

Abstract
Mutational and biophysical analysis suggests that an intracellular COOH-terminal domain of the large conductance Ca2+-activated K+ channel (BK channel) contains Ca2+-binding site(s) that are allosterically coupled to channel opening. However the structural basis of Ca2+ binding to BK channels is unknown. To pursue this question, we overexpressed the COOH-terminal 280 residues of the Drosophila slowpoke BK channel (Dslo-C280) as a FLAG- and His6-tagged protein in Escherichia coli. We purified Dslo-C280 in soluble form and used a 45Ca2+-overlay protein blot assay to detect Ca2+ binding. Dslo-C280 exhibits specific binding of 45Ca2+ in comparison with various control proteins and known EF-hand Ca2+-binding proteins. A mutation (D5N5) of Dslo-C280, in which five consecutive Asp residues of the “Ca-bowl” motif are changed to Asn, reduces 45Ca2+-binding activity by 56%. By electrophysiological assay, the corresponding D5N5 mutant of the Drosophila BK channel expressed in HEK293 cells exhibits lower Ca2+ sensitivity for activation and a shift of ≈+80 mV in the midpoint voltage for activation. This effect is associated with a decrease in the Hill coefficient (N) for activation by Ca2+ and a reduction in apparent Ca2+ affinity, suggesting the loss of one Ca2+-binding site per monomer. These results demonstrate a functional correlation between Ca2+ binding to a specific region of the BK protein and Ca2+-dependent activation, thus providing a biochemical approach to study this process.
Large conductance Ca2+-activated K+ channels (BK channels) are synergistically activated by membrane depolarization and intracellular Ca2+ (1). They are widely distributed in neurons and smooth muscle, where they respectively function in regulating the release of neurotransmitter and cell contractility. Although BK channels can be activated by high positive voltage (>+100 mV) in the virtual absence of Ca2+ (<1 nM) (2, 3), an increase in local Ca2+ concentration shifts the voltage activation threshold toward the physiological voltage range. The BK channel protein is structurally related to tetrameric voltage-gated K+ channels by homology within an S1–S6 region that contains the pore-forming domain, S5-P-S6, and an S4 voltage-sensing element (4); however, it also includes an additional NH2-terminal transmembrane span, S0 (5), and a unique COOH-terminal sequence of ≈850 residues (see Fig. 1). Sequence analysis identified four hydrophobic segments in the C-terminal region, S7–S10, as potential transmembrane spans (4), but recent evidence shows that S9 and S10 are contained within cytoplasmic domain(s) (5).
Figure 1.

Schematic overview of Drosophila BK channel and expressed fragment. (A) Topology diagram of Dslo α subunit showing the location of Core and Tail domains. (B) Construction of Dslo-C280 protein (and D5N5 mutant) expressed in E. coli. (C) Sequence of Ca-bowl motif (Dslo residues T952-Q979) of Dslo-C280 and D5N5 mutant.
Current models of BK channel gating propose that consecutive binding of one Ca2+ ion per monomer of a homotetrameric complex promotes channel opening in a cooperative reaction that is allosterically coupled to a cooperative voltage-dependent gating mechanism (6, 7). This complex mechanism shares basic features in common with the Monod-Wyman-Changeux model for allosteric activation of a tetrameric enzyme (8). Structure-function analysis indicates that the BK channel protein can be dissected into conserved “Core” and “Tail” domains that are separated by a nonconserved linker region (9). The Core corresponds to the NH2-terminal part of the protein containing S0–S6 plus a portion of the COOH terminus, whereas the ≈400 residue sequence following the linker comprises the Tail (Fig. 1). Experiments with chimeric Core-Tail constructs by using Slo1 genes of different species suggest that the Ca2+-sensing function of the channel is associated with the Tail domain (9). More recent studies making use of the Ca2+-insensitive Slo3 K+ channel gene verified that specific regions of protein sequence within the Tail confer the property of Ca2+-dependent gating (10). Complementary mutational studies identified an Asp-rich sequence motif in the Tail domain (Fig. 1), called the “Calcium Bowl” (Ca-bowl), as a possible site(s) of Ca2+ binding (11).
Despite this progress, the structural basis of Ca2+ binding to the BK channel protein and the mechanism by which Ca2+ binding is coupled to channel opening are poorly understood. To pursue this problem, we adopted a biochemical approach to assay and localize Ca2+ binding sites. This assay employs a 45Ca2+-overlay technique that has been used to identify other Ca2+-binding proteins: e.g., proteins that contain EF-hand motifs such as calmodulin (12, 13). We overexpressed in Escherichia coli the COOH-terminal 280 residues of the Drosophila BK channel (Dslo-C280) containing the Ca-bowl region. This protein was purified and found to exhibit 45Ca2+ binding activity comparable to calmodulin. The functional significance of such activity was examined by mutating five consecutive Asp residues in the Ca-bowl to Asn and comparing the 45Ca2+-signal with Ca2+-activation of the channel by electrophysiological analysis. The results demonstrate a direct correlation between Ca2+ binding and Ca2+ sensitivity for channel activation. This soluble Ca2+-binding domain thus represents a promising candidate for structural analysis of ligand binding central to the mechanism of BK channel gating.
Materials and Methods
Construction of the Ca-Bowl Mutant D5N5 and Expression of Dslo.
A pRc/cytomegalovirus (CMV) vector (Invitrogen) containing the coding sequence of an alternatively spliced variant (A1C2E1G3I0) of the Drosophila BK channel (Dslo) described by Adelman et al. (14) was generously supplied by Dr. Irwin Levitan (University of Pennsylvania). In sequencing this clone, we noted the presence of one conservative mutation, D964E (see Fig. 1C) that varied from the reported wild-type sequence (14). Silent mutations were used to introduce two unique restriction sites flanking the Ca-bowl motif (11). These sites were used to generate the mutant Dslo channel, D5N5, in which five consecutive Asp residues in the Ca-bowl region D966-D970 were changed to Asn by PCR mutagenesis (15). All constructions were verified by DNA sequencing. The restriction fragment containing the D5N5 mutant was subcloned into the pcDNA3.1 mammalian cell expression vector (Invitrogen) and used to transfect HEK293 cells with the Effectene Kit from Qiagen (Chatsworth, CA). Methods for producing a stably transfected HEK293 cell line expressing the parent Dslo clone were reported previously (16).
Construction and Bacterial Expression of COOH-Terminal Protein Fragments Dslo-C280 and C280-D5N5.
A restriction fragment coding for the last 280 aa of Dslo was inserted in frame into the bacterial expression vectors pFLAG-ATS and pH6FLAG-ATS. pFLAG-ATS is a commercial (Sigma) vector for FLAG-tagged expression of foreign proteins in E. coli. This vector adds an NH2-terminal OmpA signal sequence, followed by an eight-residue FLAG epitope sequence (DYKDDDDK). The related pH6FLAG-ATS vector was constructed by adding a hexahistidine (His6) coding sequence to pFLAG-ATS inserted after the OmpA signal sequence and directly before the FLAG sequence, as illustrated in Fig. 1. For protein production, vector-transformed E. coli strain BL21 (Stratagene) was grown in LB media at 22°C, and induced by adding 0.2 mM isopropyl β-d-thiogalactoside (IPTG).
Fractionation of Dslo-C280 and C280-D5N5.
Analytical fractionation of FLAG-tagged Dslo fusion proteins followed protocols in the Sigma FLAG E. coli Expression System Manual. A sucrose-based osmotic shock procedure was used for isolation of periplasmic proteins. Whole cells were lysed with lysozyme by using extraction buffers containing DNAaseI, and ovomucoid or aprotinin protease inhibitors. The whole cell soluble fraction was collected after centrifugation at 20,000 × g for 0.5 h. The pellet, containing whole cell insoluble protein, was washed thoroughly in 50 mM Tris⋅HCl (pH 8.0), 500 mM NaCl, 0.2% Tween 20, 10 mM EDTA, and finally in 50 mM Tris⋅HCl (pH 8.0) and 150 mM NaCl. Washed inclusion body protein was solubilized by denaturation in 6 M guanidine-HCl for 12 h at 4°C with shaking. Refolding was achieved by dialysis in 50 mM Tris⋅HCl (pH 8.0), 150 mM NaCl, 10% glycerol, and stepwise reduction of guanidine HCl (4 M to 0 M) and DTT (5 mM to 0 mM) over the course of 3 days at 4°C. Chemiluminescent detection of FLAG-tagged Dslo protein on immunoblots probed with M2 anti-FLAG antibodies (Sigma) was performed according to the supplier's (Pierce) protocol.
Purification of Dslo-C280 and C280-D5N5.
Both FLAG-based and His6-based affinity methods were used in purifying soluble and refolded Dslo protein. M2 anti-FLAG agarose was used according to the manufacturer's (Sigma) instructions. In the final step, the M2 column was eluted with 5 column volumes of 100 μg/ml FLAG peptide. The His6-based method, applied to purifying Dslo protein from inclusion bodies, combines purification and renaturation in one step on a Ni2+-nitrilotriacetic acid (NTA; Amersham Pharmacia) column at 4°C. The start buffer, composed of 5 mM imidazole, 40 mM Tris⋅HCl (pH 8.0), 6 M guanidine-HCl, 0.5 M NaCl, and 1 mM β-mercaptoethanol, was used for solubilizing inclusion bodies and loading the protein sample onto a 5-ml Ni2+-NTA column. The column was then eluted with 20 bed volumes of wash buffer (15 mM imidazole/20 mM Tris⋅HCl, pH 8.0/6 M guanidine-HCl/0.5 M NaCl/1 mM β-mercaptoethanol) to remove extraneous protein. The refolding process was performed over 20 h with the application of a gradient starting from wash buffer (150 ml) mixed linearly with an equal volume of refolding buffer (15 mM imidazole/40 mM Tris⋅HCl, pH 6.8/0.5 M NaCl/1 mM β-mercaptoethanol). Refolded His6-tagged protein was finally eluted from the Ni2+-NTA column with 0.5 M imidazole, 0.5 M NaCl, 2 mM β-mercaptoethanol, adjusted to pH 8.0 with acetic acid. Either zwittergent 3–12 (2 mM) or dodecylmaltoside (5 mM) was added before protein concentration and storage to reduce aggregation. High performance size exclusion chromatography (SEC) used a TSK G3000SW column (7.5 × 600 mm; Tosoh Biosep). The isocratic elution buffer was 20 mM Hepes-NaOH (pH 7.2), 300 mM ammonium acetate, 10 mM CHAPS detergent, and 10 mM DTT.
45Ca2+ Overlay Assay.
After SDS/PAGE, protein bands were electroblotted onto poly(vinylidene difluoride) membrane. The blot was extensively washed for 2 × 10 min and then 3 × 15 min by using 20 ml of wash buffer each time (10 mM imidazole-HCl, pH 6.7/70 mM KCl/0.5 mM MgCl2). 45Ca2+ overlay assay (14, 15) was performed by incubating the blot with 2 μM 45CaCl2 (21 mCi/mg, NEN) in 20 ml wash buffer at 22°C for 1 h, followed by 3 × 2 min washes in deionized H2O and 1 × 2 min in 50% ethanol. After the blot was air dried, it was exposed to Hyperfilm MP (Amersham Pharmacia) for 48 h. After developing the film, the poly(vinylidene difluoride) blot was stained for all proteins with Coomassie blue R250. Bands detected by 45Ca2+- autoradiography and Coomassie blue were recorded on an IS-1000 digital imaging system (Alpha Innotech, San Leandro, CA). Relative peak areas of bands were measured by using densitometry software. For quantitative comparison, Dslo-C280 and C280-D5N5 protein was measured with the BCA protein assay (Pierce).
Patch Clamp Experiments.
HEK293 cells were cultured at 37°C in DMEM media (GIBCO/BRL) supplemented with 10% FBS, 45 units/ml penicillin, and 45 mg/ml streptomycin. Cells stably transfected with the parent Dslo channel (16) were grown in the presence of 0.9 mg/ml G418 (GIBCO/BRL). Gigaohm patch recordings in the inside-out mode were performed by using an EPC-9 amplifier and pulse-pulsefit software (HEKA Electronics, Lambrecht/Pfalz, Germany). Firepolished micropipettes pulled from Corning 7052 capillary glass had a resistance of ≈8 MΩ in recording solution. The pipette solution (extracellular) was 100 mM K+-gluconate, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 1.3 mM EGTA, 10 mM Hepes-KOH (pH 7.3). The bath solution (intracellular) was 103 mM K+-gluconate, 5 mM KCl, 10 mM Hepes-KOH (pH 7.3). Bath solution was treated with Chelex 100 resin (Sigma) to reduce Ca2+ to trace levels, and solutions containing 10 to 1,000 μM Ca2+ were prepared by adding CaCl2.
Results
Expression and Purification of Dslo-C280.
Sequence coding for 280 COOH-terminal residues of Dslo, the Drosophila slowpoke gene product, was cloned into the pFLAG-ATS bacterial expression vector. As illustrated in Fig. 1B, this vector directs synthesis of a protein that has an NH2-terminal OmpA secretory signal peptide (21 residues), followed by an optional (His)6 tag, the FLAG epitope sequence (DYKDDDDK), eight residues (VKLLENSR) from the multiple cloning site, and 280 residues of DSlo. When vector-transformed E. coli BL21 cells were grown and fractionated, the Dslo-C280 protein was found in both soluble and insoluble fractions. Immunoblot detection by using M2 monoclonal antibody specific for the Flag-epitope verified that expression of a ≈34-kDa FLAG-tagged band was observed only when the cells were induced with IPTG (Fig. 2A, lane 1 vs. lanes 2–6). This FLAG-tagged protein was present in the pellet from lysed cells as well as in the high-speed supernatant (Fig. 2A, lanes 2 and 3), and was also present in soluble fractions of periplasmic protein released from osmotically shocked cells (Fig. 2A, lanes 5 and 6). These results indicate that E. coli secretes a certain amount of DSlo-C280 into the periplasm as a soluble protein, but that much of this protein is retained within the cell in the form of insoluble inclusion bodies, as typically observed for overexpression of many eukaryotic proteins in E. coli.
Figure 2.

Expression and purification of Dslo-C280. (A) Immunoblot detection of Dslo-C280 band by anti-FLAG M2 antibody in various fractions of E. coli before (lane 1) or after (lanes 2–6) induction with IPTG. Lanes: 1, whole cells before IPTG; 2, insoluble protein pellet from lysed cells; 3, soluble protein from lysed cells; 4, whole cells; 5, soluble fraction after high sucrose treatment; and 6, soluble fraction after osmotic shock. (B) Purification via FLAG epitope on M2 anti-FLAG affinity column. SDS/PAGE of various fractions. Lanes: 1, molecular mass markers (K = kDa); 2, crude soluble extract; 3, crude inclusion body sample; 4, fraction from soluble extract after M2 column purification; and 5, fraction of renatured inclusion protein after M2 column purification. (C) Purification by SEC. SDS/PAGE of fractions from TSK G3000SW column. Lanes: 1, molecular mass markers; 2, SEC peak fraction at ≈68 kDa; and 3, SEC peak fraction at ≈34 kDa. (D) Purification via His6-tag of renatured inclusion protein. SDS/PAGE of various fractions from Ni2+-NTA column. The arrow in B, C, and D points to the Dslo-C280 band.
Dslo-C280 protein was purified both from the soluble bacterial extract and from inclusion protein first denatured in guanidine-HCl and refolded during dialysis. Figure 2B illustrates immuno-affinity purification by using an anti-Flag M2 column and specific elution with FLAG peptide. A FLAG-tagged band of the expected size (≈34 kDa) was observed for both types of samples, in addition to a few contaminant bands, for the originally soluble (Fig. 2B, lane 4) and refolded preparations (Fig. 2B, lane 5). In general, the inclusion protein fraction yielded a greater amount of pure DSlo-C280 (e.g., Fig. 2B, lane 5).
The purified FLAG-tagged Dslo-C280 protein is quite soluble; however, we found that concentrated samples tend to form a large aggregate (>200 kDa) as indicated by its appearance in the void volume when injected onto a high performance size exclusion column. By testing various detergents, we found that this aggregate could be disrupted into smaller complexes. Dslo-C280 protein pretreated with 20 mM Zwittergent C3–12 or 15 mM dodecylmaltoside was found to migrate on the SEC column as three distinct protein peaks (not shown) at elution volumes that nominally correspond to monomer (≈34 kDa), dimer (≈68 kDa), and a larger complex (≈140 kDa). SDS/PAGE analysis of Dslo-C280 taken from the dimer and monomer peaks of the SEC fractionation is illustrated in Fig. 2C, lanes 2 and 3, respectively. This experiment confirmed that Dslo-C280 can exist as a monomeric species in the presence of a mild detergent, but it also showed that this protein has a tendency to aggregate by forming dimers and higher oligomers.
A (His)6 tag sequence was introduced before the FLAG tag sequence in another version of Dslo-C280. This protein and a corresponding (His)6-FLAG-tagged Ca-bowl mutant (D5N5) were separately expressed in E. coli and purified by metal chelate affinity chromatography. This method allowed us to efficiently combine purification and refolding of the denatured inclusion body protein into one step (17). Purification of the (His)6-tagged protein by elution of the Ni2+-NTA column with imidazole is shown by SDS/PAGE analysis in Fig. 2C. Together, studies of Fig. 2 demonstrate that a soluble protein corresponding to the C-terminal 280 residues of Dslo can be synthesized in E. coli and purified in soluble form after refolding from inclusion protein.
45Ca2+-Binding Activity of Dslo-C280 and a D5N5 Mutant.
Many Ca2+-binding proteins can be qualitatively identified by autoradiographic 45Ca2+-overlay assay performed in a protein blot format (12, 13). This technique is especially useful for identification of proteins containing EF hand Ca2+-binding sites such as calmodulin, troponin C, TCBP-49, and calexcitin (12, 13, 18, 19). It has also been used to identify putative Ca2+ binding sites in NH2- and COOH-terminal domains of Ca2+-ATPase (13, 20) and Ca2+-release channel/ryanodine receptor (21). We find that the DSlo-C280 protein exhibits 45Ca2+ binding activity in this assay under conditions that selectively label known Ca2+-binding proteins. Fig. 3 compares the 45Ca2+ signal (Right) from two different protein blots with the same blots stained for protein with Coomassie blue (Left). In Fig. 3A, it can be seen that both purified Dslo-C280 (lane 2) and a corresponding band from the crude E. coli extract (lane 7) retain 45Ca2+ with a signal comparable to calmodulin (lane 3) and calpain (lane 4). Both calmodulin and the large subunit of calpain, a Ca2+-activated proteinase, contain four EF hand Ca2+-binding motifs. In contrast, other proteins on the same blot, MW markers (lane 1, BSA, chicken ovalbumin, rabbit glyceraldehyde-3-phosphate dehydrogenase, bovine carbonic anhydrase, bovine trypsinogen, soybean trypsin inhibitor, and bovine α-lactalbumin), FLAG-tagged alkaline phosphatase of E. coli (lane 5), and bovine trypsin (lane 6) are not labeled by 45Ca2+ under these conditions. Because Dslo-C280 is an acidic protein with a calculated pI of 4.1, such binding could simply reflect an electrostatic interaction with divalent metal cations rather than specific localized Ca2+-binding sites like those of calmodulin. The experiment of Fig. 3B compares the 45Ca2+ signal of DSlo-C280 (lane 1) with another EF hand protein, troponin C (an ≈18-kDa component of the troponin complex in lane 2), and acidic proteins such as annexin V (pI = 4.8, lane 3), glucose oxidase (pI = 4.2, lanes 4 and 5), and chicken ovomucoid protease inhibitor (pI ≈ 4.3, lane 6). This experiment shows that Dslo-C280 exhibits a stronger 45Ca2+-binding signal than other acidic proteins tested under the same conditions, further suggesting that the BK channel fragment binds Ca2+ in a specific manner.
Figure 3.

Protein blot assays demonstrating specificity of 45Ca2+ binding. Purified Dslo-C280 and various control proteins were subjected to SDS/PAGE, electroblotted onto poly(vinylidene difluoride) membrane, and processed for 45Ca2+-overlay assay: (Right) image of exposed film; (Left) same blot stained with Coomassie Blue. (A) Lanes: 1, molecular mass markers; 2, Dslo-C280 (≈7 μg); 3, bovine calmodulin (4 μg); 4, rabbit calpain (6 μg); 5, FLAG-tagged E. coli alkaline phosphatase (4 μg); 6; bovine trypsin (4 μg); and 7, crude extract from E. coli expressing Dslo-C280. (B) Lanes: 1, Dslo-C280 (3 μg); 2, chicken troponin complex (6 μg); 3, human annexin V (3 μg); 4, Aspergillus niger glucose oxidase (3 μg); 5, glucose oxidase (6 μg); and 6, chicken ovomucoid protease inhibitor (3 μg).
To investigate the function of the Ca-bowl, we purified a D5N5 mutant of the Dslo-280 fragment in which five consecutive Asp residues of the Ca-bowl region (Fig. 1C) were changed to Asn. Schreiber and Salkoff (11) previously found that this same mutation of mouse BK channel (mSlo) reduced the sensitivity to Ca2+-activation by shifting the midpoint of the voltage activation curve (V0.5) by an average of +56 mV over the range of 4 to 1,000 μM internal Ca2+. Fig. 4 compares the 45Ca2+-binding signal of Dslo-C280 and the D5N5 mutant for 1, 3, and 8 μg of protein in a parallel assay on the same blot. The results indicate that the 45Ca2+ signal is proportional to the amount of both proteins but significantly lower for D5N5 vs. control Dslo-C280. Such data were quantitated by densitometric analysis of the 45Ca2+ and Coomassie blue protein signals (Fig. 4 B and C). The results show that the 45Ca2+-binding signal of the D5N5 mutant was reduced to 44 ± 6% (±SD, n = 3) of control (Fig. 4D). Thus, neutralization of five Asp residues of the Ca-bowl to Asn substantially reduces Ca2+ binding to a fragment of the Tail domain that is proposed to function as a Ca2+ sensor for BK channel activation (11).
Figure 4.

Comparison of 45Ca2+-binding activity of Dslo-C280 and corresponding D5N5 mutation. (A) Purified Dslo-C280 and D5N5 mutant (1, 3, and 8 μg) were subjected to SDS/PAGE, electroblotted onto PDVF, and assayed by 45Ca2+-overlay method: (Upper) image of exposed film; (Lower) Coomassie blue-stained protein bands. (B and C) Densitometric measurement of protein bands (▴), 45Ca2+ signal (●), and 45Ca2+/Coomassie ratio (♦) for Dslo-C280 (B) and D5N5 mutant (C). (D) Protein-normalized ratio of 45Ca2+-binding activity of D5N5 mutant relative to Dslo-C280.
Correlation of Ca2+-Binding with Ca2+-Dependent Activation.
To determine how the D5N5 mutation affects activation of BK channel opening, we expressed the A1C2E1G3I0 splice version of Dslo and the corresponding D5N5 mutant channel in HEK293 cells and characterized their voltage- and Ca2+-dependence in excised membrane patches. Fig. 5 shows families of macroscopic K+ currents evoked by a series of depolarizing voltage pulses for the parent Dslo channel with 60 μM internal Ca2+ (Fig. 5A) and the D5N5 mutant with 100 μM Ca2+ (Fig. 5B). At a given Ca2+ concentration, the D5N5 mutant requires higher positive voltage than the parent channel to reach maximal activation. This result is shown more directly in Fig. 5 C and D, which compare averaged voltage activation curves of the two channels at Ca2+ concentrations ranging from 10 to 1,000 μM. These data were obtained by normalizing the steady-state conductance (G) at a given voltage to the maximal conductance (Gmax) measured at high Ca2+ for the same patch.
Figure 5.

Analysis of the effect of the D5N5 Ca-bowl mutation on BK channel activation. Dslo and D5N5 mutant BK channels were expressed in HEK293 cells and characterized by patch clamp analysis in inside-out patches. (A and B) Current traces evoked by consecutive voltage steps of +10 mV from a holding potential of −140 mV for Dslo (A) and −120 mV for D5N5 (B) with 60 μM and 100 μM Ca2+ in the bath solution, respectively. (C and D) Normalized G-V curves representing the average of 5 to 12 patches at indicated Ca2+ concentrations for parent Dslo (C) and D5N5 mutant (D). Error bars denote ±SEM. Solid curves are fits to a Boltzmann relationship (Eq. 1). (E and F) Comparison of V0.5 (E) and Q (F) vs. [Ca2+]. Data points are the mean ±SD of Boltzmann fit parameters from 5–12 patches. (G and H) Mean values of steady-state Popen (G/Gmax) as a function of [Ca2+] at various voltages compared for Dslo (G) and D5N5 mutant (H). Solid curves indicate fits to Hill equation (Eq. 2). (I and J) Comparison of Hill parameters, N (I) and Kapp (J) vs. voltage.
Individual conductance-voltage curves for each patch were fit with the Boltzmann relationship:
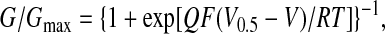 |
1 |
where V is applied voltage, V0.5 is midpoint voltage, Q is apparent gating charge, F is Faraday's constant, R is the gas constant, and T is absolute temperature. V0.5 for activation of the D5N5 channel is shifted by an average of +82 mV relative to the control Dslo channel over the accessible range of Ca2+ concentration (Fig. 5E). This behavior is similar to that observed by Schreiber and Salkoff (11) for a collection of Ca-bowl mutants of mSlo described as a “(+)-shifted” phenotype. Average values of Q derived from the Boltzmann fit typically fell in the range of 0.9–1.3 and did not show a significant or systematic difference between the mutant and control channel (Fig. 5F).
To directly examine the Ca2+ dependence of the two types of channels, activation data were also plotted as Popen (G/Gmax) vs. log[Ca2+] over the accessible voltage range and fit to the following modified form of the Hill equation:
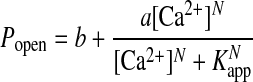 |
2 |
In accord with a Monod-Wyman-Changeux (8) theory for Ca2+-activation, Eq. 2 accounts for the fact that, in the limit of zero Ca2+, there is a minimum value of Popen, described by the b parameter, to which the channel is activated by positive voltage alone (22). Eq. 2 also incorporates a maximum value of Popen, given by the quantity b + a, that occurs in the limit of high Ca2+ and accounts for the observation that the maximum open state probability can still be reduced by negative voltage, even when the Ca2+-binding sites are fully occupied (22). Plots of the [Ca2+] dependence of Popen and fits to Eq. 2 are compared in Fig. 5 G and H for control Dslo and D5N5 mutant channels, respectively. The results show that Popen for the D5N5 mutant titrates over a greater and broader range of [Ca2+] than the parent Dslo. This finding can be summarized by comparison of the Hill coefficient, N, and the apparent dissociation constant for Ca2+, Kapp, as shown in Fig. 5 I and J, respectively. In Fig. 5I, we see that N for the parent Dslo channel ranges from 3.2 to 5.8, whereas N for the D5N5 channel is shifted to lower values ranging from 1.3 to 3.9 over the accessible voltage range. Figure 5J shows that Kapp for Ca2+ increases with negative voltage and is shifted to higher values for the mutant channel.
Discussion
This work demonstrates that a COOH-terminal portion of the Drosophila BK channel binds 45Ca2+ in a protein blot assay. Under defined conditions (ionic strength, pH, Mg2+ concentration, and washing), this assay reliably identifies many different Ca2+-binding proteins, particularly those with EF-hand motifs (12, 13). The fact that Dslo-C280 exhibits a strong 45Ca2+-binding signal relative to control proteins (Fig. 3) supports the idea that this part of the Dslo Tail domain contains Ca2+-binding site(s) discretely localized within its linear sequence. The molecular basis of Ca2+ binding to calmodulin and other EF-hand proteins in the protein blot format is unknown; however, it has been suggested that discretely localized Ca2+-binding sites can partially refold and reconstitute Ca2+-coordination within the blotted protein band in the same way that denaturation-resistant epitopes are recognized by certain antibodies after transfer of antigen proteins from SDS/PAGE gels (12).
Although the BK channel protein does not contain a canonical EF-hand motif (4), a conserved region of sequence rich in Asp residues in the Tail domain, known as the Ca-bowl, has emerged as a candidate Ca2+-binding site (10, 11). Based on studies of the interaction of BK channels with certain serine protease inhibitors, Moss et al. (23) previously suggested that the Ca-bowl region may be structurally related to the relatively low affinity (KD ≈ 100 μM) Ca2+-binding loop of serine proteinases such as trypsin, coagulation Factors VII, IX, X, and protein C (24). Such proposals for a Ca2+-binding function of the Ca-bowl are supported by results of Fig. 4, which show that mutation of five consecutive Asp residues in this region to Asn result in a 56% reduction in 45Ca2+-signal in the overlay assay.
Structure-function analyses of the Ca-bowl region of the mammalian BK channel (mSlo) have been interpreted to suggest that the Tail domain contains at least two distinct Ca2+-binding sites (10, 11). In addition to showing that the Ca-bowl region is functionally important for Ca2+-binding and Ca2+-dependent activation of an insect BK channel, the present work supports the idea that there is more than one Ca2+-binding site per Tail domain. This possibility is suggested by the fact that the D5N5 mutation does not completely eliminate 45Ca2+-binding (Fig. 4) and by the particular effect of this mutation on the Hill coefficient for Ca2+ activation. As shown in Fig. 5 G and I, the Hill coefficient of the control Dslo channel increases as a function of positive voltage, increasing from N ≈ 3 at −40 mV to N ≈ 6 at +80 mV. A systematic increase in the Hill coefficient of activation by Ca2+ (and Sr2+) over a similar voltage range has been previously observed in other studies of BK channels from diverse species (22, 25–28). This behavior can be simulated by a Monod-Wyman-Changeux type of model of Ca2+ activation and arises from the allosteric coupling of Ca2+- and voltage-dependent activation (S.B. and E.M., unpublished results). Here, we observed that Hill coefficient values of the D5N5 channel are sharply reduced from the parent Dslo channel (Fig. 5I). One way to account for the results of Fig. 5 is by postulating that the Dslo BK channel contains at least two distinct Ca2+-binding sites per Tail domain, for a minimum of eight sites per tetramer. This relationship gives a molecularity of 8 to the Ca2+-activation process and leads to the theoretical possibility of Hill coefficients ≥4 observed here and in many previous studies of BK channels (29–32). If the D5N5 mutation essentially eliminates one of two Ca2+-binding sites in the Tail domain, this mutation would result in a channel in which the allowable range of the Hill coefficient is reduced to less than 4, as observed in Fig. 5I.
In conclusion, this set of experiments provides a direct correlation between a biochemical measurement of Ca2+ binding to the Tail domain and Ca2+-dependent activation of a BK channel. The results support the idea (but do not yet prove) that the Ca-bowl sequence motif directly participates in Ca2+ binding and that BK channels contain at least two Ca2+-binding sites per monomer of a homotetrameric complex. Further quantitative biochemical and structural studies of the Tail domain produced in E. coli ought to reveal more details relevant to the mechanism of BK channel gating.
Acknowledgments
We thank Dr. Chien-Jung Huang for assistance with electrophysiology experiments. This work was supported by a grant from the National Institutes of Health (GM51172) and by fellowship awards from the American Heart Association to S.B. and I.F.
Abbreviations
- BK channel
large conductance Ca2+-activated K+ channel
- Dslo
Drosophila BK channel
- Dslo-C280
COOH-terminal 280 residues of Dslo
- IPTG
isopropyl β-d-thiogalactoside
- NTA
nitrilotriacetic acid
- SEC
size exclusion chromatography
References
- 1.Toro L, Wallner M, Meera P, Tanaka Y. News Physiol Sci. 1998;13:112–117. doi: 10.1152/physiologyonline.1998.13.3.112. [DOI] [PubMed] [Google Scholar]
- 2.Meera P, Wallner M, Jiang Z, Toro L. FEBS Lett. 1996;382:84–88. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- 3.Cui J, Cox D H, Aldrich R W. J Gen Physiol. 1997;109:647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler A, Tsunoda S, McCobb D P, Wei A, Salkoff L. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- 5.Meera P, Wallner M, Song M, Toro L. Proc Natl Acad Sci USA. 1997;94:14066–14071. doi: 10.1073/pnas.94.25.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horrigan F T, Cui J, Aldrich R W. J Gen Physiol. 1999;114:277–304. doi: 10.1085/jgp.114.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothberg B S, Magleby K L. J Gen Physiol. 1999;114:93–124. doi: 10.1085/jgp.114.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monod J, Wyman J, Changeux J P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 9.Wei A, Solaro C, Lingle C, Salkoff L. Neuron. 1994;13:671–681. doi: 10.1016/0896-6273(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber M, Yuan A, Salkoff L. Nat Neurosci. 1999;2:416–421. doi: 10.1038/8077. [DOI] [PubMed] [Google Scholar]
- 11.Schreiber M, Salkoff L. Biophys J. 1997;73:1355–1363. doi: 10.1016/S0006-3495(97)78168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maruyama K, Mikawa T, Ebashi S. J Biochem. 1984;95:511–519. doi: 10.1093/oxfordjournals.jbchem.a134633. [DOI] [PubMed] [Google Scholar]
- 13.Garrigos M S, Deschamps S, Veil A, Lund S, Champeil P, Moller J V, le Maire M. Anal Biochem. 1991;194:82–88. doi: 10.1016/0003-2697(91)90154-l. [DOI] [PubMed] [Google Scholar]
- 14.Adelman J P, Shen K-Z, Kavanaugh M P, Warren R A, Wu Y-N, Lagrutta A, Bond C T, North R A. Neuron. 1991;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- 15.Nelson R M, Long G L. Anal Biochem. 1989;180:147–151. doi: 10.1016/0003-2697(89)90103-6. [DOI] [PubMed] [Google Scholar]
- 16.Moss G W J, Marshall J, Morabito M, Howe J R, Moczydlowski E. Biochemistry. 1996;35:16024–16035. doi: 10.1021/bi961452k. [DOI] [PubMed] [Google Scholar]
- 17.Colangeli R, Heijbel A, Williams A M, Manca C, Chan J, Lyashchenko K, Gennaro M L. J Chromatogr B. 1998;714:223–235. doi: 10.1016/s0378-4347(98)00094-2. [DOI] [PubMed] [Google Scholar]
- 18.Dodds D, Schlimgen A K, Lu S-Y, Perin M S. J Neurochem. 1995;64:2339–2344. doi: 10.1046/j.1471-4159.1995.64052339.x. [DOI] [PubMed] [Google Scholar]
- 19.Nelson T J, Cavallaro S, Yi C-L, McPhie D, Schreuers B G, Gusev P A, Favit A, Zohar O, Kim J, Beushausen S, Ascoli G, Old J, Neve R, Alkon D L. Proc Natl Acad Sci USA. 1996;93:13808–13813. doi: 10.1073/pnas.93.24.13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann F, James P, Vorherr T, Carafoli E. J Biol Chem. 1993;268:10252–10259. [PubMed] [Google Scholar]
- 21.Chen S R, MacLennan D H. J Biol Chem. 1994;36:22698–22704. [PubMed] [Google Scholar]
- 22.Cox D H, Cui J, Aldrich R W. J Gen Physiol. 1997;110:257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moss G W J, Marshall J, Moczydlowski E. J Gen Physiol. 1996;108:473–484. doi: 10.1085/jgp.108.6.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Persson E, Hogg P J, Stenflo J. J Biol Chem. 1993;268:22531–22539. [PubMed] [Google Scholar]
- 25.Moczydlowski E, Latorre R. J Gen Physiol. 1983;82:511–542. doi: 10.1085/jgp.82.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugihara I. J Physiol (London) 1994;476:373–390. doi: 10.1113/jphysiol.1994.sp020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugihara I. J Gen Physiol. 1998;111:363–379. doi: 10.1085/jgp.111.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui J, Cox D H, Aldrich R W. J Gen Physiol. 1997;109:647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McManus O B, Magleby K L. J Physiol (London) 1991;443:739–777. doi: 10.1113/jphysiol.1991.sp018861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carl A, Sanders K M. Am J Physiol. 1989;257:C470–C480. doi: 10.1152/ajpcell.1989.257.3.C470. [DOI] [PubMed] [Google Scholar]
- 31.Wu J V, Shuttleworth T J, Stampe P. J Membr Biol. 1996;154:275–282. doi: 10.1007/s002329900152. [DOI] [PubMed] [Google Scholar]
- 32.Golowasch J, Kirkwood A, Miller C. J Exp Biol. 1986;124:5–13. doi: 10.1242/jeb.124.1.5. [DOI] [PubMed] [Google Scholar]