

Abstract
Autosomal-recessive exfoliative ichthyosis presents shortly after birth as dry, scaly skin over most of the body with coarse peeling of nonerythematous skin on the palms and soles, which is exacerbated by excessive moisture and minor trauma. Using whole-genome homozygosity mapping, candidate-gene analysis and deep sequencing, we have identified loss-of-function mutations in the gene for protease inhibitor cystatin A (CSTA) as the underlying genetic cause of exfoliative ichthyosis. We found two homozygous mutations, a splice-site and a nonsense mutation, in two consanguineous families of Bedouin and Turkish origin. Electron microscopy of skin biopsies from affected individuals revealed that the level of detachment occurs in the basal and lower suprabasal layers. In addition, in vitro modeling suggests that in the absence of cystatin A protein, there is a cell-cell adhesion defect in human keratinocytes that is particularly prominent when cells are subject to mechanical stress. We show here evidence of a key role for a protease inhibitor in epidermal adhesion within the lower layers of the human epidermis.
Main Text
Cystatins form a superfamily of protease inhibitors, which could be further subdivided into three groups on the basis of their different distributions and molecular structure.1,2 Cystatin A (also known as stefin A) is a member of the type 1 cystatins (comprising stefin A and B) that have a mainly intracellular localization, although cystatin A has also been detected in sweat and in the medium of cultured keratinocytes.3 The cystatins are tight, reversible inhibitors of the papain-like lysosomal cysteine proteases, such as cathepsins B, H, and L.4–6 Furthermore, an imbalance of these proteases and their cystatin inhibitors is found in many cancers and other diseases.7–9
Cystatin A was originally identified as a component of the cornified cell envelope in the upper layers of the skin10 and has also been shown to be a potent inhibitor of exogenous proteases, such as the dust mite allergens Der p 1 and Der f 1,3 which can break down the epidermal barrier of the skin. In addition, cystatin A has been associated with atopic dermatitis (AD [MIM 603165]),11,12 a chronic inflammatory skin disease often associated with a defective epidermal barrier.13 We present here a role for cystatin A in the normal physiology of the skin within the lower levels of the epidermis and show that loss-of-function mutations in cystatin A underlie a peeling skin phenotype in the absence of an obvious epidermal barrier defect.
We previously reported a large, consanguineous, Bedouin family with autosomal-recessive exfoliative ichthyosis (MIM 607936) presenting in five individuals as circumscribed peeling of the skin on palms and soles associated with dry and scaly skin (Figures 1A and 1B). Initial linkage analysis with microsatellites showed suggestive linkage to chromosomal region 12q13.14 In view of the fact that the unaffected mother was uninformative for all the markers in this region of the genome, we decided to reassess the linkage analysis in this family. The study was approved by the South East National Health Service research ethics committee and by the institutional review board of the University Hospital of Münster, and all patients enrolled gave their informed consent. High-resolution homozygosity mapping was performed by genotyping two affected individuals (Figure 1C, individuals IV-10 and V-3) with the Affymetrix GeneChip Human Mapping 250K Nsp SNP array (Affymetrix, Santa Clara, CA, USA) (see Table S1, available online). GeneChip DNA Analysis Software (Affymetrix, Santa Clara, CA, USA) was used for calling SNP genotypes and a large block of homozygosity (9333.1 kb), containing approximately 60 genes and shared by both affected individuals, was identified on chromosomal region 3q21 between rs6783609 and rs6438966. Using PLINK to analyze the SNP data,15 we observed no significant blocks of shared homozygosity on chromosomal region 12q13, and the next largest stretches of homozygosity on chromosomal regions 4p15 and 5q23 were 1976.4 kb and 1892.8 kb, respectively (see Table S2). The new region of homozygosity on 3q21 lies between an uninformative microsatellite marker, D3S2460, and a heterozygous microsatellite marker, D3S1764, used in our previous study. Linkage to 3q21 was confirmed by genotyping microsatellite markers (D3S3709 [LOD 2.67], D3S3720 [LOD 3.27] and D3S3645 [LOD 3.13]) across the region (Figure 1C). CSTA (MIM 184600), located within the block of homozygosity on 3q21, was chosen as a likely candidate gene because it encodes a protein that is known to be expressed in keratinocytes.10 We sequenced all three coding exons and the flanking exon/intron boundaries of CSTA (NM_005213.3) by using standard Sanger sequencing methods in all available members of the Bedouin family. All affected individuals were homozygous for an A>T change affecting the 3′ splice-consensus sequence of intron 1, c.67-2A>T (p.Val23_Gln26del), whereas the parents were heterozygous carriers for c.67-2A>T, and unaffected individuals were either heterozygous or wild-type (WT) (Figure 1D). c.67-2A>T was not detected in 300 chromosomes from normal controls and is not present in the dbSNP or 1000 Genomes databases.
Figure 1.

Identification of CSTA Mutations
(A) The left foot of an affected individual showing marked hyperkeratosis with an aspect of water-sensitive palmoplantar keratoderma and superficial exfoliation of skin.
(B) The lower right arm of an affected individual showing slight hyperkeratosis and exfoliation.
(C) Linkage to chromosomal region 3q21 in a large consanguineous Bedouin family with autosomal-recessive exfoliative ichthyosis confirmed by genotyping microsatellite markers D3S3709, D3S3720, and D3S3645.
(D) Candidate-gene screening in the Bedouin family identified a 3′ splice-site variant, c.67-2A>T, in CSTA, a cysteine protease inhibitor found in the skin (left panel). Affected individuals are homozygous for c.67-2A>T, whereas the unaffected parents are both carriers of c.67-2A>T; this indicates that the variant segregates with the disease. Analysis of CSTA in a Turkish family with a similar phenotype identified the homozygous nonsense mutation c.256C>T (right panel).
Because we no longer have access to material from affected members of the Bedouin family to confirm the effect of the putative splice-site mutation on the in vivo processing of CSTA, we included all the exons found in the block of homozygosity on chromosomal region 3q21 in a next-generation sequencing project to verify that there were no other potentially disease-causing mutations within this region in these individuals. All the exons were captured and sequenced in one affected individual (V-3 in Figure 1C) from the Bedouin family.16 Briefly, all the exons located within the region of interest for each disease (i.e., 121,613,170 – 124,440,035 bp on chromosomal region 3q21) were extracted from the UCSC Genome Browser database and probes to all the exons designed and included on a custom capture microarray NimbleGen (Roche Nimblegen, Madison, WI, USA). DNA from the individuals of interest was randomly fragmented with a Bioruptor sonicator (Diagenode, Denville, NJ, USA) and the library of adaptor-ligated DNA fragments was prepared by following the Illumina protocol. The DNA library was then hybridized to the custom designed microarray from NimbleGen for 72 hr, after which time any unbound DNA was washed off and the captured DNA was eluted with sodium hydroxide and amplified in a PCR with primers against the common adaptor sequences. The captured, amplified DNA fragments were then sequenced as paired-end reads on the Illumina GAIIx (Illumina, San Diego, CA, USA). Raw 76 bp paired-end reads were aligned to the human reference sequence (hg19) with novoalign, including the soft clipping, adaptor trimming, and base-call quality calibration options. Filtering for clonal reads, pileup generation, and SNP calling on the basis of allele counts and read-depth were performed with custom Perl/C++ scripts. We filtered the variants against dbSNP and 1000 Genomes to identify previously unreported variants. The 3′ splice-site change in CSTA, c.67-2A>T, was identified by next-generation sequencing. The only other coding change identified in the region was a predicted missense change (c.1058A>G, p.Asn224Ser) in SEMA5B (NM_001031702.2), a gene that encodes a protein involved in axonal guidance during neural development and, hence, does not represent an obvious candidate gene for exfoliative ichthyosis. In parallel, we identified, by standard Sanger sequencing in a Turkish family with a very similar phenotype of exfoliative ichthyosis, a homozygous nonsense mutation in CSTA, c.256C>T resulting in the premature termination codon p.Gln86stop (Figure 1D, right panel). We did not have access to materials suitable for testing a potential synthesis of a truncated protein; however, the altered glutamine residue is highly conserved (ConSeq17 score 7) and the termination codon is located within the conserved cystatin domain of cystatin A, thus clearly indicating that p.Gln86stop is a deleterious mutation. This finding provides strong support for mutations in CSTA as the underlying cause of exfoliative ichthyosis.
To assess the potential effect of c.67-2A>T on the splicing of CSTA, we compared WT and mutated DNA sequences by using in silico splice-site predictor programs. The splice-site predictor software Neural Network Splice Site Prediction Tool18 predicts that the CSTA c.67-2A>T mutation would abolish the 3′ splice-acceptor site (Figure 2A). Likewise, the online program for scoring 3′ splice sites, MaxEntScan::score3ss,19 predicts a much lower maximum entropy score for the mutant splice site (4.89) when compared to the WT splice site (13.26). To confirm the impairment of the c.67-2A>T splice site in vitro, we analyzed expression of minigene constructs in HEK293T cells. Briefly, two CSTA minigene constructs were prepared by cloning each of the three CSTA exons, along with approximately 100 bp of surrounding intron sequence, into the pcDNA3 vector. The WT minigene construct contained the normal CSTA sequence as found in the human genome database, whereas the mutant minigene construct contained the c.67-2A>T splice-site change. Both minigene constructs were transfected into human HEK293T cells, which do not express CSTA with FuGENE 6 transfection reagent (Roche Diagnostics, Burgess Hill, West Sussex, UK). Forty-eight hours after transfection, RNA was collected from the transfected cells with the QIAGEN RNeasy minikit (QIAGEN, Crawley, West Sussex, UK). cDNA was made with a mixture of Oligo dT and random hexamer primers and SuperScript II Reverse Transcriptase (Invitrogen, Paisley, UK). The cDNA was then amplified with a forward primer in exon 1 of CSTA and a reverse primer in exon 3 of CSTA, and the PCR products were sequenced. Analysis of splicing of the CSTA minigene construct revealed that the 3′ splice-site mutation c.67-2A>T leads to skipping of the first 12 base pairs of exon 2 of CSTA, which translates to an in-frame deletion of four amino acid residues in the cystatin A protein (p.Val23_Gln26del) (Figure 2B). Immunoblotting of lysates collected from cells transfected with the minigene constructs showed greatly reduced levels of protein expression from the mutant construct (Figure 2C), which we predict to be due to the utilization of the much weaker splice-acceptor site within exon 2, as predicted by the Neural Network Splice Site Prediction Tool (Figure 2A). Furthermore, in silico modeling of the WT and mutated cystatin A proteins revealed clear structural differences in the mutant cystatin A protein, including: (1) the division of the first β sheet into two shorter β sheets, (2) the loss of a complete turn and change in orientation of the α-helix, and (3) the introduction of a second twist in the fourth β sheet (Figures 2D and 2E). These changes in the tertiary structure of the mutated cystatin A protein might alter the relative location of vital residues involved in the binding of cystatin A to potential cathepsin substrates6 and, hence, could detrimentally affect the ability of cystatin A to bind. Therefore, the splice-site mutation c.67-2A>T in all likelihood represents a nonfunctional allele and, hence, together with the finding of nonsense mutations in the Turkish family reveals that loss-of-function mutations in CSTA underlie congenital autosomal-recessive exfoliative ichthyosis.
Figure 2.
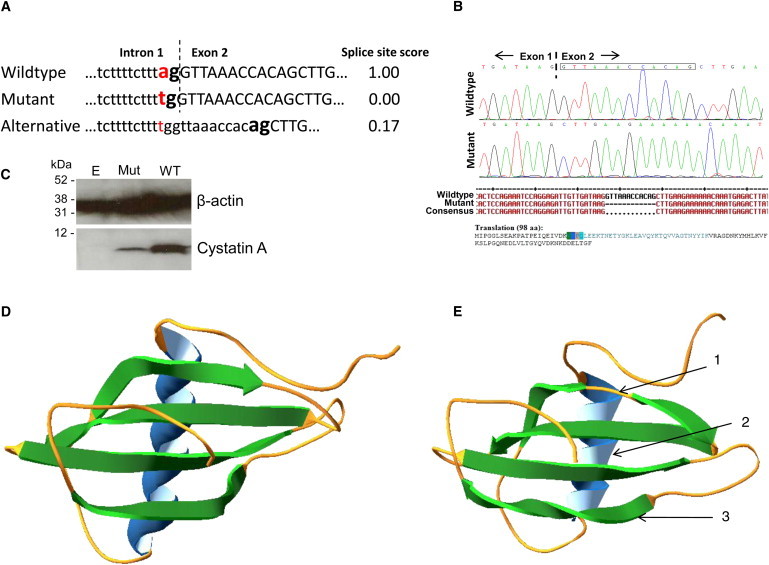
Evaluation of the c.67-2A>T Mutant Cystatin A
(A) With the Neural Network Splice Site Prediction software18 the WT CSTA splice-site scores a maximum score of 1, whereas the mutated splice site, c.67-2A>T, is predicted to score 0. A predicted alternative splice site that is 12 bp into exon 2 has a very weak score of 0.17.
(B) An in vitro splice assay showed that in the presence of the c.67-2A>T mutation, the splicing machinery uses an alternative splice site that leads to skipping of the first 12 bp of exon 2 of CSTA (boxed nucleotides on sequence trace), predicted to result in an in-frame deletion of the four amino acids: Val-Lys-Pro-Gln (residues 23-26) in the cystatin A protein.
(C) Immunoblotting of lysates collected from HEK293T cells transfected with empty pcDNA3 vector, the mutant CSTA minigene construct with the c.67-2A>T splice-site change (Mut), and the WT CSTA minigene construct showed greatly reduced levels of expression from the mutant construct, probably because of utilization of the much weaker splice-acceptor site within exon 2.
(D) In silico modeling shows the predicted ribbon structure of WT cystatin A.
(E) In silico modeling of the splice-site mutant, c.67-2A>T, cystatin A with the four amino acid deletion; the model shows the possible structural differences between the mutant and WT proteins: (1) the first β sheet is split by a random coil giving two shorter β sheets, (2) the α-helix has lost one complete turn and changed its orientation, and (3) the fourth β sheet has a second twist. Structures determined from amino acid sequence with i-Tasser online server29 and figures produced with Swiss-PdbViewer 4.0.30
The Turkish and Bedouin families both have a very similar phenotype that presents shortly after birth as a dry, scaly skin over most of the body and coarse peeling of nonerythematous skin on the palms and soles, which is exacerbated by excessive moisture (aquagenic) and minor trauma. Electron microscopy of skin biopsies from both families revealed that the upper layers of the epidermis appear mostly normal, whereas there is prominent intercellular edema of the basal and suprabasal cell layers and aggregates of tonofilaments in the basal keratinocytes (Figure 3A) demonstrating that the skin peeling initiates by weakness in keratinocyte attachment at the basal and lower suprabasal level in the affected individuals. A detailed view of the cell-cell contacts showed a loss in number and tightness of the desmosomes in the basal layer as compared to the upper epidermal layers and a partial loosening of their intercellular interaction (Figure 3A). Immunostaining of normal skin sections revealed that cystatin A is localized throughout all suprabasal layers of the epidermis with a diffuse cytoplasmic distribution and the strongest synthesis in the granular layer (Figure 3B). To mimic the phenotype observed in these families in an in vitro model, we have first used ON-TARGETplus SMARTpool siRNA (Dharmacon, Chicago, IL, USA) to knock down cystatin A expression in the HaCaT human keratinocyte cell line and studied the effect of mechanical stress on these cells. Cells were seeded at 2 × 105 cells/35 mm well and transfected with 100 nM (final concentration) of either CSTA siRNA (CSTA KD) or nontargeting pool siRNA (NTP) (Dharmacon). siRNA was prepared by mixing 10 μl of 20 mM stock concentration with 190 μl of serum-free and antibiotic-free Dulbecco's Modified Eagle's Medium/Nutrient F-12 Ham (DMEM-F12) (Sigma-Aldrich) and transfected with 6 μl DharmaFECT 1 (Dharmacon) diluted in 194 μl of serum-free and antibiotic-free DMEM-F12 media. Seventy-two hours after transfection, the efficiency of the knockdown was analyzed by immunoblotting of cell lysates and by immunocytochemistry (Figure 3C). CSTA KD cells and NTP cells were both seeded on BioFlex 6-well plates coated with pronectin, which contain a flexible, rubber membrane in each 35 mm well (Flexplates, Flexcell International, Hillsborough, NC, USA), grown to ∼80% confluency, and then the monolayers were stressed mechanically with a Flexcell FX-4000 Tension System (Flexcell International, Hillsborough, NC, USA)—a computer-regulated bioreactor that uses vacuum pressure to apply cyclic or static strain to cells cultured on flexible-bottomed culture plates. Each plate was placed over the loading station containing six planar-faced cylinders or posts. Each post (25 mm) is centered beneath the rubber membrane of each 35 mm well. Cells were subject to cyclic mechanical stretch with a frequency of 5 Hz (i.e., five cycles of stretch and relaxation per second) and an elongation of amplitude ranging from 8% to 14% (i.e., increase in diameter across the silicone membrane from 8% to 14%). Cells were stretched for 0 hr (unstretched), 1 hr, and 4 hr, and then fixed and stained for keratin 14 (Figure 3D). Upon intense stretching (4 hr) thickening of the keratin filaments was observed in both CSTA KD and NTP cells; however, the CSTA KD monolayer had split into many fragments, whereas the NTP monolayer of cells was still intact (Figure 3D, 20×). At higher magnification, breakage of keratin filaments and widened intercellular spaces could be seen in the CSTA KD cells (Figure 3D, 100×); in contrast there are no obvious cell-cell adhesion defects in the stretched NTP cell monolayer.
Figure 3.

Loss of Cystatin A Protein Affects Cell-Cell Adhesion
(A) Electron microscopy of a skin biopsy of ridged skin from palm and sole, respectively, from an affected member of the Turkish family. The upper layers of the epidermis appear mostly normal (1), whereas there is prominent intercellular edema of the suprabasal and basal cell layers (2) and aggregates of tonofilaments in the basal keratinocytes (3, arrow), suggesting that the skin peeling initiates at the basal-suprabasal level in the affected individuals. In higher magnification, cell-cell adhesion looks normal with a regular structure of desmosomes in upper layers (4) but there is loosening of the keratinocyte structure with irregularities in desmosomal interaction in the basal layer (5, arrow). The following abbreviations are used: SG, stratum granulosum; BL, basal layer.
(B) Staining of normal skin shows that cystatin A is localized throughout the epidermis and has a diffuse cytoplasmic distribution.
(C) The efficiency of cystatin A knockdown (CSTA KD) compared to cells treated with a nontargeting pool (NTP) of siRNA was analyzed by immunocytochemistry and immunoblotting of cell lysates. An approximately 85% knockdown was achieved.
(D) In an in vitro model of exfoliative ichthyosis, monolayers of CSTA KD cells were stressed mechanically and then stained for keratin 14 (green). Nuclei are stained with DAPI (blue). Upon intense stretching (4 hr) the CSTA KD monolayer broke into many fragments, whereas the NTP monolayer of cells remained intact (20×). At higher magnification (100×), breakage of keratin filaments and widened intercellular spaces could be seen between the CSTA KD cells, in contrast to no obvious cell-cell adhesion defects in the stretched NTP cell monolayer. The scale bar represents 20μm.
(E) A dispase-based dissociation assay was utilized to assess intercellular adhesion by the degree of cell monolayer integrity upon mechanical stress. CSTA KD cells showed an increased number of monolayer fragments in contrast to the small number of monolayer fragments observed for the NTP cells. These observations show a statistically significant decrease in cell-cell adhesion in monolayers lacking cystatin A protein. Error bars show the standard error of the mean.
In addition, a dispase-based dissociation assay was utilized to assess intercellular adhesion by the degree of cell monolayer integrity upon mechanical stress. Cells were seeded with a density of 2 × 105 cells/35 mm well and treated, as before, with NTP siRNA or cystatin A siRNA. Postknockdown, the monolayers were detached by incubating cells with 5 ml/60 mm dish of dispase for 40 min at 37°C. After all monolayers have detached as an intact sheet, they were subjected to agitation by inversion and all fragments obtained were counted under a dissection microscope. CSTA KD cell monolayers showed a high increase in the number of fragments, demonstrating a significant decrease in cell-cell adhesion, in contrast to the very few fragments obtained for the NTP cell monolayers (Figure 3E). These data further support the observations made for the cell stretch assay showing a cell-cell adhesion defect in cells lacking cystatin A.
To further study the effects of loss of cystatin A on differentiating keratinocytes, we have generated 3D skin models of exfoliative ichthyosis by knockdown of CSTA in normal primary keratinocytes, which were then used for organotypic tissue culture (Figure 4). 3D models were generated as described recently.20 In brief, 1 × 105 fibroblasts and 1.0–1.18 × 106 keratinocytes per cm2 were used in a 3 μm tissue-culture insert (BD Biosciences, Heidelberg, Germany). A mixture of ice-cold bovine collagen I, fibroblasts, and serum was poured into each filter insert. Medium was added after incubation at 37°C for 2 hr. Keratinocytes were seeded on the matrix in a total volume of 1 ml KGM per insert. After 24 hr the system was raised to the air-liquid interface and keratinocyte conditioned medium was used as differentiation medium for the next 8 days. For model preparation, cells from healthy female donors aged 46 and 30 years, respectively, and from the foreskin of a 9-year-old male were used. For preparing disease models, knockdown in primary keratinocytes was performed exactly 24 hr before model set up with a set of three siRNAs (740 pmol each) and 150 μl Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) in a T225 cell-culture flask. Full-thickness skin models were cultured until day 8 at the air-liquid interface. All models used here were generated from the same sources of cells.
Figure 4.
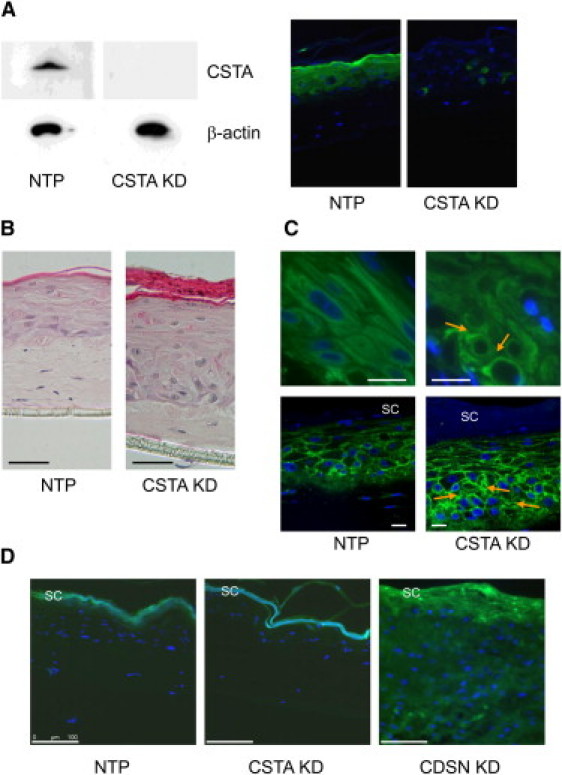
Organotypic Cultures
(A) Full skin models were generated with NTP and CSTA KD. Loss of cystatin A was shown by immunoblot analysis in protein extracts from skin models and by immunostaining against cystatin A.
(B) Hematoxylin and eosin staining showing completed epidermal differentiation, hyperkeratosis, and parakeratosis, and a disturbance of basal epidermal architecture in CSTA KD organotypic culture compared to NTP organotypic cultures. The scale bar represents 50 μm.
(C) Immunostaining against keratin 14 (upper panel) and E-cadherin (lower panel) demonstrated grossly disorganized structure, widening of intercellular spaces (arrows), and partial breakdown of keratin intermediate filaments in basal and lower suprabasal layers of disease models depleted of cystatin A. The scale bar represents 20 μm. SC is an abbreviation of stratum corneum.
(D) Barrier function appears to be largely maintained in the CSTA KD organotypic culture in a Lucifer Yellow dye penetration assay (shown in green) compared to the NTP organotypic culture and in contrast to the loss of barrier function observed in the corneodesmosin knockdown (CDSN KD) organotypic culture, which is a model of the generalized peeling skin disease. The scale bar represents 100 μm.
Loss of cystatin A in the 3D models was confirmed by immunoblot analysis with proteins extracted from model biopsy specimens and by immunostaining against cystatin A (Figure 4A). The CSTA KD model demonstrated hyperkeratosis, parakeratosis, and moderate epidermal hyperplasia, and a disturbance of basal epidermal architecture (Figure 4B). Epidermal differentiation and basement membrane interaction were confirmed by staining of epidermal marker proteins (data not shown). Immunostaining against keratin 14 and E-cadherin showed a widening of intercellular spaces and major disorganization of keratinocytes in the lower suprabasal and basal layers (Figure 4C). Circular structures, in contrast to the stretched filaments in the normal skin models, are indicative of a partial breakdown of intermediate filaments. To analyze epidermal barrier properties in exfoliative ichthyosis, we made use of a Lucifer Yellow penetration assay. A 1 mM Lucifer Yellow solution (Sigma, Munich, Germany) in PBS was applied onto control and knockdown skin models. After 1 hr at 37°C, the solution was washed off and models were snap-frozen. Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and visualized with a fluorescence microscope (LMD6000, Leica, Wetzlar, Germany). Control full-thickness skin models (WT) showed an active epidermal barrier. Barrier function, as assessed by this assay, is largely maintained in the absence of cystatin A compared with WT organotypic cultures and in contrast to organotypic cultures grown with corneodesmosin (CDSN) KD cells (Figure 4D).
We have identified inactivating mutations in CSTA in two cases of autosomal-recessive exfoliative ichthyosis. Our data on the mechanically stressed monolayer of CSTA KD cells, in combination with the organotypic tissue culture with CSTA KD keratinocytes and electron microscopy of biopsies from affected individuals, reveal an unexpected and important role for cystatin A in desmosome-mediated cell-cell adhesion in the lower levels of the epidermis. A variant of cystatin A (CSTA, c.287T>C), predicted to encode a mRNA transcript with reduced stability, has been associated with AD and is hypothesized to contribute to the defective epidermal barrier seen in AD.12 However, the data from our organotypic 3D culture model demonstrate that there is no gross barrier defect associated with CSTA KD, unlike the generalized peeling skin disease with atopic manifestations, which is caused by mutations in corneodesmosin and characterized by a severe epidermal barrier defect.21 Accordingly, no atopic component is observed in the phenotype of individuals with exfoliative ichthyosis. This is also in contrast to Netherton's syndrome (MIM 256500), where the loss of a protease inhibitor, LEKTI, leads to hyperactivity of kallikrein 5 and increased corneodesmosomal degradation, resulting in ichthyosiform erythroderma and atopic manifestations.22,23 Similarly, inactivation of the serine protease matriptase leads to autosomal-recessive ichthyosis accompanied by disturbance of the stratum corneum,24,25 but also a defect in the mouse ortholog of the matriptase inhibitor, SPINT1, leads to an ichthyosis-like phenotype.26,27 It should be noted that in a mouse model with a chromosomal deletion encompassing Csta, no obvious phenotype was observed,28 which could reflect differences between human and mouse skin. In contrast to phenotypes associated with generalized skin peeling, exfoliative ichthyosis is caused by lesions in the basal layers of the epidermis characterized by expression of KRT14. This difference might contribute to the pattern of the phenotype, which is clearly pronounced on the palmar and plantar skin. As in other disorders of cornification with a palmoplantar expression pattern, the defect in the basal and lower suprabasal layers might thus be particularly susceptible to mechanical stress, which is strongly present on palmoplantar skin. In summary, our data describe mutations in CSTA associated with a monogenic skin disease and reveals a previously unknown key role for cystatin A in basal to suprabasal keratinocyte adhesion.
Acknowledgments
We would like to thank the families for their participation in this study, and we would like to acknowledge the members of The Genome Centre, Queen Mary University of London, for their assistance. This work was funded by Barts and The London Charity and supported in part by grants from the German Federal Ministry for Education and Research as part of the Networks for Rare Diseases (01GM0902) and the Deutsche Forschungsgemeinschaft (HE3119/5-1).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
MaxEntScan::score3ss, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html
Neural Network Splice Site Prediction Tool, http://www.fruitfly.org/seq_tools/splice.html
Novoalign, http://www.novocraft.com
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/omim
UCSC Genome Browser database, http://genome.ucsc.edu/
References
- 1.Turk V., Bode W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett. 1991;285:213–219. doi: 10.1016/0014-5793(91)80804-c. [DOI] [PubMed] [Google Scholar]
- 2.Wallin H., Bjarnadottir M., Vogel L.K., Wassélius J., Ekström U., Abrahamson M. Cystatins—Extra- and intracellular cysteine protease inhibitors: High-level secretion and uptake of cystatin C in human neuroblastoma cells. Biochimie. 2010;92:1625–1634. doi: 10.1016/j.biochi.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Kato T., Takai T., Mitsuishi K., Okumura K., Ogawa H. Cystatin A inhibits IL-8 production by keratinocytes stimulated with Der p 1 and Der f 1: Biochemical skin barrier against mite cysteine proteases. J. Allergy Clin. Immunol. 2005;116:169–176. doi: 10.1016/j.jaci.2005.03.044. [DOI] [PubMed] [Google Scholar]
- 4.Jenko S., Dolenc I., Gunčar G., Doberšek A., Podobnik M., Turk D. Crystal structure of Stefin A in complex with cathepsin H: N-terminal residues of inhibitors can adapt to the active sites of endo- and exopeptidases. J. Mol. Biol. 2003;326:875–885. doi: 10.1016/s0022-2836(02)01432-8. [DOI] [PubMed] [Google Scholar]
- 5.Pavlova A., Björk I. The role of the second binding loop of the cysteine protease inhibitor, cystatin A (stefin A), in stabilizing complexes with target proteases is exerted predominantly by Leu73. Eur. J. Biochem. 2002;269:5649–5658. doi: 10.1046/j.1432-1033.2002.03273.x. [DOI] [PubMed] [Google Scholar]
- 6.Renko M., Požgan U., Majera D., Turk D. Stefin A displaces the occluding loop of cathepsin B only by as much as required to bind to the active site cleft. FEBS J. 2010;277:4338–4345. doi: 10.1111/j.1742-4658.2010.07824.x. [DOI] [PubMed] [Google Scholar]
- 7.Gelb B.D., Shi G.P., Chapman H.A., Desnick R.J. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273:1236–1238. doi: 10.1126/science.273.5279.1236. [DOI] [PubMed] [Google Scholar]
- 8.Pennacchio L.A., Lehesjoki A.E., Stone N.E., Willour V.L., Virtaneva K., Miao J., D'Amato E., Ramirez L., Faham M., Koskiniemi M. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science. 1996;271:1731–1734. doi: 10.1126/science.271.5256.1731. [DOI] [PubMed] [Google Scholar]
- 9.Toomes C., James J., Wood A.J., Wu C.L., McCormick D., Lench N., Hewitt C., Moynihan L., Roberts E., Woods C.G. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat. Genet. 1999;23:421–424. doi: 10.1038/70525. [DOI] [PubMed] [Google Scholar]
- 10.Steven A.C., Steinert P.M. Protein composition of cornified cell envelopes of epidermal keratinocytes. J. Cell Sci. 1994;107:693–700. [PubMed] [Google Scholar]
- 11.Samuelsson L., Stiller C., Friberg C., Nilsson C., Inerot A., Wahlström J. Association analysis of cystatin A and zinc finger protein 148, two genes located at the psoriasis susceptibility locus PSORS5. J. Invest. Dermatol. 2004;122:1399–1400. doi: 10.1046/j.0022-202X.2004.12604.x. [DOI] [PubMed] [Google Scholar]
- 12.Vasilopoulos Y., Cork M.J., Teare D., Marinou I., Ward S.J., Duff G.W., Tazi-Ahnini R. A nonsynonymous substitution of cystatin A, a cysteine protease inhibitor of house dust mite protease, leads to decreased mRNA stability and shows a significant association with atopic dermatitis. Allergy. 2007;62:514–519. doi: 10.1111/j.1398-9995.2007.01350.x. [DOI] [PubMed] [Google Scholar]
- 13.Cork M.J., Robinson D.A., Vasilopoulos Y., Ferguson A., Moustafa M., MacGowan A., Duff G.W., Ward S.J., Tazi-Ahnini R. New perspectives on epidermal barrier dysfunction in atopic dermatitis: Gene-environment interactions. J. Allergy Clin. Immunol. 2006;118:3–21. doi: 10.1016/j.jaci.2006.04.042. quiz 22–23. [DOI] [PubMed] [Google Scholar]
- 14.Hatsell S.J., Stevens H., Jackson A.P., Kelsell D.P., Zvulunov A. An autosomal recessive exfoliative ichthyosis with linkage to chromosome 12q13. Br. J. Dermatol. 2003;149:174–180. doi: 10.1046/j.1365-2133.2003.05386.x. [DOI] [PubMed] [Google Scholar]
- 15.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaydon D.C., Biancheri P., Di W.-L., Plagnol V., Cabral R.M., Brooke M.A., van Heel D.A., Ruschendorf F., Toynbee M., Walne A. Inflammatory Skin and Bowel disease Linked to Deletion in ADAM17. N. Engl. J. Med. 2011 doi: 10.1056/NEJMoa1100721. in press. [DOI] [PubMed] [Google Scholar]
- 17.Berezin C., Glaser F., Rosenberg J., Paz I., Pupko T., Fariselli P., Casadio R., Ben-Tal N. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20:1322–1324. doi: 10.1093/bioinformatics/bth070. [DOI] [PubMed] [Google Scholar]
- 18.Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 19.Yeo G., Burge C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 20.Eckl K.M., Alef T., Torres S., Hennies H.C. Full-thickness human skin models for congenital ichthyosis and related keratinization disorders. J. Invest. Dermatol. 2011;131:1938–1942. doi: 10.1038/jid.2011.126. [DOI] [PubMed] [Google Scholar]
- 21.Oji V., Eckl K.M., Aufenvenne K., Nätebus M., Tarinski T., Ackermann K., Seller N., Metze D., Nürnberg G., Fölster-Holst R. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: Unraveling the peeling skin disease. Am. J. Hum. Genet. 2010;87:274–281. doi: 10.1016/j.ajhg.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavanas S., Bodemer C., Rochat A., Hamel-Teillac D., Ali M., Irvine A.D., Bonafé J.L., Wilkinson J., Taïeb A., Barrandon Y. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 23.Descargues P., Deraison C., Bonnart C., Kreft M., Kishibe M., Ishida-Yamamoto A., Elias P., Barrandon Y., Zambruno G., Sonnenberg A., Hovnanian A. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet. 2005;37:56–65. doi: 10.1038/ng1493. [DOI] [PubMed] [Google Scholar]
- 24.Alef T., Torres S., Hausser I., Metze D., Türsen U., Lestringant G.G., Hennies H.C. Ichthyosis, follicular atrophoderma, and hypotrichosis caused by mutations in ST14 is associated with impaired profilaggrin processing. J. Invest. Dermatol. 2009;129:862–869. doi: 10.1038/jid.2008.311. [DOI] [PubMed] [Google Scholar]
- 25.Basel-Vanagaite L., Attia R., Ishida-Yamamoto A., Rainshtein L., Ben Amitai D., Lurie R., Pasmanik-Chor M., Indelman M., Zvulunov A., Saban S. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am. J. Hum. Genet. 2007;80:467–477. doi: 10.1086/512487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagaike K., Kawaguchi M., Takeda N., Fukushima T., Sawaguchi A., Kohama K., Setoyama M., Kataoka H. Defect of hepatocyte growth factor activator inhibitor type 1/serine protease inhibitor, Kunitz type 1 (Hai-1/Spint1) leads to ichthyosis-like condition and abnormal hair development in mice. Am. J. Pathol. 2008;173:1464–1475. doi: 10.2353/ajpath.2008.071142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szabo R., Kosa P., List K., Bugge T.H. Loss of matriptase suppression underlies spint1 mutation-associated ichthyosis and postnatal lethality. Am. J. Pathol. 2009;174:2015–2022. doi: 10.2353/ajpath.2009.090053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bilodeau M., MacRae T., Gaboury L., Laverdure J.P., Hardy M.P., Mayotte N., Paradis V., Harton S., Perreault C., Sauvageau G. Analysis of blood stem cell activity and cystatin gene expression in a mouse model presenting a chromosomal deletion encompassing Csta and Stfa2l1. PLoS ONE. 2009;4:e7500. doi: 10.1371/journal.pone.0007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy A., Kucukural A., Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.