

Summary
Linking synaptic plasticity with behavioral learning requires understanding how synaptic efficacy influences postsynaptic firing in neurons whose role in behavior is understood. Here we examine plasticity at a candidate site of motor learning: vestibular nerve synapses onto neurons that mediate reflexive movements. Pairing nerve activity with changes in postsynaptic voltage induced bidirectional synaptic plasticity in vestibular nucleus projection neurons: long-term potentiation relied on calcium-permeable AMPA receptors and postsynaptic hyperpolarization, while long-term depression relied on NMDA receptors and postsynaptic depolarization. Remarkably, both forms of plasticity uniformly scaled synaptic currents evoked by pulse trains, and these changes in synaptic efficacy were translated into linear increases or decreases in postsynaptic firing responses. Synapses onto local inhibitory neurons were also plastic but expressed only long-term depression. Bidirectional, linear gain control of vestibular nerve synapses onto projection neurons provides a plausible mechanism for motor learning underlying adaptation of vestibular reflexes.
Introduction
Activity-dependent changes in synaptic strength are assumed to underlie various forms of learning and memory, but connecting synaptic plasticity to behavioral consequences requires knowing both how plasticity affects the output of postsynaptic neurons and how those neurons influence behavior. The majority of behavioral circuits comprise neurons that encode information in the patterns and/or rates of action potentials, and understanding how changes in synaptic strength affect postsynaptic firing is a critical step for linking cellular mechanisms of plasticity with learning. Most studies of synaptic plasticity, however, have focused exclusively on how activity alters synaptic currents in neurons within complex circuits where the relationship between neuronal firing and behavioral performance is poorly understood.
Here we investigate synaptic plasticity in a system that is exceptionally well suited for linking experience-dependent changes in synaptic strength with postsynaptic firing outputs and their consequences for learning and memory. Direct control of well-defined movements and simple circuitry make motor learning in the vestibulo-ocular reflex (VOR) a tractable model for assessing the behavioral consequences of cellular plasticity. The VOR generates eye movements that stabilize images on the retina during self-motion; motor learning in the VOR is triggered by persistent image motion during head movements and results in increases or decreases in the gain of evoked eye movements (Ito, 1984; Miles and Lisberger, 1981). Decades of studies in awake, behaving animals have provided the requisite information about how neuronal firing in vestibular and cerebellar circuits affects VOR performance and learning (du Lac et al., 1995; Hirata and Highstein, 2001). Remarkably, although the central vestibular nerve synapse has long been a candidate site of behavioral modification of the VOR (Miles and Lisberger, 1981), plasticity has not been demonstrated at this synapse.
Vestibular nerve afferents and postsynaptic medial vestibular nucleus (MVN) neurons fire tonically at high rates in vivo, and synaptic transmission at central vestibular nerve synapses drives remarkably linear increases in postsynaptic firing rates (Bagnall et al., 2008). How might these constraints influence the existence and nature of plasticity at this synapse? The activity patterns required to modify synaptic efficacy in neurons that fire at high baseline rates differ from those that gate plasticity in quiescent circuits (Jorntell and Hansel, 2006; Pugh and Raman, 2009). Synaptic inhibition or hyperpolarization can induce long-lasting potentiation of intrinsic excitability in MVN neurons (Nelson et al., 2003) and can trigger synaptic plasticity in analogous deep cerebellar nucleus neurons (Pugh and Raman, 2006). Therefore, we reasoned that postsynaptic membrane potential might similarly influence vestibular nerve synaptic plasticity. In this study, we examine whether vestibular nerve synaptic activity in the presence or absence of postsynaptic hyperpolarization has long-term effects on both postsynaptic currents and evoked firing responses in MVN neurons. The results demonstrate that vestibular nerve synapses are bidirectionally plastic and the direction of plasticity depends on postsynaptic voltage. Long-term potentiation (LTP) and long-term depression (LTD) of postsynaptic currents are translated into linear changes in postsynaptic firing rates across a wide range of stimulus frequencies. Thus, plasticity at the vestibular nerve synapse functions as a cellular mechanism of linear gain control.
Results
Projection neurons can be distinguished from local inhibitory neurons with transgenic mouse lines
The MVN contains two broad classes of neurons with different physiological, neurochemical, and anatomical properties (Bagnall et al., 2007; Straka et al., 2005). These complementary populations can be identified in transgenic mouse lines: glutamatergic and glycinergic neurons are fluorescently labeled in the YFP-16 line, and a subset of GABAergic neurons is fluorescently labeled in the GIN line (Bagnall et al., 2007; Feng et al., 2000; Oliva et al., 2000). To determine whether these distinct neuronal classes correspond to known MVN cell types or mediate different circuit functions, which would constrain the consequences of synaptic plasticity, we evaluated their projections to other brain areas. Fluorescent dextran conjugates were injected in vivo into previously identified targets of the MVN (Highstein and Holstein, 2006), including the oculomotor nucleus, thalamus, medullary reticular formation, and cerebellum. In YFP-16 mice, YFP-expressing neurons were retrogradely labeled from all targets (Figure 1A–C). In contrast, GFP-expressing neurons in GIN mice were never retrogradely labeled from injections targeted outside of the vestibular nuclei. GFP-positive synaptic terminals in GIN mice were, however, observed on the somata and proximal dendrites of neurons in the MVN retrogradely labeled from injections to the cerebellum, thalamus, and reticular formation (Figure 1D). These results indicate that fluorescently labeled MVN neurons in the YFP-16 line are predominantly projection neurons, while those in the GIN line provide local inhibition within the bilateral MVN.
Figure 1.
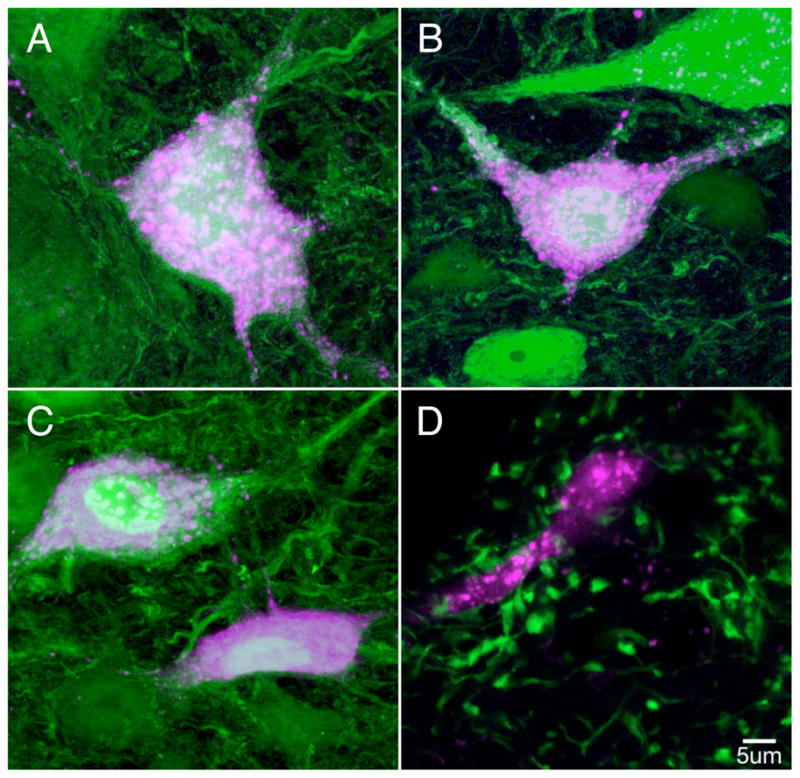
Two transgenic mouse lines define populations of projection and interneurons in the MVN. A-C: Neurons expressing fluorescent protein in the YFP-16 mouse line (green) were retrogradely labeled from stereotaxic dye injections (purple) into the thalamic parafascicular nucleus (A), the medullary reticular formation (B), and the oculomotor nucleus (C). The neurons shown are representative cases; several neurons were double labeled in multiple sections from 2–3 injections in each target structure. In panel D, fluorescent terminals in the GIN mouse line are labeled in green and are in close apposition to the proximal dendrite of a neuron retrogradely labeled from the cerebellar flocculus (purple).
The vestibular nerve synapse is bidirectionally plastic
Vestibular nerve afferents provide the major excitatory drive to the MVN and synapse directly onto both YFP-16 and GIN neurons (Bagnall et al., 2008). We sought to determine whether these synapses were capable of activity-dependent plasticity that regulated postsynaptic firing responses and whether modifications of synaptic strength depended on postsynaptic cell type. To investigate whether vestibular nerve synapses were plastic, we devised two protocols based on those used to induce LTD (Zhang and Linden, 2006) and LTP (Pugh and Raman, 2006) at the analogous excitatory mossy fiber synapse onto deep cerebellar nucleus neurons. Because MVN neurons receive a tonic barrage of both excitatory and inhibitory synaptic inputs, the protocols mimic epochs of either enhanced excitation or inhibition. The “100 Hz stim” protocol comprised 30 repetitions of 550-msec vestibular nerve stimulation at 100 Hz, which elevated the firing rate of postsynaptic neurons (Figure 2A). The “100 Hz stim plus hyperpolarization” protocol comprised 30 repetitions of 550-msec vestibular nerve stimulation at 100 Hz, paired with hyperpolarization of the postsynaptic neuron by ~15–25 mV for 250 msec to simulate strong, coincident inhibition (Figure 2B).
Figure 2.

Bidirectional, non-Hebbian plasticity of vestibular nerve synapses onto YFP-16 neurons in the MVN. (A) Representative response to protocol consisting of 100 Hz synaptic stimulation for 550 msec (left). Mean EPSC peak amplitude before and after protocol, which was applied at time= 0 min (middle, n=11). EPSC values are normalized to the mean baseline value in this and related figures. Representative EPSC before (black) and after (grey) LTD induction (right). (B) Representative response to protocol consisting of 100 Hz synaptic stimulation for 550 msec paired with 250 msec hyperpolarization (left). Mean EPSC peak amplitudes before and after protocol (middle, n=21). Representative EPSC before (black) and after (grey) LTP induction. In this and subsequent figures, error bars = SEM.
We initially examined synaptic plasticity in projection neurons highlighted in the YFP-16 line. Fluorescent neurons were targeted for whole-cell patch recordings in oblique coronal brainstem slices. Neurons were recorded primarily in current clamp to enable spontaneous firing (11 ± 2 Hz) and physiological baseline Ca2+ dynamics. Electrical stimulation of the vestibular nerve elicited synaptic responses in most MVN neurons, as previously reported (Bagnall et al., 2008). EPSCs evoked by nerve stimulation were measured during brief voltage clamp epochs every 15 sec, and a stable 10 min baseline was established prior to protocol application. The 100 Hz stim protocol evoked postsynaptic firing that averaged 38 ± 10 Hz and induced a short-lasting post-tetanic depression, followed by a robust LTD of the vestibular nerve synapse that reduced the peak EPSC amplitude to 0.82 ± 0.06 of the baseline 15 mins post-protocol (Figure 2A, n=11, p=0.03 vs. no-protocol control recordings: 0.98 ± 0.01, n=9). Thus, the vestibular nerve synapse displays activity-dependent, long-term strength modifications, and coincident pre- and post-synaptic activity results in LTD.
Given the extensive role of inhibition in the control of sensory processing and plasticity in the vestibular system (Buttner et al., 1992; Gittis and du Lac, 2006; Ito et al., 1970; Kato et al., 2003; Shimazu and Precht, 1966), we examined whether long-term synaptic efficacy was influenced by postsynaptic hyperpolarization. Remarkably, hyperpolarization of postsynaptic YFP-16 neurons during high-frequency vestibular nerve stimulation reversed the direction of long-term plasticity and resulted in LTP, which increased the peak EPSC amplitude to 1.31 ± 0.13 of the baseline (Figure 2B, n=21, p=0.05 vs. no-protocol control; average hyperpolarization: −18.2 ± 1.3 mV). The stimulation patterns that induced synaptic plasticity did not induce concomitant changes in the intrinsic properties of postsynaptic neurons (Supplemental Table 1). These data show that the first central synapse in the vestibular system is capable of robust, bidirectional plasticity and that postsynaptic voltage controls the direction of plasticity.
Plasticity modulates the gain of evoked firing in projection neurons
What are the consequences of vestibular nerve synaptic plasticity for postsynaptic neuronal output? Although synaptic efficacy is typically assessed with either single stimuli or pairs of stimuli, vestibular nerve afferents fire at high baseline rates (~30–50 Hz in vivo) and rarely fall silent for extended periods (Hullar and Minor, 1999; Lasker et al., 2008). Consequently, we assessed the effects of synaptic plasticity by stimulating with trains of pulses at rates across the physiologically relevant operating range of the vestibular nerve.
At the vestibular nerve to MVN neuron synapse, stimulus trains evoke EPSCs that depressed rapidly, over the first ~10 pulses, to a steady-state amplitude that is independent of frequency (Bagnall et al., 2008). Neither the time course nor the relative magnitude of short-term synaptic dynamics were altered following the induction of LTD (n=9, p= 0.57) or LTP (n= 8, p= 0.08)(Figures 3A, B). Importantly, a linear relationship between the rate of stimulation (5–100 Hz) and the normalized synaptic charge transfer was also preserved following the induction of LTD (R2= 0.97 ± 0.01, n=11) and LTP (R2= 0.99 ± 0.01, n= 8, Figure 3C). These results demonstrate that LTD and LTP influenced EPSC amplitude uniformly across stimulus rates without affecting the short-term synaptic dynamics. Thus, vestibular nerve plasticity scales evoked postsynaptic currents without altering the rate-independence of synaptic transmission and corresponding linear charge transfer.
Figure 3.

Synaptic plasticity uniformly scales synaptic currents evoked by stimulus trains. (A) EPSCs measured during vestibular nerve stimulation at 10 Hz rapidly reach a steady-state plateau of 49 ± 4% (EPSC11–20) of the EPSC amplitude response to a single stimulus (EPSC1) before LTD induction and 50 ± 4% after. (B) Short-term synaptic dynamics also did not change following LTP, in which the steady-state plateau was 59 ± 4% before and 55 ± 3% after induction. (C) Steady-state EPSCs evoked during stimulus trains (5–100 Hz) were measured following LTD and LTP induction to assess short-term dynamics over the physiological range of the synapse. The total synaptic charge transfer per unit time increased linearly with vestibular nerve stimulation rate. Steady-state charge transfer was calculated as the integrated area under the average steady-state EPSC, normalized to the charge transfer of the first EPSC in the train.
For synaptic plasticity to have functional consequences, it must ultimately influence firing in postsynaptic neurons. The role of firing is exceptionally well characterized in the vestibular system, where head motion modulates the firing rate of MVN neurons. Applying stimulus trains to the vestibular nerve produced increases in MVN firing rates that were proportional to the stimulation rate (Figure 4A, B). We quantified the firing response as synaptic gain: the slope of the relationship between postsynaptic firing rate and presynaptic stimulus rate. Firing responses varied considerably across neurons; baseline synaptic gains ranged from 0.11 to 0.87, corresponding to firing rates ranging from 17 to 102 Hz in response to 100 Hz stimulation.
Figure 4.

LTD and LTP control the gain of synaptically-evoked postsynaptic firing. (A) Representative responses of a YFP-16 neuron to 1-sec stimulation of the vestibular nerve at 25, 50, and 100 Hz. (B) Example synaptic gain recorded in a YFP-16 neuron before (grey) and after (black) LTD induction, and (C) before (grey) and after (black) LTP induction. Data points in B and C are the average of 5 trials. Error bars are smaller than the symbols. (D) Summary of normalized firing response gains of synaptic transmission following LTD and LTP induction. (E) Summary of goodness of linear fits following LTD and LTP induction.
To assess the effects of plasticity on firing, synaptic gain was measured in response to 25–100 Hz trains before and after induction of synaptic plasticity, while DC hyperpolarizing or depolarizing current was injected to maintain the baseline firing rate at ~ 7 Hz to facilitate comparisons across cells. Following the induction of LTD, firing responses to vestibular nerve stimulation decreased (Figure 4B). Conversely, the induction of LTP resulted in increases in synaptic gain (Figure 4C). Across the population, LTD decreased the synaptic gain to 0.83 ± 0.06 of the baseline gain (n=10, p=0.02), and LTP increased the synaptic gain to 1.49 ± 0.16 (n=10, p=0.004)(Figure 4D). The linear relationship between postsynaptic firing and stimulation rate was preserved following plasticity induction (Figure 4E). The baseline firing rate, from which increases in firing rate were evoked, was constant during LTD (Pre: 6.7 ± 0.9 Hz, Post: 6.7± 0.9 Hz) and LTP experiments (Pre: 7.2 ± 0.9 Hz, Post: 7.6 ± 0.9 Hz), and neither protocol altered the intrinsic excitability of postsynaptic neurons (Supplemental Table 1), suggesting that changes in postsynaptic firing responses were driven completely by changes in synaptic strength. In control experiments, in which 30 repetitions of 250-msec hyperpolarizing steps were applied in the absence of synaptic stimulation, synaptic gain was unchanged (1.00 ± 0.02 of the baseline, n=5, p=0.42, data not shown).
The magnitude of the changes in firing rates produced by vestibular nerve synaptic plasticity varied across stimulation rates and neurons (Figure 4D). The gain changes evoked by LTP corresponded to an average firing rate increase of 13.2 ± 3.3 Hz in response to 100 Hz stimulation, but an increase of 0.8 ± 1.6 Hz in response to 25 Hz stimulation. Similarly, gain changes following LTD decreased the average firing response by 7.2 ± 3.3 Hz during 100 Hz stimulation and by 1.8 ± 1.5 Hz during 25 Hz stimulation. Synaptic gain was unaffected in 2/10 neurons following LTP induction and in 4/10 neurons following LTD induction, despite accompanying changes in EPSC amplitude. The absolute changes in firing responses at any stimulus rate were not correlated with initial firing rate or baseline synaptic gain. These results indicate that although the relationship between synaptic strength and postsynaptic firing responses is influenced by several factors, plasticity at the vestibular nerve synapse functions as a bidirectional, linear gain control mechanism that regulates the strength of signal throughput.
Vestibular nerve synapses contain NMDA and calcium-permeable AMPA receptors
The induction of synaptic plasticity in many systems requires Ca2+ influx through postsynaptic receptors. Although several studies have indicated that vestibular nerve synapses are glutamatergic and contain both NMDA and AMPA receptors (Babalian et al., 1997; Doi et al., 1990; Kinney et al., 1994; Lewis et al., 1989; Straka et al., 1996; Takahashi et al., 1994), relatively little is known about the Ca2+ permeability or voltage dependence of the postsynaptic receptors. We measured current-voltage (IV) relationships of the AMPA and NMDA receptors in YFP-16 neurons in the presence of specific pharmacological antagonists to identify receptor subtypes (Figure 5A). The AMPA-R IV relationship strongly rectified (Rectification Index, +45 mV/−45 mV: 0.44 ± 0.06; range: 0.18–0.79, n=11) (Figure 5B, D), suggesting the presence of GluA2-lacking, Ca2+-permeable AMPA-Rs (CP-AMPA-Rs) (Bochet et al., 1994; McBain and Dingledine, 1993). The selective blocker of GluA2-lacking AMPA-Rs, Philanthotoxin-433 (PhTx-433, 10μM) reduced EPSC amplitude by 55.9 ± 10.1% (range: 7.4–86.9%, n=9, data not shown), indicating that vestibular nerve synapses onto projection neurons contain a composite of GluA2-lacking and GluA2-containing AMPA-Rs.
Figure 5.

LTD and LTP require postsynaptic calcium and distinct classes of ionotropic glutamate receptors. (A) Evoked vestibular nerve EPSCs measured at membrane potentials from −65 to +45 mV in the presence of 10 μM NBQX (top) or 100 μM D-APV (bottom). (B) Mean current-voltage relation of AMPA-R-mediated EPSCs in YFP-16 neurons, normalized to the amplitude at −65 mV (n=11). (C) Mean current-voltage relation of NMDA-R-mediated EPSCs in YFP-16 neurons, normalized to the amplitude at +45 mV (n=9). (D) Summary of rectification indices (+45/−45 mV) measured in individual YFP-16 and GIN neurons. (E) Summary of NMDA/AMPA ratio (+45/−65 mV) measured in YFP-16 and GIN neurons. (F) Inclusion of 5 mM BAPTA in the recording pipette abolished LTD and (I) LTP. (G) Bath application of 100 μM D-APV blocked LTD but (J) not LTP. (H) Bath application of 10 μM Philanthotoxin-433 did not affect LTD but (K) abolished LTP and unmasked an underlying LTD.
The NMDA-R contribution to vestibular nerve synaptic transmission was small relative to that of AMPA-Rs (NMDA +45 mV/AMPA −65 mV: 0.48 ± 0.10; range: 0.18–0.89, n=9) (Figure 5E). The NMDA-R current duration, 104 ± 18 msec, and voltage dependence (Figure 5A, C) suggested that NMDA-Rs predominantly contain the NR2A subunit (Cull-Candy and Leszkiewicz, 2004). These data show that two complementary Ca2+-permeable glutamate receptors support vestibular nerve transmission: CP-AMPA-Rs, which pass Ca2+ maximally at relatively hyperpolarized postsynaptic membrane potentials, and NMDA-Rs, which pass Ca2+ maximally at relatively depolarized potentials.
LTD and LTP depend differentially on postsynaptic glutamate receptors
Given the presence of two distinct Ca2+-permeable glutamate receptors at this synapse, we tested whether postsynaptic Ca2+ elevations were required for either type of long-term synaptic plasticity. Indeed, both LTD and LTP were blocked by the inclusion of the Ca2+ chelator BAPTA (5 mM) in the recording pipette (100 Hz stim protocol: 1.00 ± 0.03, n=7, p=0.03; 100 Hz stim plus hyperpolarization protocol: 0.98 ± 0.03, n=6, p=0.02, Figure 5F, I).
Does Ca2+ influx through NMDA-Rs or CP-AMPA-Rs contribute to the induction of long-term plasticity at the vestibular nerve synapse? We first examined the role of NMDA-Rs. Blockade of NMDA-Rs via bath application of D-APV (100 μM) or intracellular inclusion of MK-801 (1 mM) abolished 100 Hz stim-induced LTD (1.01 ± 0.03, n=6, p=0.02, Figure 5G), without blocking LTP (1.41 ± 0.13, n=9, p=0.19, Figure 5J). Thus, postsynaptic NMDA-Rs are required for LTD, but not LTP, at the vestibular nerve synapse.
To test the role of CP-AMPA-Rs, we bath applied the specific blocker PhTx-433 (10 μM). In the presence of PhTx-433, LTD induced by 100 Hz stimulation was unaltered (0.79 ± 0.07, n=6, p=0.59, Figure 5H). Interestingly, the 100 Hz stim plus hyperpolarization protocol resulted in synaptic depression (0.81 ± 0.10, n=5, p=0.008, Figure 5K) in the presence of Phtx-433, rather than potentiation, indicating that CP-AMPA-Rs are required for LTP induction. The unmasked LTD indicates that the 100 Hz stim plus hyperpolarization protocol simultaneously recruits LTP and LTD mechanisms, with the balance in favor of LTP under normal conditions. Consistent with this interpretation, applying the 100 Hz plus hyperpolarization protocol in the presence of D-APV resulted in enhanced LTP (Figure 5J), while applying same protocol in the presence of reduced extracellular Mg2+ (0.3 mM) to augment Ca2+ influx via NMDA-Rs suppressed the induction of LTP (0.95 ± 0.4, n=5, Figure S1). Thus, LTD and LTP at the vestibular nerve synapse operate in parallel and require distinct Ca2+-permeable glutamate receptors.
To determine whether CP-AMPA-Rs are additionally expressed presynapically, 10 μM PhTx-433 was washed onto slices and the paired-pulse ratio (PPR) at the 20-msec interval was measured. The presence of PhTx-433 did not alter the PPR (Baseline: 93.1 ± 4.2%, PhTx-433: 96.9 ± 3.9%, p=0.20, data not shown), suggesting that CP-AMPA-Rs predominantly exerted their effects on LTP postsynaptically. We conclude that LTP required postsynaptic Ca2+ passed by CP-AMPA-Rs and that LTD requires postsynaptic Ca2+ passed by NMDA-Rs. This complementary dependence on postsynaptic ionotropic glutamate receptors enables membrane voltage to gate the direction of synaptic plasticity.
Postinhibitory rebound depolarization contributes to LTP induction
What are the essential postsynaptic requirements for the induction of LTP? Postsynaptic hyperpolarization produces several distinct effects: it prevents firing, modulates the amplitude of voltage-dependent currents, and recruits low voltage-activated currents. Many neurons in the MVN exhibit rebound firing, a transient elevation in firing rate following relief from hyperpolarization, and the magnitude of rebound firing varies across individual neurons (du Lac and Lisberger, 1995; Sekirnjak and du Lac, 2002; Serafin et al., 1991). During the 100 Hz stim plus hyperpolarization plasticity protocol, 15 of 21 neurons (Figure 2) exhibited rebound firing (42 ± 8 Hz), quantified as the average firing rate increase over the spontaneous rate during the initial 300 msec post-hyperpolarization. LTP was induced in 11 of these 15 neurons (EPSC post-protocol: 1.45 ± 0.16, n=15, p=0.002 vs. control). In contrast, LTP was not induced in any of the remaining 6 neurons that did not exhibit rebound firing post-hyperpolarization (EPSC post-protocol: 0.95 ± 0.03, n=6, p=0.57 vs. control; Rebound vs. Non-rebound: p=0.004, Fisher Exact Test).
To assess whether rebound currents merely distinguished two subtypes of YFP-16 neurons or, additionally, played a causal role in LTP induction, we manipulated the timing of rebound relative to synaptic stimulation in the 100 Hz stim plus hyperpolarization. We first extended the duration of the hyperpolarization step to 550 msec, such that the rebound occurred immediately after, rather than during, the 550-msec stimulus train. This protocol induced LTP of 1.27 ± 0.12 (Figure 6A, n=7, p=0.05), comparable to LTP induced by hyperpolarization lasting 250 msec (p=0.81). We then extended the hyperpolarization step duration to 1550 msec so that rebound followed synaptic stimulation by 1000 msec. This protocol, which temporally dissociated synaptic input and intrinsic rebound, did not induce LTP (Figure 6B, 0.96 ± 0.09 of baseline, n=7, p=0.61 vs. control). These results suggest that intrinsic rebound is a critical component of LTP induction that must to be temporally linked to synaptic stimulation.
Figure 6.

LTP of synapses onto projection neurons depends on the relative timing of synaptic stimulation and hyperpolarization offset. (A) LTP is induced by pairing 550-msec, 100 Hz synaptic stimulation with coincident 550-msec hyperpolarization. (B) Extending the hyperpolarization step to 1550 msec, so that the rebound follows synaptic stimulation by 1 sec, prevented the induction of LTP.
To test whether this rebound requirement was mediated by enhanced firing following synaptic stimulation, we applied the 100 Hz stim plus hyperpolarization protocol to neurons lacking intrinsic rebound firing and injected a 300 msec suprathreshold depolarizing current following hyperpolarization (Figure S2). The addition of this mimicked rebound was inadequate to induce LTP in non-rebounding neurons (0.88 ± 0.07 of baseline, n=7, p=0.005 vs. 100 Hz stim plus hyperpolarization, Figure S2). Furthermore, the magnitude of EPSC potentiation did not correlate with the rate of rebound firing in YFP-16 neurons displaying intrinsic rebound (R2=0.08). Thus, while rebound firing distinguishes two populations of YFP-16 neurons and is predictive of LTP induction, hyperpolarization offset and rebound currents contribute to LTP via mechanisms beyond simple increases in postsynaptic firing rate.
Vestibular nerve synapses onto GABAergic interneurons are plastic
Thus far we have focused on projection neurons. Are vestibular nerve synapses onto local inhibitory neurons (Figure 1D) also plastic? To investigate this potential additional site of plasticity for head movement-driven behaviors, we applied the stimulus protocols that induced LTD and LTP in YFP-16 neurons to GIN neurons. The 100 Hz stim protocol evoked LTD in GIN neurons (0.74 ± 0.05 of baseline, n=6, p=0.001, Figure 7A). The induction of LTD in interneurons was blocked by intracellular, postsynaptic BAPTA (5 mM) (0.99 ± 0.08, n=6, p=0.02), as well as by the NMDA-R antagonist D-APV (100 μM) (1.01 ± 0.07, n=6, p=0.02, Figures 8A, B). These data demonstrate that LTD in vestibular nucleus interneurons requires Ca2+ influx through NMDA-Rs, as is the case for projection neurons (Figure 5G).
Figure 7.

Vestibular nerve synapses onto MVN interneurons exhibit LTD. (A) Representative response evoked by protocol consisting of 100 Hz synaptic stimulation for 550 msec (left). Mean EPSC amplitude before and after protocol (right). (B) Representative response evoked by protocol consisting of 100 Hz synaptic stimulation for 550 msec paired with 250 msec hyperpolarization (left). Mean EPSC amplitude before and after protocol (right).
Figure 8.

Plasticity in interneurons is dominated by NMDA-R-mediated LTD. (A) Inclusion of 5 mM BAPTA in the recording pipette abolished LTD induced by the 100 Hz stim protocol, as did (B) bath application of 100 μM D-APV. (C) Bath application of 100 μM D-APV during the 100 Hz stim plus hyperpolarization protocol unmasked an underlying LTP. (D) 10 μM Philanthotoxin-433 blocked the unmasked LTP.
In striking contrast, the stimulus protocol that induced LTP in projection neurons did not result in potentiation of synaptic strength onto interneurons but instead induced a weak LTD (0.90 ± 0.05 of baseline, n=9, p=0.08, Figure 7B). Interestingly, blocking NMDA-Rs with bath application of D-APV during the 100 Hz stim plus hyperpolarization protocol unmasked an underlying synaptic potentiation (1.25 ± 0.11 of baseline, n=7, p=0.04, Figure 8C). This D-APV-unmasked LTP in interneurons was sensitive to PhTx-433 (0.95 ± 0.07, n=8, p=0.04, Figure 8D). These results indicate that, as in projection neurons, CP-AMPA-R activation paired with postsynaptic hyperpolarization can induce LTP in interneurons, but LTP is typically masked by NMDA-R-mediated LTD.
Several factors might underlie LTD dominance in interneurons, including differential expression of glutamate receptors or post-hyperpolarization rebound currents. Indeed, the AMPA-R IV relationship in GIN neurons was less rectifying than that measured in YFP-16 neurons (Rectification Index GIN: 0.69 ± 0.06, n=13, p=0.01, Figure 5D), while the NMDA/AMPA ratio in GIN neurons was not significantly different from YFP-16 neurons (GIN: 0.47 ± 0.09, n=13, p=0.67, Figure 5E). The inclusion of a relatively smaller proportion of CP-AMPA-Rs at synapses onto GIN neurons may limit the recruitment of LTP. All GIN neurons exhibited post-hyperpolarization rebound firing, although its magnitude was less than half that of the YFP-16 neurons that rebounded (GIN: 20 ± 3 Hz, YFP-16: 42 ± 8 Hz, p=0.02). Thus, CP-AMPA-R and rebound currents are differentially expressed according to circuit function in the MVN, such that both are minimized in local inhibitory GIN neurons.
Discussion
This study demonstrates that the first central synapse in the vestibular system is bidirectionally plastic. Vestibular nerve synaptic plasticity is governed by non-Hebbian induction rules and produces linear changes in postsynaptic neuronal firing responses to vestibular nerve activity across a wide range of presynaptic rates. Vestibular nerve LTD and LTP depend on Ca2+ influx through different postsynaptic ionotropic glutamate receptors: LTD requires activation of NMDA-Rs, while LTP requires activation of CP-AMPA-Rs. Although LTD can be induced by nerve afferent activity alone, the induction of LTP requires a specific temporal conjunction of vestibular nerve synaptic activity and post-synaptic hyperpolarization. Synapses onto both MVN projection neurons and local interneurons are plastic, and differences in synaptic and intrinsic conductances across cell types shape the balance of LTD and LTP. Given the powerful influence of the vestibular nerve on MVN neuronal firing, plasticity at this synapse is optimally suited to regulate the gain of vestibular behaviors while preserving the speed and computation of the underlying circuits.
Linear scaling of postsynaptic currents and firing rates via synaptic plasticity
Synaptic efficacy has typically been probed by measuring postsynaptic, subthreshold responses to stimuli consisting of either single shocks or pairs of shocks. Given that many synapses exhibit short-term plasticity (changes in efficacy as a function of recent activity), it is not possible to infer synaptic responses to multiple stimuli from responses to individual stimuli. For example, although LTP uniformly potentiates synaptic currents elicited by brief presynaptic bursts in hippocampal neurons (Pananceau et al., 1998; Selig et al., 1999), LTP expression depends strongly on stimulus rate and history in cortical neurons (Markram and Tsodyks, 1996). Our results demonstrate that at central vestibular nerve synapses, the induction of either LTD or LTP results in a uniform scaling of synaptic efficacy, such that steady-state synaptic transmission remains rate-invariant over a wide range of stimulus frequencies (Figure 3). What mechanisms could account for these proportionate changes in synaptic efficacy? Given that changes in the probability of transmitter release are typically accompanied by changes in short-term synaptic dynamics (Zucker and Regehr, 2002) and that postsynaptic properties do not contribute to short-term dynamics at the vestibular nerve synapse (Bagnall et al., 2008), our findings are most consistent with a postsynaptic locus of expression of vestibular nerve synaptic plasticity.
The effects of bidirectional synaptic plasticity on postsynaptic firing have not been previously reported. Several studies report how changes in synaptic strength translate into changes in postsynaptic firing (Bliss and Lomo, 1973; Fetz and Gustafsson, 1983; Mittmann and Hausser, 2007; Reyes and Fetz, 1993; Smith and Otis, 2005; Walter and Khodakhah, 2006), but none have probed responses over the entire range of behaviorally relevant stimulus rates. The transformation of synaptic inputs into action potentials is influenced by both active and passive dendritic processes as well as by somatic filtering, complicating the relationship between synaptic strength and resultant postsynaptic firing. Vestibular nucleus neurons differ markedly from the much more extensively studied pyramidal cells in the hippocampus and cortex, both in the rate-invariance of primary excitatory synaptic transmission (Bagnall et al., 2008) and in the wide range of firing rates over which spike generation remains linear (Sekirnjak and du Lac, 2002; Serafin et al., 1991). Whether synaptic plasticity produces scaling of postsynaptic firing responses in other neurons that exhibit linear spike generation—such as fast-firing hippocampal, cortical, and basal ganglia interneurons, as well as other brainstem and cerebellar neurons—remains to be examined.
Activity requirements and mechanisms of vestibular nerve synaptic plasticity
Distinct mechanisms and postsynaptic Ca2+ sources underlie LTD and LTP at the vestibular nerve synapse. LTD is induced by high-frequency synaptic activation paired with postsynaptic depolarization. The ability of BAPTA and D-APV to prevent LTD (Figure 5) implies that LTD is triggered by Ca2+ influx through postsynaptic NMDA-Rs, whose Mg2+ sensitivity enables Ca2+ influx to reflect postsynaptic voltage. The NMDA-R component at the vestibular nerve synapse is relatively small (Figure 5E) and contributes minimally to synaptically-evoked firing in postsynaptic neurons (Bagnall et al., 2008), suggesting a predominant function of NMDA-Rs at this synapse may thus be to regulate synaptic depression.
It is remarkable that the direction of plasticity induced by high frequency synaptic stimulation is reversed by postsynaptic membrane hyperpolarization (Figure 2). The effects of BAPTA, PhTx-433, and delayed rebound indicate that LTP is triggered by a combination of Ca2+ influx through postsynaptic AMPA-Rs and signals which accompany the offset of hyperpolarization. The dependence of LTP and LTD on distinct glutamate receptors with complementary voltage dependences (Figure 5B,C) enables postsynaptic membrane potential to bidirectionally bias the induction of plasticity.
Our findings indicate that the relative timing of synaptic activity and postsynaptic hyperpolarization is critical for the induction of LTP (Figure 6B). The correlation between rebound firing and LTP suggests that underlying rebound currents may play an important role in LTP induction. Hyperpolarization affects several sources of Ca2+ that could contribute to both rebound and LTP induction, including low-voltage activated T-type Ca2+ channels (Aizenman and Linden, 1999; Alvina et al., 2009; Molineux et al., 2006; Sekirnjak and du Lac, 2002; Zheng and Raman, 2009). Our results are consistent with a model in which Ca2+ influx via AMPA-Rs primes synapses and then subsequent Ca2+ influx during rebound triggers LTP.
Alternatively, hyperpolarization offset could influence LTP by restoring, rather than elevating, Ca2+ levels. In addition to recruiting low-voltage activated Ca2+ channels, hyperpolarization also deactivates high-voltage activated Ca2+ channels that are tonically active in spontaneously firing neurons (Nelson et al., 2003; Zheng and Raman, 2009). A recent study of the analogous mossy fiber to cerebellar nucleus synapse demonstrated that LTP induction there requires transient decreases in the baseline Ca2+ levels of postsynaptic neurons (Person and Raman, 2010). Further experiments are required to define both the critical currents modulated by hyperpolarization and the Ca2+ dynamics required for LTP induction at the vestibular nerve synapse.
Synaptic plasticity in brainstem and cerebellar circuits
Several forms of plasticity in brainstem and cerebellar circuits depend on inhibition (Pugh and Raman, 2009), including intrinsic excitability in the MVN (Nelson et al., 2003) and synaptic plasticity in the cerebellar nuclei (CN) (Pugh and Raman, 2008). Plasticity mechanisms in the MVN and CN are thought to be coordinated by the cerebellar cortex, which directly inhibits neurons in both nuclei. Interestingly, although the stimulus protocols that induced bidirectional plasticity in this study are quite similar to those that induce plasticity at the analogous mossy-fiber synapse in the CN, the underlying mechanisms differ. In CN neurons, high-frequency presynaptic activity paired with postsynaptic depolarization induces mGluR1-dependent LTD, whereas presynaptic activity paired with postsynaptic hyperpolarization induces NMDA-R-dependent LTP (Pugh and Raman, 2006, 2008; Zhang and Linden, 2006). While the intrinsic firing properties of neurons in the cerebellar and vestibular nuclei are largely equivalent (Bagnall et al., 2007; Bagnall et al., 2009; Sekirnjak and du Lac, 2002; Uusisaari et al., 2007), the two nuclei are functionally distinct: vestibular nucleus neurons can influence motoneurons directly via projections to ocular motor and spinal nuclei, whereas CN neurons modulate motor and cortical function via multiple intervening synapses. Furthermore, vestibular nerve axons convey primary sensory signals, while most cerebellar mossy fibers convey signals that have already been processed extensively by central neurons. The finding that hyperpolarization gates potentiation of excitatory synapses similarly in the two different nuclei, albeit via distinct mechanisms, implies that such plasticity rules may be a general strategy used by spontaneously firing neurons in cerebellar and brainstem circuits.
The activity patterns and Ca2+ signals required to induce synaptic plasticity are thought to be inverted in the cerebellum relative to the hippocampus and cerebral cortex (Jorntell and Hansel, 2006), reflecting differing circuit functions and baseline firing rates. While vestibular nerve synaptic plasticity is similar to many forms of plasticity in cerebellar circuits in its non-Hebbian induction requirements, it differs in its Ca2+ dependence. Low intracellular BAPTA concentrations (0.1mM) had no effect on vestibular nerve LTD (0.80 ± 0.13, n=4) but interfered with LTP induction (0.98 ± 0.01, n=4, data not shown), suggesting that, as in forebrain synapses (Bear et al., 1987; Cummings et al., 1996; Hansel et al., 1997), LTP requires a higher postsynaptic Ca2+ concentration than does LTD. Thus, while the high baseline firing rates in cerebellar and vestibular circuits constrain the activity patterns that induce synaptic plasticity, the underlying mechanisms are tuned specifically to each synapse.
Cell type specific regulation of synaptic plasticity
Although repeated high frequency stimulation elicits LTD at vestibular nerve synapses onto all MVN neurons, the addition of postsynatpic hyperpolarization induces LTP in YFP-16 neurons but not in GIN neurons (Figs 2, 8). Retrograde labeling experiments revealed that YFP-16 neurons, which are glutamatergic or glycinergic (Bagnall et al., 2007), are a major source of projections to motor, cerebellar, and thalamic targets (Figure 1). In contrast, GIN neurons, which are GABAergic (Bagnall et al., 2007), do not appear to project outside of the vestibular nuclei but instead provide local and commissural inhibition of projection neurons (Figure 1D). The differential recruitment of plasticity onto projection neurons and interneurons, coupled with extensively intermingled cell types within the vestibular nucleus, complicate drawing parallels with the extensive dataset on plasticity of field potentials evoked by synaptic stimulation within the vestibular complex (Grassi et al., 2009; Grassi et al., 2002; Grassi and Pettorossi, 2001; Grassi et al., 1996; Puyal et al., 2003). The cell-type specificity of LTP induction reported here highlights the importance of targeting recordings to identified neuronal populations (McBain, 2008; Tzounopoulos et al., 2004).
The mechanisms of bidirectional plasticity are conserved across YFP-16 and GIN neurons. Blockade of NMDA-Rs (Figure 8C) revealed that synapses onto GIN neurons can be potentiated by pairing vestibular nerve stimulation with hyperpolarization but that concomitant depression typically masks this potentiation. The bias against LTP in interneurons appears to be regulated by both intrinsic and synaptic mechanisms: rebound currents and CP-AMPA-Rs at the vestibular nerve synapse are both minimized in GIN interneurons relative to YFP-16 projection neurons. Thus, the balance of LTD and LTP at vestibular nerve synapses appears to be influenced by two distinct mechanisms that are both regulated according to cell type.
Functional Implications
The vestibular system, which is plastic throughout life, is critical for a wide range of functions, including stability during movement, spatial orientation, and autonomic regulation (Angelaki and Cullen, 2008). Our results demonstrate that vestibular signaling is plastic at the first stages of central transmission. The firing of MVN neurons influences several modifiable reflexes via projections onto ocular and spinal motor neurons, most notably the vestibulo-ocular reflex (VOR), which has long served as a model for cerebellar-dependent learning (Ito, 1984; Miles and Lisberger, 1981). Our results support the Miles-Lisberger hypothesis of learning in the VOR (Miles and Lisberger, 1981), which proposes that MVN firing responses to head movements could be regulated by plasticity of the primary sensory input onto a subset of vestibular nucleus neurons.
Motor learning in the VOR requires the cerebellum, but results of cerebellar inactivation experiments indicate that the expression of learned changes resides downstream of the cerebellar Purkinje cells, in the MVN (Kassardjian et al., 2005; Shutoh et al., 2006). Recordings from vestibular nucleus neurons, together with behavioral analyses of motor learning, indicate that parallel modifiable and unmodifiable pathways mediate performance and adaptation of the VOR (Lisberger, 1984; Lisberger and Pavelko, 1986). The modifiable pathway includes neurons that are powerfully inhibited by cerebellar Purkinje cells and project to ocular motoneurons, and the faster, unmodifiable pathway is thought to include neurons that do not receive cerebellar inhibition but that also project to ocular motoneurons (Lisberger and Pavelko, 1988; Ramachandran and Lisberger, 2008; Scudder and Fuchs, 1992). Increases or decrease in the gain of the VOR are associated with bidirectional changes in the firing responses to head movements of MVN neurons receiving cerebellar inhibition. Our findings that bidirectional plasticity of vestibular nerve synapses can only be evoked in a distinct subset of projection neurons is consistent with the notion of modifiable and unmodifiable VOR pathways.
That the LTD and LTP demonstrated here cause bidirectional, linear changes in postsynaptic firing responses to vestibular nerve stimulation make them plausible candidate mechanisms for contributing to VOR learning. An attractive hypothesis is that the YFP-16 neurons which exhibit bidirectional plasticity comprise the modifiable pathway, and that the relative timing of Purkinje cell inhibition and vestibular nerve activity modulates the gain of the VOR via bidirectional vestibular nerve synaptic plasticity. Given direct projections to ocular motoneurons, LTP or LTD of the vestibular nerve synapse onto YFP-16 neurons would result in increases or decreases in VOR gain, respectively. Relating cellular mechanisms in vitro with behavioral function in vivo, however, requires extrapolation across different physiological conditions. The range of evoked firing rates in our study was lower than that observed in intact animals (Beraneck and Cullen, 2007; Lasker et al., 2008), reflecting limitations in the number of vestibular afferents that can be stimulated in a viable brain slice and lower in vitro baseline firing rates. Given the remarkably wide range of linear spike generation in vestibular nucleus neurons that project to ocular motor neurons, however, it is likely that the linear scaling of synaptic currents observed in this study apply to the wider range of synaptic drive experienced by neurons in vivo.
Several types and loci of plasticity are likely to contribute to motor learning in the vestibular system. The robust depression onto interneurons described here is a regulatory site that could be utilized independently or in conjunction with plasticity at other sites to influence vestibular processing. Local inhibition in the MVN includes feedforward (Straka et al., 1997) and commissural connections (Bagnall et al., 2007; Malinvaud et al., 2010), whose specific roles in signal processing and circuit throughput are only beginning to be dissected (Biesdorf et al., 2008). Plasticity of the primary excitatory drive onto interneurons could regulate vestibular behaviors by modulating tonic inhibition of ipsi- or contralateral projection neurons. Behavioral evidence suggests that the learning of VOR gain increases and decreases may not be mediated by mechanisms that are direct opposites of one another (Boyden and Raymond, 2003; Miles and Eighmy, 1980), and the multiple forms and sites of synaptic plasticity described here provide the circuit with several potential regulatory mechanisms. The straightforward relationship between synaptic efficacy and postsynaptic firing responses, coupled with extensive knowledge of the functional significance of firing in identified cell types (Angelaki and Cullen, 2008; du Lac et al., 1995), makes linking synaptic plasticity with circuit and, ultimately, behavioral consequences tractable in the vestibular system.
Experimental Procedures
Tracer Injections
The oculomotor nucleus, thalamus, cerebellum, and medullary reticular formation were stereotaxically targeted for microinjection of Texas Red dextran crystals (3,000 molecular weight; Invitrogen) in mice aged 45–65 days postnatal. Animals were deeply anesthetized with isoflurane and then placed on a stereotaxic apparatus (Benchmark Angle Two; http://MyNeuroLab.com). A custom-made injector needle (0.2 mm outer diameter, 0.1 mm inner diameter; Creative Instruments Development Company) was loaded with dextran crystals, and the tip was sealed with melted bone wax. The needle was lowered into the targeted brain region, and the interior plunger was repeatedly depressed (~100 μm) with calibrated air pressure (30 psi, 30 msec) to deliver the crystals into the tissue. After waiting 1–2 min, the needle was withdrawn and the skin was sutured. 4–6 days post injection, mice were anesthetized with Nembutal and perfused transcardially with phosphate buffered saline (PBS), followed by 4% paraformaldehyde in PBS (PFA) for 5 min. After removal of the brain from the skull, the tissue was postfixed for 30–60 min in PFA and then stored overnight at 4°C in 30% sucrose in PBS. 25 μm coronal sections were cut on a freezing microtome (Microm) and washed in PBS. Sections were then wet-mounted and coverslipped with 2.5% DABCO (1,4-diazabicyclo-[2.2.2]octane).
Anatomical analyses
Confocal images were acquired in 1.0 μm steps on a Olympus BX60 microscope using laser lines of 488 and 561 nm, with a 100x objective and 2.5x digital zoom. Fluoview software was used to z-stack images. Images were transferred to Adobe Photoshop (Adobe Systems) for whole-image brightness/contrast adjustment and image overlay. GFP/YFP expression and the presence of Texas Red in retrogradely labelled neurons were detected with fluorescein isothiocyanate and rhodamine filters, respectively. The presence of double-labeled neurons was assessed in 12–20 sections containing the rostral half of the MVN from each of 2–3 YFP-16 or GIN mice. The presence of GIN+ terminals opposed to somata or proximal dendrites was assessed in at least 5 neurons retrogradely labeled from each target.
Physiology
Oblique coronal brainstem slices were prepared from YFP-16 (Feng et al., 2000) and GIN mice (Oliva et al., 2000), backcrossed 10 generations onto a C57BL/6 background. Mice aged 18–24 days postnatal were deeply anesthetized with Nembutal and then decapitated. The brain was dissected in ice cold Ringer’s solution (in mM: 124 NaCl, 5 KCl, 1.3 MgSO4, 26 NaHCO3, 2.5 CaCl2, 1 NaH2PO4, and 11 dextrose). 300-μm slices were cut on a DSK DTK-1500E or Leica VT1000S vibratome and allowed to recover in 34 oC Ringer’s for 30 min. Slices rested at room temperature before being transferred to a recording chamber and perfused with carbogenated Ringer’s containing 1–10 μM strychnine and 100 μM picrotoxin at 34 °C. All experiments were carried out in accordance with the standards of the Salk Institute IACUC.
Patch pipettes were pulled from flame-polished glass (Warner) with resistances of 2–4 MΩ. Pipette internal solution contained (in mM): 140 K gluconate, 20 HEPES, 8 NaCl, 0.1 EGTA, 0.1 Spermine, 2 Mg-ATP, 0.3 Na2-GTP. Where indicated in the text, 1,2-bis(2-animophenoxy)ethane-N,N,N′,N′-tetraacitic acid (BAPTA, 5 mM) replaced EGTA in the internal solution. For recordings of synaptic current-voltage relationships, the internal solution contained (in mM): 120 CsMeSO4, 10 HEPES, 8 NaCl, 10 QX-314, 5 TEA-Cl, 5 BAPTA, 0.1 Spermine, 2 Mg-ATP, 0.3 Na2-GTP. Membrane potential is corrected for measured junction potentials.
Neurons were visualized with epifluorescence through a GFP filter, as well as under infrared differential interference contrast illumination with Nomarski optics. The vestibular nerve tract was identifiable under 10X magnification and was confirmed by anterograde transport of fluorescently labeled dextran conjugates injected into the vestibular ganglion. Synaptic transmission at the nerve synapse was evoked by a bipolar concentric stimulating electrode (FHC, Maine) placed on the vestibular nerve lateral to the vestibular complex (approximate location: 6–6.4 mm caudal to bregma, 4.4 mm ventral to the horizontal plane passing through bregma and lambda, and 1.7–1.8 mm lateral to the midline) and controlled via two Isoflex stimulus isolation units (AMPI, Israel). A biphasic pulse, consisting of two 100 μsec pulses of opposite polarity with a 100 μsec interval, was delivered to the electrode to avoid charge buildup.
Data were acquired with a Multiclamp 700B low-pass filtering at 6–10 kHz for voltage clamp and 10 kHz for current clamp. Data were digitized at 40 kHz with an ITC-16 or 18 (InstruTECH). House-written code in Igor 6 was used for acquisition and analysis. Series resistance was monitored continuously with small hyperpolarizing square pulses, and experiments showing a series resistance above 16 MΩ or a change greater than 20% were excluded.
Neurons were held in current clamp between 200 msec voltage clamp epochs, when synaptic currents were measured. EPSCs were measured at −75 mV every 15 sec before and after induction protocols. EPSCs were normalized to the pre-protocol baseline, which lasted 10–12 min. EPSC changes reported in the text are the averages of minutes 10–20 post-protocol. Firing rates are reported as the average of the reciprocal of the inter-spike interval. For LTP mechanism experiments, only neurons with intrinsic rebound were included for analysis and statistics.
Data are reported as mean ± SEM, and statistical significance was evaluated with paired (where possible) or unpaired Wilcoxon signed-rank tests in KaleidaGraph 3.6 (Synergy Software). Chemicals were purchased from Sigma (St. Louis MO), with the exception of D-APV (Tocris, Bristol, UK) and BAPTA tetrapotassium salt (Invitrogen).
Supplementary Material
Acknowledgments
This work was supported by the Howard Hughes Medical Institute, National Institutes of Health grant EY-11027, an NSF graduate fellowship (M.W.B), and The Aginsky Research Scholar Award (L.E.M). The authors thank Kristine Kolkman for the data shown in Figure 1D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Alvina K, Ellis-Davies G, Khodakhah K. T-type calcium channels mediate rebound firing in intact deep cerebellar neurons. Neuroscience. 2009;158:635–641. doi: 10.1016/j.neuroscience.2008.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelaki DE, Cullen KE. Vestibular system: the many facets of a multimodal sense. Annu Rev Neurosci. 2008;31:125–150. doi: 10.1146/annurev.neuro.31.060407.125555. [DOI] [PubMed] [Google Scholar]
- Babalian A, Vibert N, Assie G, Serafin M, Muhlethaler M, Vidal PP. Central vestibular networks in the guinea-pig: functional characterization in the isolated whole brain in vitro. Neuroscience. 1997;81:405–426. doi: 10.1016/s0306-4522(97)00069-9. [DOI] [PubMed] [Google Scholar]
- Bagnall MW, McElvain LE, Faulstich M, du Lac S. Frequency-independent synaptic transmission supports a linear vestibular behavior. Neuron. 2008;60:343–352. doi: 10.1016/j.neuron.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall MW, Stevens RJ, du Lac S. Transgenic mouse lines subdivide medial vestibular nucleus neurons into discrete, neurochemically distinct populations. J Neurosci. 2007;27:2318–2330. doi: 10.1523/JNEUROSCI.4322-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnall MW, Zingg B, Sakatos A, Moghadam SH, Zeilhofer HU, du Lac S. Glycinergic projection neurons of the cerebellum. J Neurosci. 2009;29:10104–10110. doi: 10.1523/JNEUROSCI.2087-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- Beraneck M, Cullen KE. Activity of vestibular nuclei neurons during vestibular and optokinetic stimulation in the alert mouse. J Neurophysiol. 2007;98:1549–1565. doi: 10.1152/jn.00590.2007. [DOI] [PubMed] [Google Scholar]
- Biesdorf S, Malinvaud D, Reichenberger I, Pfanzelt S, Straka H. Differential inhibitory control of semicircular canal nerve afferent-evoked inputs in second-order vestibular neurons by glycinergic and GABAergic circuits. J Neurophysiol. 2008;99:1758–1769. doi: 10.1152/jn.01207.2007. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochet P, Audinat E, Lambolez B, Crepel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Raymond JL. Active reversal of motor memories reveals rules governing memory encoding. Neuron. 2003;39:1031–1042. doi: 10.1016/s0896-6273(03)00562-2. [DOI] [PubMed] [Google Scholar]
- Buttner U, Straube A, Kurzan R. Oculomotor effects of gamma-aminobutyric acid agonists and antagonists in the vestibular nuclei of the alert monkey. Ann N Y Acad Sci. 1992;656:645–659. doi: 10.1111/j.1749-6632.1992.tb25242.x. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Doi K, Tsumoto T, Matsunaga T. Actions of excitatory amino acid antagonists on synaptic inputs to the rat medial vestibular nucleus: an electrophysiological study in vitro. Exp Brain Res. 1990;82:254–262. doi: 10.1007/BF00231245. [DOI] [PubMed] [Google Scholar]
- du Lac S, Lisberger SG. Membrane and firing properties of avian medial vestibular nucleus neurons in vitro. J Comp Physiol A. 1995;176:641–651. doi: 10.1007/BF01021584. [DOI] [PubMed] [Google Scholar]
- du Lac S, Raymond JL, Sejnowski TJ, Lisberger SG. Learning and memory in the vestibulo-ocular reflex. Annu Rev Neurosci. 1995;18:409–441. doi: 10.1146/annurev.ne.18.030195.002205. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Methods. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fetz EE, Gustafsson B. Relation between shapes of post-synaptic potentials and changes in firing probability of cat motoneurones. J Physiol. 1983;341:387–410. doi: 10.1113/jphysiol.1983.sp014812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, du Lac S. Intrinsic and synaptic plasticity in the vestibular system. Curr Opin Neurobiol. 2006;16:385–390. doi: 10.1016/j.conb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Grassi S, Frondaroli A, Dieni C, Scarduzio M, Pettorossi VE. Long-term potentiation in the rat medial vestibular nuclei depends on locally synthesized 17beta-estradiol. J Neurosci. 2009;29:10779–10783. doi: 10.1523/JNEUROSCI.1697-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi S, Frondaroli A, Pettorossi VE. Different metabotropic glutamate receptors play opposite roles in synaptic plasticity of the rat medial vestibular nuclei. J Physiol. 2002;543:795–806. doi: 10.1113/jphysiol.2002.023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi S, Pettorossi VE. Synaptic plasticity in the medial vestibular nuclei: role of glutamate receptors and retrograde messengers in rat brainstem slices. Prog Neurobiol. 2001;64:527–553. doi: 10.1016/s0301-0082(00)00070-8. [DOI] [PubMed] [Google Scholar]
- Grassi S, Pettorossi VE, Zampolini M. Low-frequency stimulation cancels the high-frequency-induced long-lasting effects in the rat medial vestibular nuclei. J Neurosci. 1996;16:3373–3380. doi: 10.1523/JNEUROSCI.16-10-03373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, Artola A, Singer W. Relation between dendritic Ca2+ levels and the polarity of synaptic long-term modifications in rat visual cortex neurons. Eur J Neurosci. 1997;9:2309–2322. doi: 10.1111/j.1460-9568.1997.tb01648.x. [DOI] [PubMed] [Google Scholar]
- Highstein SM, Holstein GR. The anatomy of the vestibular nuclei. Prog Brain Res. 2006;151:157–203. doi: 10.1016/S0079-6123(05)51006-9. [DOI] [PubMed] [Google Scholar]
- Hirata Y, Highstein SM. Acute adaptation of the vestibuloocular reflex: signal processing by floccular and ventral parafloccular Purkinje cells. J Neurophysiol. 2001;85:2267–2288. doi: 10.1152/jn.2001.85.5.2267. [DOI] [PubMed] [Google Scholar]
- Hullar TE, Minor LB. High-frequency dynamics of regularly discharging canal afferents provide a linear signal for angular vestibuloocular reflexes. J Neurophysiol. 1999;82:2000–2005. doi: 10.1152/jn.1999.82.4.2000. [DOI] [PubMed] [Google Scholar]
- Ito M. The Cerebellum and Neural Control. New York: Raven; 1984. [Google Scholar]
- Ito M, Highstein SM, Fukuda J. Cerebellar inhibition of the vestibulo-ocular reflex in rabbit and cat and its blockage by picrotoxin. Brain Res. 1970;17:524–526. doi: 10.1016/0006-8993(70)90261-1. [DOI] [PubMed] [Google Scholar]
- Jorntell H, Hansel C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 2006;52:227–238. doi: 10.1016/j.neuron.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Kassardjian CD, Tan YF, Chung JY, Heskin R, Peterson MJ, Broussard DM. The site of a motor memory shifts with consolidation. J Neurosci. 2005;25:7979–7985. doi: 10.1523/JNEUROSCI.2215-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato R, Iwamoto Y, Yoshida K. Contribution of GABAergic inhibition to the responses of secondary vestibular neurons to head rotation in the rat. Neurosci Res. 2003;46:499–508. doi: 10.1016/s0168-0102(03)00161-5. [DOI] [PubMed] [Google Scholar]
- Kinney GA, Peterson BW, Slater NT. The synaptic activation of N-methyl-D-aspartate receptors in the rat medial vestibular nucleus. J Neurophysiol. 1994;72:1588–1595. doi: 10.1152/jn.1994.72.4.1588. [DOI] [PubMed] [Google Scholar]
- Lasker DM, Han GC, Park HJ, Minor LB. Rotational responses of vestibular-nerve afferents innervating the semicircular canals in the C57BL/6 mouse. J Assoc Res Otolaryngol. 2008;9:334–348. doi: 10.1007/s10162-008-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MR, Phelan KD, Shinnick-Gallagher P, Gallagher JP. Primary afferent excitatory transmission recorded intracellularly in vitro from rat medial vestibular neurons. Synapse. 1989;3:149–153. doi: 10.1002/syn.890030206. [DOI] [PubMed] [Google Scholar]
- Lisberger SG. The latency of pathways containing the site of motor learning in the monkey vestibulo-ocular reflex. Science. 1984;225:74–76. doi: 10.1126/science.6610214. [DOI] [PubMed] [Google Scholar]
- Lisberger SG, Pavelko TA. Vestibular signals carried by pathways subserving plasticity of the vestibulo-ocular reflex in monkeys. J Neurosci. 1986;6:346–354. doi: 10.1523/JNEUROSCI.06-02-00346.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisberger SG, Pavelko TA. Brain stem neurons in modified pathways for motor learning in the primate vestibulo-ocular reflex. Science. 1988;242:771–773. doi: 10.1126/science.3142040. [DOI] [PubMed] [Google Scholar]
- Malinvaud D, Vassias I, Reichenberger I, Rossert C, Straka H. Functional organization of vestibular commissural connections in frog. J Neurosci. 2010;30:3310–3325. doi: 10.1523/JNEUROSCI.5318-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Tsodyks M. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 1996;382:807–810. doi: 10.1038/382807a0. [DOI] [PubMed] [Google Scholar]
- McBain CJ. Differential mechanisms of transmission and plasticity at mossy fiber synapses. Prog Brain Res. 2008;169:225–240. doi: 10.1016/S0079-6123(07)00013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Dingledine R. Heterogeneity of synaptic glutamate receptors on CA3 stratum radiatum interneurones of rat hippocampus. J Physiol. 1993;462:373–392. doi: 10.1113/jphysiol.1993.sp019560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles FA, Eighmy BB. Long-term adaptive changes in primate vestibuloocular reflex. I. Behavioral observations. J Neurophysiol. 1980;43:1406–1425. doi: 10.1152/jn.1980.43.5.1406. [DOI] [PubMed] [Google Scholar]
- Miles FA, Lisberger SG. Plasticity in the vestibulo-ocular reflex: a new hypothesis. Annu Rev Neurosci. 1981;4:273–299. doi: 10.1146/annurev.ne.04.030181.001421. [DOI] [PubMed] [Google Scholar]
- Mittmann W, Hausser M. Linking synaptic plasticity and spike output at excitatory and inhibitory synapses onto cerebellar Purkinje cells. J Neurosci. 2007;27:5559–5570. doi: 10.1523/JNEUROSCI.5117-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineux ML, McRory JE, McKay BE, Hamid J, Mehaffey WH, Rehak R, Snutch TP, Zamponi GW, Turner RW. Specific T-type calcium channel isoforms are associated with distinct burst phenotypes in deep cerebellar nuclear neurons. Proc Natl Acad Sci U S A. 2006;103:5555–5560. doi: 10.1073/pnas.0601261103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pananceau M, Chen H, Gustafsson B. Short-term facilitation evoked during brief afferent tetani is not altered by long-term potentiation in the guinea-pig hippocampal CA1 region. J Physiol. 1998;508(Pt 2):503–514. doi: 10.1111/j.1469-7793.1998.503bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AL, Raman IM. Deactivation of L-type Ca current by inhibition controls LTP at excitatory synapses in the cerebellar nuclei. Neuron. 2010;66:550–559. doi: 10.1016/j.neuron.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron. 2006;51:113–123. doi: 10.1016/j.neuron.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Mechanisms of potentiation of mossy fiber EPSCs in the cerebellar nuclei by coincident synaptic excitation and inhibition. J Neurosci. 2008;28:10549–10560. doi: 10.1523/JNEUROSCI.2061-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. Nothing can be coincidence: synaptic inhibition and plasticity in the cerebellar nuclei. Trends Neurosci. 2009;32:170–177. doi: 10.1016/j.tins.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyal J, Grassi S, Dieni C, Frondaroli A, Dememes D, Raymond J, Pettorossi VE. Developmental shift from long-term depression to long-term potentiation in the rat medial vestibular nuclei: role of group I metabotropic glutamate receptors. J Physiol. 2003;553:427–443. doi: 10.1113/jphysiol.2003.051995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Lisberger SG. Neural substrate of modified and unmodified pathways for learning in monkey vestibuloocular reflex. J Neurophysiol. 2008;100:1868–1878. doi: 10.1152/jn.90498.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes AD, Fetz EE. Effects of transient depolarizing potentials on the firing rate of cat neocortical neurons. J Neurophysiol. 1993;69:1673–1683. doi: 10.1152/jn.1993.69.5.1673. [DOI] [PubMed] [Google Scholar]
- Scudder CA, Fuchs AF. Physiological and behavioral identification of vestibular nucleus neurons mediating the horizontal vestibuloocular reflex in trained rhesus monkeys. J Neurophysiol. 1992;68:244–264. doi: 10.1152/jn.1992.68.1.244. [DOI] [PubMed] [Google Scholar]
- Sekirnjak C, du Lac S. Intrinsic firing dynamics of vestibular nucleus neurons. J Neurosci. 2002;22:2083–2095. doi: 10.1523/JNEUROSCI.22-06-02083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig DK, Nicoll RA, Malenka RC. Hippocampal long-term potentiation preserves the fidelity of postsynaptic responses to presynaptic bursts. J Neurosci. 1999;19:1236–1246. doi: 10.1523/JNEUROSCI.19-04-01236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafin M, de Waele C, Khateb A, Vidal PP, Muhlethaler M. Medial vestibular nucleus in the guinea-pig. II. Ionic basis of the intrinsic membrane properties in brainstem slices. Exp Brain Res. 1991;84:426–433. doi: 10.1007/BF00231465. [DOI] [PubMed] [Google Scholar]
- Shimazu H, Precht W. Inhibition of central vestibular neurons from the contralateral labyrinth and its mediating pathway. J Neurophysiol. 1966;29:467–492. doi: 10.1152/jn.1966.29.3.467. [DOI] [PubMed] [Google Scholar]
- Shutoh F, Ohki M, Kitazawa H, Itohara S, Nagao S. Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience. 2006;139:767–777. doi: 10.1016/j.neuroscience.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Smith SL, Otis TS. Pattern-dependent, simultaneous plasticity differentially transforms the input-output relationship of a feedforward circuit. Proc Natl Acad Sci U S A. 2005;102:14901–14906. doi: 10.1073/pnas.0505028102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straka H, Biesdorf S, Dieringer N. Canal-specific excitation and inhibition of frog second-order vestibular neurons. J Neurophysiol. 1997;78:1363–1372. doi: 10.1152/jn.1997.78.3.1363. [DOI] [PubMed] [Google Scholar]
- Straka H, Debler K, Dieringer N. Size-related properties of vestibular afferent fibers in the frog: differential synaptic activation of N-methyl-D-aspartate and non-N-methyl-D-aspartate receptors. Neuroscience. 1996;70:697–707. doi: 10.1016/s0306-4522(96)83008-9. [DOI] [PubMed] [Google Scholar]
- Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol. 2005;76:349–392. doi: 10.1016/j.pneurobio.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Tsumoto T, Kubo T. N-methyl-D-aspartate receptors contribute to afferent synaptic transmission in the medial vestibular nucleus of young rats. Brain Res. 1994;659:287–291. doi: 10.1016/0006-8993(94)90895-8. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Uusisaari M, Obata K, Knopfel T. Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol. 2007;97:901–911. doi: 10.1152/jn.00974.2006. [DOI] [PubMed] [Google Scholar]
- Walter JT, Khodakhah K. The linear computational algorithm of cerebellar Purkinje cells. J Neurosci. 2006;26:12861–12872. doi: 10.1523/JNEUROSCI.4507-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. Long-term depression at the mossy fiber-deep cerebellar nucleus synapse. J Neurosci. 2006;26:6935–6944. doi: 10.1523/JNEUROSCI.0784-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Raman IM. Ca currents activated by spontaneous firing and synaptic disinhibition in neurons of the cerebellar nuclei. J Neurosci. 2009;29:9826–9838. doi: 10.1523/JNEUROSCI.2069-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.