

Abstract
This investigation was pursued to test the use of intracellular antibodies (intrabodies) as a means of blocking the pathogenesis of Huntington's disease (HD). HD is characterized by abnormally elongated polyglutamine near the N terminus of the huntingtin protein, which induces pathological protein–protein interactions and aggregate formation by huntingtin or its exon 1-containing fragments. Selection from a large human phage display library yielded a single-chain Fv (sFv) antibody specific for the 17 N-terminal residues of huntingtin, adjacent to the polyglutamine in HD exon 1. This anti-huntingtin sFv intrabody was tested in a cellular model of the disease in which huntingtin exon 1 had been fused to green fluorescent protein (GFP). Expression of expanded repeat HD-polyQ-GFP in transfected cells shows perinuclear aggregation similar to human HD pathology, which worsens with increasing polyglutamine length; the number of aggregates in these transfected cells provided a quantifiable model of HD for this study. Coexpression of anti-huntingtin sFv intrabodies with the abnormal huntingtin-GFP fusion protein dramatically reduced the number of aggregates, compared with controls lacking the intrabody. Anti-huntingtin sFv fused with a nuclear localization signal retargeted huntingtin analogues to cell nuclei, providing further evidence of the anti-huntingtin sFv specificity and of its capacity to redirect the subcellular localization of exon 1. This study suggests that intrabody-mediated modulation of abnormal neuronal proteins may contribute to the treatment of neurodegenerative diseases such as HD, Alzheimer's, Parkinson's, prion disease, and the spinocerebellar ataxias.
Huntington's disease (HD) is a genetic disorder that derives from expanded CAG repeats in the huntingtin gene (1), which then encodes pathological huntingtin protein with abnormally long polyglutamine sequences (polyQ, or Qn). Huntingtin is expressed ubiquitously by human cells, with high levels in the brain, particularly the cortex and striatum. Murine model studies of HD have shown it occurs through dominant gain of function when a single abnormal allele is present (2–6). Polyglutamine lengths ranging up to 35 residues (Q35) are present in healthy individuals, whereas Q40 or longer are associated with HD pathogenesis (1, 7). Larger polyQ expansions are correlated with earlier onset and more severe symptoms. The polyglutamine length-dependent aggregation of huntingtin has been reported to involve interaction with other proteins as well as self-association. Intracellular protein aggregates are found in human HD brains at autopsy and in tissues of mice carrying transgenes with expanded-repeat huntingtin (8–11). Here we report on the use of intracellular antibodies (intrabodies) as a potential therapeutic strategy on the basis of their ability to inhibit aberrant protein aggregate formation in a cellular model for HD. This approach may be applicable to other diseases as well, because HD is a paradigm for several adult-onset neurodegenerative diseases that exhibit pathological interactions by neuronal proteins.
Antibody engineering has made it practical to genetically select specific antibody combining sites from antibody phage display libraries. The minimal intact binding species consists of the 25-kDa Fv region; this is most often manipulated in the form of single-chain Fv (sFv) antibodies, having the VH and VL variable domains bridged by a linker peptide such as (Gly4Ser)3. The present study takes advantage of sFv intrabodies, which are expressed intracellularly against intracellular targets (12). The huntingtin analogue used in most of our studies was developed by Kazantsev et al. (13), who genetically fused the huntingtin exon 1 fragment to green fluorescent protein (HD-Qn-GFP). When this HD model is examined in transfected cells, the huntingtin analogues with expanded polyglutamine (polyQ) repeats exhibit visible aggregate formation that increases proportionately with the number of glutamine residues in the polyQ segment. Using this model, we have found that intrabody association with huntingtin via its N-terminal residues (1–17) can significantly reduce in situ aggregation of expanded-repeat exon 1 analogues.
Materials and Methods
Construction, expression, and purification of glutathione S-transferase (GST) and GFP-fusion proteins were conducted according to previously described procedures (13, 14). The purification of proteins generally used affinity chromatography on immobilized metal chelate columns to purify C4 sFv-His6 protein and glutathione Sepharose 4B to isolate GST-HD analogues for in vitro analysis of antibody specificity.
The selections of human sFv antibodies specific to the N-terminal HD peptide (1–17) were carried out with a large, naïve, human sFv-phage display library of 7 × 109 diversity (15), according to selection scheme 1 from Schier et al. (16). The antigen consisted of N-terminal residues 1–17 of huntingtin biotinylated at its C terminus (synthesized at the Protein Core Facility, Tufts University, Boston, MA); plasmon resonance studies of sFv binding affinity were conducted with a BIACORE 2000, according to ref. 17.
Immunopurification, Western blot, and protein turnover experiments were conducted by standard methods described previously by our group (18). Please refer to the supplemental material, which is published on the PNAS web site (www.pnas.org), to obtain more detailed discussions of the materials and methods used in this investigation.
Transfection, Immunofluorescence Microscopy, and Aggregate Quantitation.
COS-7 cells grown on glass cover slips (105 cells/well) were cotransfected at an sFv-to-HD antigen plasmid ratio of 3:1. Intrabody plasmids coded for either anti-huntingtin C4 sFv-hemagglutinin (HA)-nls or anticaspase-7 C8 sFv-HA-nls (18), as a negative control. The hemagglutinin epitope sequence (HA tag) was fused to the C termini of all sFv constructs to facilitate immunolocalization. In some experiments, the simian virus 40 nuclear localization sequence (nls) was fused as the C terminus of the entire protein to effect nuclear retargeting in certain control experiments. In addition, plasmids coding for HD-Qn-myc-His6 or for GFP fusion proteins were used as listed in Table 1, were transfected, where noted, by using 5 μl of SuperFect transfection reagent (Qiagen, Chatsworth, CA). The remainder of the transfections were performed in COS-7, HEK 293, and BHK-21 cells by using either the calcium phosphate method or TransFast transfection reagent (Promega) with plasmids coding for C4 sFv-HA or negative control anti-erbB-2 ML3–9 sFv-HA, with or without the antigen, HD-polyQ-GFP. Forty-eight hours after transfection, cells were prepared for immunofluorescence as described previously by Zhu et al. (18).
Table 1.
Amino acid sequences of fusion proteins
| Abbreviation | Sequence |
|---|---|
| Bacterial expression | |
| GST-HD-Q25 | GST-lvprgsMATLEKLMKAFESLKSF(Q)25lqpgstraaas |
| GST-HD-Q42 | GST-lvprgsMATLEKLMKAFESLKSF(Q)42lqpgstraaas |
| GST-DRPLA-Qn | GST-lvprgsVSTHHHHH(Q)nHHGNSGPPefpgrlerphrd (n = 0, 35) |
| Mammalian expression | |
| HD-Qn-EGFP | MATLEKLMKAFESLKSF(Q)n-EGFP (n = 25, 72, 103) (ref. 13) |
| HD-Qn-myc-His6 | MATLEKLMKAFESLKSF(Q)nlqpggstmsrgpfeqkliseedl- nmhte(h)6 |
| GFP-HD-Q25 | GFP-idgggggkgpvtgtgsMATLEKLMKAFESLKSF(Q)25lqpriltn |
| GFP-HD-Q103 | GFP-idgggggkgpvtgtgsMATLEKLMKAFESLKSF(Q)103lqpriltn |
| GFP-DRPLA-Q81 | GFP-idgggggkgpvtgtgsVSTHHHHH(Q)81HHSGPPef |
GFP is the same as EGFP of ref. 13. The atrophin-1 and huntingtin protein sequences are underlined. The flanking sequences cited in lowercase letters are part of the expression vectors.
Aggregates were counted 48 h posttransfection after lysis with a detergent solution consisting of 2% SDS and 2% Triton X-100 in 50 mM Tris buffer (pH 7.6), as described by Kazantsev et al. (13). Six or eight random fields were counted per cotransfection, each performed in sextuplicate. Relative luminescence for the luciferase assay was determined by the Wallac 1409 scintillation counter in sextuplicate by using Luciferase Assay Reagent (Promega) and 20 μl cell lysate. Means were compared by using t-test statistics.
Results
Selection from the sFv-Phage Display Library.
Specific phage antibodies were selected for binding to HD residues (1–17) having biotin incorporated at the C terminus during peptide synthesis, against a large, naive human sFv-phage display library (15). After the third round of selection, 90 clones were analyzed by ELISA, by using the peptide adsorbed as the primary layer on the plate. Twenty clones showed binding to HD (1–17). Eight sFv clones showing the highest OD in ELISA were chosen for preparative scale sFv expression. Bacterial supernatants containing sFv-myc were assayed on microtiter plates coated with GST-HD-Q42, GST-HD-Q65, and GST-DRPLA-Q35 [DRPLA, dentatorubral-pallidoluysian atrophy (atrophin-1), N-terminal sequence], which was used as a negative control (14) (sequences of constructs are noted in Table 1). Bound sFv-myc fusion protein was detected with anti-c-myc 9E10 IgG followed by alkaline phosphatase-conjugated goat anti-mouse IgG. Single-chain Fv clone C4 expressed well in bacterial culture and reacted only with protein antigens containing HD residues (1–17) (data not shown).
Characterization of the Anti-HD C4 sFv Protein in Vitro.
The cDNA encoding the VH-linker-VL form of C4 sFv was transferred from the pHEN1 to the pSYN-1 vector, which secretes sFv-myc-His6, where the His6 peptide facilitates isolation of the expressed protein by immobilized metal ion affinity chromatography. The specificity of the purified C4 sFv was first analyzed by assessing its binding to several immunoadsorbant resins; glutathione–Sepharose was complexed with a series of GST fusion proteins, which are included in Table 1. The C4 sFv was mixed with each resin, and eluates were analyzed by SDS/PAGE, as presented in Fig. 1A. The C4 sFv bound strictly to GST fusion proteins that contained the HD (1–17) peptide sequence (bound and eluted from the GST-HD-Q25 and Q42 resins) and was devoid of adsorption to the GST-DRPLA-Q35 resin. Western blot analysis confirmed the identity of C4 sFv protein in bands observed by Coomassie blue staining (data not shown).
Figure 1.
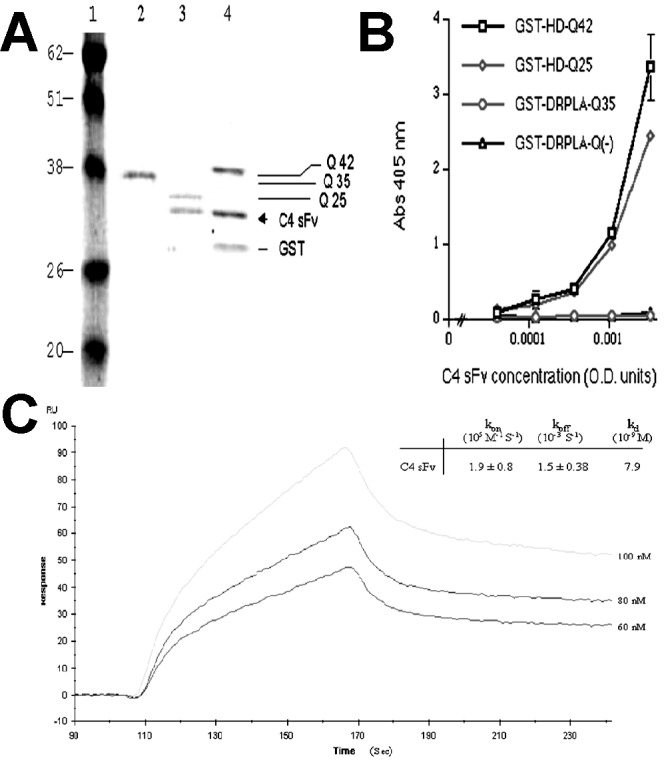
Characterization of anti-HD C4 sFv. (A) Coomassie blue staining of affinity-purified GST-fusion proteins GST-DRPLA-Q35 (lane 2), GST-HD-Q25 (lane 3), and GST-HD-Q42 (lane 4), mixed with anti-HD C4 sFv. Molecular weight markers are in lane 1. (B) Direct binding of C4 sFv to immobilized GST-fusion proteins analyzed by ELISA. C4 sFv-myc-His6 antibodies were quantitated in microtiter plates coated with 70 nM of GST-fusion proteins and blocked with 1% BSA in PBS. Bound sFv were detected with anti-myc antibodies (9E10, 1 μg/ml), followed by alkaline phosphatase-conjugated goat anti-mouse IgG. (C) Kinetic binding affinity of C4 sFv analyzed by BIAcore. A range of concentration (60–100 nM) of C4 sFv was used to measure the association rate (kon) on 50 resonance units of biotinylated-HD peptide bound to a streptavidin sensor chip.
ELISA studies demonstrated qualitatively that C4 sFv bound specifically to the HD (1–17) sequence (Fig. 1B). Plasmon resonance studies with the BIACORE 2000 provided quantitative measures of sFv-antigen binding and dissociation kinetics. The biotinylated HD-peptide was loaded at very low concentrations (50 resonance units) onto a streptavidin-coated sensor chip for kinetics measurements (Fig. 1C). The Kd was calculated to be 7.9 nM, based on the measured ka and kd rate constants by using a 1:1 Langmuir model for simple bimolecular interactions (biaevaluation 3.0 software, Biacore International, Uppsala, Sweden). As negative controls, C4 sFv was tested at higher concentration (4.6 μM) on a flow cell that contained streptavidin alone, and it was also tested for association with an irrelevant 16 residue biotinylated peptide bound to the streptavidin-coated sensor chip (170 resonance units). No significant sFv binding was observed in either control (data not shown). The C4 sFv association constant (Ka) of 1.3 × 108 M−1 is typical of antibodies binding to structurally well-defined protein epitopes, and it represents a rather high affinity for a binding site to a short peptide. This affinity would be consistent with the C4 sFv combining with a relatively rigid HD (1–17) epitope rather than an ensemble of conformers that would significantly reduce the apparent binding constant.
Analysis of sFv Antibody and Antigen Protein Turnover in Transfected Cells.
COS-7 cells were transfected with plasmids coding for either C4 sFv-HA or GFP-HD-Q103 or cotransfected with both plasmids. Average half-lives of the proteins were quantitated by PhosphorImager (Molecular Dynamics) analysis at different chase times. Whether in singly or doubly transfected cells, the C4 intrabody showed a half-life that was greater than 20 h, whereas the transfected GFP-HD-Q103 antigen displayed a half-life of between 8 and 20 h (data not shown). The C4 sFv half-life was in the range needed for well-behaved intrabodies, as was shown in a previous study for an anticaspase-7 intrabody, C8 sFv (18). The Q103 antigen half-life values in singly or cotransfected cells showed no significant differences, given the experimental error. In all these studies, the measured radiolabeled proteins were in the soluble fraction of the cell lysate. No contribution to the measured counts came from HD aggregates, which were pelleted during centrifugation; the aggregates would be expected to act like bacterial inclusion bodies in being significantly protected from intracellular degradation.
Characterization of the Anti-HD C4 sFv Protein in Situ.
The final stage of sFv specificity analysis involved immunofluorescence microscopy of intrabody-transfected COS-7 cells. In Fig. 2, the HD-Q103-myc antigen was used for a retargeting study to avoid ambiguities caused by the slight propensity of GFP fusions for nuclear localization. The negative control C8 intrabodies (Fig. 2 a, c, and e) and the anti-HD C4 intrabodies (Fig. 2 b, d, and f) were constructed as sFv-HA-nls fusions, where nls was the simian virus 40 large T-antigen nuclear localization signal. Given the results of our earlier study (Zhu et al., ref. 18), intrabody-bound antigens should likewise be retargeted from the cytosol to the nucleus. Fig. 2 a and b show the C8 and C4 intrabodies to localize to the cell nuclei of cotransfected cells. In Fig. 2 c and e, the anti-myc staining shows that the negative sFv control (C8 sFv-HA-nls) is ineffective at retargeting the HD-Q103-myc antigen to cell nuclei, whereas the C4 intrabody does so efficiently (Fig. 2 d and f). Additionally, the control C8 intrabody did not prevent aggregation of the Q103 antigen (shown by arrowheads in Fig. 2 c and e), unlike the C4 sFv in these preliminary transfection experiments.
Figure 2.
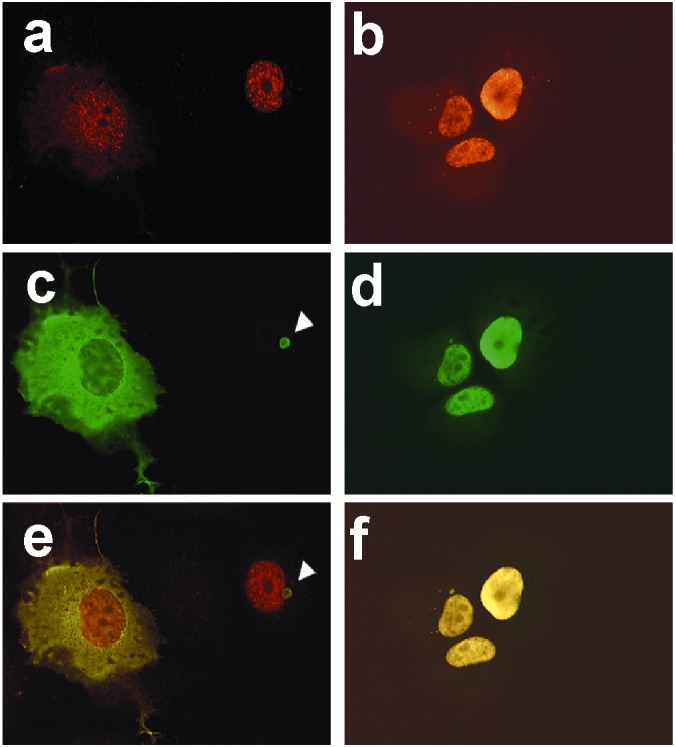
Retargeting of GFP-fusion HD fragments to the nucleus by anti-HD C4 sFv-HA-nls. Immunofluorescence of HD-Q103-myc-His6 with C4 sFv-HA-nls (b, d, f) or a negative control sFv-HA-nls intrabody (a, c, e), after 48-h cotransfection in COS-7. HD-Q103-myc-His6 was detected by 9E10 mAb, followed by FITC-labeled goat anti-mouse IgG antibodies (c, d). The intrabodies were detected with polyclonal anti-HA antibodies, followed by rhodamine-labeled goat anti-rabbit IgG antibodies (a, b). (e, f) Dual staining of the same fields.
COS-7 cells stably expressing C4 sFv-HA-nls were further transfected with different GFP-labeled antigens for immunofluorescence microscopy. Cells transfected with GFP-DRPLA-Q81 displayed both diffuse cytoplasmic antigen and large fluorescent aggregates, whereas in cells transfected with GFP-HD-Q103, the antigen was fully retargeted to the cell nuclei and aggregates were less apparent. These results provides in situ evidence for the HD (1–17) specificity of C4 sFv intrabodies and also suggests that the intrabody–antigen complex might counteract aggregate formation by the HD antigen.
Quantitation of Pathological HD Aggregate Formation in Cells Transfected with HD-polyQ-GFP, With or Without Anti-HD C4 sFv Intrabody.
COS-7, BHK-21, and HEK 293 cells were cotransfected with plasmids that express the C4 or a control intrabody and HD-polyQ-GFP fusion proteins containing Q25, Q72, or Q103. Aggregates were counted after treatment by the detergent method described previously (13). Aggregate formation by HD-Q72-GFP or HD-Q103-GFP was observed to qualitatively decrease at an intrabody-to-antigen plasmid ratio of 3:1, but for quantitative analysis, the effects were counted at a ratio of 5:1. Up to 86% fewer aggregates were present in intrabody cotransfectants, compared with control levels (note BHK-21 cells, Fig. 4A). In contrast, control transfections with nonspecific intrabody plasmids (anti-erbB-2 ML3–9 sFv) showed little or no statistically significant reduction in aggregate formation compared with the parent empty vector. Cells transfected with the Q25 plasmid alone never showed SDS-insoluble aggregates (data not shown).
Figure 4.

Inhibition of aggregate formation of pathogenic HD-polyQ-GFP in the presence of cotransfected anti-HD C4 sFv. (A) Quantitation of aggregate numbers at 48 h after cotransfection (5:1 ratio) with anti-HD C4 sFv-HA, negative control ML3–9 sFv-HA, or parent vector (pcDNA) and HD-Q72-GFP in COS-7, BHK-21, or HEK 293 cells, as indicated. After lysis with 2% SDS/2% Triton X-100/50 mM Tris, the number of insoluble fluorescent aggregates in six or eight random fields was counted. Bars represent means of six cotransfections ± SEM. C4 sFv-HA significantly reduced the number of aggregates when compared with control ML3–9 sFv-HA or pcDNA (P < 0.005). Similar results seen with HD-Q103-GFP (not shown). (B) Western blot analysis of HD-Q72-GFP protein expression in cotransfections. Cell lysates were collected from 5:1 cotransfections (sFv/HD-72Q-GFP) in duplicate at 24 h, before prominent aggregate formation and loss of HD-72Q-GFP to the insoluble compartment, and subjected to SDS/PAGE. The membrane was probed with 1:1,000 anti-AFP primary antibody (Quantum Biotechnologies, Montreal, PQ, Canada), stripped and reprobed with 1:500 anti-actin antibody (Sigma). HD-Q72-GFP bands were of similar intensity after detection by HRP-conjugated secondary antibody and chemiluminescence. (C) Relative luminescence of cell lysates from cotransfections of pGL2-Control (a luciferase-expressing reporter construct) with C4 sFv, control intrabody or parental vector. Mean luminescence (±SEM) as determined by Wallac 1409 scintillation counter was not different.
Double-label immunofluorescence studies confirmed that BHK-21 cells expressing HD-Q72-GFP or HD-Q103-GFP (green label, Fig. 3 A and B) plus anti-HD C4 sFv (red label, Fig. 3B) showed substantial diffuse GFP label, with only an occasional fluorescent aggregate (usually labeled with HA as well; note arrow in Fig. 3B). Those cells that failed to express the intrabody showed evidence of large aggregates (Fig. 3B, arrowheads). The diffuse labeling was also characteristic of the normal length Q25 construct, with or without intrabody cotransfection (Fig. 3 A and B). The reduction of aggregates in dual transfectants did not appear to derive from suppression of HD protein expression per se. Western blots showed the levels of expression under different cotransfection combinations to be unchanged (Fig. 4B) as did expression of a cotransfected heterologous luciferase reporter (Fig. 4C).
Figure 3.

Immunofluorescence of HD-polyQ-GFP in the presence of cotransfected anti-HD C4 sFv. (A) BHK-21 cells transfected with HD-polyQ-GFP (normal control, Q25; pathogenic, Q72 or Q103) alone or cotransfected with C4 intrabody or control (ML3–9 sFv-HA) at an sFv to antigen plasmid ratio of 5:1. Live cells are shown photographed at 48 h after transfection, showing aggregates as intense foci of fluorescence. Similar results were seen with HD-Q72-GFP cotransfections (not shown). (B) Double labeling of 5:1 C4/HD-polyQ-GFP cotransfections; HD-polyQ-GFP (green), C4 sFv (red, detected by anti-HA antibody and Alexa 568-conjugated secondary antibody) and merged image. Some aggregates are found in Q103 cotransfectants but never in Q25 transfectants; cells not expressing C4 intrabody (arrowheads) often display aggregates, and to a lesser degree, in some cotransfected cells (arrow).
Discussion
We have selected a human sFv antibody specific to the N-terminal 17 residues of huntingtin protein. When cotransfected with expanded-repeat HD-polyQ-GFP fragments, this C4 intrabody counteracts most of the aggregate formation seen in cells that have been singly transfected with the HD expanded repeat exon 1 constructs. These experiments suggest this intrabody offers clinical potential to alleviate the toxic effects of abnormal huntingtin aggregation. The C4 sFv bound to huntingtin form soluble complexes that undergo normal protein turnover. The C4 intrabody is distinct from other approaches that target polyglutamine epitopes. It specifically binds to the HD (1–17) N-terminal sequence unique to huntingtin, rather than to the expanded polyglutamine segment that appears to be directly responsible for self-aggregation in a variety of diseases, but that is also present in normal mammalian proteins such as TATA-binding protein (19, 20). We found that the soluble antigen–intrabody complexes exhibit turnover rates that are similar to those of uncomplexed sFv and soluble exon 1-GFP analogue. These results suggest that the intrabody counteracts aggregation by a previously unrecognized mechanism: the C4 intrabody appears to act as a polyelectrolyte drug that, when bound to the pathological huntingtin, maintains the HD exon 1 protein in a soluble state with little propensity for aggregate formation. This complex is then subjected to the normal mechanisms of protein turnover, which dispose of the abnormal protein complexes, largely precluding aggregate formation.
Assuming that no additional degradation signals are added to the intrabody, C4 sFv binding to exon 1 should allow the native HD protein to maintain its normal functions, which are still unknown. Although the C4 intrabody should crossreact with the normal mouse huntingtin sequence, we have seen no evidence of toxicity from overexpression of C4 intrabodies in any rodent or human cell lines. A new study suggests that the domains of the huntingtin protein that are C-terminal to exon 1 have antiapoptotic properties (21). Thus, C4 may be able to react with the extreme N-terminal region of huntingtin without affecting functional properties of the remainder of the molecule. Recently, a peptide was selected against polyglutamine by using a phage display library (22), and this peptide could reduce the number of aggregates and the number of cells with condensed nuclear DNA when cotransfected into COS-7 cells with polyQ81-GFP. A monoclonal antibody specific for elongated polyQ can also inhibit exon 1 fibrillogenesis in filter assay (23). However, there are sufficient numbers of normal proteins containing polyQ repeats in the 30–40 range that an approach based on polyglutamine targeting alone would appear to lack the protein specificity necessary for in vivo applications. Rather, significant specificity for a single protein should ideally be a component of any clinical treatment, especially because it might continue for decades with a given patient.
The precise relationship remains unclear between pathogenesis, the neuronal inclusions found in both human and animal models of expanded-repeat disease, and the aggregates seen in cells overexpressing fragments of polyQ proteins. There are many studies showing accumulation of abnormal material as both neuronal intranuclear inclusions and neuropil aggregates in transgenic (9, 24), and in human postmortem studies (10, 11). Early stages of pathogenesis may involve the full-length protein, whereas later stages require only N-terminal proteolytic fragments that contain exon 1 (25, 26). Both aggregates and cellular toxicity of N-terminal huntingtin fragments can be suppressed by overexpression of nonspecific chaperone proteins (27, 28). In an important experiment, an HD transgene expressed under a tetracycline-inducible promoter was turned off after induction of the disease. Most dramatically, a reduction in histologically observed aggregates was seen to correlate with improved behavioral parameters over a course of weeks (29).
The aggregates in this test system are likely to be surrogates for a set of abnormal hydrophobic protein–protein interactions, rather than a strict model of the inclusions found in aging brains in vivo. The aggregates that are formed in cells appear to differ in structure, depending on the length of the flanking sequence (30). Regardless of the particular subcellular localization of pathological aggregates, the ability to retarget them to intracellular degradation pathways is a long-term goal that gains considerable credence from the present investigation. Several studies have shown the validity of using intrabodies as immunotherapeutic agents (31–34). However, this investigation has identified a solubilizing property of intrabodies that offers a unique means for counteracting the pathogenesis of neurodegenerative diseases mediated by protein aggregation. We have discovered that huntingtin-specific intrabodies can reduce aggregate formation in cellular models of HD. This work is being extended to the animal level as a further step in preclinical development. Taking a broader perspective, this approach should hold promise for the treatment of other genetic expanded CAG repeat diseases, for neurological diseases that derive from the accumulation of pathological aggregates and for other diseases that involve pathogenic protein–protein interactions.
Supplementary Material
Acknowledgments
This work was inspired by Dr. Helen Molinari and is dedicated to her memory. We thank Alan DuPuis III and Cheryl Moorhead for excellent technical assistance and Dr. Robert Murphy for helpful discussions. This investigation was supported by the Hereditary Disease Foundation (Cure HD Initiative) and National Institutes of Health (NIH) Small Business Technology Transfer Research grants NS38002 (A.M.) and P01-CA42063 (D.H.).
Abbreviations
- intrabodies
intracellular antibodies
- sFv
single-chain Fv
- GST
glutathione S-transferase
- HA
hemagglutinin
- HD
Huntington's disease (huntingtin), N-terminal sequence
- nls
nuclear localization sequence
- DRPLA
dentatorubral-pallidoluysian atrophy (atrophin-1), N-terminal sequence
References
- 1.The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Duyao M P, Auerbach A B, Ryan A, Persichetti F, Barnes G T, McNeil S M, Ge P, Vonsattel J P, Gusella J F, Joyner A L, et al. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 3.Nasir J, Floresco S B, O'Kusky J R, Diewert V M, Richman J M, Zeisler J, Borowski A, Marth J D, Phillips A G, Hayden M R. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 4.Zeitlin S, Liu J P, Chapman D L, Papaioannou V E, Efstratiadis A. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 5.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies S W, et al. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 6.Hodgson J G, Agopyan N, Gutekunst C A, Leavitt B R, LePiane F, Singaraja R, Smith D J, Bissada N, McCutcheon K, Nasir J, et al. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 7.Gusella J F, MacDonald M E. Semin Cell Biol. 1995;6:21–28. doi: 10.1016/1043-4682(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 8.Davies S W, Turmaine M, Cozens B A, Difiglia M, Sharp A H, Ross C A, Scherzinger E, Wanker E E, Mangiarini L, Bates G P. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Li S H, Cheng A L, Mangiarini L, Bates G P, Li X J. Hum Mol Genet. 1999;8:1227–1236. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- 10.Difiglia M, Sapp E, Chase K O, Davies S W, Bates G P, Vonsattel J P, Aronin N. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 11.Gutekunst C A, Li S H, Yi H, Mulroy J S, Kuemmerle S, Jones R, Rye D, Ferrante R J, Hersch S M, Li X J. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marasco W A, Haseltine W A, Chen S Y. Proc Natl Acad Sci USA. 1993;90:7889–7893. doi: 10.1073/pnas.90.16.7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Proc Natl Acad Sci USA. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onodera O, Roses A D, Tsuji S, Vance J M, Strittmatter W J, Burke J R. FEBS Lett. 1996;399:135–139. doi: 10.1016/s0014-5793(96)01301-4. [DOI] [PubMed] [Google Scholar]
- 15.Sheets M D, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, Hemingsen G, Wong C, Gerhart J C, Marks J D, et al. Proc Natl Acad Sci USA. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schier R, Bye J, Apell G, McCall A, Adams G P, Malmqvist M, Weiner L M, Marks J D. J Mol Biol. 1996;255:28–43. doi: 10.1006/jmbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 17.Karlsson R, Michaelsson A, Mattsson L. J Immunol Methods. 1991;145:229–240. doi: 10.1016/0022-1759(91)90331-9. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Q, Zeng C, Huhalov A, Yao J, Turi T G, Danley D, Hynes T, Cong Y, DiMattia D, Kennedy S, et al. J Immunol Methods. 1999;231:207–222. doi: 10.1016/s0022-1759(99)00158-1. [DOI] [PubMed] [Google Scholar]
- 19.Peterson M G, Tanese N, Pugh B F, Tjian R. Science. 1990;248:1625–1630. doi: 10.1126/science.2363050. [DOI] [PubMed] [Google Scholar]
- 20.Rubinsztein D C, Leggo J, Crow T J, DeLisi L E, Walsh C, Jain S, Paykel E S. Am J Med Genet. 1996;67:495–498. doi: 10.1002/(SICI)1096-8628(19960920)67:5<495::AID-AJMG12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 21.Rigamonti D, Bauer J H, De Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden M R, Li Y, et al. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagai Y, Tucker T, Ren H, Kenan D J, Henderson B S, Keene J D, Strittmatter W J, Burke J R. J Biol Chem. 2000;275:10437–10442. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- 23.Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, Lehrach H, Wanker E E. Proc Natl Acad Sci USA. 2000;97:6739–6744. doi: 10.1073/pnas.110138997. . (First Published May 30, 2000; 10.1073/pnas.110138997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ordway J M, Tallaksen-Greene S, Gutekunst C A, Bernstein E M, Cearley J A, Wiener H W, Dure L S, Lindsey R, Hersch S M, Jope R S, et al. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 25.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim S U, et al. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 26.Kegel K B, Kim M, Sapp E, McIntyre C, Castano J G, Aronin N, Difiglia M. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jana N R, Tanaka M, Wang G, Nukina N. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 28.Carmichael J, Chatellier J, Woolfson A, Milstein C, Fersht A R, Rubinsztein D C. Proc Natl Acad Sci USA. 2000;97:9701–9705. doi: 10.1073/pnas.170280697. . (First Published August 1, 2000; 10.1073/pnas.170280697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto A, Lucas J J, Hen R. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 30.Persichetti F, Trettel F, Huang C C, Fraefel C, Timmers H T, Gusella J F, MacDonald M E. Neurobiol Dis. 1999;6:364–375. doi: 10.1006/nbdi.1999.0260. [DOI] [PubMed] [Google Scholar]
- 31.Richardson J H, Sodroski J G, Waldmann T A, Marasco W A. Proc Natl Acad Sci USA. 1995;92:3137–3141. doi: 10.1073/pnas.92.8.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshane J, Siegal G P, Wang M, Wright M, Bucy R P, Alvarez R D, Curiel D T. Gynecol Oncol. 1997;64:378–385. doi: 10.1006/gyno.1996.4566. [DOI] [PubMed] [Google Scholar]
- 33.Graus-Porta D, Beerli R R, Hynes N E. Mol Cell Biol. 1995;15:1182–1191. doi: 10.1128/mcb.15.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mhashilkar A M, Bagley J, Chen S Y, Szilvay A M, Helland D G, Marasco W A. EMBO J. 1995;14:1542–1551. doi: 10.1002/j.1460-2075.1995.tb07140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.