (0.1 N, 16 mL) for bromination or HCl (0.1 N, 4 mL) for the chlorination, and the N-halosuccinimide (0.37 mmol) was added in one portion. The reaction mixture was stirred until TLC indicated the starting material had been consumed, (approx. 3h), and the mixture was then poured into a separatory funnel containing dichloromethane (17 mL). Sufficient aqueous sodium hydroxide (10%) solution was added to raise the pH of the aqueous layer to ca. 10, the organic layer was separated and the aqueous layer was extracted further with three portions of 9:1 dichloromethane-methanol (10 mL). The organic extracts were combined and dried over anhydrous sodium sulfate, and the solvent removed under reduced pressure. The residue was purified by column chromatography over silica gel eluting with a gradient from 5% to 10% ethyl acetate in hexane.
General procedure B, Grignard addition
The Grignard reagents were prepared from the corresponding bromides (6 mmol) by reaction with magnesium (218 mg, 9 mmol) in anhydrous THF (6 mL) containing a crystal of iodine. The Grignard reagents were titrated prior to use by adding 1 mL of the Grignard solution to a flask containing 1,10-phenanthroline (~2 mg) in anhydrous THF (2 mL) (purple solution) and titrating with 1M 2-butanol (anhydrous) in THF (end point pale yellow solution).
A solution of the appropriate Grignard reagent (1 M in THF, 1.2 mL, 1.2 mmol) was treated dropwise at room temperature with a solution of N-cyclopropylmethyl-6,14-endo-ethanonorthevinone (8) (500 mg, 1.18 mmol) in anhydrous toluene (12 mL). After stirring at room temperature for 20 h, the reaction was quenched by addition of saturated aqueous ammonium chloride solution (20 mL). The phases were separated and the aqueous phase extracted with EtOAc. The combined organic phases were washed with saturated aqueous sodium bicarbonate, dried over MgSO4, filtered and evaporated in vacuo. The residue was purified by column chromatography over silica gel eluting with a gradient from 10% to 30% ethyl acetate in hexane. Rf values are recorded from TLC eluted with 30:1:69 ethyl acetate/ammonia solution/hexane.
General Procedure C, 3-O-demethylation
A solution of the appropriate methyl ether (0.1 mmol) in anhydrous HMPA (0.5 mL) under an inert atmosphere was treated with sodium hydride (8.5 mg, 0.35 mmol) followed by 1-propanethiol (32 μl, 0.35 mmol). After the addition was complete, the reaction mixture was heated to 120°C and stirred until completion (~ 3 h). On cooling to room temperature, NH4Cl (sat, aq) was added and the mixture extracted with diethyl ether. The organic extracts were washed with water (3×) and brine. The organic phase was dried over MgSO4 filtered and evaporated to dryness. The residue was purified by column chromatography over silica gel.
The HCl salts were prepared by the addition of 5N HCl in isopropanol (2 equiv.) to a solution of the orvinol in diethyl ether. The white precipitate which formed was collected by filtration, washed with ether and dried under high vacuum.
(2′R, 5α, 6R, 7R, 14α)-2′-(1-Bromo-17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-3′,3′-dimethylbutan-2′-ol (6a)
5a was treated as described in General Procedure C and 6a isolated as a white solid (52 mg, 89%). Rf 0.20, 1H NMR (270 MHz, CDCl3) δ 0.10–0.14 (2H, d, J=8.0 Hz), 0.48–0.50 (2H, t, J=7.8, 8.9 Hz), 0.73–0.75 (1H, m), 1.22 (9H, s), 1.22–1.27 (1H, m), 1.34 (3H, s), 1.62–2.17 (6H, m), 2.19 –2.23 (3H, m), 2.30–2.32 (2H, d, J=6.4 Hz), 2.58–2.60 (1H, m), 2.75–2.82 (1H, d, J= 18.1 Hz), 2.85–2.86 (1H, m), 3.01–3.03 (1H, d, J= 5 Hz), 3.50 (3H, s), 4.46 (1H, s), 5.84 (1H, s), 6.90 (1H, s); ESIMS m/z: 547 [M + 1]+. Anal (C29H40BrNO4·0.75H2O) CHN.
(2′R, 5α, 6R, 7R, 14α)-2′-(1-Chloro-17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-3′,3′-dimethylbutan-2′-ol (6b)
5b was treated as described in General Procedure C and 6b isolated as a white solid (8 mg, 56%). Rf 0.15, 1H NMR (270 MHz, CDCl3) δ 0.12–0.13 (2H, d, J=8.0 Hz), 0.47–0.49 (2H, m), 0.49–0.51 (1H, m), 0.78–0.83 (4H, m), 1.03 (9H, s), 1.25–1.36 (4H, m), 1.56 (3H, brs), 1.68–1.97 (4H, m), 2.04–2.14 (3H, m), 2.32–2.34 (1H, d, J=6.3 Hz), 2.61–2.65 (1H, m), 2.81–2.82 (2H, m), 3.02–3.03 (1H, d, J= 6.3 Hz), 3.51 (3H, s), 4.47 (1H, s), 5.77 (1H, s), 6.75 (1H, s); ESIMS m/z: 503 [M + 1]. Anal (C 29H40ClNO4·HCl·0.75H2O) CHN.
(2′R, 5α, 6R, 7R, 14α)-2′-(2-Bromo-17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-3′,3′-dimethylbutan-2′-ol (7)
From buprenorphine (1a) using the general halogenation procedure A yielding 7 as a white solid (70 mg, 60%). Rf 0.25, 1H NMR (270 MHz, CDCl3) δ 0.08–0.14 (2H, d, J=8.0 Hz), 0.47–0.50 (2H, t, J=7.8, 8.9 Hz), 0.99–1.07 (1H, m), 1.22 (9H, s), 1.23–1.27 (3H, m), 1.35 (3H, s), 1.61–2.12 (5H, m), 2.19 –2.30 (3H, m), 2.30–2.33 (2H, d, J=6.4 Hz), 2.58–2.60 (1H, m), 2.75–2.82 (1H, d, J= 18.1 Hz), 2.85–2.86 (1H, m), 3.00–3.03 (1H, d, J= 5 Hz), 3.53 (3H, s), 4.45 (1H, s), 5.80 (1H, s), 6.90 (1H, s); ESIMS m/z: 547 [M + 1]+. Anal (C29H40BrNO4·0.45H2O) CHN.
(1′RS, 5α, 6R, 7R, 14α)-2′-(17-Cyclopropylmethyl-7,8-dihydro-3,6-dimethoxy-4,5-epoxy-6,14-ethano-morphinan-7-yl)- 3′,3′-dimethylpentan-2′-ol (9b)
General Procedure B and 9b isolated as a clear oil (428 mg, 15%). Rf 0.58, 1H NMR (270 MHz, CDCl3) δ 0.08–0.11 (2H, m), 0.44–0.53 (2H, m), 0.81–0.86 (3H, m), 0.83–0.86 (3H, m), 0.89 (6H, s), 1.24–1.31 (2H, m), 1.34 (1H, s), 1.41 (3H, s), 1.55–1.80 (3H, m), 1.95–2.03 (2H, m), 2.27–2.34 (5H, m), 0.84 (1H, m), 2.79–2.84 (1H, t, J=9 Hz), 2.94–2.98 (1H, m), 3.53 (3H, s), 3.86 (3H, s), 4.41 (1H, s), 5.96 (1H, s), 6.53 (1H, d, J=8.0 Hz), 6.67 (1H, d, J=8.0 Hz, m); ESIMS m/z: 496 [M + 1]+.
(2′R, 5α, 6R, 7R, 14α)-2′-(17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-3′,3′-dimethylpentan-2′-ol (1b)
Procedure C to isolate (1b), as a clear oil (182 mg, 80%). Rf 0.23, 1H NMR (400 MHz, CDCl3) δ 0.08–0.10 (2H, d, J=8.0 Hz), 0.44–0.49 (2H, t, J=7.8, 8.9 Hz), 0.65–0.69 (1H, m), 0.72–0.80 (1H, m), 0.86–0.90 (3H, m), 0.91 (6H, s), 1.03–1.06 (1H, m), 1.31–1.34 (2H, m), 1.35 (3H, s), 1.40–1.45 (1H, m), 1.60–1.75 (3H, m), 1.80–1.86 (1H, m), 1.93–2.00 (1H, m), 2.15–2.36 (5H, m), 2.57–2.62 (1H, dd, J=5 Hz), 2.79–2.86 (1H, m), 2.93–2.96 (1H, d, J= 17.0 Hz), 2.96–2.98 (1H, d, J=7.0 Hz), 3.51 (3H, s), 4.43 (1H, s), 5.95 (1H, s), 6.49 (1H, d, J=8.1 Hz), 6.67 (1H, d, J=8.1 Hz); 13C NMR (100.6 MHz, CDCl3) δ 3.2, 4.1, 9.1, 9.4, 18.3, 20.6, 21.4, 21.7, 22.8, 28.5, 29.5, 33.3, 35.5, 35.8, 42.7, 43.0, 43.6, 46.3, 52.4, 58.2, 59.4, 80.4, 80.8, 96.9, 116.3, 119.5, 128.3, 132.6, 137.2, 145.3; ESIMS m/z: 482 [M + 1]+. Anal (C30H43NO4·0.3 H2O) CHN.
(2′R, 5α, 6R, 7R, 14α)-2′-(17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-4′,4′-dimethylpentan-2′-ol (1c)
Isolated as a white solid, by using General procedure C to yield (1c) (20 mg, 52%). Rf 0.46, 1H NMR (400 MHz, CDCl3) δ 0.10–0.11 (2H, d, J=8.0 Hz), 0.47–0.50 (2H, m), 0.72–0.75 (1H, m), 0.81–0.88 (2H, m), 1.08 (9H, s), 1.24 (2H, s), 1.44 (6H, m), 1.63–1.66 (1H, d, J= 3 Hz) 1.73–1.81 (2H, m), 1.84–2.04 (2H, m), 2.17–2.40 (4H, m), 2.61–2.65 (1H, m), 2.81–2.87 (1H, m), 2.94–2.99 (1H, d, J= 18.1 Hz), 2.99–3.00 (1H, d, J= 5 Hz), 3.50 (3H, s), 4.42 (1H, s), 5.14 (1H, s), 6.48–6.50 (1H, d, J= 8.0 Hz), 6.66–6.68 (1H, d, J= 8.0 Hz); 13C NMR (100.6 MHz, CDCl3) δ 3.4, 4.1, 9.4, 18.0, 22.6, 24.0, 29.8, 31.9, 32.0, 32.6, 35.5, 36.0, 47.1, 48.4, 52.0, 52.6, 58.2, 59.8, 78.2, 80.9, 97.5, 116.2, 119.4, 128.4, 132.4, 137.1, 145.4; ESIMS m/z: 482 [M + 1]. Anal (C30H43NO4·HCl·0.3H2O) CHN
(2′R, 5α, 6R, 7R, 14α)-2′-(17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-4′-methyl-4′-phenylpentan-2′-ol (1d)
Procedure C to prepare (1d). Isolated as a clear oil (60 mg, 80%). Rf 0.34, 1H NMR (400 MHz, CDCl3) δ 0.13–0.14 (2H, d, J=8.1 Hz), 0.52–0.53 (2H, m), 0.55–0.56 (1H, m), 0.85–0.96 (6H, m), 1.41 (3H, s), 1.52 (5H, m), 1.62–1.64 (2H, t, J=6.8 Hz), 1.73–1.85 (3H, m), 2.03–2.07 (1H, d, J= 14.5 Hz), 2.16–2.35 (4H, m), 2.58–2.71 (2H, m), 2.92–2.96 (1H, d, J= 18.4 Hz), 2.95–2.96 (1H, m), 3.43 (3H, s), 4.27 (1H, s), 5.18 (1H, brs), 6.46–6.48 (1H, d, J= 8.1 Hz), 6.64–6.66 (1H, d, J= 8.1 Hz), 7.15–7.17 (1H, t, J=7.1 Hz), 7.27–7.29 (2H, d, J=7.1 Hz), 7.44–7.45 (2H, d, J=7.2 Hz); 13C NMR (100.6 MHz, CDCl3) δ 3.5, 4.0, 9.4, 18.3, 22.6, 23.8, 29.6, 32.6, 33.4, 35.3, 35.9, 37.9, 43.5, 46.6, 46.8, 52.4, 52.7, 58.3, 59.9, 80.8, 96.9, 116.3, 119.4, 125.0, 126.1, 127.9, 128.1, 132.4, 137.1, 145.4, 150.5; ESIMS m/z: 544 [M + 1]+. Anal (C35H45NO4·HCl·1.5H2O) CHN
(2′R, 5α, 6R, 7R, 14α)-1′-(4 ′-T-butyl-phenyl)-1′-(17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-ethan-1′-ol (1e)
(1e) was prepared using General Procedure C. Isolated as a white solid (50 mg, 79%). Rf 0.33, 1H NMR (400 MHz, CDCl3) δ -0.07–0.00 (2H, m), 0.31–0.33 (2H, d, J= 8.0 Hz), 0.68–0.72 (1H, m), 0.73–0.86 (1H, m), 0.86–0.92 (1H, m), 0.99–1.03 (1H, m), 1.31 (9H, s), 1.52–1.55 (1H, d, J= 12.5 Hz), 1.77 (6H, m), 1.81–1.84 (1H, m), 2.03–2.10 (1H, m), 2.13–2.20 (5H, m), 2.46–2.48 (1H, m), 2.77–278 (1H, m), 2.87–2.91 (1H, d, J=18.3 Hz), 3.54 (3H, s), 4.43 (1H, s), 5.44 (1H, s), 6.44–6.46 (1H, d, J= 8.0 Hz), 6.61–6.63 (1H, d, J=8.0 Hz), 7.32–7.33 (2H, d, J=4.7 Hz), 7.34–7.35 (2H, d, J=4.7 Hz); 13C NMR (100.6 MHz, CDCl3) δ 3.4, 3.5, 9.1, 17.8, 23.0, 23.4, 29.8, 31.3, 32.4, 34.3, 35.5, 36.0, 43.2, 47.0, 48.3, 52.7, 58.5, 59.2, 80.7, 97.2, 116.3, 119.4, 124.6, 125.6, 128.1, 132.3, 137.2, 144.0, 145.4, 149.3; ESIMS m/z: 544 [M + 1]+. Anal (C35H45NO4) CHN.
(2′R, 5α, 6R, 7R, 14α)-2′-(17-cyclopropylmethyl-7,8-dihydro-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxy-morphinan-7-yl)-1′-(bicyclo[2.2.1]heptan-1-yl)-propan-2′-ol (1f)
General Procedure C was used to prepare (1f). Isolated as a white solid (55 mg, 62%). Rf 0.23, 1H NMR (400 MHz, MeOD) δ 0.16–0.18 (2H, m), 0.51–0.53 (2H, d, J=8.1 Hz), 0.71–0.82 (3H, m), 1.14–1.35 (3H, m), 1.37–1.38 (15 H, m), 1.54–1.86 (7H, m), 2.22–2.39 (4H, m), 2.69 (1H, m), 2.99–3.00 (1H, m), 3.03–3.05 (1H, d, J=18.1 Hz), 3.58 (3H, s), 4.36 (1H, s), 6.47–6.50 (1H, d, J=8.0 Hz), 6.62–6.64 (1H, d, J=8.0 Hz); 13C NMR (100.6 MHz, CDCl3) δ 4.0, 4.4, 10.2, 19.1, 23.8, 24.8, 29.6, 30.7, 32.9, 36.0, 37.2, 38.6, 39.8, 40.9, 43.5, 44.8, 46.9, 53.0, 60.8, 67.1, 69.2, 81.9, 97.9, 117.9, 120.4, 128.5, 133.6, 139.5, 147.3; ESIMS m/z: 520 [M + 1]+. Anal (C33H45NO4·HCl·0.5H2O) CHN
Supplementary Material
Scheme 2.

(i) RMgBr, THF/toluene, 15–45% (ii) PrSNa, HMPA, 120°C, 52–80%
Acknowledgments
This work was funded by the National Institutes of Health National Institute on Drug Abuse grant DA023281.
Abbreviations
- MOP receptor
mu opioid receptor
- NOP receptor
nociceptin/orphanin FQ receptor
- MC-CAM
methoclocinnamox
- CHO
Chinese hamster ovary
- KOP receptor
kappa opioid receptor
- DOP
delta opioid receptor
Footnotes
Supporting Information Available: Full experimental details (spectra and microanalysis data) plus figures detailing antagonist activity of 6b in vitro. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Marsch LA, Stephens MAC, Mudric T, Strain EC, Bigelow GE, Johnson RE. Predictors of Outcome in LAAM, Buprenorphine, and Methadone Treatment for Opioid Dependence. Experimental and Clinical Psychopharmacology. 2005;13:293–302. doi: 10.1037/1064-1297.13.4.293. [DOI] [PubMed] [Google Scholar]
- 2.Cowan A, Doxey JC, Harry EJ. The animal pharmacology of buprenorphine, an oripavine analgesic agent. Br J Pharmacol. 1997a;60:547–554. doi: 10.1111/j.1476-5381.1977.tb07533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowan A, Lewis JW, Macfarlane IR. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol. 1997b;60:537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Negus SS, Woods JH. Reinforcing effects, discriminative stimulus effects and physical dependence liability of buprenorphine. In: Cowan A, Lewis JW, editors. Buprenorphine – combating drug abuse w ith a unique opioid. Wiley-Liss; New York: 1995. pp. 71–101. [Google Scholar]
- 5.Cowan A. Update on the general pharmacology of buprenorphine. In: Cowan, Lewis, editors. Buprenorphine: Combating drug abuse with a unique opioid. Wiley-Liss; New York: 1995. pp. 31–47. [Google Scholar]
- 6.Cowan A, Friderichs E, Strasburger W, Raffa RB. Basic pharmacology of buprenorphine. In: Budd K, Raffa R, editors. Buprenorphine – the unique opioid analgesic. Thieme; New York: 2005. [Google Scholar]
- 7.Zaveri N, Polgar WE, Olsen CM, Kelson AB, Grundt P, Lewis JW, Toll L. Characterization of opiates, neuroleptics, and synthetic analogs at ORL1 and opioid receptors. Eur J Pharmacol. 2001;428:29–36. doi: 10.1016/s0014-2999(01)01282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wnendt S, Kruger T, Janocha E, Hildebrandt D, Englberger W. Agonistic effect of buprenorphine in a nociceptin/OFQ receptor-triggered reporter gene assay. Mol Pharmacol. 1999;56:334–338. doi: 10.1124/mol.56.2.334. [DOI] [PubMed] [Google Scholar]
- 9.Bloms-Funke P, Gillen C, Schuettler AJ, Wnendt S. Agonistic effects of the opioid buprenorphine on the nociceptin/OFQ receptor. Peptides. 2000;21:1141–1146. doi: 10.1016/s0196-9781(00)00252-7. [DOI] [PubMed] [Google Scholar]
- 10.Huang P, Kehner GB, Cowan A, Liu-Chen LY. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther. 2001;297:688–695. [PubMed] [Google Scholar]
- 11.Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, Kieffer BL, Takeshima H, Carroll FI, Maidment NT, Evans CJ. Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J Neurosci. 2003;23:10331–10337. doi: 10.1523/JNEUROSCI.23-32-10331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciccocioppo R, Economidou D, Rimondini R, Sommer W, Massi M, Heilig M. Buprenorphine Reduces Alcohol Drinking Through Activation of the Nociceptin/Orphanin FQ-NOP Receptor System. Biol Psychiatry. 2007;61:4–12. doi: 10.1016/j.biopsych.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dum JE, Herz A. In vivo receptor binding of the opiate partial agonist, buprenorphine, correlated with its agonistic and antagonistic actions. Br J Pharmacol. 1981;74:627–633. doi: 10.1111/j.1476-5381.1981.tb10473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P, Ronzoni S, Giardina GA, Scheideler MA. Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (−)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]- 6,7,8,9- tetrahydro-5H-benzocyclohepten-5-ol (SB-612111) J Pharmacol Exp Ther. 2004;308:454–461. doi: 10.1124/jpet.103.055848. [DOI] [PubMed] [Google Scholar]
- 15.Ozaki S, Kawamoto H, Itoh Y, Miyaji M, Azuma T, Ichikawa D, Nambu H, Iguchi T, Iwasawa Y, Ohta H. In vitro and in vivo pharmacological characterization of J-113397, a potent and selective non-peptidyl ORL1 receptor antagonist. Eur J Pharmacol. 2000;402:45–53. doi: 10.1016/s0014-2999(00)00520-3. [DOI] [PubMed] [Google Scholar]
- 16.Chang AC, Takemori AE, Portoghese PS. 2-(3, 4-dichlorophenyl)-N-methyl-N-[(1S)-1-(3-isothiocyanatophenyl)-2-(1-pyrrolidinyl)ethyl]acetamide: an opioid recptor affinity label that produces selective long-lasting κ antagonism in mice. J Med Chem. 1994;37:1547–1549. doi: 10.1021/jm00037a001. [DOI] [PubMed] [Google Scholar]
- 17.Husbands SM, Lewis JW. Opioid ligands having delayed long-term antagonist activity: Potential pharmacotherapies for opioid abuse. Mini-Reviews in Med Chem. 2003;3:137–144. doi: 10.2174/1389557033405395. [DOI] [PubMed] [Google Scholar]
- 18.Jiang Q, Seyed-Mozaffari A, Sebastian A, Archer S, Bidlack JM. Preventing morphine antinociceptive tolerance by irreversible mu opioid antagonists before the onset of their antagonism. J Pharmacol Exp Ther. 1995;273:680–688. [PubMed] [Google Scholar]
- 19.McLaughlin JP, Hill KP, Jiang Q, Sebastian A, Archer S, Bidlack JM. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of μ-selective agonist and antagonist activity. J Pharmacol Exp Ther. 1999;289:304–311. [PubMed] [Google Scholar]
- 20.Zaveri N, Jiang F, Olsen C, Polgar W, Toll L. Small-Molecule Agonists Antagonists of the Opioid Receptor-Like Receptor (ORL1, NOP): Ligand-Based Analysis of Structural Factors Influencing Intrinsic Activity at NOP. Aaps J. 2005;7:E345–352. doi: 10.1208/aapsj070234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenck F, Wichmann J, Dautzenberg FM, Moreau JL, Ouagazzal AM, Martin JR, Lundstrom K, Cesura AM, Poli SM, Roever S, Kolczewski S, Adam G, Kilpatrick G. A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: anxiolytic profile in the rat. Proc Natl Acad Sci (USA), G. 2000;97:4938–4943. doi: 10.1073/pnas.090514397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson ML, Carroll PJ, Dalton DR. Decoration of the Aromatic Ring of Dihydrocodeinone (Hydrocodone) and 14-Hydroxydihydrocodeinone (Oxycodone) J Org Chem. 2005:6492–6495. doi: 10.1021/jo050264+. [DOI] [PubMed] [Google Scholar]
- 23.Debrabandere L, Vanboven M, Daenens P. Synthesis of iodobuprenorphine for use in radioimmunoassay. J Label Compd Radiopharm. 1992;31:575–588. [Google Scholar]
- 24.Bentley KW, Hardy DG, Crocker HP, Haddlesey DI, Mayor PA. Novel analgesics and molecular rearrangements in the morphine-thebaine group. VI. Base-catalyzed rearrangements in the 6, 14-endo-ethenotetrahydrothebaine series. J Am Chem Soc. 1967;89:3312–3321. doi: 10.1021/ja00989a035. [DOI] [PubMed] [Google Scholar]
- 25.Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzetei-Gurske I, Khroyan TV, Husbands SM, Lewis JW, Toll L, Zaveri NT. Activities of mixed NOP and mu-opioid receptor ligands. Br J Pharmacol. 2008;153:609–619. doi: 10.1038/sj.bjp.0707598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khroyan TV, Polgar WE, Jiang F, Zaveri NT, Toll L. NOP Receptor Activation Attenuates Antinociception Induced by Mixed NOP/Mu-Opioid Receptor Agonists. J Pharmacol Exp Ther. 2009;331:946–953. doi: 10.1124/jpet.109.156711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- 28.Lewis JW. Buprenorphine – Medicinal Chemistry. In: Cowan A, Lewis JW, editors. Buprenorphine – combating drug abuse with a unique opioid. Wiley-Liss; New York: 1995. pp. 3–16. [Google Scholar]
- 29.Husbands SM, Lewis JW. Structural Determinants of Efficacy for κ-Opioid Receptors in the Orvinol Series: 7,7-Spiro Analogues of Buprenorphine. J Med Chem. 2000;43:139–141. doi: 10.1021/jm991165p. [DOI] [PubMed] [Google Scholar]
- 30.Lewis JW, Husbands SM. The Orvinols and Related Opioids – High Affinity Ligands with Diverse Efficacy Profiles. Current Pharmaceutical Design. 2004;10:717–732. doi: 10.2174/1381612043453027. [DOI] [PubMed] [Google Scholar]
- 31.Khroyan TV, Polgar WE, Cami-Kobeci G, Husbands SM, Zaveri NT, Toll L. The First ‘Universal Opioid Ligand’ (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): Characterization of the In Vitro Profile and In Vivo Behavioral Effects in Mouse Models of Acute Pain and Cocaine-induced Reward. J Pharmacol Exp Ther. 2011;336:952–961. doi: 10.1124/jpet.110.175620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.