

The synthesis of fatty acids is an essential primary metabolic process for energy storage and cellular structural integrity. Assembly of saturated fatty acids is achieved by fatty acid synthases (FASs) that combine acetyl- and malonyl-CoAs by repetitive decarboxylative Claisen condensations with subsequent reduction and dehydration steps.[1] In mammals seven catalytic domains are encoded by a single gene, giving rise to an α2-homodimeric protein. An acyl-carrier protein (ACP) that is post-translationally modified with a CoA derived arm by a phosphopantetheinyltransferase (PPT), tethers the growing fatty acid via a thioester linkage during the iterative catalytic cycle. Such attachment to the holo-ACP arm fosters high effective substrate concentrations during the synthesis. In fungi, however, eight catalytic domains are divided between two subunits, and an architecturally distinct α6β6 canonical complex releases the final product as a CoA ester rather than as a free-acid, as occurs with animal FASs.
Several examples are known in fungi where dedicated FASs have evolved to interact with polyketide synthases (PKSs) in secondary metabolic pathways.[2] For example, norsolorinic acid synthase (NorS) is comprised of a pair of fatty acid subunits, HexA and HexB,[3] that synthesize a C6-fatty acid starter unit to prime the associated PKS, PksA, in the formation of the aflatoxin (1) precursor, norsolorinic acid (2). (Scheme 1) These subunits associate into an approximately 1.4 MDa species as estimated by size exclusion chromatography, and they are thought to form an α2β2γ2 complex that is quite distinct from the FAS of primary fungal metabolism.[4] Hexanoyl-CoA was not detected as a free intermediate in in vitro assays, suggesting, but not proving, that a direct transfer could take place between the FAS and PKS subunits. A starter unit:ACP transacylase (SAT) domain in the accompanying PKS was identified that exhibited C6-chain length specificity and catalyzed transfer to the PksA ACP to bridge fatty acid and polyketide synthesis.[5] Such drastic differences in the protein organization of primary and secondary metabolic FASs reflect different evolutionary histories.
Scheme 1.

Major advances have been made recently in elucidating the structures of FASs from primary fungal metabolism.[6–9] The α6β6 complex harbors six distinct reaction compartments where the ACPs lie inside each compartment and are positioned to deliver the elongating substrate to the deep-seated active sites of the remaining catalytic domains, which are organized in an accessible circular fashion.[7, 8] The apo-ACPs must be activated by a PPT to the holo-form containing the approximately 18 Å 4′-phosphopantetheine arm derived from CoA to extend the acyl intermediates down into these active sites. In addition, the ACP domain itself has flexible upstream and downstream linker sequences that allow effective substrate transfer of the tethered intermediates among the catalytic domains. The PPT domain, however, is external to the rigid, tightly associated complex and is unable to contact its cognate ACP for activation.[9] In agreement with biochemical studies this quaternary organization necessitates activation prior to complex association.[10] Consequently, the ACP and PPT reside in the same α-subunit. Such organization would be expected to facilitate efficient intramolecular activation. (Figure 1) It is not clear, however, which ACP/PPT activation partnerships occur in FAS/PKS hybrid complexes where multiple ACPs are present, but only one PPT resides on the FAS.
Figure 1. Domain organization of FAS α- and β-subunits.
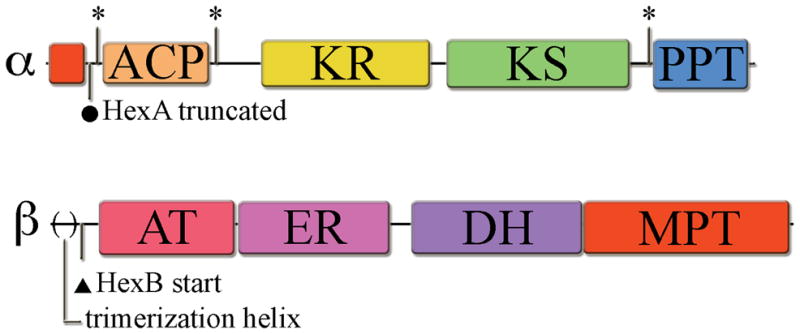
Domains include: acyl-carrier protein (ACP), ketoreductase (KR), ketosynthase (KS), phosphopantetheinyltransferase (PPT), acetyl transacylase (AT), enoyl reductase (ER), dehydrase (DH), and malonyl-palmitoyl transacylase (MPT). (*) indicates cloning sites used within the flexible linker regions surrounding the catalytic domains. (●)indicates the old predicted start site for hexA, which resulted in a truncated gene product. (▲) indicates the predicted hexB start site downstream of the N-terminal structural helix involved in β-subunit trimerization in the primary metabolism FAS. The Aspergillus parasiticus HexA (revised) and HexB proteins (GenBank accession # AY371490) share significant sequence homology to the recently determined crystal structure of similar subunits from Thermomyces lanuginosus (α-subunit, 2UV9_A, 39.4/56.7/11.5 % id/sim/gap, global alignment, http://www.ebi.ac.uk/emboss/align/, and β-subunit, 2UVA_G, 38.1/54.2/11.5, respectively). For a detailed view of domain organization including sequence expansion segments see the T. lanuginosus crystal structure reference [7].
The structures of the 2.6 MDa α6β6 fungal primary metabolic FASs have yielded deeper understanding of the mechanism of fungal fatty acid biosynthesis. The N-terminus of the fatty acid α subunit (HexA homolog, 56.7% global similarity) complements the malonyl/palmitoyl transacylase (MPT) catalytic domain and acts as the upstream ACP anchor.[7, 8] In the current description of the HexA protein sequence, such a domain is not present, raising concern that the HexA encoding sequences as currently described are incorrect. Alignment of the primary metabolic β-subunit and HexB revealed that the latter described sequence lacks the N-terminal trimerization helix. This helix forms extensive β3 interactions in the primary metabolic FAS likely essential for α6β6 dodecamer formation.[7] Contrary to the HexA encoded sequence, loss of this structural element in HexB plausibly contributes to the marked difference in structural organization between primary and secondary metabolic FASs. Inspection of the small intergenic region between hexA and hexB revealed that two very small exons could potentially code for the additional region in HexA. Previously, a 5′-RACE experiment carried out on RNA isolated at 48 hours supported the presence of only the truncated version of hexA that is missing these exons.[3] Other descriptions of hexA orthologs annotated in GenBank similarly lack these N-terminal coding sequences. Early genes in aflatoxin production are known to be turned on during early growth phase,[11] so an earlier 24-hour RNA time point was selected for mRNA analysis in the present study. Reverse transcription near the 5′-end of hexA from A. flavus and A. parasiticus was carried out, and the results revealed that alternative splicing than previously described, indeed, is occurring. (Figure 2) 5′-RACE analysis on hexA in A. parasiticus further demonstrated that the 24-hour RNA has a transcriptional start point 58 bp upstream to the new predicted translational start point. By homology to the primary FASs, this region encodes the complementary part of the MPT domain and the ACP anchor sequence. However, the 48-hour RNA time point did not yield any PCR product in this study, suggesting that mRNA is either not made at the later time or, if made, is in insufficient amount to be detected by the rather sensitive RACE PCR techniques that were used. By sequence comparison, it is probable that other hexA homologs in other organisms that produce products related to aflatoxin biosynthetic precursors, for example, A. nidulans and A. nomius, share this transcription pattern.
Figure 2. Splicing pattern for hexA. A) Revised transcript pattern B) 5′-transcript end.

The 5′-transcript sequence 58 bp upstream of the new predicted translational start site for A. parasiticus is shown. (#1) = the transcriptional start point as determined by RACE analysis in this study. C) Splicing pattern for the first three exons. A. parasiticus and A. flavus transcript splicing were determined in this study. The first ATG downstream from the revised transcriptional start point begins the sequence listed. A. nomius and A. nidulans were inferred by sequence comparison. (#2) = former transcriptional start point (truncated).
RACE analysis on hexB in A. parasiticus corroborated earlier assignments, which lack the trimerization helix. Furthermore, clones of the N-terminal acetyl-transacylase (AT) domain in HexB utilizing the currently described translational start point yielded a soluble, competent monodomain that exhibited specificity for acetyl-CoA, but not malonyl-CoA (unpublished). In addition to the role of AT domains to introduce acetyl starter units, as in primary metabolic FASs, proper protein folding of this N-terminal domain supports the currently described translational start point for HexB.
The high degree of homology among secondary metabolic FAS α-subunits and the incorporation of this revised N-terminal coding sequence implies that this sequence is required for FAS function. During the release of palmitoyl-CoA in primary metabolic FASs, the MPT domain is occupied by the palmitoyl-chain, blocking malonyl binding.[9] This allows the AT to bind the acetyl starter unit to initiate another cycle of synthesis. Averufin, an intermediate in the aflatoxin biosynthetic pathway, was determined to exhibit the acetyl starter unit effect in 13C-labeling studies.[12, 13] Combined with the AT specificity as described above suggests that a similar level of cooperativity likely occurs in secondary metabolic FAS subunits as well. The homologous MPT-like domain in the secondary metabolic FAS subunits could be restricted to malonyl transfer for chain extension and a direct transfer of the C6-ACP starter unit to the PksA SAT domain could occur. Or, this domain could have dual functionality, like the primary metabolic FASs, and act as a malonyl-hexanoyl transacylase (MHT), shuttling hexanoyl-CoA as a substrate for the PKS SAT domain.[5]
In aflatoxin-producing species the genes involved in biosynthesis predominantly reside in a 75 kb cluster.[14] Association of HexA, HexB, and PksA in the NorS complex led us to investigate in more detail the ACP/PPT partnerships required for initiation of aflatoxin biosynthesis. From the revised hexA cDNA sequence as described above, the ACPHexA and PPTHexA monodomains were cloned, expressed in E. coli, and purified using Ni NTA-Sepharose chromatography for in vitro activation studies. Cloning sites were selected based upon application of the Udwary-Merski algorithm (UMA [15]) to the revised HexA homologs in conjunction with the primary metabolic FAS α-subunits. UMA combines primary sequence similarity, local hydrophobicity, and structural similarity to assess probable linker regions that occur between domains. Sequence expansion segments fused to fungal FAS catalytic domains revealed in the recently determined crystal structures complicated analysis to parse apart the linkers between all of the domains in the primary FASs. Selection of truncation sites for the HexA ACP and PPT, however, were in good agreement with the structures, and fall directly within the flexible linker regions found in the homologous primary metabolic FASs. Notably, the upstream ACP linker occurred just downstream of the predicted ACP anchoring attachment point in the revised HexA sequence. The ACPHexA was purified as a mixture of two inactive apo-species as determined by MALDI-TOF mass spectrometry. (Table 1) Both species lacked the starter fMet, although partial modification by α-N-6-phosphogluconoylation occured, which often accompanies N-terminally His6x-tagged proteins in E. coli.[16] The dephosphorylated α-N-D-gluconoyl-group shows an additional mass of 178 Da. The purified ACPHexA species could not be activated by the promiscuous bacterial PPT Svp isolated from the bleomycin gene cluster in Streptomyces verticillus.[17] The PPTHexA monodomain, however, was able to transfer the 4′-phosphopantetheine moiety (340 Da) onto its cognate apo-ACPHexA monodomains to generate the active holo-ACPHexA forms. The dissected fragments were not subject to any restricted motions that could occur in the associated NorS complex, and, thus, were freely able to carry out activation. Earlier attempts to analyze ACP activation of dissected domains from the primary metabolic yeast FAS were made to no avail.[18] Due to the sequence similarity between primary and secondary FASs, this was likely due to the selection of alternative truncation sites.
Table 1.
ACPHexA MALDI-TOF results.
| Species | calculated | m/z |
|---|---|---|
| (−)fMet apo-ACP | 25024 | 25022 |
| (−)fMet apo-ACP modified | 25202 | 25202 |
| (−)fMet holo-ACP | 25364 | 25368 |
| (−)fMet holo-ACP modified | 22542 | 22543 |
The PPTHexA was not able to transfer the 4′-phosphopantetheine moiety onto the partner PKS ACPPksA. Although, the FAS and PKS proteins form a complex, the PKS must be activated by an alternative PPT. Deletion of a discretely expressed PPT (npgA) in A. nidulans, which produces sterigmatocystin, an advanced intermediate in aflatoxin biosynthesis, destroys sterigmatocystin production.[19] This led us to seek the native npgA homolog in A. parasiticus. A single PPT, NpgA, in Aspergillus nidulans was shown to be responsible for ACP activation of both primary and secondary metabolic pathways including pigment, lysine,[20] siderophore,[21] and penicillin,[22] in addition to sterigmatocystin biosynthesis. The discretely expressed, promiscuous enzyme has been proposed to be the sole PPT required for all PKS and non-ribosomal peptide synthetase (NRPS) metabolism, and, consequently, controls asexual development in A. nidulans.[23] The A. parasiticus homolog of A. nidulans npgA was cloned, expressed, and purified to determine if it has a role in ACP activation in the aflatoxin biosynthetic pathway. While we were searching for the A. parasiticus npgA gene, the genome sequence of the similar fungus A. oryzae was released. Consequently, the full-length A. parasiticus npgA was amplified based on this genome sequence information.[24] The one exon coding sequence was inserted into pET24a to create the C-terminal His6x-tag fusion construct, pET-NpgA. The full-length npgA was expressed in BL21(DE3) E. coli cells and purified by Ni NTA-Sepharose chromatography to examine its role in ACP activation.
In order to easily analyze all ACP/PPT combinations, a biotin-CoA conjugate [25, 26] was used to visualize the reactions in a far-Western blot with a streptavidin-horseradish peroxidase conjugate. (Figure 3) The promiscuous bacterial PPT, Svp, was only able to activate the PKS apo-ACPPksA, a finding that had been demonstrated previously.[5] The universal fungal PPT, NpgA, was more broadly active and could activate both the PKS apo-ACPPksA, and the FAS apo-ACPHexA. The FAS PPTHexA, however, could only activate its cognate ACPHexA, supporting the MALDI-TOF results described above. This result suggests that specific ACP/PPT interactions are required in the secondary metabolic fungal FASs, and the promiscuity of NpgA is not limited to PKS and NRPS enzymes in vitro. Mutagenesis of the C-terminal PPT domain in the homologous yeast FAS α-subunit led to undetectable incorporation of the CoA derived arm in the N-terminal apo-ACP domain.[10] By analogy, NpgA activation of the FAS ACPs is likely not the primary activation pathway in vivo.
Figure 3. Western blot showing ACP/PPT activation partnerships. Top) PksA ACP activation.

Lanes 1. molecular mass references 2. Svp 3. PPTHexA 4. NpgA 5. apo-ACPPksA 6. apo-ACPPksA / Svp 7. apo-ACPPksA / PPTHexA 8. apo-ACPPksA / NpgA. Bottom) HexA ACP activation. Lanes 1. molecular mass references 2. Svp 3. PPTHexA 4. NpgA 5. apo-ACPHexA 6. apo-ACPHexA / Svp 7. apo-ACPHexA / PPTHexA 8. apo-ACPHexA / NpgA. The blot was treated with streptavidin-horseradish peroxidase conjugate to detect biotinylated-ACPs due to PPT activation.
Fatty acid biosyntheses are critical cellular processes both for cell survival, and the production of intermediates dedicated to secondary metabolic pathways. Determination of the transcription pattern for hexA revealed that an alternative translated protein sequence was produced to afford an N-terminus similar to that of the primary metabolic FASs, which is necessary for protein function and ACP anchoring. Bioinformatics analysis of the revised sequences led to the production and isolation of individual dissected ACPHexA and PPTHexA monodomains. The discretely expressed PPT NpgA was found to be capable of activating both the FAS and the PKS ACP domains in vitro, further expanding its known roles in carrier protein activation. The universal activity of NpgA provides a route for PksA ACP activation in the biosynthesis of aflatoxin (1). The FAS PPT domain, however, exhibited specificity only for its cognate ACP domain. This specific activation partnership demonstrates that fungal FASs evolved to self-pantetheinylate to ensure proper ACP activation in both primary, and secondary metabolic pathways.
Experimental
Full experimental details can be found online in Supporting Information.
Supplementary Material
Acknowledgments
We thank Dr. D. M. Bartley for the synthesis of the biotin-CoA conjugate. We also thank Drs. M. Leibundgut, S. Jenni, and N. Ban for alerting us to the possible alternative transcription pattern for hexA. This work was supported by the National Institutes of Health Grant ES 001670 and the United States Department of Agriculture Grant USDA-SCA-58-6435-6-020.
References
- 1.Smith S, Tsai SC. Nat Prod Rep. 2007;24:1041. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown DW, Adams TH, Keller NP. Proc Natl Acad Sci U S A. 1996;93:14873. doi: 10.1073/pnas.93.25.14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hitchman T, Schmidt E, Trail F, Rarick M, Linz J, Townsend C. Bioorg Chem. 2001;29:293. doi: 10.1006/bioo.2001.1216. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe C, Townsend C. Chem Biol. 2002;9:981. doi: 10.1016/s1074-5521(02)00213-2. [DOI] [PubMed] [Google Scholar]
- 5.Crawford JM, Dancy BCR, Hill EA, Udwary DW, Townsend CA. Proc Natl Acad Sci U S A. 2006;103:16728. doi: 10.1073/pnas.0604112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jenni S, Leibundgut M, Maier T, Ban N. Science. 2006;311:1263. doi: 10.1126/science.1123251. [DOI] [PubMed] [Google Scholar]
- 7.Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Science. 2007;316:254. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- 8.Leibundgut M, Jenni S, Frick C, Ban N. Science. 2007;316:288. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- 9.Lomakin IB, Xiong Y, Steitz TA. Cell. 2007;129:319. doi: 10.1016/j.cell.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Fichtlscherer F, Wellein C, Mittag M, Schweizer E. Eur J Biochem. 2000;267:2666. doi: 10.1046/j.1432-1327.2000.01282.x. [DOI] [PubMed] [Google Scholar]
- 11.Klich MA, Montalbano B, Ehrlich K. Appl Microbiol Biotechnol. 1997;47:246. [Google Scholar]
- 12.Chandler M, Simpson TJ. Chem Commun. 1987:17. [Google Scholar]
- 13.Brobst SW, Townsend CA. Can J Chem. 1994;72:200. [Google Scholar]
- 14.Yu J, Chang PK, Ehrlich KC, Cary JW, Bhatnagar D, Cleveland TE, Payne GA, Linz JE, Woloshuk CP, Bennett JW. Appl Environ Microbiol. 2004;70:1253. doi: 10.1128/AEM.70.3.1253-1262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Udwary D, Merski M, Townsend C. J Mol Biol. 2002;323:585. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geoghegan KF, Dixon HBF, Rosner P, Hoth LR, Lanzetti AJ, Borzilleri KA, Marr ES, Pezzullo LH, Martin LB, LeMote PK, McColl AS, Kamath AV, Stroh JG. Anal Biochem. 1999;267:169. doi: 10.1006/abio.1998.2990. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez C, Du L, Edwards DJ, Toney MD, Shen B. Chem Biol. 2001;8:725. doi: 10.1016/s1074-5521(01)00047-3. [DOI] [PubMed] [Google Scholar]
- 18.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chem Biol. 1996;3:923. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 19.Guzman-de-Pena D, Aguirre J, Ruiz-Herrera J. Antonie Van Leeuwenhoek. 1998;73:199. doi: 10.1023/a:1000820221945. [DOI] [PubMed] [Google Scholar]
- 20.Mootz HD, Schorgendorfer K, Marahiel MA. FEMS Microbiol Lett. 2002;213:51. doi: 10.1111/j.1574-6968.2002.tb11285.x. [DOI] [PubMed] [Google Scholar]
- 21.Oberegger H, Eisendle M, Schrettl M, Graessle S, Haas H. Curr Genet. 2003;44:211. doi: 10.1007/s00294-003-0434-z. [DOI] [PubMed] [Google Scholar]
- 22.Keszenman-Pereyra D, Lawrence S, Twfieg ME, Price J, Turner G. Curr Genet. 2003;43:186. doi: 10.1007/s00294-003-0382-7. [DOI] [PubMed] [Google Scholar]
- 23.Marquez-Fernandez O, Trigos A, Ramos-Balderas JL, Viniegra-Gonzalez G, Deising HB, Aguirre J. Eukaryot Cell. 2007;6:710. doi: 10.1128/EC.00362-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H. Nature. 2005;438:1157. doi: 10.1038/nature04300. [DOI] [PubMed] [Google Scholar]
- 25.Johnsson K, George N. WO 2004 / 104588 A1. patent. 2004
- 26.La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Chem Biol. 2004;11:195. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.