

Abstract
Intrinsically disordered proteins (IDPs), also known as intrinsically unstructured proteins (IUPs), lack a well-defined 3D structure in vitro and, in some cases, also in vivo. Here, we discuss the question of proteolytic sensitivity of IDPs, with a view to better explaining their in vivo characteristics. After an initial assessment of the status of IDPs in vivo, we briefly survey the intracellular proteolytic systems. Subsequently, we discuss the evidence for IDPs being inherently sensitive to proteolysis. Such sensitivity would not, however, result in enhanced degradation if the protease-sensitive sites were sequestered. Accordingly, IDP access to and degradation by the proteasome, the major proteolytic complex within eukaryotic cells, are discussed in detail. The emerging picture appears to be that IDPs are inherently sensitive to proteasomal degradation along the lines of the “degradation by default” model. However, available data sets of intracellular protein half-lives suggest that intrinsic disorder does not imply a significantly shorter half-life. We assess the power of available systemic half-life measurements, but also discuss possible mechanisms that could protect IDPs from intracellular degradation. Finally, we discuss the relevance of the proteolytic sensitivity of IDPs to their function and evolution.
Keywords: intrinsically disordered proteins, IDPs, intrinsically unstructured proteins, IUPs, proteolytic sensitivity, protein half-lives, evolution
Introduction
Initial considerations
The long accepted dogma has been that functional proteins, such as enzymes, antibodies, and receptors for hormones and neurotransmitters, exist as compact structures in which the polypeptide backbone and its sidechains are folded into a unique conformation that is crucial for the protein's function. However, it has recently become apparent that some amino acid (aa) sequences, usually characterized by high net charge and low hydrophobicity,1 adopt in solution an ensemble of extended, flexible conformations with little or no structure propensity. This phenomenon is known as intrinsic disorder. Intrinsically disordered proteins (IDPs), that is, proteins containing localized intrinsically disordered regions (IDRs) or completely devoid of structure along their whole lengths, are highly abundant, especially in eukaryotes. Furthermore, they are enriched in proteins associated with such functions as signaling and regulation.2,3 Although IDPs can resemble folding intermediates of globular proteins in shape, they appear to be qualitatively different from them, as indicated by their unusual aa composition and biophysical properties, such as resistance to temperature and chemical denaturation. It is thus considered likely that even in vivo, where macromolecular crowding, known to enhance refolding of globular proteins,4 is much higher than in vitro, IDPs generally retain the disorder observed in vitro. Already in the 1970s, proton NMR experiments hinted that proteins can possess a random coil-like state inside vesicles and intact cells5; the recent advent of in-cell NMR has confirmed this suggestion. Although the situation may vary from protein to protein, a study of the neuronal tau protein at near native concentration in X. laevis oocytes6 suggests that disorder is sustainable under the conditions prevailing in a eukaryotic cell. However, it can still be argued that if IDPs are expressed in the environment of their native cell, where all their cognate binding partners are present, they may exist solely in a bound and folded state. This folding-upon-binding model may indeed be true for some IDRs, for example, for certain ribosomal proteins, but in other cases IDRs appear to be disordered or not fully folded, even when bound. This is exemplified by the phenomenon of “fuzziness,”7 and, even more strikingly, by the case of Sic1-Cdc4 binding, in which Sic1 still populates a dynamic ensemble even when bound, with the ensemble as a whole contributing to binding.8 Furthermore, IDPs often function in a compartment different from that in which they are synthesized, and are frequently involved in low-affinity interactions; thus they are likely to exist, at least transiently, in an unbound state. Finally, disorder can contribute to protein function, for example, by allowing for a broad spectrum of interactions,9 for faster association rates10,11 and even facilitating catalysis.12 Some tasks, such as entropic chain functions, actually require disorder,13 with entropic bristles formed by Phe-Gly repeats in nucleoporins serving as an example.14,15 Importance of disorder for function is underscored by the existence of regions for which the disordered conformation, but not the sequence, is conserved throughout evolution.16 In summary, it is likely that there is less protein disorder in vivo than in vitro, mainly due to binding—but it is present inside the cell and is of functional importance.
One of the features commonly associated with IDPs is their susceptibility to proteolysis.17 Limited proteolysis is widely used as a tool for detecting disorder,18,19 and problems are often encountered in production of recombinant IDPs due to their sensitivity to bacterial proteases. Extrapolation to eukaryotic cells suggests that eukaryotic IDPs are easy targets for proteases in vivo. In the following, we discuss both theoretical considerations and experimental evidence relating to the susceptibility of IDPs to proteolysis.
Sensitivity of IDPs to Proteolysis In Vivo
Intracellular proteases
Genes coding for proteases are found in large numbers in both prokaryotic and eukaryotic genomes. Broad-specificity intracellular proteases capable of cleaving most sequences are used by organisms for removing misfolded and damaged proteins. Levels of native proteins are regulated at the level of their synthesis,20,21 but proteolysis is also important, especially in slowly dividing cells. Selective degradation of regulatory proteins plays an important role in dynamic processes such as cell cycle control and signal transduction. To fulfill these roles efficiently, without causing off-target damage, the broad-specificity proteases must be tightly regulated. This is generally achieved by sequestration in a separate compartment, that is, a subcellular organelle or a protein chamber, and by controlled substrate delivery. Compartmentalization brings together different proteolytic activities, thus enhancing degradative efficiency. Autophagy and proteasome-dependent degradation are two major proteolytic mechanisms in eukaryotic cells.22
Autophagy
Many intracellular proteases are enclosed in acidic organelles, such as the mammalian lysosome and the analogous yeast and plant vacuoles. Degradation by these organelles23 (autophagy) is important during nutrient-starvation, and is also involved in removing damaged organelles or proteins, especially large aggregates. Since little is known about degradation of IDPs by this route, we will not discuss it further.
The proteasome
The proteasome,24,25 a protein complex found in both the cytoplasm and nucleus of eukaryotes, is responsible for selective degradation of regulatory proteins, as well as of misfolded or damaged proteins. The 20S core particle of the proteasome (20S CP) is composed of 28 subunits, two copies each of 14 different, but related, α- and β-subunits (α1-7 and β1-7). It contains six active sites, of three different types, all associated with β-subunits, which, taken together, make it a broad-specificity protease capable of degrading most aa sequences. These subunits are organized as four rings (two inner β rings and two outer α rings), which delimit three chambers: two antechambers on either side of the proteolytic chamber. It has been demonstrated (for archeal 20S CP) that the antechamber has the capacity to maintain substrates in an unfolded state.26 Each antechamber is separated from the outside by a narrow channel known as the α-annulus, closed by a gate composed of N-terminal residues of the α-subunits. The free form of 20S CP is present at a high intracellular concentration; but part of the intracellular 20S CP is tightly associated with activators, viz., the 19S regulatory particle (19S RP) and the 11S complex, both of which stabilize the open conformation of the gate.27–30 The relative abundance of these populations is influenced by intracellular conditions, such as ATP concentration and temperature. The 26S proteasome (26SP) consists of 20S CP bound, at either or both of its endplates, to the 19S RP. The relative abundance of singly and doubly capped 26SPs differs from one cell type to another.31 The 19S RP, which includes six ATPase subunits, recognizes proteins tagged with a poly-ubiquitin (poly-Ub) chain,32 and subsequently deubiquitinates, unfolds, and translocates them into the core particle in an ATP-dependent process.33
When referring below to degradation by 20S CP, this means that the core particle alone is capable of hydrolyzing a given substrate in the absence of ubiquitination. Association of 20S CP with its activators does not necessarily abolish this capability. Degradation by 26SP means predominantly (Ub)- and ATP-dependent degradation, which requires association with the 19S RP.
Proteases in bacteria and archea
The proteasome is also found in some bacteria (actinomycetes) and archea. E. coli contains different but similar nano-compartmentalized systems, including the cytoplasmic Lon and Clp and the membrane-associated FtsH.34 Like 26SP, they are barrel-like structures associated with ATPases. It appears that substrates require specific sequential signals, which are recognized either directly by the ATPases, or by an adaptor, which then brings the substrate in contact with the protease.35 For example, the C-terminal residues of SulA, the N-terminal residues of SoxS, the ssrA tag, and stretches of aromatic residues all have been implicated in targeting to Lon.36 A degradation tag functionally analogous to Ub, called prokaryotic Ub-like protein (Pup), has recently been identified in M. tuberculosis.37 Interestingly, Pup is an IDP which, unlike Ub, appears to be degraded together with the substrate it is attached to, serving as a degradation initiation site.
Sensitivity of disordered regions to proteolytic cleavage
The substrate specificity of proteases is influenced by aa residues of the substrate extending in both the N- and C-terminal directions.38,39 These sequence preferences alone, however, cannot explain the extraordinary selectivity often observed in proteolysis experiments, especially when broad-specificity proteases are used, such as subtilisin, thermolysin, or proteinase K. Rates of proteolysis are well known to be enhanced by substrate denaturation.19 Thus, for example, the rate of hydrolysis of one particular bond in ribonuclease T1 increases 1700-fold upon unfolding.40 In 1980, Neurath41 remarked that proteolysis of folded proteins occurs preferentially “at hinges and fringes.” Indeed, it was subsequently shown that preferred subtilisin cleavage sites in thermolysin correspond to peaks in X-ray-derived B-factor values, which are known to be higher for flexible regions devoid of secondary structure.42 It was later demonstrated that cleavage sites also tend to overlap with regions for which electron density in crystal structures is completely absent, and are thus predicted to be intrinsically disordered.18 Hubbard et al.43 suggested that to interact with a protease, a protein segment must adopt a conformation similar to that seen for inhibitor molecules in crystal structures, viz., an extended conformation that requires a propensity for local unfolding of at least 12 residues around the scissile bond. A recent survey was made of all PDB-deposited structures of proteases, including the proteasome, in complex with substrates and inhibitors.44 This survey confirmed that polypeptides and other ligands bound within the protease active site adopt an extended conformation almost exclusively. It has also been argued that IDPs have greater capture radii,10 which should enhance their rates of interaction with binding partners, including proteases. It is possible to use in vitro display technology45 to design short linear peptides relatively resistant to a broad spectrum of proteases; but to the best of our knowledge no naturally occurring IDR of significant length has been demonstrated to be unusually resistant to proteolysis in vitro. Intracellular eukaryotic IDPs do not normally encounter proteases used in limited proteolysis experiments. However, their sensitivity to such proteases suggests generally high susceptibility to cleavage by protease binding sites, without the requirement for the prior local or global unfolding required of folded regions (Fig. 1).
Figure 1.
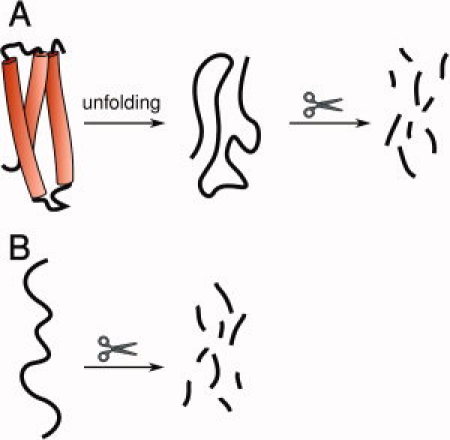
A: Folded proteins require denaturation prior to efficient proteolytic degradation, for example, by subtilisin, under mild conditions. B: Intrinsically disordered regions are inherently sensitive to proteolysis without the need for prior denaturation.
Degradation by bacterial proteases
Problems with susceptibility of IDPs to degradation are often encountered during over-expression in bacteria and subsequent purification46 (e.g., in the cases of BH3-only proteins47 and neuroligin 348). However, there are also examples of IDPs that can be successfully produced in E. coli without taking any special measures to avoid degradation. One such example is the cytoplasmic domain of gliotactin (gli-cyt). Despite being resistant to proteolysis in E. coli, gli-cyt is readily degraded by both proteinase K and 20S CP in vitro.49 This suggests that gli-cyt is not inherently insensitive to hydrolysis, but merely protected from the proteolytic sites of compartmentalized bacterial proteases such as Lon and Clp, which grant access to substrates displaying particular sequential signals (see section “Proteases in bacteria and archea”).
20S CP-dependent degradation of IDPs and the 20S CP gate
Many IDPs have been shown to undergo degradation by 20S CP both in vivo and in vitro, including α-synuclein,50 c-Jun,51 eIF4F and eIF3,52 HIF-1α,53 p16 and p21,54 Rb,55 p53, and p73.56 Indeed, we have suggested that susceptibility to Ub-independent proteolysis by 20S CP is a general property of IDPs.49 Baugh et al.57 subjected protein fractions from a rabbit reticulocyte lysate purification protocol to proteasomal degradation in vitro. Two hundred fifty-eight out of 1167 detectable protein species were found to be cleaved by both 20S CP and 26SP. Cleavage occurred at the IDRs, and proteolysis was in most cases incomplete, yielding trimmed folded domains. This suggests that 20S CP may be involved in processing rather than in degradation. However, we favor the hypothesis that 20S CP-mediated degradation normally occurs to completion in vivo, but is less efficient in vitro, perhaps due to lack of some additional, as yet unidentified, cellular factors. Extrapolating these results to the entire rabbit proteome, >20% of all cellular proteins are potential targets of 20S CP.
The difference between degradation by subtilisin or proteinase K and degradation by nano-compartmentalized bacterial and eukaryotic proteases is that for the latter, not only do the substrates have to be able to bind efficiently to the active sites but also have first to access the sites by passing through the gate to the inner compartment. In X-ray structures of eukaryotic (S. cerevisiae) 20S CP particles, both without58,59 and with inhibitors,60–62 the gate was seen to be closed, with the N-terminal tails of α-subunits blocking the entry channel. The structures displayed an open gate conformation either when proteasome activators were bound63,64 or when the α3 subunit N-terminal tail was deleted.59 Furthermore, low concentrations of denaturants, dialysis against distilled water, heat treatment,65,66 and addition of specific hydrophobic peptides67 are all known to “switch on” 20S CP, presumably also by gate opening. Thus, two states of 20S CP can be distinguished: latent (gate closed) and activated (gate open). However, latent 20S CP does display detectable, albeit low, peptidase activity.68,69 Atomic force microscopy (AFM) studies suggest that latent eukaryotic 20S CP is in equilibrium between the open and closed conformations,70,71 consistent with a stochastic model of 20S CP gate opening.69 A 1:3 ratio of open to closed conformers was observed for S. cerevisiae 20S CP, with intraconversion occurring on a millisecond timescale. Changes in the distribution of open and closed conformers, for example, after adding degradable peptides, suggest an allosteric coupling between the active site and the gate. This coupling may lead to enhanced degradation and product release. However, bearing in mind that two substrates can access 20S CP at the same time from the opposite ends,72 it may also result in cooperativity in degradation, whereby cleavage of one substrate molecule promotes entry of the next one. Furthermore, these studies show that the free energy difference between the open and closed states is small, and that the open conformation is relatively stable, even in the absence of any stabilizing mechanism. The stochastic model of gate opening is confirmed by NMR analysis of archeal 20S CP gate dynamics,73 and appears to be consistent with experimental evidence. Although many instances have been reported of substrates interacting with α-subunits, which could potentially lead to active gate opening, for other substrates such information is lacking. It seems that unfolded substrates can randomly enter through the gate when it is in the open state, and subsequently prevent its closing. This mechanism of random rather than targeted recognition of 20S CP seems plausible, bearing in mind its very high intracellular concentration. For some substrates, binding to 20S CP may be conjectured to further stabilize the open conformation. No direct evidence is available that this indeed occurs, but it would be consistent with the stimulatory action of some peptides on 20S CP.67 For other substrates, binding may simply serve to recruit the substrate to the vicinity of the 20S CP gate, where it can “wait” for gate opening to occur.
The open gate is not wide; as a consequence, it grants access only to unfolded substrates. When a gold particle around 2 nm in diameter is attached to a denatured polypeptide chain, this otherwise degradable substrate can no longer be processed by archeal 20S CP.74 The X-ray structures of 20S CPs from various species suggest that the gate has a diameter of ∼17 Å (∼13 Å when van der Waals radii are taken into account). Thus, a single, extended polypeptide chain can easily pass through it, and so can two adjacent chains in a β-strand conformation.75
Because of the narrow dimensions of the gate, not only is flexibility of the polypeptide chain required to permit productive interaction with the active sites of the protease subunits (function i in Fig. 2), but also the physical ability to access them (function ii in Fig. 2). Both elements are crucial for efficient degradation.74 This is consistent with our observation that a molten globule species was rapidly degraded by proteinase K, but not by 20S CP.49
Figure 2.
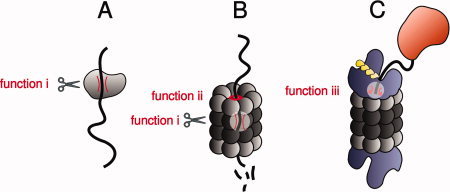
Role of intrinsic disorder in different types of proteolytic degradation. A: Disordered regions are preferentially cut by small broad-specificity proteases, such as proteinase K, subtilisin, or thermolysin, because they can easily adopt an extended conformation required by the protease active site (function i). B: For the same reason (also function i), disordered regions are efficiently degraded by the 20S proteasome without the need for prior catalyzed unfolding. More importantly, however, they can initiate degradation by accessing the narrow 20S proteasome gate, which opens and closes in a stochastic manner (function ii). C: Intrinsically disordered initiator regions are likely also required for 26S proteasome-dependent degradation, but in such cases it has been suggested that they function by interacting with the ATPase loops in the 19S subunit and triggering unfolding (function iii). Although intrinsic disorder serves as a sufficient signal for proteinase K- or 20S proteasome-mediated degradation, it does not do so for 26S proteasome-mediated degradation. In the latter case, a polyubiquitin chain or a specific mechanism of recruitment to the proteasome is also required.
Proteasomal activators in Ub-independent IDP degradation
26SP, with its gate stabilized in the open conformation and enhanced peptidase activity,30,57 should be capable of degrading IDPs, especially if binding to the 20S CP surface is not necessary for entry into the antechamber. Indeed, there is experimental evidence suggesting that 26SP can degrade IDRs in vitro57,76 without a requirement for ATP hydrolysis. However, this might be due to the 26SP preparations studied having been partially singly capped, that is, possessing one “26SP-like” and one “20S CP-like” endplate.57 Interestingly, the in vivo degradation of the IDP, p21, is enhanced by siRNA against a 19S RP subunit.54 This suggests that the 20S CP or the “20S CP-like” endplate of 26SP is more efficient in p21 degradation than the “26SP-like” endplate.
Another activator, REGγ, has been implicated in degradation of several IDPs, including p21, p16,54,77 SRC-1/AIB1,78 Smurf1,79 and hepatitis C virus core particle.80 However, c-Fos was shown to be degraded by 20S CP, but not by REGγ-activated 20S CP81; thus, REGγ may be an important, but not a universal, activator of IDP degradation by 20S CP.
Initiator regions and processivity of degradation by 20S CP
IDPs degradable by 20S CP include both wholly disordered polypeptide chains and proteins containing localized IDRs (Fig. 3). We have shown that IDPs, which are largely disordered, are readily degraded by 20S CP in vitro49 [Fig. 3(A)]. p53, which contains two IDRs at its termini, and a folded region in the middle,82 is also completely degraded by 20S CP both in vivo and in vitro. The region necessary for initiating this process corresponds to the N-terminal IDR.83 During degradation, the folded domain must be gradually unraveled and fed into the narrow entry channel without the help of ATP-dependent unfoldases. The C-terminal IDR of p53, although readily degraded in vitro by 20S CP when expressed in isolation, is not capable of initiating the degradation of p53 if the N-terminal domain is deleted. This is consistent with the hypothesis that different proteins are degraded from either the N- or the C-terminus, the direction being determined by some sequence-related or structural feature(s), but with a requirement for an IDR at the relevant end. Alternatively, it is feasible that the directionality of degradation is not pre-determined by any specific factors, but that proteolysis simply proceeds from wherever it can be efficiently initiated.84 In the latter scenario, the C-terminal domain of p53, despite its disorder, would lack some of the features necessary for a good initiation site. Efficiency of protein unraveling by ATP-dependent proteases does not depend on global protein thermal stability, but rather on the local structure adjacent to the site of degradation initiation.85,86 The same may also be true for 20S CP-mediated degradation.
Figure 3.

Degradation by the 20S proteasome. A: Fully disordered proteins are subject to proteolytic degradation by the 20S proteasome. Partially disordered proteins possessing disordered initiator regions at (B) N- or (C) C-termini can also access the 20S proteasome and undergo degradation. D: It is still uncertain if internal disordered regions can also play an initiator role, but the current consensus is that they can. Folded domains are prevented from accessing the 20S proteasome and are generally not subject to degradation by the ubiquitin-independent pathway.
In other 20S CP-degradable IDPs it is also often possible to map particular IDRs, at either the N- or C-terminus,87 that are responsible for initiation of degradation [Fig. 3(B,C)]. Such initiator regions are often capable of destabilizing a stable GFP reporter when fused to it, as is the case for a 40 aa C-terminal region of Fra-1.88 Both yeast and human ornithine decarboxylases (ODCs) are targeted to ubiquitin-independent degradation by IDRs, the initiator regions being at the N- and C-terminus, respectively. One difference is that while 20CP is sufficient for human ODC degradation, efficient removal of its yeast homologue requires at least some subunits of the 19S complex. Generally, however, it appears that although the initiator region of ODC “moved” from the N- to the C-terminus in the course of evolution, the intrinsic susceptibility for degradation, whether by 20S CP or 26SP, was maintained.89 Whether an internal IDR can be an efficient degradation initiator is still not certain [Fig. 3(D)]; but the open gate of 20S CP seems to be wide enough to accommodate two adjacent protein chains, as mentioned above, and circular protein constructs without N- or C-termini have been observed to be cleaved in vitro by the 20S CP.76
Summing up, many different studies suggest that IDRs are capable of constitutively destabilizing proteins by targeting them to 20S CP-dependent degradation without the need for prior modification. In other words, wholly disordered proteins are generally unstable by default.90 In proteins with both disordered and tightly ordered regions, the situation is more complicated. A single disordered region at one of the termini can target the whole protein for degradation, without the need for ATP-dependent unfolding. However, IDRs incapable of initiating degradation of a partially folded protein are also known, such as the C-terminal IDR of p53.
The role of intrinsic disorder in ATP- and Ub-dependent degradation by 26SP
Recent studies suggest a two-component degradation signal (degron) model for Ub-dependent degradation. Thus, not only a poly-Ub tag, but also an N- or C-terminal IDR of at least 20–34 aa,91–96 are both required for efficient targeting to 26SP. A long internal IDR may also be effective.97 A disordered conformation of such regions appears to be sufficient, but some sequence requirements have also been postulated.96 In some substrates, disordered initiator regions may be created by local unfolding triggered by the poly-Ub tag.98 Endoplasmic reticulum (ER) quality control substrates are probably denatured before 26SP-mediated degradation by p97 AAA+ ATP-dependent unfoldase, enabling them to bypass this requirement. However, when such a protein possesses a C-terminal IDR, it can be degraded in the absence of p97,99 further underscoring the importance of the initiator IDR. The poly-Ub tag and the IDR can be located on two different proteins bound to each other, but in this case only the protein possessing an IDR appears to be degraded.100
It was estimated that 24–34 aa in a random coil conformation span 50–70 Å, which is similar to the predicted distance between the entrance to the ATPase ring and the pore loops (60–70 A).101 Homologous loops in bacterial and archeal proteolytic complexes recognize linear sequence motifs of substrates, leading to unfolding and degradation. This suggests that the initiator IDR fulfills different roles in degradation by 26SP and 20S CP: for 26SP, interaction with the ATPase initiating unfolding is crucial, whereas for 20S CP the ability to pass through the gate is required [function iii in Fig. 2(C) and function ii in 2B, respectively]. Additionally, the IDR may contribute to the affinity of binding to the 26SP.94 Certain low-complexity IDRs (e.g., glycine-alanine repeats in EBNA1), when appropriately located in a protein, hinder degradation by a mechanism that is not fully understood.33,102,103
Although disorder is important for ATP-dependent degradation by the 26SP, it remains a tightly controlled process that is regulated by specific tagging of substrates. Substrates degraded by this pathway are stable by default, and are only destabilized upon poly-ubiquitination. It is true that, like other post-translational modifications, poly-ubiquitination sites are often observed within disordered regions.104,105 Examples are provided by cyclin B and securin, which are degraded during mitosis in a Ub-dependent fashion.106 But in these cases, and perhaps in general, disorder is not sufficient for polyubiquitination. These proteins are ubiquitinated primarily owing to specific sequential elements that they possess; and this only occurs when the E3 ligase capable of recognizing them, the APC/C complex, is assembled and active towards these particular substrates.107 IDRs (especially at N- or C-termini) may, however, suffice to target proteins to Ub-independent degradation by 26SP (See section “Proteasomal activators in Ub-independent IDP degradation”).
Intracellular half-lives of IDPs
A traditional approach to measuring protein half-lives in vivo, not so far applied to large protein sets, is the pulse-chase method: After pulse-labeling of the proteins, the decay of radioactivity of a particular protein is monitored using its cognate antibody or by measuring mass shifts of specific tryptic fragments.108 Another approach consists of tagging proteins, inhibiting translation (which has significant side effects), and monitoring the abundance of the tag over time. Belle et al.109 thus analyzed >3,750 S. cerevisiae proteins. Yet another method compares turnover of two fluorescently labeled proteins expressed from a single transcript, one of which is measured, and the other used as a reference, separated by an internal ribosome entry site. Using this technique, Yen et al.110 measured protein stability index values of ∼8,000 human proteins labeled with enhanced GFP. These values can be approximately correlated with protein half-lives. Both these large data sets (yeast109 and human110) have been used to analyze the correlation between protein half-life and intrinsic disorder. It was observed, using the yeast data set, that the fraction of residues predicted to be intrinsically disordered correlates with protein half-life, albeit very weakly, provided the data are normalized to protein length.111 A different analysis of the same data set examined the average protein half-life in three groups of proteins: highly structured (0–10% disordered residues), moderately unstructured (10–30%), and highly unstructured (30–100%). A significantly different distribution of half-lives between these three groups was observed, with highly unstructured proteins having, on average, shorter half-lives.12 The authors also examined the abundance of PEST motifs in the same three groups of proteins, and found more profound differences; but the relevance of PEST motifs to degradation is controversial.111 Yen et al.110 reported that they failed to find a correlation between protein instability and length or number of IDRs. Surprisingly, amino acids identified to be enriched in longer lived proteins partially overlap with disorder-promoting amino acids.105 In another study, all human proteins were divided into five bins according to the percentage of disordered residues ([0–20], [20–40], [40–60], [60–80], and [80–100]%); the average protein stability score in each bin was then analyzed. No correlation was observed, but the most highly disordered proteins [80–100%] had a significantly higher average protein stability index values than the others, suggesting that they have on average longer half-lives. In summary, no strong correlation between disorder and short in vivo half-lives, suggestive of a deterministic role of disorder in protein degradation, was observed in these analyses. This is in surprising contrast with the almost deterministic role of disorder observed for in vitro 20S CP-dependent degradation: indeed, we proposed sensitivity to this mode of degradation as a possible operational definition of IDPs.49
This discrepancy may in part stem from the bias introduced into half-life measurements by globular tags fused to one of the termini of the proteins analyzed. The authors of both the yeast and human studies rigorously validated their procedures by comparing their data with those obtained by other techniques, as well as with studies comparing the dynamics of tagged and untagged proteins in vivo; they mostly found relatively good agreement (see Supporting Information by Eden et al.21). However, these comparative studies were very limited in scope, and did not focus specifically on IDPs. Bearing in mind the importance of disordered regions at the termini of many known substrates of 20S CP, the effects of tagging might be expected to be more significant for IDPs than for other proteins. Upon tagging, a terminal IDR becomes an internal one and, as mentioned above, whether internal IDRs can interact productively with 20S CP is still controversial. It is also possible that in some cases incomplete degradation occurred, yielding the free tag domain, which could still be detected. Furthermore, the apparent special role of disordered termini in initiating degradation suggests that one should perhaps take into account only disorder in terminal regions, especially at the ends not blocked by the tag, not overall disorder, as was done in the available analyses.
Although it is important to rule out these different possibilities before reaching final conclusions, one should treat seriously the analyses performed so far. IDPs appear surprisingly longer-lived in vivo than expected bearing in mind their “default instability” observed in vitro, suggesting that in their native environments they are protected from degradation.
Protection of IDPs from degradation
Inside the cell, IDPs probably exist largely in a bound state. We were able to show49 that the cytoplasmic domain of neuroligin 3 could be prevented from 20S CP-mediated degradation in vitro by either S-SCAM or PSD95, with both of which it is known to form a complex.112,113 Forming a functional complex in vivo [Fig. 4(A)] thus provides a way to avoid degradation, either by promoting order in or masking IDRs. Stabilization is thus often a “by-product” of functional interactions in which IDPs are involved. However, some binding partners interact with IDPs solely or principally to prevent their degradation. Such proteins can be roughly divided into: (1) proteasome gatekeepers [Fig. 4(B)], which interact with both 20S CP and many IDPs to protect the latter; (2) nannies [Fig. 4(C)], which are usually more specific for particular IDPs. The term gatekeeper best fits NQO1, an ubiquitous enzyme involved in redox reactions. Its main function with respect to several IDPs with which it interacts in an NADH-dependent manner, is to protect them from degradation by 20S CP.114 NQO1 interacts with 20S CP, but not with 26SP.56 A yeast ortholog of NQO1, Lot6, serves a similar function. It has been suggested that degradation of the transcription factor, Yap4, is regulated by the redox state of the flavin cofactor of Lot6, which was thus termed a “redox switch” of the 20S yeast proteasome.115
Figure 4.
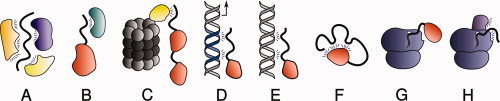
Mechanisms preventing disordered proteins from degradation by default. Proteins can be stabilized when involved in intermolecular interactions with (A) other members of the same stable complex (e.g., the cytoplasmic domain of neuroligin 3 with S-SCAM and PSD95); (B) a nanny protein (e.g., p53 with Hdmx); (C) a proteasome gatekeeper (e.g., p53 with NQO1); (C) a nanny protein (e.g., p53 with Hdmx); (D) a functional DNA binding site; (E) a nonfunctional “decoy” DNA binding site. F: Proteins can also be protected by intramolecular interactions (e.g., calcinin N). The nascent chain of an intrinsically disordered protein can be protected from degradation by (G) local folding, by interaction with a partner or (H) interacting with the ribosome or with ribosome-associated proteins.
An insight into the nanny model [Fig. 4(C)] is provided by the MDM2 and Hdmx proteins, which are homologous, and can both interact with the N termini of p53 and p73. MDM2, unlike Hdmx, displays E3 Ub ligase activity towards p53, but not towards p73. Binding of MDM2 to p53 leads to increased degradation of this protein via a Ub-dependent pathway, whereas its binding to p73 results in stabilization of the latter, by preventing its Ub-independent degradation. A mutation in MDM2 that prevents it from poly-ubiquitinating p53 results in its stabilizing both p53 and p73. Hdmx, like this inactive MDM2 mutant, also stabilizes the two proteins. Both Hdmx and MDM2 serve as nannies by virtue of interacting with the initiator IDR of p53; but MDM2 also displays Ub ligase activity towards p53 that overrides its protective role.116 The nanny action of MDM2 is specific for p53. Its interaction with both p21 and pRb enhances their degradation.55,117
IDPs can bind not only to other proteins, but also to nucleic acids. Indeed, many IDPs function as transcription factors. One well-studied example, Ets-1, is known to become more structured upon interaction with its cognate DNA.118,119 Many transcription factors are protected when bound to their binding regions on promoter or enhancer elements and actively involved in regulation of transcription [Fig. 4(D)]. Eukaryotic genomes also contain many “decoy” binding sites that can compete with functional regions for binding to transcription factors. It was recently proposed that one of their functions is to protect DNA-interacting IDPs from degradation120 [Fig. 4(E)]. There is some analogy between these “decoy” sites and nanny proteins since the main function of both appears to be regulation of the half-lives of IDPs.
Another mechanism for stabilizing IDPs involves intramolecular interactions that allow the protein to largely retain both its disorder (if only in the form of “floppy” loops) and the functional advantages associated with disorder. However, at the same time, this mechanism minimizes the number of relatively unconstrained long IDRs capable of accessing 20S CP [Fig. 4(F)]. If only the termini can function as initiation sites, then it would be sufficient if only they were blocked by such interactions. An antibiotic protein, colicin N, which contains a functionally important disordered T-domain, is sensitive to proteolytic degradation,121,122 yet can survive in the protease-rich environment of the animal host gut. It has been proposed that stabilization is achieved by intramolecular interaction of the T-domain with folded parts of colicin N.122 IDPs capable of being stabilized in such ways should not, perhaps, be called unstable by default.
If a protein has a disordered N-terminus, its degradation can potentially be initiated when it starts to emerge from the ribosomal channel, even if it becomes stabilized by an intramolecular interaction in the full-length protein. Simister and coworkers recently proposed that some IDPs might be stabilized by an N-terminal folded domain [Fig. 4(G)] that avoids degradation, and subsequently provides a platform on which the disordered remainder of the protein can gradually self-organize into a loopy structure [Fig. 4(F)].123 These authors used bioinformatics tools to identify over 50 human proteins that fitted their model. Nascent N-terminal IDRs could also, in principle, be protected by interactions with ribosomal proteins [Fig. 4(H)] until a longer sequence were synthesized that could be stabilized by a suitable nanny.
It is unlikely that chaperones are involved in protecting IDPs from degradation. On the basis of pairwise interactions recorded in the IntAct database, IDPs do not show preferential binding to chaperones, despite having on average a higher number of interactions relative to ordered proteins.124 Furthermore, chaperones might actually destabilize IDPs, consistent with their recently recognized role in chaperone-assisted degradation (both by 26SP and autophagy).125 It appears that folding-incompetent proteins, which are repetitively bound by chaperones, are more likely to encounter a degrading chaperone complex, and thus be degraded. Such degrading complexes contain E3 Ub ligases, including CHIP, Cul5, parkin, and E6-AP. Indeed, chaperones have been implicated in promoting degradation of several IDPs, including p53, Tau, and HIF-1α.125 In a recent study, an interaction between a C-terminal IDR of RGS9-2 and Hsc70 was reported to be responsible for RGS9-2 degradation following its dissociation from its functional binding partner, R7BP, which, unlike the chaperone, stabilizes RGS9-2.126
Summary and implications
Within eukaryotic cells, proteolytic active sites are sequestered from the intracellular environment by either a lipid membrane or a protein chamber; but one of the most abundant proteases, 20S CP, appears to be inherently accessible to IDPs, especially those with an IDR at one of their termini. Such proteins can enter through a narrow gate that opens in a stochastic manner. Thus IDPs are unstable by default, as opposed to globular proteins. The latter can only be degraded by 26SP when tagged with a poly-Ub chain, and are, therefore, stable by default. Despite this distinction, no strong correlation between intrinsic protein disorder and shorter protein half-lives has been reported. Although this may, in part, be due to limitations of half-life measurements, it may also reflect the existence of protective mechanisms.
Even if ordered and disordered proteins do not differ, on average, with respect to their half-lives, they do differ with respect to the mechanisms by which they are regulated: IDPs (unstable by default) need to be stabilized to remain undegraded, while globular proteins (stable by default) need to be destabilized to be degraded. This permits these two categories of proteins to serve different functions. Single-subunit housekeeping proteins, that are required at relatively constant levels during normal cell function, are more likely to be stable by default. By contrast, members of stable protein complexes can be expected to often be unstable by default. This results in their being degraded when not part of the complex (thus avoiding off-target effects), but being stable within it. As to proteins involved in highly dynamic processes, such as cell cycle control, signaling, apoptosis and transcription regulation, both “stable” and “unstable” proteins may be employed. This permits the function of various feedback loops and of other regulatory elements that contribute to the intricate networks operating within the living cell. For example, cell cycle regulators are degraded by various mechanisms. These may be 20S CP-mediated on the one hand, and 26SP-mediated, dependent on APC/C, SCF, and other Ub E3 ligases, on the other.
The sensitivity to proteolysis of IDPs in the absence of protective elements has a possible implication for protein evolution. It has been suggested that an evolutionary pathway from one rigid fold to another might, in some cases, involve a flexible or disordered intermediate state with a propensity for different conformations.127 However, if such a putative transition were to occur within eukaryotic cells, it would have to cope with the potentially increased proteolytic susceptibility of the intermediate.
Acknowledgments
The authors acknowledge support from the European Commission VIth Framework Research and Technological Development Program, “SPINE2-COMPLEXES” Project, under contract No. 031220, and “Teach-SG” Project, under contract No. ISSG-CT-2007-037198, and from the Divadol Foundation. They thank Maria Gaczynska and Pawel A. Osmulski for valuable discussions in the course of preparing the manuscript. J.L.S. is the incumbent of the Morton and Gladys Pickman Professorial Chair and Y.S. is the incumbent of the Oscar and Emma Getz Professorial Chair.
References
- 1.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 2.Dunker AK, Silman I, Uversky VN, Sussman JL. Function and structure of inherently disordered proteins. Curr Opin Struct Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 4.Zhou H. Protein folding in confined and crowded environments. Arch Biochem Biophys. 2008;469:76–82. doi: 10.1016/j.abb.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daniels AJ, Williams RJ, Wright PE. The character of the stored molecules in chromaffin granules of the adrenal medulla: a nuclear magnetic resonance study. Neuroscience. 1978;3:573–585. doi: 10.1016/0306-4522(78)90022-2. [DOI] [PubMed] [Google Scholar]
- 6.Bodart J, Wieruszeski J, Amniai L, Leroy A, Landrieu I, Rousseau-Lescuyer A, Vilain J, Lippens G. NMR observation of Tau in Xenopus oocytes. J Magn Reson. 2008;192:252–257. doi: 10.1016/j.jmr.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Mittag T, Marsh J, Grishaev A, Orlicky S, Lin H, Sicheri F, Tyers M, Forman-Kay JD. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol Cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Y, Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J Mol Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Bemporad F, Gsponer J, Hopearuoho HI, Plakoutsi G, Stati G, Stefani M, Taddei N, Vendruscolo M, Chiti F. Biological function in a non-native partially folded state of a protein. EMBO J. 2008;27:1525–1535. doi: 10.1038/emboj.2008.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tompa P. Structure and function of instrinsically disordered proteins. Boca Raton-London: CRC Press; 2010. [Google Scholar]
- 14.Patel SS, Belmont BJ, Sante JM, Rexach MF. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 2007;129:83–96. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 15.Denning DP, Patel SS, Uversky V, Fink AL, Rexach M. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc Natl Acad Sci USA. 2003;100:2450–2455. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellay J, Han S, Michaut M, Kim T, Costanzo M, Andrews BJ, Boone C, Bader GD, Myers CL, Kim PM. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011;12:R14. doi: 10.1186/gb-2011-12-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 18.Fontana A, Polverino de Laureto P, Spolaore B, Frare E, Zambonin M. Detecting disordered regions in proteins by limited proteolysis. In: Uversky V, Longhi S, editors. Assessing structures and conformations of intrinsically disordered proteins (Wiley Series in Protein and Peptide Science) New York: John Wiley and Sons; 2010. pp. 569–626. [Google Scholar]
- 19.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 20.Schimke RT, Doyle D. Control of enzyme levels in animal tissues. Annu Rev Biochem. 1970;39:929–976. doi: 10.1146/annurev.bi.39.070170.004433. [DOI] [PubMed] [Google Scholar]
- 21.Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, Cohen L, Mayo A, Alon U. Proteome half-life dynamics in living human cells. Science. 2011;331:764–768. doi: 10.1126/science.1199784. [DOI] [PubMed] [Google Scholar]
- 22.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 23.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 24.Groll M, Clausen T. Molecular shredders: how proteasomes fulfill their role. Curr Opin Struct Biol. 2003;13:665–673. doi: 10.1016/j.sbi.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Förster F, Lasker K, Nickell S, Sali A, Baumeister W. Toward an integrated structural model of the 26S proteasome. Mol Cell Proteomics. 2010;9:1666–1677. doi: 10.1074/mcp.R000002-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruschak AM, Religa TL, Breuer S, Witt S, Kay LE. The proteasome antechamber maintains substrates in an unfolded state. Nature. 2010;467:868–871. doi: 10.1038/nature09444. [DOI] [PubMed] [Google Scholar]
- 27.Tanahashi N, Murakami Y, Minami Y, Shimbara N, Hendil KB, Tanaka K. Hybrid proteasomes. Induction by interferon-gamma and contribution to ATP-dependent proteolysis. J Biol Chem. 2000;275:14336–14345. doi: 10.1074/jbc.275.19.14336. [DOI] [PubMed] [Google Scholar]
- 28.Brooks P, Fuertes G, Murray RZ, Bose S, Knecht E, Rechsteiner MC, Hendil KB, Tanaka K, Dyson J, Rivett J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem J. 2000;346(Part 1):155–161. [PMC free article] [PubMed] [Google Scholar]
- 29.Shibatani T, Carlson EJ, Larabee F, McCormack AL, Früh K, Skach WR. Global organization and function of mammalian cytosolic proteasome pools: implications for PA28 and 19S regulatory complexes. Mol Biol Cell. 2006;17:4962–4971. doi: 10.1091/mbc.E06-04-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 2011;41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tai H, Besche H, Goldberg AL, Schuman EM. Characterization of the brain 26S proteasome and its interacting proteins. Front Mol Neurosci. 2010;3:12. doi: 10.3389/fnmol.2010.00012. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deveraux Q, Ustrell V, Pickart C, Rechsteiner M. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem. 1994;269:7059–7061. [PubMed] [Google Scholar]
- 33.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Striebel F, Kress W, Weber-Ban E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol. 2009;19:209–217. doi: 10.1016/j.sbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Inobe T, Matouschek A. Protein targeting to ATP-dependent proteases. Curr Opin Struct Biol. 2008;18:43–51. doi: 10.1016/j.sbi.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gur E, Sauer RT. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008;22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burns KE, Darwin KH. Pupylation versus ubiquitylation: tagging for proteasome-dependent degradation. Cell Microbiol. 2010;12:424–431. doi: 10.1111/j.1462-5822.2010.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berger A, Schechter I. Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos Trans R Soc Lond B Biol Sci. 1970;257:249–264. doi: 10.1098/rstb.1970.0024. [DOI] [PubMed] [Google Scholar]
- 39.Keil B. Specificity of proteolysis. New York: Springer-Verlag, Berlin-Heidelberg; 1992. [Google Scholar]
- 40.Pace CN, Barrett AJ. Kinetics of tryptic hydrolysis of the arginine-valine bond in folded and unfolded ribonuclease T1. Biochem J. 1984;219:411–417. doi: 10.1042/bj2190411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neurath H. Limited proteolysis, protein folding and physiological regulation. In: Jaenicke R, editor. Protein folding. New York: Elsevier/North Holland Amsterdam; 1980. pp. 501–524. [Google Scholar]
- 42.Fontana A, Fassina G, Vita C, Dalzoppo D, Zamai M, Zambonin M. Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry. 1986;25:1847–1851. doi: 10.1021/bi00356a001. [DOI] [PubMed] [Google Scholar]
- 43.Hubbard SJ, Eisenmenger F, Thornton JM. Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 1994;3:757–768. doi: 10.1002/pro.5560030505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madala PK, Tyndall JDA, Nall T, Fairlie DP. Update 1 of: Proteases universally recognize beta strands in their active sites. Chem Rev. 2010;110:PR1–PR31. doi: 10.1021/cr900368a. [DOI] [PubMed] [Google Scholar]
- 45.Eldridge B, Cooley RN, Odegrip R, McGregor DP, Fitzgerald KJ, Ullman CG. An in vitro selection strategy for conferring protease resistance to ligand binding peptides. Protein Eng Des Sel. 2009;22:691–698. doi: 10.1093/protein/gzp052. [DOI] [PubMed] [Google Scholar]
- 46.Paz A, Zeev-Ben-Mordehai T, Sussman JL, Silman I. Purification of intrinsically disordered proteins. In: Uversky V, Longhi S, editors. Instrumental analysis of intrinsically disordered proteins: assessing structure and conformation (Wiley Series in Protein and Peptide Science) New York: John Wiley and Sons; 2010. pp. 695–704. [Google Scholar]
- 47.Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DCS, Day CL. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 48.Paz A, Zeev-Ben-Mordehai T, Lundqvist M, Sherman E, Mylonas E, Weiner L, Haran G, Svergun DI, Mulder FAA, Sussman JL, et al. Biophysical characterization of the unstructured cytoplasmic domain of the human neuronal adhesion protein neuroligin 3. Biophys J. 2008;95:1928–1944. doi: 10.1529/biophysj.107.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70:1357–1366. doi: 10.1002/prot.21614. [DOI] [PubMed] [Google Scholar]
- 50.Tofaris GK, Layfield R, Spillantini MG. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 51.Jariel-Encontre I, Pariat M, Martin F, Carillo S, Salvat C, Piechaczyk M. Ubiquitinylation is not an absolute requirement for degradation of c-Jun protein by the 26 S proteasome. J Biol Chem. 1995;270:11623–11627. doi: 10.1074/jbc.270.19.11623. [DOI] [PubMed] [Google Scholar]
- 52.Baugh JM, Pilipenko EV. 20S proteasome differentially alters translation of different mRNAs via the cleavage of eIF4F and eIF3. Mol Cell. 2004;16:575–586. doi: 10.1016/j.molcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 53.Kong X, Alvarez-Castelao B, Lin Z, Castaño JG, Caro J. Constitutive/hypoxic degradation of HIF-alpha proteins by the proteasome is independent of von Hippel Lindau protein ubiquitylation and the transactivation activity of the protein. J Biol Chem. 2007;282:15498–15505. doi: 10.1074/jbc.M700704200. [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–852. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sdek P, Ying H, Chang DLF, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao ZJ. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 56.Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19:316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baugh JM, Viktorova EG, Pilipenko EV. Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J Mol Biol. 2009;386:814–827. doi: 10.1016/j.jmb.2008.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 59.Groll M, Bajorek M, Köhler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome core particle. Nat Struct Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 60.Loidl G, Groll M, Musiol HJ, Ditzel L, Huber R, Moroder L. Bifunctional inhibitors of the trypsin-like activity of eukaryotic proteasomes. Chem Biol. 1999;6:197–204. doi: 10.1016/S1074-5521(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 61.Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 62.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomic20S proteasome reveals a molecular basis for selectivity of α′,β′-epoxyketone proteasome inhibitors. J Am Chem Soc. 2000;122:1237–1238. [Google Scholar]
- 63.Smith DM, Chang S, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's alpha ring opens the gate for substrate entry. Mol Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Förster A, Whitby FG, Hill CP. The pore of activated 20S proteasomes has an ordered 7-fold symmetric conformation. EMBO J. 2003;22:4356–4364. doi: 10.1093/emboj/cdg436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rivett AJ. Proteasomes: multicatalytic proteinase complexes. Biochem J. 1993;291(Part 1):1–10. doi: 10.1042/bj2910001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dahlmann B, Becher B, Sobek A, Ehlers C, Kopp F, Kuehn L. In vitro activation of the 20S proteasome. Enzyme Protein. 1993;47:274–284. doi: 10.1159/000468685. [DOI] [PubMed] [Google Scholar]
- 67.Kisselev AF, Kaganovich D, Goldberg AL. Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the alpha-rings. J Biol Chem. 2002;277:22260–22270. doi: 10.1074/jbc.M112360200. [DOI] [PubMed] [Google Scholar]
- 68.Leibovitz D, Koch Y, Pitzer F, Fridkin M, Dantes A, Baumeister W, Amsterdam A. Sequential degradation of the neuropeptide gonadotropin-releasing hormone by the 20 S granulosa cell proteasomes. FEBS Lett. 1994;346:203–206. doi: 10.1016/0014-5793(94)00472-2. [DOI] [PubMed] [Google Scholar]
- 69.Bajorek M, Finley D, Glickman MH. Proteasome disassembly and downregulation is correlated with viability during stationary phase. Curr Biol. 2003;13:1140–1144. doi: 10.1016/s0960-9822(03)00417-2. [DOI] [PubMed] [Google Scholar]
- 70.Osmulski PA, Gaczynska M. Atomic force microscopy reveals two conformations of the 20 S proteasome from fission yeast. J Biol Chem. 2000;275:13171–13174. doi: 10.1074/jbc.c901035199. [DOI] [PubMed] [Google Scholar]
- 71.Osmulski PA, Hochstrasser M, Gaczynska M. A tetrahedral transition state at the active sites of the 20S proteasome is coupled to opening of the alpha-ring channel. Structure. 2009;17:1137–1147. doi: 10.1016/j.str.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hutschenreiter S, Tinazli A, Model K, Tampé R. Two-substrate association with the 20S proteasome at single-molecule level. EMBO J. 2004;23:2488–2497. doi: 10.1038/sj.emboj.7600262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science. 2010;328:98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- 74.Wenzel T, Baumeister W. Conformational constraints in protein degradation by the 20S proteasome. Nat Struct Biol. 1995;2:199–204. doi: 10.1038/nsb0395-199. [DOI] [PubMed] [Google Scholar]
- 75.Förster A, Hill CP. Proteasome degradation: enter the substrate. Trends Cell Biol. 2003;13:550–553. doi: 10.1016/j.tcb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 76.Liu C, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O'Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell. 2007;26:831–842. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 78.Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai M, O'Malley BW. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;124:381–392. doi: 10.1016/j.cell.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 79.Nie J, Wu M, Wang J, Xing G, He F, Zhang L. REGgamma proteasome mediates degradation of the ubiquitin ligase Smurf1. FEBS Lett. 2010;584:3021–3027. doi: 10.1016/j.febslet.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 80.Suzuki R, Moriishi K, Fukuda K, Shirakura M, Ishii K, Shoji I, Wakita T, Miyamura T, Matsuura Y, Suzuki T. Proteasomal turnover of hepatitis C virus core protein is regulated by two distinct mechanisms: a ubiquitin-dependent mechanism and a ubiquitin- independent but PA28gamma-dependent mechanism. J Virol. 2009;83:2389–2392. doi: 10.1128/JVI.01690-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adler J, Reuven N, Kahana C, Shaul Y. c-Fos proteasomal degradation is activated by a default mechanism, and its regulation by NAD(P)H:quinone oxidoreductase 1 determines c-Fos serum response kinetics. Mol Cell Biol. 2010;30:3767–3778. doi: 10.1128/MCB.00899-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bell S, Klein C, Müller L, Hansen S, Buchner J. p53 contains large unstructured regions in its native state. J Mol Biol. 2002;322:917–927. doi: 10.1016/s0022-2836(02)00848-3. [DOI] [PubMed] [Google Scholar]
- 83.Tsvetkov P, Reuven N, Prives C, Shaul Y. Susceptibility of p53 unstructured N terminus to 20 S proteasomal degradation programs the stress response. J Biol Chem. 2009;284:26234–26242. doi: 10.1074/jbc.M109.040493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Navon A, Goldberg AL. Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell. 2001;8:1339–1349. doi: 10.1016/s1097-2765(01)00407-5. [DOI] [PubMed] [Google Scholar]
- 85.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- 86.Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- 87.Peña MMO, Melo SP, Xing Y, White K, Barbour KW, Berger FG. The intrinsically disordered N-terminal domain of thymidylate synthase targets the enzyme to the ubiquitin-independent proteasomal degradation pathway. J Biol Chem. 2009;284:31597–31607. doi: 10.1074/jbc.M109.038455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Basbous J, Chalbos D, Hipskind R, Jariel-Encontre I, Piechaczyk M. Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabilizer. Mol Cell Biol. 2007;27:3936–3950. doi: 10.1128/MCB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gödderz D, Schäfer E, Palanimurugan R, Dohmen RJ. The N-terminal unstructured domain of yeast ODC functions as a transplantable and replaceable ubiquitin-independent degron. J Mol Biol. 2011;407:354–367. doi: 10.1016/j.jmb.2011.01.051. [DOI] [PubMed] [Google Scholar]
- 90.Asher G, Reuven N, Shaul Y. 20S proteasomes and protein degradation “by default”. Bioessays. 2006;28:844–849. doi: 10.1002/bies.20447. [DOI] [PubMed] [Google Scholar]
- 91.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 92.Takeuchi J, Chen H, Coffino P. Proteasome substrate degradation requires association plus extended peptide. EMBO J. 2007;26:123–131. doi: 10.1038/sj.emboj.7601476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Verhoef LGGC, Heinen C, Selivanova A, Halff EF, Salomons FA, Dantuma NP. Minimal length requirement for proteasomal degradation of ubiquitin-dependent substrates. FASEB J. 2009;23:123–133. doi: 10.1096/fj.08-115055. [DOI] [PubMed] [Google Scholar]
- 94.Shabek N, Herman-Bachinsky Y, Ciechanover A. Ubiquitin degradation with its substrate, or as a monomer in a ubiquitination-independent mode, provides clues to proteasome regulation. Proc Natl Acad Sci USA. 2009;106:11907–11912. doi: 10.1073/pnas.0905746106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shabek N, Iwai K, Ciechanover A. Ubiquitin is degraded by the ubiquitin system as a monomer and as part of its conjugated target. Biochem Biophys Res Commun. 2007;363:425–431. doi: 10.1016/j.bbrc.2007.08.185. [DOI] [PubMed] [Google Scholar]
- 96.Zhao M, Zhang N, Zurawel A, Hansen KC, Liu C. Degradation of some polyubiquitinated proteins requires an intrinsic proteasomal binding element in the substrates. J Biol Chem. 2010;285:4771–4780. doi: 10.1074/jbc.M109.060095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat Commun. 2011;2:192. doi: 10.1038/ncomms1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hagai T, Levy Y. Ubiquitin not only serves as a tag but also assists degradation by inducing protein unfolding. Proc Natl Acad Sci USA. 2010;107:2001–2006. doi: 10.1073/pnas.0912335107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aviram S, Kornitzer D. The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol Cell Biol. 2010;30:985–994. doi: 10.1128/MCB.00909-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prakash S, Inobe T, Hatch AJ, Matouschek A. Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol. 2009;5:29–36. doi: 10.1038/nchembio.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Inobe T, Fishbain S, Prakash S, Matouschek A. Defining the geometry of the two-component proteasome degron. Nat Chem Biol. 2011;7:161–167. doi: 10.1038/nchembio.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Piwko W, Jentsch S. Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat Struct Mol Biol. 2006;13:691–697. doi: 10.1038/nsmb1122. [DOI] [PubMed] [Google Scholar]
- 103.Daskalogianni C, Apcher S, Candeias MM, Naski N, Calvo F, Fåhraeus R. Gly-Ala repeats induce position- and substrate-specific regulation of 26 S proteasome-dependent partial processing. J Biol Chem. 2008;283:30090–30100. doi: 10.1074/jbc.M803290200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Edwards YJK, Lobley AE, Pentony MM, Jones DT. Insights into the regulation of intrinsically disordered proteins in the human proteome by analyzing sequence and gene expression data. Genome Biol. 2009;10:R50. doi: 10.1186/gb-2009-10-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cox CJ, Dutta K, Petri ET, Hwang WC, Lin Y, Pascal SM, Basavappa R. The regions of securin and cyclin B proteins recognized by the ubiquitination machinery are natively unfolded. FEBS Lett. 2002;527:303–308. doi: 10.1016/s0014-5793(02)03246-5. [DOI] [PubMed] [Google Scholar]
- 107.van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786:49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 108.Pratt JM, Petty J, Riba-Garcia I, Robertson DHL, Gaskell SJ, Oliver SG, Beynon RJ. Dynamics of protein turnover, a missing dimension in proteomics. Mol Cell Proteomics. 2002;1:579–591. doi: 10.1074/mcp.m200046-mcp200. [DOI] [PubMed] [Google Scholar]
- 109.Belle A, Tanay A, Bitincka L, Shamir R, O'Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci USA. 2006;103:13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yen HS, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- 111.Tompa P, Prilusky J, Silman I, Sussman JL. Structural disorder serves as a weak signal for intracellular protein degradation. Proteins. 2008;71:903–909. doi: 10.1002/prot.21773. [DOI] [PubMed] [Google Scholar]
- 112.Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y. A novel multiple PDZ domain-containing molecule interacting with N-methyl-D-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem. 1998;273:21105–21110. doi: 10.1074/jbc.273.33.21105. [DOI] [PubMed] [Google Scholar]
- 113.Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Südhof TC. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- 114.Tsvetkov P, Reuven N, Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010;17:103–108. doi: 10.1038/cdd.2009.67. [DOI] [PubMed] [Google Scholar]
- 115.Sollner S, Schober M, Wagner A, Prem A, Lorkova L, Palfey BA, Groll M, Macheroux P. Quinone reductase acts as a redox switch of the 20S yeast proteasome. EMBO Rep. 2009;10:65–70. doi: 10.1038/embor.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tsvetkov P, Reuven N, Shaul Y. The nanny model for IDPs. Nat Chem Biol. 2009;5:778–781. doi: 10.1038/nchembio.233. [DOI] [PubMed] [Google Scholar]
- 117.Xu H, Zhang Z, Li M, Zhang R. MDM2 promotes proteasomal degradation of p21Waf1 via a conformation change. J Biol Chem. 2010;285:18407–18414. doi: 10.1074/jbc.M109.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pufall MA, Lee GM, Nelson ML, Kang H, Velyvis A, Kay LE, McIntosh LP, Graves BJ. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 119.Levy Y, Onuchic JN, Wolynes PG. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J Am Chem Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 120.Burger A, Walczak AM, Wolynes PG. Abduction and asylum in the lives of transcription factors. Proc Natl Acad Sci USA. 2010;107:4016–4021. doi: 10.1073/pnas.0915138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vetter IR, Parker MW, Tucker AD, Lakey JH, Pattus F, Tsernoglou D. Crystal structure of a colicin N fragment suggests a model for toxicity. Structure. 1998;6:863–874. doi: 10.1016/s0969-2126(98)00088-4. [DOI] [PubMed] [Google Scholar]
- 122.Hecht O, Ridley H, Boetzel R, Lewin A, Cull N, Chalton DA, Lakey JH, Moore GR. Self-recognition by an intrinsically disordered protein. FEBS Lett. 2008;582:2673–2677. doi: 10.1016/j.febslet.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 123.Simister PC, Schaper F, O'Reilly N, McGowan S, Feller SM. Self-organization and regulation of intrinsically disordered proteins with folded N-termini. PLoS Biol. 2011;9:e1000591. doi: 10.1371/journal.pbio.1000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hegyi H, Tompa P. Intrinsically disordered proteins display no preference for chaperone binding in vivo. PLoS Comput Biol. 2008;4:e1000017. doi: 10.1371/journal.pcbi.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kettern N, Dreiseidler M, Tawo R, Höhfeld J. Chaperone-assisted degradation: multiple paths to destruction. Biol Chem. 2010;391:481–489. doi: 10.1515/BC.2010.058. [DOI] [PubMed] [Google Scholar]
- 126.Posokhova E, Uversky V, Martemyanov KA. Proteomic identification of Hsc70 as a mediator of RGS9-2 degradation by in vivo interactome analysis. J Proteome Res. 2010;9:1510–1521. doi: 10.1021/pr901022m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bornberg-Bauer E, Huylmans A, Sikosek T. How do new proteins arise? Curr Opin Struct Biol. 2010;20:390–396. doi: 10.1016/j.sbi.2010.02.005. [DOI] [PubMed] [Google Scholar]