

Abstract
Cortical spreading depression (CSD) has been suggested to underlie migraine visual aura. However, it has been challenging to test this hypothesis in human cerebral cortex. Using high-field functional MRI with near-continuous recording during visual aura in three subjects, we observed blood oxygenation level-dependent (BOLD) signal changes that demonstrated at least eight characteristics of CSD, time-locked to percept/onset of the aura. Initially, a focal increase in BOLD signal (possibly reflecting vasodilation), developed within extrastriate cortex (area V3A). This BOLD change progressed contiguously and slowly (3.5 ± 1.1 mm/min) over occipital cortex, congruent with the retinotopy of the visual percept. Following the same retinotopic progression, the BOLD signal then diminished (possibly reflecting vasoconstriction after the initial vasodilation), as did the BOLD response to visual activation. During periods with no visual stimulation, but while the subject was experiencing scintillations, BOLD signal followed the retinotopic progression of the visual percept. These data strongly suggest that an electrophysiological event such as CSD generates the aura in human visual cortex.
Migraine is a very common and debilitating disorder. In 20% of cases (1), the migraine headache is preceded by a visual hallucination/illusion known as an aura. Typically, the aura is a serrated arc of scintillating, shining, crenelated shapes, beginning adjacent to central vision and expanding peripherally over 5–20 min, within one visual field, usually followed by headache. The scintillations are followed temporarily by a blind region, after the same retinotopic progression from central to peripheral visual fields.
Leao (2) first suggested a relationship between cortical spreading depression (CSD) and migraine aura, based on the uniquely slow spread of clinical and electrophysiological events. CSD is a wave of neuronal and glial depolarization, followed by long-lasting suppression of neural activity, which is easily evoked in mammals with lissencephalic (2, 3) or folded cortex (4).
Numerous human neuroimaging studies have indirectly suggested that CSD underlies migraine (5). These include planar Xenon (6–9), single photon emission tomography (8, 10–14), positron-emission tomography (15, 16), magnetoencephalography (17, 18), and MRI (19–21). Each demonstrated one or more aspects of CSD associated with migraine aura. However, subjects never experienced symptoms of typical visual auras in studies showing spreading hypoperfusion (15) or blood oxygenation level-dependent (BOLD) signal changes (19), and the initial hyperemia characteristics of CSD have never been directly demonstrated in human cortex. This uncertainty about the mechanisms of migraine visual aura has seriously limited the development of therapeutic drugs that could act directly on CSD.
To detect and follow analogous temporal and spatial events in human visual cortex, we used high-field functional MRI to map the progression of the BOLD events during migraine aura. The BOLD signal is not directly equated with blood flow, rather it reflects the balance between oxygen delivery and oxygen consumption. Nevertheless, changes in BOLD signal intensity have been successfully used in experimental animals to detect the presence of CSD, the rate of CSD propagation, and attendant changes in apparent diffusion coefficients (4). To clarify the topography of the activity spread across cortex, these BOLD events were mapped onto flattened cerebral cortex, in relationship to the retinotopy, in each of three subjects. By so doing, we could identify at least eight fundamental and distinguishing characteristics of CSD in visual cortex during migraine aura in humans, which could not be revealed with previously available techniques.
Patients and Methods
Subjects.
We studied three male patients (36 ± 9 years), each fulfilling the International Headache Society criteria for the diagnosis of migraine with aura. A total of five migraine attacks with visual aura were studied. Two were induced in a single patient (P.R.), and three spontaneous migraine episodes were scanned in two other patients (S.R. and M.C.). None were taking prophylactic medication at that time, and none had taken any acute antimigraine drug for at least 1 week before the scan. Informed written consent was obtained for each subject before each scanning session, and all procedures were approved by Massachusetts General Hospital Human Studies Protocol numbers 96–7464 and 93–7253.
General Procedure.
Magnetic resonance (MR) data were acquired in a 3-Tesla scanner, using echoplanar imaging. Subjects were scanned by using a bilateral quadrature surface coil. Sixteen contiguous slices, 4 mm thick, were collected in an oblique coronal plane, oriented perpendicular to the calcarine sulcus. The functional scans used the following parameters: gradient echo, echo time = 50 ms, repetition time = 4,000 ms, matrix 128 × 64, in-plane resolution 3.1 × 3.1 mm. Visual stimuli were generated on a Silicon Graphics O2 computer and projected onto the center of a rear projection screen located about 20 cm from the subject's eyes. Face and fixation stimuli subtended 48 × 36o of visual angle. Subjects viewed the screen in a darkened room through a mirror, and corrective lenses were used if necessary.
Subjects fixated the center of a radial flickering checkerboard (2 Hz), which was presented in 16-s epochs (on period), alternating with 16-s presentation of a uniform black stimulus (off period), during two consecutive runs. A central fixation point was present at all times. A squeeze bulb was used to record the initiation and termination of the visual aura and headache.
In addition to interictal scans, retinotopic maps of polar angle and eccentricity were generated in the same subjects by using stimuli as described (22–26). To improve topographic clarity, all data were analyzed and displayed in cortical surface format as described (22–26).
Perfusion weighted imaging scans were done at the end of the BOLD imaging during a migraine attack to assess late hemodynamic changes. A standard head coil was used for the perfusion weighted imaging [echoplanar imaging spin echo, 10 axial slices, 51 images/slice, repetition time 1500, echo time 75, 0.2 mmol/kg of Gd-pentaacetic acid at a rate of 5 cc/sec by using a MedRad power injector (MedRad, Pittsburgh, PA)].
Experimental Design.
Triggered case.
Immediately before each of the imaging studies, our subject (P.R.) reproduced those conditions known in the past to induce his migraine attack. After ≈80 min of continuous basketball playing, when the patient considered that the level of exercise was adequate to trigger an attack, he was taken to the MRI facility, and BOLD imaging was started before any visual symptom was present. The patient was instructed to press a squeeze bulb on three different occasions to indicate (i) the beginning of the visual aura, (ii) the end of the visual aura, and (iii) the beginning of headache.
Each scan consisted of 512 images/slice, lasting 34 min, 8 s. Two scans were done per session, and a total of three scanning sessions were done. In two scanning sessions, the subject developed stereotypical migraine with visual aura. On a third scan session done under the same conditions, the patient did not have any symptoms (visual aura or pain).
Subject P.R. described the visual aura as a scintillating white noise (“like TV snow”) beginning in the paracentral left visual field. The boundary of the scintillations was well defined. The aura had an expanding crescent shape, progressing slowly outward, with a minor clockwise component, affecting the lower quadrant to a greater extend early on. The white noise was closely followed by a scotoma that persisted in the center of the visual field for a few minutes. The scintillations plus the scotoma moved from the center of vision toward the periphery over 22–27 min. While the vision was still abnormal a mild throbbing headache began contralateral to the hemifield defect.
The other two spontaneous cases described similar visual symptoms lasting between 25 and 35 min, followed by either a bilateral occipital (subject S.R.) or frontotemporal headache contralateral to the hemifield defect (subject M.C.).
Spontaneous cases.
Patients suffering from migraine with visual aura contacted us as soon as possible after the initiation of visual symptoms. Because the subjects worked in the same building as the MRI facility, they could be placed in the magnet within 15–20 min after onset. The protocol included a BOLD study followed by perfusion weighted imaging scans. Patients were instructed to indicate to indicate first the end of the visual aura, and then the beginning of headache, using the squeeze bulb.
Duration of each of the imaging BOLD scans for subject S.R. and subject M.C. (one/attacks) was 34 min, 8 s. The second attack of subject M.C. was imaged during three scans of 8 min, 34 s.
Image Processing and Statistical Analysis.
MR time courses (Figs. 1–3) were analyzed by using a standard t-statistic, computing the difference between the activation amplitude during the off period preceding the aura, compared with each of the on (Fig. 2) and off periods throughout the scanning session. MR time courses were extracted independently from voxels within specific visual areas, and/or at specific eccentricities (Figs. 3 and 5).
Figure 1.

Ictal and interictal BOLD responses in human visual cortex. A representative functional MRI slice is shown (A). The slice plane was oriented near-perpendicular to the calcarine fissure, so that cerebellum occupies the lower portion of the figure, and occipital lobe occupies the upper portion. (B–E) Representative BOLD responses over time, taken from single voxels within homologous areas of the occipital lobe (B and D, Right vs. C and E, Left), as designated by green arrows. Time is shown on the x axis, and levels of MR modulation are shown on the y axis. The stimulus-driven signal oscillation in B–E is the BOLD responses to 16-s presentations of the checkerboard visual stimulus (on response), relative to the intervening 16-s presentations of a black screen with a fixation point (off response). (D and E) Normal BOLD modulation during an interictal period for each hemisphere. (B and C) The BOLD responses during a migraine aura affecting only the right hemisphere (B) (see Fig. 2). Perturbations did not appear in the left hemisphere during the ictal (C), or interictal scans (D and E).
Figure 3.

Spreading suppression of cortical activation during migraine aura. (A) A drawing showing the progression over 20 min of the scintillations and the visual field defect affecting the left hemifield, as described by the patient (P.R.). The fixation point appears as a small white cross. The red line shows the overall direction of progression of the visual percept. The front of the scintillation at different times within the aura is indicated by a white line. (B) A reconstruction of the same patient's brain (P.R.), based on anatomical MR data. The posterior medial aspect of occipital lobe is shown in an inflated cortex format. In this format, the cortical sulci and gyri appear in darker and lighter gray, respectively, on a computationally inflated surface. MR signal changes over time are shown to the right. Each time course was recorded from one in a sequence of voxels that were sampled along the calcarine sulcus, in the primary visual cortex (V1), from the posterior pole to more anterior location, as indicated by arrowheads. A similar BOLD response was found within all of the extrastriate areas, differing only in the time of onset of the MR perturbation The MR perturbations developed earlier in the foveal representation, compared with more eccentric representations of retinotopic visual cortex. This finding was consistent with the progression of the aura from central to peripheral eccentricities in the corresponding visual field (A and C). (C) The MR maps of retinotopic eccentricity from this same subject, acquired during interictal scans. As shown in the logo in the upper left, voxels that show retinotopically specific activation in the fovea are coded in red (centered at 1.5° eccentricity). Parafoveal eccentricities are shown in blue, and more peripheral eccentricities are shown in green (centered at 3.8° and 10.3°, respectively).
Figure 2.
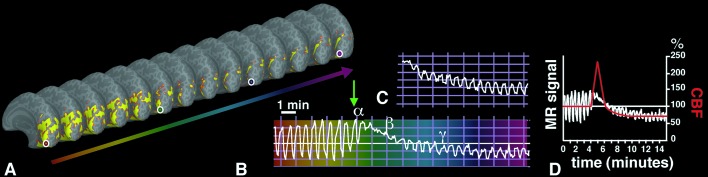
Time-dependent BOLD activity changes from a single region of interest in VI, acquired before and during episodes of either spontaneous (C) or induced (B) visual aura. (A) A series of anatomical images, including BOLD activity on “inflated” cortical hemispheres showing the medial bank (similar to a conventional midsagittal view). Images were sampled at 32-s intervals, showing the same region of interest (circles) in V1. (B) The MR signal perturbation over time from the circled region of interest; the perturbation is similar to that in Fig. 1B. Variations in time are color-coded (deep red to magenta), and the four colored circles match corresponding times within the V1 region of interest. The slice prescription failed to include a few mm in the most posterior part of the occipital pole in that induced attack, so activation is not revealed in any of these images. B shows that before the onset of the aura, the BOLD response to visual stimulation shows a normal, oscillating activation pattern. After the onset of aura (green arrow), the BOLD response showed a marked increase in mean level (α), a marked suppression to light modulation (β), followed by a partial recovery of the response to light modulation at decreased mean level (γ; −3% to −6%). (C) Data from a spontaneous attack (subject M.C.), captured ≈18 min after the onset of the visual symptoms affecting the right hemifield. The data represent the time course in left visual area V1, at an eccentricity of ≈20° of visual angle. (D) A superimposition of CBF changes seen in the rat during CSD (as described by Lauritzen et al. in ref. 46) with the MR signal data shown in A. Note that the timing of the hyperemia (3–4.5 min in CSD vs. 3.3 ± 1.9 min in migraine aura) is remarkably similar in these two quite different data sets. The amplitude of the hyperemia is different in the two conditions, presumably because of differences in the blood flow measurement techniques used (laser doppler versus BOLD) and the nonlinear relationship between blood flow and BOLD signal.
Figure 5.

Time-course evidence for a migraine origin in visual area V3A, taken from the same subject (P.R.) illustrated in Fig. 6, but in a different migraine attack. The slice prescription did include the most posterior part of the occipital pole. Each of the four panels shows the BOLD signals from a voxel in either area V3A, or V3, or V2 or V1. All voxels were sampled from approximately equivalent (parafoveal) retinotopic eccentricities. Time is represented along the x axis, and the range is equal and synchronized in each of the four panels. MR amplitude (indicated on the y axis) varied slightly in the different pixels and visual areas. Area V3A showed the first BOLD perturbation. Within a few minutes, the perturbation spread into adjacent area V3. The perturbations then spread further posteriorly into areas V2 and V1—but in those areas, the perturbations occurred at times following the time range shown.
To detect the source of the signal change, we analyzed the time course of each voxel individually, analyzing only the voxels that were significantly (P > 0.01) activated. A reference baseline (mean MR signal amplitude) and SD were computed on the six first cycles (before the beginning of the aura). Then, pixels which exhibited, at a given latency, (i) a mean MR signal higher than the reference mean plus the reference SD and (ii) a SD less than the reference SD were coded as positive, if this change lasted for at least two cycles. The value represented is the beginning of the signal change in the positive voxels as defined above.
Results
Three patients were investigated during five episodes of visual aura. In one case (subject P.R.), the attacks were triggered by exercise, and data were acquired before, during the aura, and in the headache phase in two episodes. In the other two migraineurs (M.C. and S.R.), spontaneous auras were captured 15–20 min after onset, and imaging acquisition continued into the headache phase.
Interictal Studies.
Interictal studies were performed in the three migraineurs and in seven healthy, age-matched male volunteers. All subjects showed the same pattern of activation of the visual cortex, and no difference was observed between migraineurs and nonmigraineurs. During continuous BOLD imaging (scan duration = 34 min, 8 s), the amplitude of the activation elicited by the checkerboard presentation remained constant (Fig. 1 D and E).
In the inducible patient, exercise failed to trigger the migraine attack in one of the trials. The MR findings during that nonmigrainous trial were comparable to those obtained in the interictal studies in migraineurs and nonmigraineurs.
Induced Attacks.
In both attacks of migraine with visual aura, the BOLD signal changes were similar and were restricted to the occipital cortex contralateral to the visual aura (Fig. 1). Before the onset of the aura, the BOLD signal showed a stereotypical, normal response, consisting of an increase in BOLD signal during the checkerboard presentation, followed by a decrease in signal when the screen was black (Fig. 1 D and E). Only after the onset of scintillations (signaled by the subject, see above) did the BOLD response deviate from this stereotypical pattern.
The first changes consisted of an increase in the mean MR signal (5 ± 1.5%) and a decrease of the amplitude of the signal oscillation (duration = 3.3 ± 1.9 min). These initial BOLD features are consistent with the initial features of the aura percept. That is, increases in visual activity (scintillations) occur during increases in the mean BOLD levels. Furthermore, those same scintillations (which overlie existing visual stimuli) are paralleled by decreases in the stimulus-driven MR oscillations (Fig. 2B, α).
These initial changes were followed by a decrease in the mean MR signal (5 ± 0.7%) (Fig. 2B, β and γ) whereas the stimulus-induced response remained suppressed. Perceptually, this phase appears to correspond to the localized scotoma. Both the mean MR signal and the stimulus-induced response recovered slowly (to 80% of initial level by 15 ± 3 min), following the same sequence as the suppression of stimulus-induced response (see below).
Perfusion weighted images taken at 60–111 min after attack onset showed a decrease in regional cerebral blood flow, regional cerebral blood volume, and an increase in mean transit time in the occipital cortex—where the BOLD changes described above were observed, confirming the results previously published from one of our studies (21).
If these BOLD changes are mechanistically linked to the aura percept, the signal changes should progress systematically across the visual cortical gray matter in accord with the underlying retinotopic maps in the same tissue. This topographic hypothesis was tested by using a flattened cortical format. The retinotopic progression of the BOLD perturbations was systematic and consistent with the migration of visual aura from central to peripheral visual fields. After the onset of the perceptual scintillations, the BOLD signal complex progressed systematically from the posterior pole (where central visual fields are represented), toward more anterior regions representing peripheral visual fields (Figs. 2 and 3).
In a signal source analysis, we found BOLD signal changes developed first in extrastriate cortex (area V3A), in subject P.R (see Fig. 4). These early changes were confirmed during a second attack (Fig. 5). To examine the correspondence between the spreading BOLD events in this study, compared with the velocity of the CSD in previous animal experiments, the rate of signal migration was calculated precisely in the flattened cortex. The average velocity was 3.5 ± 1.1 mm/min, measured along the cortical surface from to its point of origin in V3A. Although our stimulus activated cortex as far anteriorly as the precuneus, the spreading BOLD perturbation did not cross the parieto-occipital gyrus (see Fig. 4); in this sense it matches the predictions from the CSD literature (27). However, our MR perturbations did cross cytoarchitectonic boundaries transparently, such as the prominent one between V1 and V2 (see Fig. 3). As one would expect from CSD, temporo-spatial aspects of our BOLD changes did not fit simply within any known arterial or venous macrovascular territory.
Figure 4.

Source localization and time of onset of the MR signal perturbations. (A and C) The data on (normally) folded right hemispheric cortex; (B and D) the same data on inflated cortical surface (as in Figs. 2 and 3). (E) A fully flattened view of the cortical surface, as shown in previous publications (24–26, 37, 53). (A and B) A view of the exposed medial bank from the posterior pole. (C and D) Shown is the entire hemisphere, from a posterior-medial view. Pos indicates the parieto-occipital sulcus. As described in Fig. 2, activation data were not acquired from the extreme posterior tip of the occipital pole. Cortical locations showing the first BOLD perturbations are coded in red (E). Locations showing the BOLD perturbations at progressively later times are coded by green and blue (see pseudocolor scale to the right). The aura-related changes appeared first in extrastriate cortex (V3A, closely followed by V3 and V2), then progressed into V1. The spread of the aura began, and was most systematic, in the representation of the lower visual field (upper bank), becoming less regular as it progressed into the representation of the upper visual field.
Spontaneous Attacks.
Three spontaneous attacks were captured in two subjects within 15–20 min of the onset of visual symptoms. These BOLD data revealed a decrease in the amplitude and the mean MR signal (Fig. 2A). The observed changes were similar to these in the induced attack, when comparisons were made at similar latencies after the aura onset. Because the attacks were spontaneously generated and therefore unpredictable, no baseline (BOLD signal previous to visual symptoms) was available in the spontaneous attacks. Therefore, we could not quantify the percentage decrease in the amplitude or recovery, as was possible in induced attacks. Nevertheless, similarities between the BOLD signal perturbations in five induced and spontaneous attacks (decreased amplitude and mean MR signal, temporal pattern of signal recovery, and retinotopic congruence to the visual percept) suggest that our findings are likely to be generalizable to a much larger population of migraineurs with visual aura.
Scintillation Percept in the Dark.
In the triggered case, we also examined the BOLD signal during the off periods (Fig. 6). As expected, no signal was present during these periods before the beginning of the symptoms. However, with the subjective apparition of scintillations described by the subject, one could observe a BOLD signal change appearing first in area V3A and then progressing congruently with the retinotopic percept.
Figure 6.

Progression of the scintillations in the dark, without the flickering checkerboard stimuli. (A) A series of images of the brain of the subject at different times, from the beginning of the scanning session (see the little clocks below). The primary visual cortex lies within the white line. Initially, no activation can be seen in V1. However, with the subjective apparition of the scintillations (after 20 min, see clock), activation appears in V1, that progresses from the foveal representation of the visual field to more peripheral representations, paralleling the progression of the scintillations described by the subject. (B) A medial view of the subject's brain with the MR maps of retinotopic eccentricity acquired during interictal scans. (see above, Fig. 3).
Discussion
Our data confirmed previous reports that CSD-like phenomena can be seen with neuroimaging techniques (7, 10, 12, 15, 19, 20, 28). Like those previous studies, our data indicated a slowly spreading area of abnormal blood flow in the occipital lobe during migraine aura.
Here, we extended these important studies in a number of ways. Within the same attack, and using analytical techniques that enhanced both the spatial and temporal resolution, we measured the retinotopic nature of the observed BOLD signal changes. This approach revealed at least eight neurovascular events in the occipital cortex that resemble cortical spreading depression: (i) an initial cortical gray hyperemia, with (ii) a characteristic duration, and (iii) a characteristic velocity, which (iv) is followed by hypoperfusion, and shows (v) an attenuated response to visual activation, and (vi) a recovery to baseline mean level, and (vii) a concurrent recovery of the stimulus-driven activation. Finally (viii), we found that (like CSD) the spreading phenomenon did not cross prominent sulci (e.g., the parieto-occipital sulcus).
An increase in the mean MR signal and a decrease of stimulus-driven modulation were the first BOLD signal changes in subject P.R. The most likely source of those initial responses was an increase in blood flow and volume caused by heightened neural activity reflected perceptually by the shining, scintillating migrating visual aura.
Alternative interpretations could be offered, but those seem less likely to us. For instance, the mean MR signal increase could reflect low oxygen consumption, due to decreased neural activity with (uncoupled) constant blood flow. However, the scintillating percept appears to reflect increased (rather than decreased) neural activity—so this interpretation does not seem likely.
Another possible interpretation is that venoconstriction could increase the mean BOLD level. However, by this hypothesis, the resultant blood volume decrease presumably would be outweighed by the associated increase in blood flow (29–31). The initial increase in BOLD signal is also inconsistent with a vasospasm; instead the early increase suggests flow changes driven by neural and metabolic demands.
It has not been clear whether a specific region in occipital lobe initiates the spreading event. Some have attributed the visual phenomena to primary visual cortex (32, 33), because the visual image is oriented and highly retinotopic, and striate cortex is also retinotopic and selective for oriented stimuli (34–37). However, human occipital cortex is comprised of multiple cortical areas, many of which (like V1) are also retinotopic (24–26, 37–39) and orientation-selective (e.g., ref. 37). Hence, the auras could arise from many of these extrastriate areas (e.g., V2, V3/VP, V3A, V4v), as well as V1.
We found that area V3A first developed BOLD signal changes in subject P.R. These early changes were confirmed during a second attack (Fig. 5). Such extrastriate foci are consistent with prior reports (15). V3A is especially sensitive to both motion and luminance contrast (26), and aberrant neuronal firing in this location might well produce scintillations like those described by migraineurs during a typical visual aura. Moreover, area V3A is retinotopic, and it has a continuous representation of a whole hemifield, which is again consistent with the progression of a typical migraine aura.
Because source analysis could not be performed in all attacks, additional studies will be necessary to establish the relationship between area V3A and the onset of prototypical visual auras. Other types of migraine aura, e.g., those containing corrugated lines (the “fortress illusion”) might initiate in area V1, whereas those containing colored phosphenes, might reflect a focus in area V8, a color-selective area (24). Somaesthetic auras might well originate in somatosensory cortex. Whether such cortical areas possess a unique density or distribution of calcium channels, as mutated in some patients with familial hemiplegic migraine (40), remains to be studied.
Short of electrophysiological measurements, the present high-resolution imaging suggests that CSD within human occipital cortex explains the spatial and temporal characteristics of the migraine visual aura. CSD is characterized by initial hyperemia lasting 3–4.5 min (4, 6, 7, 41–44) (see Fig. 2C). The hyperemia is then followed by a mild hypoperfusion (6, 18, 19, 45–51) lasting 1–2 h—not unlike what was described many minutes after the aura in human occipital cortex (7, 10, 15, 20, 21).
Convergent data suggest that migraineurs may be especially susceptible to slowly spreading excitable events within cerebral cortex (19). First, the slow rate of progression (2–5 mm/min) in CSD, and the topography of the cortical spread, are comparable to the evolution of the BOLD signal complex we observed during the visual aura (Fig. 6). Second, CSDs tend to stop their progression at major sulci (27)—not unlike what was observed at the parieto-occipital sulcus in subject P.R. (Fig. 4). Third, light-evoked visual responses are suppressed during CSD in rabbit, and recover in 15–30 min (2, 3). Our light-induced MR signals also were suppressed during visual aura, and they recovered with a time course very similar to that in experimental CSD. Finally, in both CSD and migraine aura, the first affected areas were the first to recover.
Our findings build on the recent observations of Cao et al. (19) in the following ways. First, we identified specific BOLD events in the visual cortex that appeared to be directly linked to the aura percept, in both space (retinotopy) and time. Throughout the progression of each aura, we found a unique BOLD perturbation in the corresponding regions of retinotopic visual cortex. Like the progression of the aura in the visual field, the BOLD perturbations progressed from the representation of paracentral to more peripheral eccentricities—and these BOLD changes were found only in the hemisphere corresponding to the aura. Second, we were able to identify a source of the aura-related BOLD changes. Surprisingly, that source was in extrastriate visual cortex (area V3A) rather than in striate cortex (V1). Third, we were able to measure the rate of spread of the BOLD perturbations across the flattened cortical gray matter, and we found it to be consistent with previous measures of CSD. Finally, it is worth noting that our migraine auras were not provoked by the visual stimuli. For example, interictal scans with identical visual stimuli did not produce auras nor migraines. Therefore our measurements were not confounded by the inducing stimulus. Evidence of spreading BOLD signal change during the light off periods also dissociated the BOLD perturbations from light stimulation.
Consistent with our findings, and as reported earlier in experimental animals (4), the spread of induced CSD was confined to the hemisphere in which it was initiated. James et al. (4) demonstrated in a gyrencephalic brain BOLD and apparent diffusion coefficient changes (ADC) due to CSD. They showed a clear temporal relation and an inverse correlation between these two measures, with an increase in BOLD signal paralleling a decrease in ADC. The magnitude of the BOLD signal change they measured (4–6% at 2 T) is comparable to that reported in our subjects.
We conclude that migraine aura is not evoked by ischemia (19, 52). More likely, it is evoked by aberrant firing of neurons and related cellular elements characteristic of CSD, and vascular changes develop due to fluctuations in neuronal activity during the visual aura. Drugs that inhibit the development and propagation of CSD provide novel treatment targets for both migraine aura, even before headache onset, as well as for stroke. Future studies using similar techniques should clarify the BOLD correlate of the onset of the headache pain, to better understand the relationship between CSD and pain.
Acknowledgments
We thank P.R. and his wife for agreeing to play basketball and triggering his attacks for us, as well as S.R. We also thank Dr. Richard Kraig, University of Chicago, and our colleague Dr. Richard Hoge, for helpful discussion, A. K. Liu for making the visual stimulus, and T. Campbell and M. Foley for technical assistance. This project was made possible by: National Eye Institute Grant 07980 (to N.H. and R.B.H.T.); American Association for the Study of Headache Research Fellowship 1998 (to M.S.d.R.); National Institutes of Health Grant K08-NS01803A (to F.M.C.); National Institutes of Health Grant P41 RR 14075 (to B.R.R.); and National Institutes of Health Grant 5 PO1 NS 35611 (to M.A.M. and A.G.S.).
Abbreviations
- CSD
cortical spreading depression
- BOLD
blood oxygenation level-dependent
- MR
magnetic resonance
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Russell M B, Olesen J. Brain. 1996;119:355–361. doi: 10.1093/brain/119.2.355. [DOI] [PubMed] [Google Scholar]
- 2.Leao A A. J Neurophysiol. 1944;7:359–390. [Google Scholar]
- 3.Leao A A. Funct Neurol. 1986;1:363–366. [PubMed] [Google Scholar]
- 4.James M F, Smith M I, Bockhorst K H, Hall L D, Houston G C, Papadakis N G, Smith J M, Williams A J, Xing D, Parsons A A, et al. J Physiol (London) 1999;519:415–425. doi: 10.1111/j.1469-7793.1999.0415m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olesen J, Tfelt-Hansen P, Welch K M A. The Headaches. Philadelphia: Lippincott; 2000. [Google Scholar]
- 6.Lauritzen M, Skyhoj Olsen T, Lassen N A, Paulson O B. Ann Neurol. 1983;13:633–641. doi: 10.1002/ana.410130609. [DOI] [PubMed] [Google Scholar]
- 7.Lauritzen M, Olesen J. Brain. 1984;107:447–461. doi: 10.1093/brain/107.2.447. [DOI] [PubMed] [Google Scholar]
- 8.Olesen J, Lauritzen M, Tfelt-Hansen P, Henriksen L, Larsen B. Headache. 1982;22:242–248. doi: 10.1111/j.1526-4610.1982.hed2206242.x. [DOI] [PubMed] [Google Scholar]
- 9.Skyhoj Olsen T, Friberg L, Lassen N A. Arch Neurol. 1987;44:156–161. doi: 10.1001/archneur.1987.00520140028013. [DOI] [PubMed] [Google Scholar]
- 10.Andersen A R, Friberg L, Skyhoj Olsen T, Olesen J. Arch Neurol. 1988;45:154–159. doi: 10.1001/archneur.1988.00520260040017. [DOI] [PubMed] [Google Scholar]
- 11.La Spina I, Vignati A, Porazzi D. Headache. 1997;37:43–47. doi: 10.1046/j.1526-4610.1997.3701043.x. [DOI] [PubMed] [Google Scholar]
- 12.Olesen J, Friberg L, Skyhoj Olsen T, Iversen H K, Lassen N A, Andersen A R, Karle A. Ann Neurol. 1990;28:791–798. doi: 10.1002/ana.410280610. [DOI] [PubMed] [Google Scholar]
- 13.Seto H, Shimizu M, Futatsuya R, Kageyama M, Wu Y, Kamei T, Shibata R, Kakishita M. Clin Nucl Med. 1994;19:215–218. [PubMed] [Google Scholar]
- 14.Soriani S, Feggi L, Battistella P A, Arnaldi C, De Carlo L, Stipa S. Headache. 1997;37:31–36. doi: 10.1046/j.1526-4610.1997.3701031.x. [DOI] [PubMed] [Google Scholar]
- 15.Woods R P, Iacoboni M, Mazziotta J C. N Engl J Med. 1994;331:1689–1692. doi: 10.1056/NEJM199412223312505. [DOI] [PubMed] [Google Scholar]
- 16.Andersson J L, Muhr C, Lilja A, Valind S, Lundberg P O, Langstrom B. Cephalalgia. 1997;17:570–579. doi: 10.1046/j.1468-2982.1997.1705570.x. [DOI] [PubMed] [Google Scholar]
- 17.Tepley N, Wijesinghe R S. Brain Topogr. 1996;8:345–353. doi: 10.1007/BF01186910. [DOI] [PubMed] [Google Scholar]
- 18.Barkley G L, Tepley N, Simkins R, Moran J, Welch K M. Cephalalgia. 1990;10:171–176. doi: 10.1046/j.1468-2982.1990.1004171.x. [DOI] [PubMed] [Google Scholar]
- 19.Cao Y, Welch K M A, Aurora S, Vikingstad E. Arch Neurol. 1999;56:548–554. doi: 10.1001/archneur.56.5.548. [DOI] [PubMed] [Google Scholar]
- 20.Cutrer F M, Sorensen A G, Weisskoff R M, Ostergaard L, Sanchez del Rio M, Lee E J, Rosen B R, Moskowitz M A. Ann Neurol. 1998;43:25–31. doi: 10.1002/ana.410430108. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez del Rio M, Bakker D, Wu O, Agosti R, Mitsikostas D D, Ostergaard L, Wells W A, Rosen B, Sorensen A G, Moskowitz M A, Cutrer F M. Cephalagia. 1999;19:1–7. doi: 10.1046/j.1468-2982.1999.019008701.x. [DOI] [PubMed] [Google Scholar]
- 22.Dale A M, Fischl B, Sereno M I. NeuroImage. 1999;9:179–194. doi: 10.1006/nimg.1998.0395. [DOI] [PubMed] [Google Scholar]
- 23.Fischl B, Sereno M I, Dale A M. NeuroImage. 1999;9:195–207. doi: 10.1006/nimg.1998.0396. [DOI] [PubMed] [Google Scholar]
- 24.Hadjikhani N, Liu A K, Dale A M, Cavanagh P, Tootell R B H. Nat Neurosci. 1998;1:235–241. doi: 10.1038/681. [DOI] [PubMed] [Google Scholar]
- 25.Sereno M I, Dale A M, Reppas J B, Kwong K K, Belliveau J W, Brady T J, Rosen B R, Tootell R B H. Science. 1995;268:889–893. doi: 10.1126/science.7754376. [DOI] [PubMed] [Google Scholar]
- 26.Tootell R B, Mendola J D, Hadjikhani N K, Ledden P J, Liu A K, Reppas J B, Sereno M I, Dale A M. J Neurosci. 1997;17:7060–7078. doi: 10.1523/JNEUROSCI.17-18-07060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bures J, Buresova O, Krivanek J. The Mechanism and Application of Leao's Spreading Depression of Electroencephalic Activity. New York: Academic; 1974. [Google Scholar]
- 28.Friberg L, Olesen J, Lassen N A, Olsen T S, Karle A. Stroke. 1994;25:974–979. doi: 10.1161/01.str.25.5.974. [DOI] [PubMed] [Google Scholar]
- 29.Bandettini P A, Wong E C. NMR Biomed. 1997;10:197–203. doi: 10.1002/(sici)1099-1492(199706/08)10:4/5<197::aid-nbm466>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 30.Grubb R L, Raichle M E, Eichling J O, Ter-Pogossian M M. Stroke. 1974;5:630–639. doi: 10.1161/01.str.5.5.630. [DOI] [PubMed] [Google Scholar]
- 31.Hoge R D, Atkinson J, Gill B, Crelier G R, Marrett S, Pike G B. Proc Natl Acad Sci USA. 1999;96:9403–9408. doi: 10.1073/pnas.96.16.9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aring C D. J Am Med Assoc. 1972;220:519–522. [Google Scholar]
- 33.Richards W. Sci Am. 1971;224:88–96. doi: 10.1038/scientificamerican0571-88. [DOI] [PubMed] [Google Scholar]
- 34.Talbot S A, Marshall W H. Am J Ophthalmol. 1941;24:1255–1271. [Google Scholar]
- 35.Daniel P M, Whitteridge D. J Physiol (London) 1961;159:203–221. doi: 10.1113/jphysiol.1961.sp006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hubel D H, Wiesel T N, Stryker M P. J Comp Neurol. 1978;177:361–380. doi: 10.1002/cne.901770302. [DOI] [PubMed] [Google Scholar]
- 37.Tootell R B, Hadjikhani N K, Vanduffel W, Liu A K, Mendola J D, Sereno M I, Dale A M. Proc Natl Acad Sci USA. 1998;95:811–817. doi: 10.1073/pnas.95.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeYoe E A, Carman G J, Bandettini P, Glickman S, Wieser J, Cox R, Miller D, Neitz J. Proc Natl Acad Sci USA. 1996;93:2382–2386. doi: 10.1073/pnas.93.6.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engel S A, Rumelhart D E, Wandell B A, Lee A T, Glover G H, Chichilnisky E J, Shadlen M N. Nature (London) 1994;369:525. doi: 10.1038/369525a0. [DOI] [PubMed] [Google Scholar]
- 40.Ophoff R A, Terwindt G M, Vergouwe M N, van Eijk R, Oefner P J, Hoffman S M, Lamerdin J E, Mohrenweiser H W, Bulman D E, Ferrari M, et al. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 41.Piper R D, Lambert G A, Duckworth J W. Am J Physiol. 1991;261:H96–H102. doi: 10.1152/ajpheart.1991.261.1.H96. [DOI] [PubMed] [Google Scholar]
- 42.Shibata M, Leffler C W, Busija D W. Brain Res. 1990;530:267–274. doi: 10.1016/0006-8993(90)91294-q. [DOI] [PubMed] [Google Scholar]
- 43.Shimazawa M, Hara H. Clin Exp Pharmacol Physiol. 1996;23:890–892. doi: 10.1111/j.1440-1681.1996.tb01139.x. [DOI] [PubMed] [Google Scholar]
- 44.Wahl M, Lauritzen M, Schilling L. Brain Res. 1987;411:72–80. doi: 10.1016/0006-8993(87)90682-2. [DOI] [PubMed] [Google Scholar]
- 45.Kraig R P, Nicholson C. Neuroscience. 1978;3:1045–1059. doi: 10.1016/0306-4522(78)90122-7. [DOI] [PubMed] [Google Scholar]
- 46.Lauritzen M, Jorgensen M B, Diemer N H, Gjedde A, Hansen A J. Ann Neurol. 1982;12:469–474. doi: 10.1002/ana.410120510. [DOI] [PubMed] [Google Scholar]
- 47.Lauritzen M. Acta Neurol Scand Suppl. 1987;113:1–40. [PubMed] [Google Scholar]
- 48.Lauritzen M. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- 49.Okada Y C, Lauritzen M, Nicholson C. Brain Res. 1988;442:185–190. doi: 10.1016/0006-8993(88)91450-3. [DOI] [PubMed] [Google Scholar]
- 50.Olesen J, Larsen B, Lauritzen M. Ann Neurol. 1981;9:344–352. doi: 10.1002/ana.410090406. [DOI] [PubMed] [Google Scholar]
- 51.Welch K M, Barkley G L, Ramadan N M, D'Andrea G. Pathol Biol (Paris) 1992;40:349–354. [PubMed] [Google Scholar]
- 52.Wolff H G. Headache and Other Head Pain. Oxford: Oxford Univ. Press; 1963. [Google Scholar]
- 53.Tootell R B, Dale A M, Sereno M I, Malach R. Trends Neurosci. 1996;19:481–489. doi: 10.1016/S0166-2236(96)10053-9. [DOI] [PubMed] [Google Scholar]