

Abstract
Two sets of ligand binding decoys have been constructed for the CSAR (Community Structure-Activity Resource) benchmark by using the MDock and DOCK programs for rigid-ligand and flexible-ligand docking, respectively. The decoys generated for each complex in the benchmark thoroughly cover the binding site and also contain a certain number of near-native binding modes. A few scoring functions have been evaluated using the ligand binding decoy sets for their abilities of predicting near-native binding modes. Among them, ITScore achieved a success rate of 86.7% for the rigid-ligand decoys and 79.7% for the flexible-ligand decoys, under the common definition of a successful prediction as RMSD < 2.0 Å from the native structure if the top-scored binding mode was considered. The decoy sets may serve as benchmarks for binding mode prediction of a scoring function, which are available at the CSAR website (http://www.csardock.org/).
Keywords: molecular docking, scoring function, CSAR benchmark, binding mode, knowledge-based
1 Introduction
Performance assessment is an important issue in the development of scoring functions for structure-based drug design.1–7 Usually, the performance can be assessed from three aspects: binding mode prediction, binding affinity prediction, and database screening.1 The binding mode prediction is to evaluate the ability of a scoring function to identify experimentally determined native structures in a set of diverse putative binding modes. The binding affinity prediction is to assess the ability to correctly predict the binding tightness between a protein and a ligand, for example, the correlation between the experimentally measured binding affinities and the computationally calculated binding energy scores. A perfect scoring function should give a correlation coefficient of 1.0. Lastly, the virtual database screening is to test the reliability of a scoring function to distinguish true binders from a set of decoy compounds for a given target protein.
These three aspects are closely related to each other and a perfect scoring function should perform equally well on all of them. Unfortunately, current scoring functions are far from perfection, and many of them are specialized or designed for only one or two of the three aspects/purposes.1,8–10 Achieving excellent performance on all the three criteria (i.e., binding mode prediction, binding affinity prediction and database screening) is challenging.
A variety of test sets have been developed for the assessment of scoring functions on mode prediction, affinity prediction or database screening.8,11–20 These test sets are valuable assets for the molecular docking community, but also pose difficulty for comparative analysis of existing scoring functions as different research groups use different test sets. There is a pressing need for the scoring community to have access to common, high-quality and publicly available scoring benchmarks so that the assessments can be performed on the same baseline. Lessons learned from scoring these benchmarks will provide valuable insights into how to improve the existing scoring methods and how to develop novel approaches. Recently, Dr. Heather Carlson at the University of Michigan, Ann Arbor has been leading an NIH-sponsored big project on constructing such benchmarks named CSAR (Community Structure-Affinity Resource, http://www.csardock.org/). The first CSAR benchmark was released in 2009, which consists of 345 diverse protein-ligand complexes with high-resolution structures and known affinities. Particularly, the crystal structures of the complexes in the benchmark were carefully examined and any inconsistencies with the electron density maps were fixed by recalculating the related atomic coordinates by the CSAR team (http://www.csardock.org/).
In an accompanying work,21 we have assessed the ability of our ITScore scoring function on binding affinity prediction for the CSAR benchmark. In the present study, we have constructed decoys of ligand binding modes for every complex in the CSAR benchmark. The motivations are as follows: First, a good performance on affinity prediction does not necessarily warrants the scoring function to be capable of discriminating near-native binding modes from non-native binding modes in the decoys.1 For example, scoring functions that are mainly based on contact scores may favor any saltbridge formation without considering the desolvation penalty and van der Waals (VDW) repulsion, leading to an unsatisfactory performance in binding mode prediction. Second, a scoring function that fails in mode prediction is expected to also fail in virtual screening. Lastly, binding mode prediction is important for mechanistic studies of ligand binding, the insight from which can provide guidelines on lead optimization. In this work, we present two sets of ligand binding decoys (up to 500 sampled decoys plus one native ligand mode per complex in each set) that we constructed for the CSAR benchmark of 345 protein-ligand complexes by rigid and flexible ligand docking with MDock9,10,22,23 and DOCK (v5.3.0)24,25 tools. These decoys may serve as a CSAR benchmark of binding mode prediction for the ligand-protein docking community.
2 Materials and Methods
2.1 Docking Protocol
We generated two sets of ligand binding mode decoys for the CSAR benchmark using a hierarchical docking protocol, one for rigid ligand treatment and the other for flexible ligand treatment. The flowchart of the protocol is illustrated in Figure 1.
Figure 1.

A flowchart of the docking protocol for construction of the ligand binding decoys for the CSAR benchmark.
Specifically, for rigid-ligand docking, the putative ligand binding modes were sampled by docking the native ligand against the binding site using MDcok for each protein-ligand complex in the CSAR benchmark. MDock is an automated molecular docking program (http://zoulab.dalton.missouri.edu/software.htm) which integrates the fast ensemble docking algorithm for multiple protein structures22,23 and efficient ITScore scoring function for protein-ligand interactions.9,10 The program suite uses the commonly-used Sybyl (Tripos, Inc.) mol2 files of the protein and the ligand as input, in which hydrogens and atomic charges are optional and their effects will be ignored when they are present during docking/scoring calculations. MDock samples ligand binding orientations by performing exhaustive matching between the ligand heavy atoms and the representative spheres points of the binding pocket (see below), which generates and scores thousands of ligand binding modes with a wide range of RMSD values within seconds, as described in ref 22. Here, RMSD refers to the root mean square deviation of the heavy atoms between the ligand decoy and the native structure. During the docking calculations with MDock, the binding site was defined as the region within 15 Å from the native ligand. The sphere points that represent the negative image of the whole protein were generated using Kuntz et al's algorithm.26 The sphere points that are within the binding site were selected to guide putative ligand orientation sampling via a matching algorithm.22,26,27 There were about 30∼100 sphere points for each complex, depending on the size of the binding pocket. The maximum number of the effective ligand orientations was set to 2000 for sufficient sampling around the binding site. Next, the ligand orientations being generated were clustered to remove the degeneracy in the decoys. If two ligand orientations had an RMSD < 1.0 Å from each other, only the one with lower binding score was kept. The top 500 ligand binding modes after clustering were retained as the decoys for each complex.
Since the scoring function ITScore in MDock considers heavy atoms only and ignores the contribution from hydrogens, MDock did not optimize the positions of the hydrogens during ligand sampling despite the hydrogens were kept in the mol2 files. Therefore, the decoys generated by MDock may contain atomic clashes from hydrogens between the protein and ligand. However, many scoring functions use all-models, and the positions of hydrogen atoms should be optimized before the assessment of those scoring functions. Therefore, all the ligand decoys generated by MDock were further optimized/refined by the all-atom force-field scoring function of DOCK v5.3.0 (http://dock.compbio.ucsf.edu/).25,28 For each complex, the native structure was also included in the decoys, yielding a set of rigid-ligand decoys of up to 501 binding modes (up to 500 sampled decoys plus one native structure).
It should be noted that there still exist a few atomic clashes in the decoys after DOCK refinement due to small-size binding pockets and improperly preassigned hydrogens. Such ligand decoys were retained deliberately in order to test whether a scoring function (e.g., a contact-based scoring function) penalizes too-close contacts. This set of decoys is recommended for the assessment of general binding mode prediction. However, if a scoring function has difficult in handling atomic clashes, a second set of decoys was prepared by removing all the decoys that contain atomic clashes (Figure 1). Specifically, a program was written in which two atoms are defined to have clashes if Å for heavy-heavy atom pairs,29 Å for heavy atom-hydrogen pairs, or rij < 0.5 Å for hydrogen-hydrogen pairs. Here, rij is the distance between atom i and atom j, and and are their van der Waals (VDW) radii, respectively. The program was used to remove the atomic clashes in the first general set of ligand decoys.
The aforementioned process considered ligand orientational sampling only. The decoys evaluate and/or help improve the non-conformational factors/terms in a scoring function on mode prediction. To further include the conformational effect, a flexible-ligand decoy set was also generated for the CSAR benchmark. The procedure was similar except that the ligand flexibility was represented by multiple conformers and each conformer was then docked as a rigid body (Figure 1). Specifically, a set of conformations were generated for each ligand using the OMEGA program (version 2.2.0, OpenEye Scientific, Santa Fe, NM) with the default parameters. Up to 400 conformers were generated for each ligand. Then, each ligand conformer (including native ligand) was docked to the protein by MDock and putative orientations were sampled. The putative binding conformations of all the conformers were combined and clustered. The final top 500 conformations were refined by DOCK. Thus, up to 501 decoy conformations (up to 500 decoys plus one native structure) were obtained for each protein-ligand complex, serving as a CSAR benchmark for flexible-ligand decoys. A second flexible-ligand decoys were also prepared by further removing the decoys that contain atomic clashes.
2.2 Preparation of the Protein and Ligand files
The protein (in pdb format) and ligand (in mol2 format) files for the CSAR benchmark (2009 release) have been well prepared by the CSAR team, which consists of set 1 and set 2 with a total of 345 diverse protein-ligand complexes. Set 1 consists of 176 complexes, most of which were deposited in the Protein Data Bank (PDB)30 in 2007 and 2008. Set 2 contains 169 complexes which were deposited in 2006 or earlier. The missing atoms and hydrogens in the protein were pre-added by the CSAR team. Water molecules were removed from the protein because the positions of structural waters are normally unknown in advance for real docking calculations. The protonation states and AMBER (Assisted Model Building with Energy Refinement)31 charges were automatically assigned for the proteins using the UCSF Chimera software.29 The metal ions, unnatural residues, cofactors, and post-translational modifications that Chimera failed to assign for charges were treated as part of the proteins and assigned with Gasteiger charges32. The Sybyl (Tripos, Inc.) mol2-format files of the ligands provided in the CSAR benchmark were directly used for our docking, in which the AM1-BCC charges 33,34 were preassigned.
3 Results and Discussion
Figure 2 shows the statistics of the ligand binding decoys generated by MDock. It can be seen from the figure that most of the complexes in the CSAR benchmark have a certain number of decoys close to their native structures (RMSD < 2.0 Å), which is necessary for the assessment of binding mode prediction. For rigid-ligand docking, there are a maximum of 31 near-native decoys for a complex, whereas for flexible ligand docking, the number is 132. The presence of more near-native modes in the flexible ligand decoys is because a ligand can adopt different conformations through OMEGA during orientational sampling in flexible ligand docking, which increased the conformational diversity of ligand binding decoys.
Figure 2.
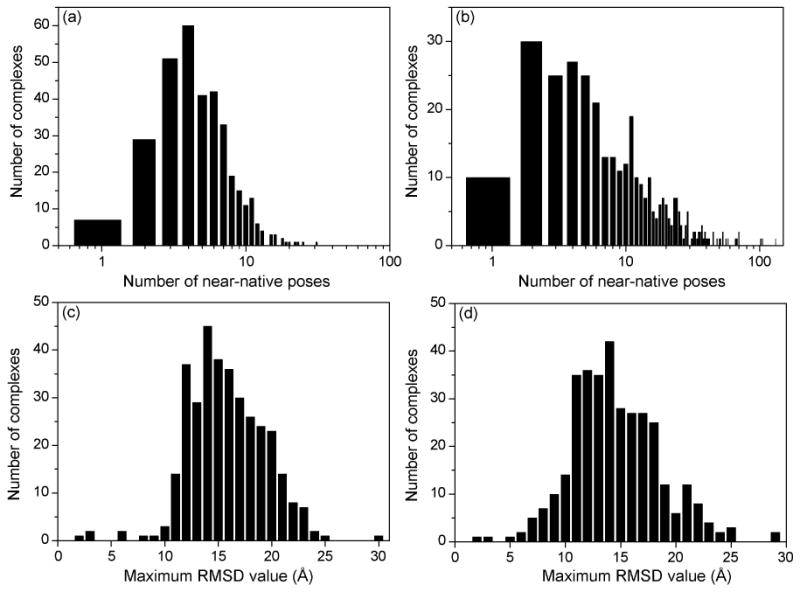
The statistics of the ligand binding decoys for the CSAR benchmark: The number distribution of the near-native binding modes (RMSD<2.0 Å) in the (a) rigid-ligand and (b) flexible-ligand decoys of a complex; The maximum RMSD distribution in the (c) rigid-ligand and (d) flexible-ligand decoys of a complex. Notice that the horizontal axes in Panel (a) and Panel (b) are in logarithmic scales.
It can also been noticed in Figure 2 that about 40 complexes have only one or two near-native binding modes in their rigid and flexible ligand decoys. There may be two reasons for the few near-native modes in these cases. The first reason may be due to our clustering procedure in ligand sampling, which removes many other near-native binding modes within 1.0 Å RMSD from the kept near-native modes in order to increase the sampling diversity and challenging level of the ligand decoys. The second possible reason is that some binding pockets are small and only the native ligand mode can fit these binding pockets well. In such cases, there was an RMSD gap between the native mode (RMSD ≃ 0 Å) and non-native binding decoys (RMSD > 2 Å). However, this fact would not affect the evaluation of scoring functions on the decoy sets as the native ligand binding modes were included in the final decoys. Furthermore, generating more near-native poses by reducing the clustering cutoff would not significantly improve the performance of a scoring function, given the fact that the native mode is already in the decoys and its position can be easily optimized near the native state by the scoring function.
In addition, Figure 2 also indicates that the sampled ligand decoys span a wide range around the binding site. For most of the complexes, the maximum RMSD values between the decoys and the native structure range from 10 Å to 25 Å. Figure 3 shows an example of the distribution of the flexible ligand binding decoys in terms of RMSD values (Set 1, #122, PDB entry: 1UTO). The corresponding binding conformations on the protein are illustrated in Figure 4, showing that the ligand decoys fully cover the binding site.
Figure 3.

The RMSD distribution observed in the ligand binding decoys for No.122 complex of set 1 (PDB code: 1UTO) constructed by (a) rigid-ligand and (b) flexible-ligand docking, respectively.
Figure 4.
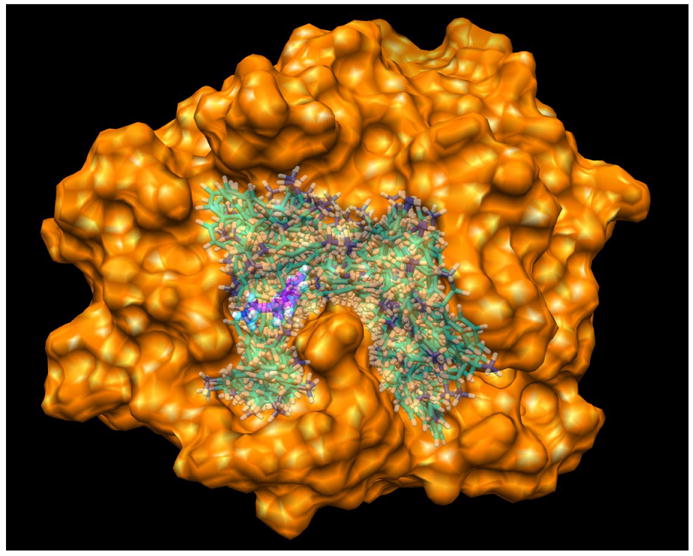
The flexible ligand binding decoys sampled by our docking method for the No.122 complex in set 1 (PDB code: 1UTO). The protein is represented by molecular surface. The ligand binding decoys are shown in semi-transparent, stick mode. The native ligand is shown in solid, stick mode and colored in magenta (C), blue (N) and gray (H). The figure was prepared using UCSF Chimera.29
Next, we assessed our scoring function ITScore in MDock with the CSAR mode prediction benchmark. ITScore is a knowledge-based scoring function of which the pairwise potentials were extracted using an iterative method from the experimentally determined protein-ligand crystal structures that are mostly different from the CSAR benchmark and the very small overlap has little effect on the scoring results.9,10,21 Specifically, we calculated the success rate of ITScore on binding mode prediction with the benchmark. By default, a prediction was defined to be a success if the top-scored binding mode has an RMSD < 2.0 Å from the native structure, unless otherwise specified. Table 1 and Figure 5 show the success rates of ITScore on identifying native binding modes for the CSAR benchmark of 345 complexes. For references, we also evaluated the performances of the force-field scoring function (DOCK/FF) and van der Waals (VDW) only 28 provided by UCSF DOCK (v5.3.0) with the same decoy sets. Minimization was done for each decoy structure during scoring calculations.
Table 1.
Success rates of ITScore, DOCK/FF and DOCK/VDW on identifying near native binding modes with the ligand decoys constructed for the CSAR benchmark, using different RMSD criteria for the top-scored binding mode.
| RMSD criteria | Success rates (%) | |||||
|---|---|---|---|---|---|---|
| Rigid ligand decoys | Flexible ligand decoys | |||||
| ITScore | FF | VDW | ITScore | FF | VDW | |
| < 0.5 Å | 76.8 | 55.7 | 43.5 | 68.7 | 48.1 | 33.9 |
| < 1.0 Å | 82.6 | 71.3 | 58.0 | 73.6 | 62.3 | 46.1 |
| < 1.5 Å | 84.6 | 77.7 | 61.7 | 75.9 | 66.7 | 50.2 |
| < 2.0 Å | 86.7 | 80.0 | 64.1 | 79.7 | 71.0 | 52.8 |
|
| ||||||
| a< 2.0 Å | 82.0 | 78.3 | 61.7 | 73.0 | 68.1 | 50.0 |
| b< 2.0 Å | 86.8 | 86.6 | 64.4 | 81.3 | 79.3 | 57.1 |
The native ligand binding modes were excluded from the decoys in the calculations for these success rates.
The ligand binding decoys were constructed based on the CSAR-NRC HiQ benchmark.
Figure 5.
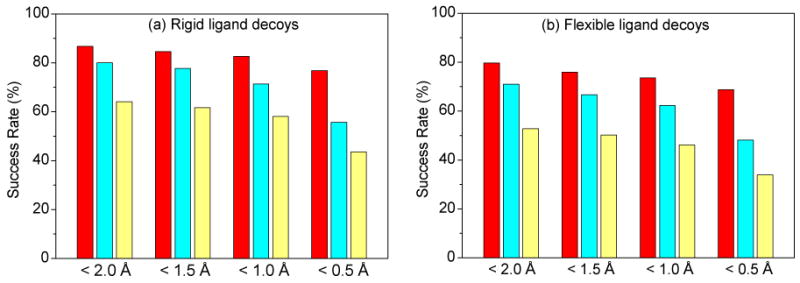
The success rates of ITScore, DOCK/FF, and DOCK/VDW on identifying native binding modes with the ligand decoys constructed for the CSAR benchmark when different criteria were used regarding the RMSD (Å) between the top-scored pose and the native structure. For each RMSD criterion, the bars correspond to ITScore, DOCK/FF and DOCK/VDW from left to right, respectively.
It can be seen from Figure 5 that ITScore achieved the highest success rates in identifying native binding modes for both rigid and flexible ligand decoys. For rigid-ligand decoys, the success rate is 86.7% for ITScore, compared to 80.0% and 64.1% for DOCK/FF and VDW scoring functions. For flexible-ligand decoys, the success rate is 79.7% for MDock, compared to 71.0% and 52.8% for DOCK/FF and VDW. The RMSD of the top predicted binding mode for each complex in the CSAR benchmark is shown in Figures 6 and 7. It can be seen that ITScore predicted the highest number of near-native binding modes (i.e., RMSD < 2.0 Å), which is consistent with Figure 5. Moreover, overall speaking, the binding modes predicted by ITScore are closer to the experimentally determined structures (i.e., with smaller RMSD values) compared to DOCK/FF and VDW.
Figure 6.

The RMSD values of the top binding modes predicted by ITScore (black, filled circle), DOCK/FF (red, filled diamond), and DOCK/VDW (blue, open circle) with set 1 of the CSAR benchmark: (a) rigid ligand decoys; (b) flexible ligand decoys. Notice that the CSAR ID numbers of the complexes are not sequential, and therefore the x axes extend up to 300, which is more than the total number of complexes.
Figure 7.

The RMSD values of the top binding modes predicted by ITScore (black, filled circle), DOCK/FF (red, filled diamond), and DOCK/VDW (blue, open circle) with set 2 of the CSAR benchmark: (a) rigid ligand decoys; (b) flexible ligand decoys. Notice that the CSAR ID numbers of the complexes are not sequential, and therefore the x axes extend up to 300, which is more than the total number of complexes.
The results are similar when different RMSD criteria were used, as shown in Table 1 and Figure 5. For example, for a more stringent criterion of RMSD < 0.5 Å, ITScore yielded relatively higher success rates of 76.8% and 68.7% for rigid and flexible ligand decoys, compared to 55.7% and 48.1% for DOCK/FF and 43.5% and 33.9% for VDW. In addition, to examine the effect of including native binding modes, we have calculated the success rates of ITScore, DOCK/FF and VDW by excluding native ligand modes from the binding decoys. The success rates showed no significantly difference for their relative performances (Table 1). These results suggest the advantage of the pairwise effective potentials of ITScore, which were extracted from the experimental structures.
It is thought that the energy landscape for protein-ligand interactions exhibits a funnel-like shape near the native state so as to facilitate rapid ligand binding in the dynamics point of view.35 This funnel-like shape may be roughly measured by the correlation between the scores and the RMSDs of ligand binding decoys of a complex. The correlation coefficients vary from −1.0 to 1.0. The higher the correlation, the more likely the energy landscape is funnel-like and thus the more reasonable the scoring function would be. A coefficient of 1.0 stands for an ideal energy funnel, 0.0 represents a random case, and −1.0 corresponds to complete anti-correlation (i.e., worse scoring). Figure 8 shows the distribution of score-rmsd correlations (i.e., Pearson coefficients) of ITScore, DOCK/FF, and VDW for the rigid and flexible ligand decoys of the 345 CSAR complexes. It can be seen from Figure 8 that ITScore achieved good score-rmsd correlations for significantly more complexes than DOCK/FF and VDW, suggesting that ITScore is also more reasonable in terms of binding dynamics.
Figure 8.
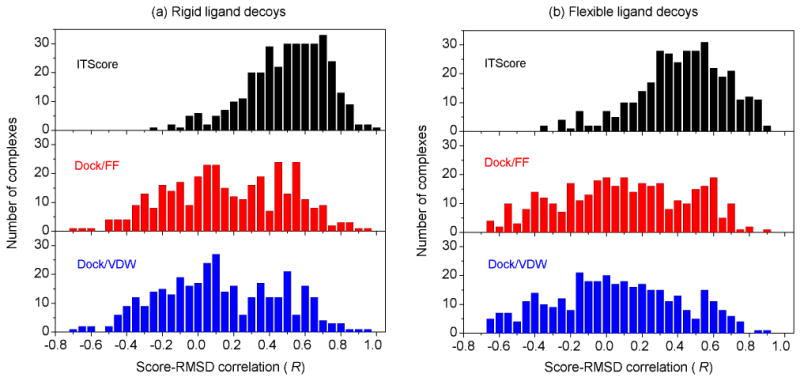
The distribution of score-RMSD correlations of ITScore, DOCK/FF, and DOCK/VDW for (a) rigid and (b) flexible ligand binding decoys of the complexes in the CSAR benchmark.
Very recently, a new version of CSAR benchmark was released, which is referred to as the CSAR-NRC HiQ benchmark. A major difference between the two versions is that the protonation states of the complexes were curated in the CSAR-NRC HiQ benchmark. Because the matching procedure for ligand orientation generation in MDock requires only the positions of ligand heavy atoms and sphere points, which are not affected by protonation states/partial charges of the atoms, it is expected that there will be no dramatic difference between the ligand decoys for the CSAR and CSAR-NRC HiQ benchmarks in terms of sampling. The statistics of the ligand decoys for the CSAR-NRC HiQ benchmark are shown in the Supporting Information, and the corresponding success rates of ITScore, DOCK/FF and VDW are listed in Table 1. It can be seen from Table 1 that there is no significant difference in the success rates of ITScore and VDW between the CSAR and CSAR-NRC HiQ benchmarks, suggesting the robustness of ITScore. It is also notable that DOCK/FF yielded improved success rates of 86.6% and 79.3% for rigid and flexible ligand decoys of the CSAR-NRC HiQ benchmark, respectively, compared to 80.0% and 71.0% for the CSAR benchmark. The finding again indicates the importance of atomic charge/protonation assignments to force filed scoring functions, even though the curation of protonation states for the CSAR-NRC HiQ version did not improve the performance of DOCK/FF in affinity prediction.21
Considering the sensitivity of force filed scoring functions to electrostatic interactions (and therefore charge assignments), users are recommended to re-assign the partial charges that are consistent to their force fields to the protein and ligand atoms for the best scoring performance when they use the ligand decoy sets presented in this work.
4 Conclusion
Binding affinity prediction and binding mode prediction are two important aspects for assessment of a scoring function. Yielding good correlation in affinity prediction does not guarantee a scoring function to perform equally well in mode prediction, and vice versa.1 The CSAR decoy test sets constructed in the present study evaluate the ability of a scoring function to discriminate near-native binding modes from non-native decoy modes for the complexes in the CSAR benchmark.
Specifically, we have constructed two sets of well-sampled ligand binding decoys (up to 500 docking decoys plus one native structure per complex in each set) for the CSAR benchmark around the binding sites. One set is the rigid-ligand decoys, in which the ligands were treated as rigid bodies and only the orientational space was sampled. The other set is the flexible ligand decoys, in which the ligand conformational space was also sampled. The two sets of ligand binding decoys may serve as a benchmark for assessing a scoring function on binding mode prediction.
The assessment of our ITScore scoring function using the CSAR rigid-ligand decoy sets achieved a success rate of 86.7% on identifying native binding modes when the top orientation was considered for each complex under the criterion of RMSD < 2.0 Å, compared to 80.0% and 64.1% for the force field (DOCK/FF) and van der Waals (DOCK/VDW) scoring functions of UCSF DOCK. With the CSAR flexible-ligand decoys, ITScore yielded a success of 79.7%, compared to 71.0% and 52.8% for DOCK/FF and DOCK/VDW. Moreover, the binding modes predicted by ITScore are closer to the experimentally determined structures (i.e., more accurate) than DOCK/FF and DOCK/VDW. The lower success rates with the flexible ligand decoys than with the rigid ligand decoys may be because none of these scoring functions explicitly account for ligand conformational energies. The two reference scoring functions, DOCK/VDW and DOCK/FF, were introduced as crude gauges of energetic contributions from nonpolar interactions and electrostatic interactions. It is not surprising that VDW yielded the lowest success rates for binding mode prediction, because this reference scoring function accounts for only the effect of geometric complementarity on binding. DOCK/FF, which is the sum of the coulombic electrostatic energy and the VDW energy, performs better than VDW on binding mode prediction because of the inclusion of electrostatic specificity. In contrast, ITScore is an iterative knowledge-based scoring function.9 Its pairwise potentials not only consider the VDW and electrostatic interactions but also implicitly incorporate the effects of desolvation and entropy,36 which may be attributed to the best performance of ITScore.
The present decoy sets consist of a reasonable number of well-sampled, diverse binding modes. Ideally, one may construct a scoring-function-independent decoy set by using exhaustive sampling around the binding site. However, exhaustive sampling is unnecessary for assessment of scoring functions because most of the generated ligand modes would be trivial binding decoys that either make no contact or have severe atomic clashes with the protein. Therefore, a scoring function such as ITScore used in this study is required to filter out those trivial binding modes so as to form a set of physicochemically reasonable ligand binding decoys. The resulting decoys would provide a good, more challenging basis to assess the performance of different scoring functions. The decoys could lead to a bias for a scoring function being assessed to outperform the filtering scoring function (e.g. ITScore in the present study), because the filtering scoring function actually ranked many more ligand binding modes during the initial ligand sampling than the decoy set which kept only the modes with relatively good scores from ITScore.
In the present decoys, the native ligand structure was included in ligand sampling for each complex. Future study may replace the native mode by a set of near-native ligand conformations (e.g., RMSD within 2.0 Å from the native mode) generated by programs like OMEGA using a small clustering cutoff. Combined with the rest ligand decoys, the new benchmark would better mimic practices like virtual screening. For the new benchmark, the scoring performance may depend not only on the tested scoring function but also on the quality of generated ligand conformations, an interesting issue to be studied in the future.
Supplementary Material
Acknowledgments
Support to XZ from OpenEye Scientific Software Inc. (Santa Fe, NM) and Tripos, Inc. (St. Louis, MO) is gratefully acknowledged. XZ is supported by NIH grant R21GM088517 and NSF CAREER Award 0953839. The computations were performed on the HPC resources at the University of Missouri Bioinformatics Consortium (UMBC).
Footnotes
Supporting Information: Additional figures are provided in the Supporting Information. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys Chem Chem Phys. 2010;12:12899–12908. doi: 10.1039/c0cp00151a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cole JC, Murray CW, Nissink JWM, Taylor RD, Taylor R. Comparing protein-ligand docking programs is difficult. Proteins. 2005;60:325–332. doi: 10.1002/prot.20497. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J Chem Inf Model. 2006;46:401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- 4.Jain AN, Nicholls A. Recommendations for evaluation of computational methods. J Comput-Aided Mol Des. 2008;22:133–139. doi: 10.1007/s10822-008-9196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hawkins PCD, Warren GL, Skillman AG, Nicholls A. How to do an evaluation: Pitfalls and traps. J Comput-Aided Mol Des. 2008;22:179–190. doi: 10.1007/s10822-007-9166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirchmair J, Markt P, Distinto S, Wolber G, Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selections What can we learn from earlier mistakes. J Comput-Aided Mol Des. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 7.Huang SY, Zou X. Advances and Challenges in Protein-Ligand Docking. Int J Mol Sci. 2010;11:3016–3034. doi: 10.3390/ijms11083016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 9.Huang SY, Zou X. An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials. J Comput Chem. 2006;27:1866–1875. doi: 10.1002/jcc.20504. [DOI] [PubMed] [Google Scholar]
- 10.Huang SY, Zou X. An iterative knowledge-based scoring function to predict protein-ligand interactions: II. Validation of the scoring function. J Comput Chem. 2006;27:1876–1882. doi: 10.1002/jcc.20505. [DOI] [PubMed] [Google Scholar]
- 11.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 12.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 13.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 14.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 15.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 16.Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57:225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- 17.Bursulaya BD, Totrov M, Abagyan R, Brooks CL., III Comparative study of several algorithms for flexible ligand docking. J Comput-Aided Mol Des. 2003;17:755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 18.Kim R, Skolnick J. Assessment of Programs for Ligand Binding Affinity Prediction. J Comput Chem. 2008;29:1316–1331. doi: 10.1002/jcc.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y. Automated docking screens: a feasibility study. J Med Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang SY, Zou X. Scoring and lessons learned with the CSAR benchmark using an improved iterative knowledge-based scoring function. J Chem Inf Model. 2011 doi: 10.1021/ci2000727. accompanying paper, same special issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang SY, Zou X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins. 2007;66:399–421. doi: 10.1002/prot.21214. [DOI] [PubMed] [Google Scholar]
- 23.Huang SY, Zou X. Efficient molecular docking of NMR structures: Application to HIV-1 protease. Protein Sci. 2007;16:43–51. doi: 10.1110/ps.062501507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewing TJA, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J Comput-Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 25.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J Comput-Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 26.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 27.Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- 28.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy approach to macromolecule-ligand interactions. J Comput Chem. 1992;13:505–524. [Google Scholar]
- 29.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera – A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 30.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasteiger J, Marsili M. Iterative Partial Equalization of Orbital Electronegativity – A Rapid Access to Atomic Charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 33.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J Comput Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 34.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 35.Meiler J, Baker D. ROSETTALIGAND: Protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 36.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J Med Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.