

Abstract
Resistance to the anti-HER2 monoclonal antibody trastuzumab is a major problem in the treatment of HER2-overexpressing metastatic breast cancer. Growth differentiation factor 15 (GDF15), which is structurally similar to TGF beta, has been reported to stimulate phosphorylation of HER2. We tested the hypothesis that GDF15-mediated phosphorylation of HER2 reduces the sensitivity of HER2-overexpressing breast cancer cell lines to trastuzumab. Gene microarray analysis, real-time PCR, and ELISA were used to assess GDF15 expression. Growth inhibition and proliferation assays in response to pharmacologic inhibitors of HER2, TGF beta receptor, or Src were performed on cells stimulated with recombinant human GDF15 or stable GDF15 transfectants. Western blotting was performed to determine effects of GDF15 on HER2 signaling. Cells were infected with lentiviral GDF15 shRNA plasmid to determine effects of GDF15 knockdown on cell survival in response to trastuzumab. Cells with acquired or primary trastuzumab resistance showed increased GDF15 expression. Exposure of trastuzumab-sensitive cells to recombinant human GDF15 or stable transfection of a GDF15 expression plasmid inhibited trastuzumab-mediated growth inhibition. HER2 tyrosine kinase inhibition abrogated GDF15-mediated Akt and Erk1/2 phosphorylation and blocked GDF15-mediated trastuzumab resistance. Pharmacologic inhibition of TGF beta receptor blocked GDF15-mediated phosphorylation of Src. Further, TGF beta receptor inhibition or Src inhibition blocked GDF15-mediated trastuzumab resistance. Finally, lentiviral GDF15 shRNA increased trastuzumab sensitivity in cells with acquired or primary trastuzumab resistance. These results support GDF15-mediated activation of TGF beta receptor-Src-HER2 signaling crosstalk as a novel mechanism of trastuzumab resistance.
Keywords: breast cancer, erbB2, Herceptin, resistance, trastuzumab, lapatinib
1. INTRODUCTION
Human epidermal growth factor receptor 2 (HER2/erbB2) is overexpressed in approximately 20% to 30% of metastatic breast cancers, and is associated with poor prognosis [1]. Trastuzumab (Herceptin™; Genentech, South San Francisco, CA) is a recombinant humanized monoclonal antibody directed against an extracellular region of the HER2 protein. Initial clinical trials of single-agent trastuzumab demonstrated overall response rates ranging from 11% to 21% in patients with HER2-overexpressing metastatic breast cancer [2,3]. Thus, almost two-thirds of patients demonstrated intrinsic resistance to trastuzumab in these initial single agent trials, although response rates dramatically improved up to 81% when combined with chemotherapy [4,5]. The median duration of response to trastuzumab given as a single agent or in combination with taxanes was less than one year [2–5], indicating that acquired trastuzumab resistance is a major clinical concern. We previously reported that acquired resistance to trastuzumab does not appear to be due to changes in the drug target, as loss of HER2 overexpression and HER2 gene mutation were not observed [6]. Published reports have implicated increased phosphatidylinositol-3 kinase (PI3K) signaling as a potential mechanism of trastuzumab resistance [7,8]. Indeed, we and others have reported that pharmacologic inhibition of PI3K improves trastuzumab sensitivity in cells that have acquired resistance [7,9]. While PI3K activation may occur by hyper-activating mutations in the catalytic subunit of PIK3CA or down-regulation of the PI3K phosphatase PTEN [7,8], additional studies have demonstrated a role for increased growth factor signaling as a mechanism of trastuzumab resistance [10,11].
Growth differentiation factor 15 (GDF15, also called MIC-1, NAG-1, PTGF-beta, and PDF) is a distant member of the transforming growth factor (TGF) beta superfamily of cytokines based on structural similarity [12]. Increased circulating levels of GDF15 have been associated clinically with disease progression and resistance to chemotherapy in breast, prostate, ovarian, and colorectal cancer [13–17], suggesting that GDF15 may serve as a biomarker of advanced disease or predictor of therapeutic resistance. GDF15 is expressed in the cytoplasm as a precursor 35-kDa protein that is cleaved to produce a mature 17-kDa secreted cytokine [12]. Functionally, GDF15 appears to mediate pleiotropic effects [13], resulting in apoptosis in pre-malignant stages and activating cell survival and anti-apoptotic pathways in advanced disease, similar to what is reported for TGF beta. Knockdown of GDF15 in malignant gliomas reduced cell proliferation in vitro and tumorigenesis in vivo [15], suggesting that GDF15 contributes to cancer progression and may serve as a novel molecular target in advanced malignancies. GDF15 was previously reported to induce Src-dependent phosphorylation of HER2 [18,19]. However, the role of TGF beta receptor and the biological effect of GDF15-mediated HER2 phosphorylation on sensitivity to HER2-targeted drugs have never been examined. Thus, in the current study, we tested the hypothesis that GDF15-mediated HER2 phosphorylation reduces sensitivity to trastuzumab in a TGF beta receptor-dependent manner.
2. MATERIALS AND METHODS
2.1 Materials
Trastuzumab (Herceptin™, Genentech, South San Francisco, CA) was purchased from the Winship Cancer Institute pharmacy and dissolved in sterile water at a stock concentration of 20 mg/mL. Recombinant human GDF15 (rhGDF15; R&D Systems, Minneapolis, MN) was dissolved to a final stock concentration of 200 μg/mL in 4mM HCl containing 0.1% BSA vehicle. HER2 kinase inhibitor AG879 (Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO at a stock concentration of 10 mM. Lapatinib (Santa Cruz, Biotech, Santa Cruz, CA) was dissolved in DMSO at a stock concentration of 10 mM. SB431542 (Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO at a stock concentration of 10 mM.
2.2 Cell culture
SKBR3, BT474, and MDA-MB-453 HER2-overexpressing breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). HCC1419 and HCC1954 HER2-overexpressing breast cancer cells were maintained in RPMI with 10% FBS and 1% P/S. MDA-MB-361 was maintained in RPMI with 20% FBS and 1% P/S. All cell lines were purchased from American Type Culture Collection, Manassas, VA. As previously reported [6,20], trastuzumab-resistant cells were derived from SKBR3 and BT474 by maintaining cells in 4 μg/ml trastuzumab for 3 months, at which point surviving pools and clones were selected; all SKBR3- and BT474-derived resistant cells were routinely maintained on 4 μg/ml trastuzumab, and trastuzumab was removed from cultures for 24 h prior to performing experiments.
2.2.1 Stable transfection
SKBR3 and BT474 cells were transfected with 3 μg of plasmid DNA (pCMV empty vector or pCMVmyc-GDF15, both from Origene, Rockville, MD) using Lipofectamine (Invitrogen, Carlsbad, CA) and DMSO shock. After 24–36 h, cells were maintained in 200 μg/mL G418 to select successfully transfected cells. After approximately 2–3 weeks, surviving clones were isolated and tested by real-time PCR for GDF15 expression.
2.2.2 Lentiviral shRNA infection
Lentiviral shRNA construct for GDF15 and lentiviral control shRNA in pLKO1 vector were purchased from Open Biosystems (RHS4078) (Huntsville, AL). Lentiviral helper plasmids (pCMV-dR8.2 dvpr and pCMV-VSV-G) were obtained from Addgene (8455 and 8454) (Cambridge, MA). Transient lentivirus stocks were prepared following the manufacturer’s protocol. Briefly, 2.5×106 293T cells were plated in 10 cm dishes. Cells were co-transfected with shRNA construct (3μg) together with 3 μg pCMV-dR8.2 dvpr and 0.3 μg pCMV-VSV-G helper constructs. Culture medium containing virus was harvested and breast cancer cells were infected with virus-containing media.
2.3 Microarray analysis
Total RNA was extracted from triplicate cultures of each cell line (RNeasy; Qiagen, Germantown, MD). RNA was then taken to the Emory University Microarray Core Facility where RNA integrity was confirmed using an Agilent 2100 Bioanalyzer. RNA was labeled using TotalPrep RNA (Ambion, Austin, TX), and hybridized onto Illumina Human Ref-8 v3 Expression BeadChip for analysis of approximately 24,500 well-annotated transcripts. Triplicate cultures were run for each cell line in order to assess the reproducibility of microarray results; for each set of triplicates, r2=0.99, confirming reproducibility of results. Transcripts that were differentially expressed in each resistant pool versus parental cell line were determined using significance analysis of microarrays (SAM) [21], with false discovery rate (FDR) < 1%.
2.4 ELISA
To quantify the amount of GDF15 released into the media from cells, human GDF15 immunoassay (R&D Systems, Minneapolis, MN) was used according to the manufacturer’s directions. Briefly, sample media was incubated in GDF15 antibody-coated microplate for 2 hours which was washed 4X and then incubated with GDF15 antibody conjugated to horseradish peroxidase for 1 hour. After washing 4X the wells were incubated with color reagent (hydrogen peroxide-chromogen mix) for 30 minutes, at which point the stop solution was added. Optical density of each well was determined using a microplate reader set to 450nm. The concentrations were calculated according to the standards supplied with the kit by creating a four parameter logistic curve-fit.
2.5 Real-time PCR analysis
Parental and trastuzumab-resistant clones from BT474 and SKBR3 were grown in standard culture conditions. Cells were seeded and allowed to grow until~85% confluent. Total RNA was then extracted using the RNeasy purification kit (Qiagen, Germantown, MD) and DNase treated (Invitrogen, Carlsbad, CA). cDNA was then prepared from total RNA using random primers and the Superscript III first strand synthesis Kit (Invitrogen, Carlsbad, CA). Relative levels of mRNA were determined by real-time quantitative PCR using an Applied Biosystems cycler and the TaqMan Universal PCR master Mix (4304437; Applied Biosystems, Carlsbad, CA); RPLPO mRNA levels were used for normalization. Primers for GDF15 (Hs00171132_m1) and RPLPO (Hs99999902_m1) were obtained from Applied Biosystems (TaqMan Gene Expression Assays) (Carlsbad, CA). After amplification, the data was normalized with RPLPO levels and analyzed by deltaCt method.
2.6 Growth inhibition and proliferation assays
For growth inhibition assays, cells were plated at 3 × 104 per well in 12-well plate format and treated with GDF15 +/− trastuzumab, AG879, lapatinib, SB431542, or PP2 for an additional 48 or 72 hours. Control cultures were treated with vehicle control for rhGDF15 +/− DMSO (solvent for AG879 and lapatinib). Cell count was measured by trypan blue exclusion assay. For proliferation assays, cells were plated at 3000 cells per well in 96-well format and treated with lapatinib for 48 hours. MTS colorimetric assays (Promega, Madison, WI) were then performed according to manufacturer protocol.
2.7 Western blotting
Cells were lysed in RIPA buffer (Cell Signaling, Danvers, MA) supplemented with protease and phosphatase inhibitors (Cell Signaling, Danvers, MA). Total protein extracts were run on SDS-PAGE and blotted onto nitrocellulose. Blots were probed overnight using the following antibodies at indicated dilutions. Total HER2 (1:1000) from Abcam (Cambridge, MA); from Cell Signaling (Danvers, MA), p-S473 Akt-XP used at 1:1000, and polyclonal antibodies against Akt (1:1000), p-Thr202/Tyr204 p42/p44 Erk1/2 (1:1000), total p42/p44 Erk1/2 (1:1000); from Santa Cruz Biotechnology (Santa Cruz, CA), polyclonal p-Tyr1248 HER2 (1:1000); and β-actin monoclonal AC-15 (Sigma-Aldrich, St. Louis, MO) at 1:10,000. Appropriate secondary antibodies were used. Protein bands were detected using the Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE).
2.8 Statistics
P-values were determined for experimental versus control treatments by Student’s t-test; *p<0.05, **p<0.005.
3. RESULTS
3.1 Trastuzumab-resistant breast cancer cell lines show increased GDF15 expression
Gene microarray analysis was performed on parental SKBR3 and BT474 HER2-overexpressing breast cancer cell lines, a trastuzumab-resistant pool of cells derived from SKBR3, and a trastuzumab-resistant pool of cells derived from BT474. Total RNA was isolated from each cell line in triplicate to ensure reproducibility. For each set of triplicates, r2=0.99, confirming reproducibility of results. Samples were hybridized onto Illumina Human Ref-8 v3 Expression BeadChip containing approximately 24,500 well-annotated transcripts. Differentially expressed transcripts in each resistant pool versus the corresponding parental line were identified using significance analysis of microarrays (SAM) [21] with a false discovery rate less than 1%. For BT474 resistant pool vs. BT474 parental, 1903 genes were differentially regulated (up or down) by 1.5-fold or more; in SKBR3 resistant cells vs. SKBR3 parental, 3207 genes were differentially regulated by 1.5-fold or more. There were 690 genes that were up-regulated by 1.5-fold or more in BT474 resistant cells vs. parental cells, and 1417 genes up-regulated in SKBR3 resistant vs. parental cells. Among these up-regulated genes, only 218 were up-regulated by 1.5-fold or more in both SKBR3 resistant and BT474 resistant vs. parental cells. Genes that were up-regulated by 4-fold or more in both SKBR3 and BT474 resistant cells vs. parental cells are listed (Table 1). The most highly over-expressed transcript in the resistant pools was PPP1R1B/Darpp-32, which was previously shown to be over-expressed in trastuzumab-resistant cells [22]. The second most highly over-expressed transcript in resistant cells versus parental cells was GDF15 (also called macrophage inhibitory cytokine 1, or MIC-1), which was over-expressed an average of 26-fold in resistant vs. parental cells.
Table 1.
Transcripts up-regulated on gene microarray in cells with acquired trastuzumab resistance versus parental cell lines
| Gene | Fold up-regulation |
|---|---|
| PPP1R1B | 30.51597422 |
| GDF15 | 26.24737319 |
| SCGB2A2 | 11.30935665 |
| CYP1A1 | 9.814828986 |
| TNFSF10 | 8.460664911 |
| SLC2A10 | 6.012329038 |
| BEX2 | 4.937933164 |
| ETV5 | 4.638589618 |
| SCGB2A1 | 4.555765328 |
| SH3PXD2A | 4.410699182 |
| HS.579631 | 4.359287911 |
| RORC | 4.223554895 |
| NUPR1 | 4.12469436 |
| P8 | 4.096969749 |
| UNC5A | 4.083438983 |
| LAMP3 | 4.029105703 |
| TCEAL8 | 3.999739961 |
Total RNA was collected from SK-par, SK-HRp2, BT-par, and BT-HRp3 cells, converted to cDNA and hybridized onto Illumina Human Ref-8 Expression BeadChip. Triplicate cultures were run for each line, and r2=0.99 for all sets of triplicates, confirming high degree of reproducibility. Differentially regulated genes were determined by significance analysis of microarrays (SAM) [21]; false discovery rate < 1%. There were 218 genes up-regulated > 1.5-fold in both SKBR3 resistant and BT474 resistant vs. parental cells. Genes that were up-regulated by 4-fold or higher in resistant cells vs. parental cells are listed. Values reflect the average fold up-regulation in SKBR3 and BT474 resistant cell line samples versus parental cell lines.
To confirm microarray data, we performed real-time PCR analysis of GDF15 transcript levels in BT474 and SKBR3 acquired trastuzumab-resistant cells versus their parental counterparts (Figure 1A). These results were consistent with microarray data, showing increased GDF15 transcript expression in cells with acquired resistance versus the parental, sensitive cell lines. Since GDF15 is believed to be bioactive as a secreted cytokine, we performed ELISAs for both the endogenous and secreted forms of GDF15 protein in parental and trastuzumab-resistant cells. GDF15-specific ELISA (R&D Systems, Inc., Minneapolis, MN) performed on whole cell protein lysates (excluding media) showed that endogenous GDF15 expression was elevated in BT474 resistant cells by 3- to 20-fold versus BT474 parental cells (depending on the resistant clone), and 50- to 400-fold in SKBR3 resistant clones versus SKBR3 parental cells (Figure 1B). In addition, media collected from cells showed that the concentration of secreted GDF15 was 4-fold higher in BT474 resistant clones vs. BT474 parental and 76- to 172-fold higher in SKBR3 resistant cells vs. SKBR3 parental (Figure 1C). Thus, both the secreted and endogenous forms of GDF15 protein were increased in trastuzumab-resistant cells versus trastuzumab-sensitive parental cells. In addition to models of acquired trastuzumab resistance, we examined GDF15 secretion by ELISA in HER2-overexpressing cell lines that have primary (intrinsic) resistance to trastuzumab. The level of secreted GDF15 in the cell culture media from primary trastuzumab-resistant HCC1419, HCC1954, MDA453, and MDA361 cell lines was significantly higher than the level of secreted GDF15 in SKBR3 and BT474 cells (Figure 1D). Thus, multiple human cell line models of acquired and primary trastuzumab resistance showed increased expression of GDF15 relative to trastuzumab-sensitive breast cancer cell lines.
Figure 1. HER2-overexpressing trastuzumab-resistant breast cancer cells express increased levels of GDF15.

(A) Total RNA was extracted from BT474 parental (Par), resistant clone 2 (c2) and clone 3 (c3), and SKBR3 parental (Par) and resistant clone 3 (c3) cells. RNA was converted to cDNA and analyzed by real-time PCR for GDF15 transcript level. Results are reported as fold increase in GDF15 transcript level versus parental counterpart. Values were normalized to RPLPO housekeeping ribosomal gene transcript levels as internal control. Each sample was run in triplicate per experiment, and 3 independent experiments were performed on separate occasions to ensure reproducibility. Values represent the average of the 3 separate experiments; error bars represent standard error between the triplicate experiments. P-values were calculated by student’s t-test for each resistant line versus the parental line; *p<0.05, **p<0.005. GDF15 levels were also determined by (B) ELISA using total protein lysates excluding media, or (C) ELISA using cell culture media only. Error bars represent standard deviation between triplicates. P-values were calculated by student’s t-test for each resistant line versus the parental line; *p<0.05, **p<0.005. Results were confirmed on three separate occasions. Acquired resistant cells showed statistically significant increase in both the endogenous and secreted forms of GDF15 versus sensitive lines. (D) Secreted GDF15 was measured by ELISA in the cell culture media of HCC1419, HCC1954, MDA-MB-453, and MDA-MB-361 cells, which have primary resistance to trastuzumab, versus BT474 and SKBR3 trastuzumab-sensitive cells. Error bars represent standard deviation between triplicates. P-values were calculated by student’s t-test for each resistant line versus BT474; **p<0.005. Results were confirmed on three separate occasions. Cells with primary trastuzumab resistance showed higher levels of secreted GDF15 versus trastuzumab-sensitive cells.
3.2 GDF15 reduces response of HER2-overexpressing breast cancer cells to trastuzumab
Next, we examined whether increased GDF15 expression results in reduced sensitivity to trastuzumab. Exposure of HER2-overexpressing SKBR3 or BT474 breast cancer cells to a physiologically relevant concentration of recombinant human GDF15 (10ng/mL) blocked growth inhibition of a clinically relevant concentration of trastuzumab (20μg/mL) (Figure 2A). In addition, we developed stable GDF15-overexpressing clones from the SKBR3 and BT474 cell lines. Real-time PCR confirmed increased expression of GDF15 in stable clones versus empty vector control clones in both SKBR3 and BT474 cells (Figure 2B). To determine if stable GDF15 over-expression reduced response to trastuzumab, we treated GDF15 stable clones or control empty vector clones with 20μg/mL trastuzumab for 72 h. Control SKBR3 and BT474 clones showed significant reduction in growth upon treatment with trastuzumab (Figure 2C). In contrast, GDF15 stable clones showed reduced sensitivity to trastuzumab. These results suggest that increased exposure to exogenous GDF15 or increased expression of endogenous GDF15 reduces the sensitivity of HER2-overexpressing breast cancer cells to trastuzumab.
Figure 2. Increased GDF15 expression reduces trastuzumab-mediated growth inhibition.

(A) SKBR3 and BT474 cells were treated with vehicle control (C), 20 μg/mL trastuzumab (T), or pre-treated with 10 ng/mL GDF15 for 48h followed by 20 μg/mL trastuzumab plus 10 ng/mL GDF15 for an additional 72 h (G+T); media plus treatments were changed each day. Cells were counted by trypan blue exclusion; cell count is shown as a percentage of the control group per cell line. Each sample was run in triplicate cultures per experiment; experiments were performed on 3 independent occasions for reproducibility. Error bars represent standard error between the 3 independent experiments. P-values were determined by student’s t-test for GDF15 plus trastuzumab (G+T) group versus trastuzumab alone (T); *p<0.05. Stimulation with recombinant human GDF15 significantly reduced trastuzumab-mediated growth inhibition. (B) SKBR3 and BT474 clones stably transfected with pCMV empty vector control or pCMV-myc-GDF15 were analyzed by real-time PCR for GDF15 transcript level. Results are reported as fold increase in GDF15 transcript level versus stable control clone per line. Values were normalized to RPLPO housekeeping ribosomal gene transcript levels as internal control. Error bars represent standard deviation between triplicates. (C) Stable clones were treated with 20 μg/mL trastuzumab (Tr) for 72h. Cells were counted by trypan blue exclusion; cell count is shown as a percentage of control untreated cells (C) per clone. Error bars represent standard error between duplicate experiments, each run in triplicate. P-values were determined by student’s t-test for trastuzumab-treated cells versus untreated cells per clone; *p<0.05. Trastuzumab significantly reduced growth of stable control clones; no significant response to trastuzumab was measured in stable GDF15-overexpressing SKBR3 clones. Stable GDF15 BT474 clone 3 showed statistically significant reduction in growth, but less than control BT474 clone cells.
3.3 GDF15-mediated phosphorylation of HER2 reduces trastuzumab sensitivity
GDF15/MIC-1 has been shown to stimulate phosphorylation of HER2 in SKBR3 HER2-overexpressing breast cancer cells [18,19]. Stimulation of BT474 cells with recombinant human GDF15 induced phosphorylation of HER2 and downstream Akt and rapid, but transient phosphorylation of Erk1/2 (Figure 3A). Tyrosine kinase inhibition of HER2 using tyrphostin AG879 reduced GDF15-mediated phosphorylation of HER2, Akt, and Erk1/2 (Figure 3B). Similarly, the dual EGFR/HER2 kinase inhibitor lapatinib blocked GDF15-mediated phosphorylation of Akt and Erk1/2 (Figure 3C). These results suggest that GDF15-mediated activation of Akt and Erk1/2 occurs downstream of HER2 activation.
Figure 3. GDF15-mediated activation of HER2 signaling reduces trastuzumab sensitivity.

(A) upper panel: BT474 cells were treated with 2 ng/mL recombinant human GDF15 for 10, 30, or 60 min or with the corresponding volume of vehicle control for 60 min. Total protein lysates were Western blotted for phosphorylated and total HER2, Akt, and Erk1/2. lower panel: BT474 cells were treated with 2 ng/mL vehicle control for 0, 10, 30, or 60 min, or with 2 ng/mL recombinant human GDF15 for 0, 10, 30, or 60 min. Total protein lysates were Western blotted for phosphorylated and total Erk1/2. (B) BT474 cells were treated with vehicle control (HCl-BSA plus DMSO), 2 ng/mL GDF15 for 10 or 30 min, or 2 ng/mL GDF15 plus 5 μM AG879 for 30 min. Total protein lysates were Western blotted for phosphorylated and total HER2, Akt, and Erk1/2. (C) BT474 cells were treated with vehicle control (HCl-BSA plus DMSO), 2 ng/mL GDF15 for 10 or 30 min, or 2 ng/mL GDF15 plus 1 μM lapatinib for 30 min. Total protein lysates were Western blotted for phosphorylated and total Akt and Erk1/2. (D) BT474 cells were treated with vehicle control (C), 20 μg/mL trastuzumab (T), pre-treated with 2 ng/mL GDF15 for 48h followed by 20 μg/mL trastuzumab plus 2 ng/mL GDF15 for an additional 72 h (GDF+T), or pre-treated with 2 ng/mL GDF15 for 48h followed by 20 μg/mL trastuzumab plus 2 ng/mL GDF15 plus 5 μM AG879 for an additional 72 h (GDF+T+AG); media plus treatments were changed each day. Cells were counted by trypan blue exclusion; cell count is shown as a percentage of the control group per cell line. Error bars represent standard error between duplicate experiments, each run in triplicate. . P-values were determined by student’s t-test for GDF+T versus T alone, and for GDF+T+AG versus GDF+T; *p<0.05, **p<0.005. (E) BT474 cells were treated with vehicle control (C), 20 μg/mL trastuzumab (T), 1 μM lapatinib (L), pre-treated with 2 ng/mL GDF15 for 48h followed by 20 μg/mL trastuzumab plus 2 ng/mL GDF15 for an additional 72 h (G+T), pre-treated with 2 ng/mL GDF15 for 48h followed by 1 μM lapatinib plus 2 ng/mL GDF15 for an additional 72 h (G+L), or pre-treated with 2 ng/mL GDF15 for 48h followed by 20 μg/mL trastuzumab plus 1 μM lapatinib plus 2 ng/mL GDF15 for an additional 72 h (G+T+L); media plus treatments were changed each day. Cells were counted by trypan blue exclusion; cell count is shown as a percentage of the control group per cell line. Error bars represent standard deviation between triplicates. P-values were determined by student’s t-test for G+T+L versus G+T; *p<0.05. (F) Stable SKBR3 control and GDF15 clones were treated with 100 nM lapatinib (Lap) or corresponding dose of DMSO control (C) for 48h. Proliferation was measured by MTS assay, and is shown as a percentage of control per clone. Error bars represent standard error between duplicate experiments, each run in triplicate. . P-values were determined by student’s t-test for lapatinib-treated cells versus control per clone; *p<0.05, **p<0.005. Stable GDF15-overexpressing clones retained sensitivity to lapatinib.
We next determined if phosphorylation of HER2 is the mechanism by which GDF15 promotes trastuzumab resistance. BT474 cells were treated with trastuzumab in the absence or presence of recombinant human GDF15. Again, GDF15 blocked the ability of trastuzumab to inhibit growth (Figure 3D). Inhibition of HER2 kinase using AG879 restored trastuzumab sensitivity to GDF15-stimulated cells. Further, the dual EGFR/HER2 kinase inhibitor lapatinib inhibited survival of BT474 cells even in the presence of GDF15 stimulation (Figure 3E). Co-treatment with lapatinib plus trastuzumab blocked survival of HER2-overexpressing breast cancer cells exposed to GDF15. Similarly, lapatinib inhibited proliferation of stable GDF15-overexpressing SKBR3 clones at a level comparable to control clone cells (Figure 3F). Collectively, these results indicate that GDF15-mediated trastuzumab resistance is dependent upon phosphorylation of HER2, as HER2 kinase inhibition overcomes GDF15-mediated resistance.
3.4 TGF beta receptor-dependent Src phosphorylation contributes to GDF15-mediated resistance
GDF15 was previously shown to induce Src-dependent phosphorylation of HER2 [19]. In addition GDF15 shares structural homology with TGF beta [12]. Thus, we examined whether TGF beta receptor and Src signaling are required for GDF15-mediated trastuzumab resistance. Stimulation of BT474 cells with recombinant human GDF15 induced phosphorylation of Smad2 (Figure 4A). Inhibition of TGF beta receptor type II using SB431542 resulted in reduced GDF15-mediated Smad2 phosphorylation. Thus, GDF15 activated TGF beta receptor signaling in HER2-overexpressing breast cancer cells. GDF15 also induced phosphorylation of Src in BT474 cells (Figure 4B). Addition of trastuzumab reduced GDF15-mediated Src phosphorylation (Figures 4B and 4C). Inhibition of TGF beta receptor with SB431542 further reduced GDF15-mediated Src phosphorylation (Figure 4B). However, lapatinib did not block GDF15-mediated Src phosphorylation (Figure 4C), suggesting that GDF15-mediated HER2 phosphorylation does not regulate Src phosphorylation. These results indicate that GDF15 activates TGF beta receptor signaling, which in turn induces Src phosphorylation.
Figure 4. TGF beta receptor-dependent Src phosphorylation contributes to GDF15-mediated resistance.

(A) BT474 cells were treated with vehicle control (HCl-BSA plus DMSO), 2 ng/mL GDF15 for 10 or 30 min, or 2 ng/mL GDF15 plus 5 μM SB431542 for 30 min. Total protein lysates were Western blotted for phosphorylated and total Smad2. (B) BT474 cells were treated with vehicle control (C) (HCl-BSA plus DMSO), 20 μg/mL trastuzumab (T), 2 ng/mL GDF15 (G), trastuzumab plus GDF15 (TG), 5 μM SB431542 (S), trastuzumab plus SB431542 (TS), or trastuzumab plus GDF15 plus SB431542 (TGS) for 30 min. Total protein lysates were Western blotted for phosphorylated and total Src. (C) BT474 cells were treated with vehicle control (C) (HCl-BSA plus DMSO), 20 μg/mL trastuzumab (T), 2 ng/mL GDF15 (G), trastuzumab plus GDF15 (TG), 1 μM lapatinib (L), trastuzumab plus lapatinib (TL), or trastuzumab plus GDF15 plus lapatinib (TGL) for 30 min. Total protein lysates were Western blotted for phosphorylated and total Src. (D) BT474 control and GDF15 stable clones 2 and 3 were treated with vehicle control (C) (HCl-BSA plus DMSO), 20 μg/mL trastuzumab (T), 5 μM SB431542 (S), trastuzumab plus SB431542 (TS), 1 μM PP2 (P), or trastuzumab plus PP2 (TP) for 72 h; media plus treatments were changed each day. Cells were counted by trypan blue exclusion; cell count is shown as a percentage of the control group per cell line. Error bars represent standard deviation between triplicates. P-values were determined by student’s t-test for each group versus T alone; *p<0.05.
Next, we examined survival of BT474 control and GDF15 stable clones in response to trastuzumab combined with TGF beta receptor inhibitor SB431542 or Src inhibitor PP2. Trastuzumab-mediated growth inhibition of BT474 control clone cells was not affected by TGF beta receptor inhibition or Src inhibition (Figure 4D). In contrast, trastuzumab sensitivity was increased in GDF15-overexpressing stable clone cells when co-treated with SB431542 or PP2. These results suggest that TGF beta receptor and Src signaling are involved in GDF15-mediated trastuzumab resistance, as pharmacologic inhibition of TGF beta receptor or Src abrogated GDF15-mediated resistance.
3.5 GDF15 knockdown increases trastuzumab sensitivity
To determine whether inhibition of GDF15 improves trastuzumab sensitivity, we knocked down GDF15 expression and measured response to trastuzumab in resistant cells. BT474 parental and trastuzumab-resistant clone 3 (BT-HRc3) cells and SKBR3 parental and trastuzumab-resistant clone 3 (SK-HRc3) cells were infected with GDF15-specific shRNA in a lentiviral backbone or with corresponding control shRNA in the same lentiviral vector backbone. After 72 h, cells were either untreated or treated with trastuzumab for an additional 72 h. GDF15-specific ELISA performed on cell culture media showed 60–90% knockdown of GDF15 in infected cells compared to non-infected cells (Figure 5A); GDF15 knockdown was maintained in the presence of trastuzumab. Knockdown of GDF15 alone reduced growth of SK-HRc3 by 20% and BT-HRc3 cells by 50% (Figure 5B). In SK-HRc3 cells, GDF15 knockdown significantly increased trastuzumab sensitivity, with growth inhibition increasing from 20% with trastuzumab alone to 50% when combined with shGDF15. Similarly, BT-HRc3 cells showed significantly reduced cell survival with combination shGDF15 and trastuzumab versus trastuzumab alone; the majority of growth inhibition in BT-HRc3 cells appeared to be due to knockdown of GDF15. These experiments suggest that inhibition of GDF15 expression reduces cell survival and improves trastuzumab sensitivity in cells that have acquired trastuzumab resistance. In addition, HCC1419 cells, which show primary resistance to trastuzumab and secrete high levels of GDF15, showed improved sensitivity to trastuzumab with knockdown of GDF15 versus control shRNA (Figure 5C). Hence, GDF15 knockdown is a potential strategy for improving response to trastuzumab in HER2-overexpressing breast cancer cells that have acquired resistance to trastuzumab or are trastuzumab-naïve but fail to respond to single agent trastuzumab.
Figure 5. GDF15 knockdown increases trastuzumab sensitivity in cells with acquired or primary trastuzumab resistance.

(A) BT-HRc3 and SK-HRc3 cells were infected with lentiviral control shRNA (shC) or GDF15 shRNA (shGDF). After 72 h, 20 μg/mL trastuzumab (Tras) was added to cultures for an additional 72 h or cells were left untreated. Cell culture media was examined using GDF15-specific ELISA to confirm knockdown of GDF15. Error bars represent standard deviation between triplicates. (B) BT-parental, BT-HRc3, SK-parental, and SK-HRc3 cells were infected with lentiviral control shRNA (shC) or GDF15 shRNA (shGDF). After 72 h, 20 μg/mL trastuzumab (Tras) was added to cultures for an additional 72 h or cells were left untreated. Cells were then counted by trypan blue exclusion; growth is shown as a percentage of shC-infected cells per line. Treatments were done in triplicate, with error bars representing standard deviation between replicates. Results were confirmed in duplicate experiments. P-values were determined by student’s t-test; *p<0.05, **p<0.005. (C) HCC1419 cells were infected with lentiviral control shRNA (shC) or GDF15 shRNA (shGDF); knockdown of GDF15 was confirmed by real-time PCR. Results are reported as fold change in GDF15 transcript level versus control shRNA. Values were normalized to RPLPO housekeeping ribosomal gene transcript levels as internal control. Error bars represent standard deviation between triplicates. Cells were infected with control shRNA or GDF15 shRNA, and after 72 h, treated with 20 μg/mL trastuzumab (Tras) for an additional 72 h or left untreated. Cells were counted by trypan blue exclusion; growth is shown as a percentage of shC-infected cells. Treatments were done in triplicate, with error bars representing standard deviation between replicates. Results were confirmed in duplicate experiments. P-values were determined by student’s t-test; *p<0.05, **p<0.005.
4. DISCUSSION
We present novel data showing that trastuzumab-resistant cell lines express increased levels of GDF15. Further, increased exposure to recombinant human GDF15 or stable over-expression of GDF15 reduced the sensitivity of HER2-overexpressing breast cancer cells to trastuzumab. GDF15-mediated trastuzumab resistance required phosphorylation of HER2, as pharmacologic inhibition of HER2 overcame the resistance conferred by GDF15. TGF beta receptor signaling was activated by GDF15, which induced phosphorylation of Src. GDF15-mediated phosphorylation of HER2 has previously been shown to be Src-dependent [19]. Inhibition of TGF beta receptor or Src overcame GDF15-mediated trastuzumab resistance. Finally, knockdown of GDF15 improved sensitivity to trastuzumab in models of acquired trastuzumab resistance. These results support GDF15-mediated activation of TGF beta receptor-Src-HER2 signaling crosstalk as a novel mechanism of trastuzumab resistance (Figure 6).
Figure 6. Proposed mechanism of GDF15-mediated trastuzumab resistance.
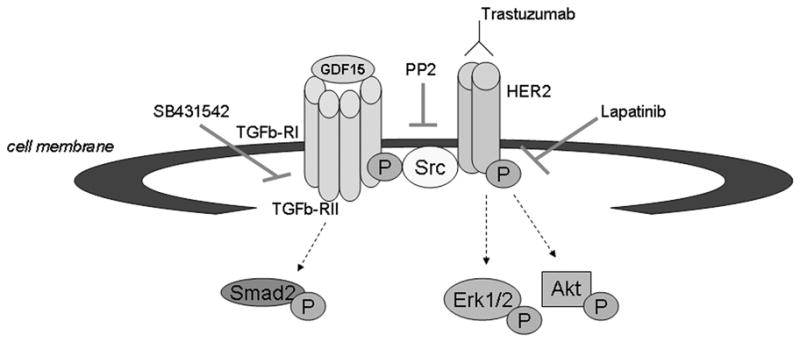
We propose that GDF15 activates TGF beta receptor-Src-HER2 signaling crosstalk as a novel mechanism of trastuzumab resistance. GDF15 appears to activate TGF beta receptor, as measured by phosphorylation of the TGF beta receptor substrate Smad2. GDF15 activates Src in a TGF beta receptor-dependent manner, which subsequently induces phosphorylation of HER2 and abrogates the growth inhibitory effects of the HER2-targeted antibody trastuzumab. Inhibition of the HER2 kinase by lapatinib restores sensitivity to trastuzumab in models of GDF15-mediated trastuzumab resistance.
We found that expression of GDF15 was increased in HER2-overexpressing breast cancer cells that displayed either acquired or primary resistance. This was a fairly generalized finding, as resistant cells showed higher levels of endogenous and secreted GDF15 versus SKBR3 and BT474 trastuzumab-sensitive cells, although levels varied from clone to clone. Real-time PCR confirmed that GDF15 transcript levels were increased, suggesting that GDF15 is up-regulated in resistant cancers at the transcriptional level or possibly that GDF15 mRNA stability is altered in resistant cancers. Multiple transcription factor binding sites are present in the GDF15 promoter, including response elements for AP-1, Sp1 [23], p53 [24], and EGR [25]. Cell lines used in this study are p53 mutant, suggesting that the mechanism of GDF15 up-regulation is likely to be p53-independent. However, other members of the p53 family may contribute to GDF15 transcription [26]. Further studies examining regulators of GDF15 transcription and GDF15 mRNA stability in trastuzumab-resistant cancer are warranted in order to determine the mechanisms by which GDF15 expression is increased. In addition to being regulated by transcription factors, GDF15 expression can be modulated by signaling molecules including Erk1/2 [27] and PI3K/Akt/GSK-3 beta [28], indicating the presence of positive feedback signaling loops since GDF15 activates these same signaling pathways that potentially regulate its expression. Thus, if future work demonstrates that increased PI3K/MAPK signaling induces GDF15 expression in trastuzumab resistance, pharmacologic inhibitors against these pathways may partially reduce GDF15 expression and reduce GDF15-mediated resistance. In addition, increased GDF15 may serve as a marker of heightened PI3K/MAPK signaling in resistant cells. However, HCC1419 cells have wild-type PIK3CA [29] and do not show hyperactive Akt signaling (not shown), yet showed increased GDF15; conversely, the BT474 cell line, which has hyper-activating mutation K111N in PIK3CA [29], showed low expression of GDF15. Thus, elevated GDF15 is not a reflection of baseline PI3K signaling in these cell lines, although acquired resistant clones derived from BT474 and SKBR3 showed increased GDF15 expression and increased PI3K signaling.
In our study, trastuzumab-sensitive cells stimulated with GDF15 or stably transfected with a GDF15 expression construct showed reduced response to trastuzumab, suggesting that GDF15 directly contributes to the development of trastuzumab resistance. Stimulation with recombinant human GDF15 partially reduced trastuzumab sensitivity, while endogenous GDF15 over-expression (stable transfection) appeared to induce a stronger (almost complete) resistance phenotype. The recombinant cytokine is a purified form of the secreted form of GDF15, whereas stable transfection incorporates the endogenous full-length precursor which can then be cleaved into the secreted form. Thus, it is possible that the precursor form possess additional activity beyond that of the secreted form, and that increased transcription of GDF15, not just increased release of the secreted form, promotes resistance. This would be consistent with our findings that GDF15 is increased at the transcript level in cells with acquired or primary trastuzumab resistance. Additional studies addressing the functional differences between full-length intracellular GDF15 versus secreted GDF15 will allow us to determine the exact mechanism of GDF15-mediated resistance.
Further, knockdown of GDF15 restored trastuzumab sensitivity to SKBR3-derived resistant cells, and reduced survival of BT474-derived trastuzumab-resistant cells. Interestingly, attempts to develop stable GDF15 shRNA clones were unsuccessful, as long-term GDF15 knockdown killed trastuzumab-resistant cells in culture. These data indicate that long-term knockdown of GDF15 is incompatible with survival of acquired trastuzumab-resistant cells, suggesting that resistant cells may have developed dependence upon GDF15. Thus, inhibition of GDF15 is a potential strategy for treating breast cancers that have progressed on trastuzumab-based therapy.
Our data indicated that the mechanism by which GDF15 promotes trastuzumab resistance involves activation of HER2 signaling. In our study, GDF15 stimulated phosphorylation of HER2, Akt, and Erk1/2. Pharmacologic inhibition of HER2 kinase blocked GDF15-stimulated Akt and Erk1/2 signaling, indicating that GDF15-mediated PI3K and MAPK activation occur downstream of HER2 activation. However, since the major induction of Erk1/2 phosphorylation preceded Her2 phosphorylation, it is likely that there is also a HER2-independent mechanism contributing to GDF15-mediated Erk1/2 phosphorylation. Since GDF15 is structurally similar to TGF beta, we also examined whether GDF15 stimulated phosphorylation of the TGF beta receptor substrate Smad2, and whether TGF beta receptor signaling contributes to GDF15-mediated trastuzumab resistance. Pharmacologic inhibition of TGF beta receptor type II blocked GDF15-mediated Smad2 phosphorylation and reduced GDF15-mediated trastuzumab resistance. Finally, since GDF15-mediated phosphorylation of HER2 was shown to involve Src [19], we examined the role of Src in GDF15-mediated resistance. GDF15-mediated Src phosphorylation in HER2-overexpressing cells was blocked by TGF beta receptor inhibition but not by HER2 kinase inhibition. Thus, GDF15 appears to activate TGF beta receptor, which in turn activates Src, which induces phosphorylation of HER2, abrogating the growth inhibitory effects of HER2-targeted trastuzumab.
In summary, we propose that GDF15 should be studied further as a potential biological predictor of trastuzumab response and putative new therapeutic target in trastuzumab-resistant breast cancer. Clinical correlative studies are needed to address whether GDF15 levels are associated with reduced response to trastuzumab. Ultimately, such studies may lead to development of a serum-based assay to measure circulating GDF15 levels as a predictor of resistance to trastuzumab. The significance of GDF15 in trastuzumab resistance is highlighted by our data showing that knockdown of GDF15 increased trastuzumab sensitivity of cells with primary or acquired resistance. Ongoing experiments are examining whether pharmacologic inhibition of GDF15 restores trastuzumab sensitivity to trastuzumab-resistant breast cancers. Ultimately, this work may lead to the development of drugs that target GDF15 as a new molecular target for improving response to trastuzumab. Increased serum levels of GDF15 have been reported in multiple solid tumor types and have been associated with progression of disease [13–17]. Our study is consistent with the concept that increased GDF15 expression is associated with advanced, drug-resistant cancer. Breast cancers that overexpress HER2 carry a particularly poor prognosis [1], and primary or acquired resistance to trastuzumab occurs in many patients. Understanding the mechanistic basis for progression of HER2-overexpressing breast cancer will allow more effective targeted treatment options to be developed. Our work suggests that elevated levels of GDF15 may contribute to trastuzumab resistance, and supports further development of GDF15 as a novel therapeutic target in HER2-overexpressing breast cancers that have progressed on trastuzumab treatment.
Acknowledgments
R. Nahta gratefully acknowledges funding from The Mary Kay Foundation, The Georgia Cancer Coalition Distinguished Cancer Scholars Program, and NIH K01CA118174.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–44. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 3.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 4.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 5.Esteva FJ, Valero V, Booser D, Guerra LT, Murray JL, Pusztai L, et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:1800–8. doi: 10.1200/JCO.2002.07.058. [DOI] [PubMed] [Google Scholar]
- 6.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64:3981–6. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 7.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 8.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 9.Ozbay T, Durden DL, Liu T, O’Regan RM, Nahta R. In vitro evaluation of pan-PI3-kinase inhibitor SF1126 in trastuzumab-sensitive and trastuzumab-resistant HER2-over-expressing breast cancer cells. Cancer Chemother Pharmacol. 2010;65:697–706. doi: 10.1007/s00280-009-1075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin) J Natl Cancer Inst. 2001;93:1852–7. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 11.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–28. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 12.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A. 1997;94:11514–9. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimeault M, Batra SK. Divergent molecular mechanisms underlying the pleiotropic functions of macrophage inhibitory cytokine-1 in cancer. J Cell Physiol. 2010;224:626–35. doi: 10.1002/jcp.22196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staff AC, Bock AJ, Becker C, Kempf T, Wollert KC, Davidson B. Growth differentiation factor-15 as a prognostic biomarker in ovarian cancer. Gynecol Oncol. 2010;118:237–43. doi: 10.1016/j.ygyno.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 15.Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, Breit SN, et al. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin Cancer Res. 2010;16:3851–9. doi: 10.1158/1078-0432.CCR-10-0705. [DOI] [PubMed] [Google Scholar]
- 16.Brown DA, Ward RL, Buckhaults P, Bälter K, Adami HO, Zheng SL, et al. MIC-1 serum level and genotype: associations with progress and prognosis of colorectal carcinoma. Clin Cancer Res. 2003;9:2642–50. [PubMed] [Google Scholar]
- 17.Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc Natl Acad Sci U S A. 2003;100:3410–5. doi: 10.1073/pnas.0530278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KK, Lee JJ, Yang Y, You KH, Lee JH. Macrophage inhibitory cytokine-1 activates AKT and ERK-1/2 via the transactivation of HER2 in human breast and gastric cancer cells. Carcinogenesis. 2008;29:704–12. doi: 10.1093/carcin/bgn031. [DOI] [PubMed] [Google Scholar]
- 19.Park YJ, Lee H, Lee JH. Macrophage inhibitory cytokine-1 transactivates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep. 2010;43:91–6. doi: 10.5483/bmbrep.2010.43.2.091. [DOI] [PubMed] [Google Scholar]
- 20.Rowe DL, Ozbay T, Bender LM, Nahta R. Nordihydroguaiaretic acid, a cytotoxic insulin-like growth factor-I receptor/HER2 inhibitor in trastuzumab-resistant breast cancer. Mol Cancer Ther. 2008;7:1900–8. doi: 10.1158/1535-7163.MCT-08-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tusher, Tibshirani, Chu Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu L, Waliany S, Kane SE. Darpp-32 and its truncated variant t-Darpp have antagonistic effects on breast cancer cell growth and herceptin resistance. PLoS One. 2009;4:e6220. doi: 10.1371/journal.pone.0006220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Böttner M, Laaff M, Schechinger B, Rappold G, Unsicker K, Suter-Crazzolara C. Characterization of the rat, mouse, and human genes of growth/differentiation factor-15/macrophage inhibiting cytokine-1 (GDF-15/MIC-1) Gene. 1999;237:105–11. doi: 10.1016/s0378-1119(99)00309-1. [DOI] [PubMed] [Google Scholar]
- 24.Osada M, Park HL, Park MJ, Liu JW, Wu G, Trink B, Sidransky D. A p53-type response element in the GDF15 promoter confers high specificity for p53 activation. Biochem Biophys Res Commun. 2007;354:913–8. doi: 10.1016/j.bbrc.2007.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baek SJ, Kim JS, Nixon JB, DiAugustine RP, Eling TE. Expression of NAG-1, a transforming growth factor-beta superfamily member, by troglitazone requires the early growth response gene EGR-1. J Biol Chem. 2004;279:6883–92. doi: 10.1074/jbc.M305295200. [DOI] [PubMed] [Google Scholar]
- 26.Ichikawa T, Suenaga Y, Koda T, Ozaki T, Nakagawara A. TAp63-dependent induction of growth differentiation factor 15 (GDF15) plays a critical role in the regulation of keratinocyte differentiation. Oncogene. 2008;27:409–20. doi: 10.1038/sj.onc.1210658. [DOI] [PubMed] [Google Scholar]
- 27.Chen YL, Lin PC, Chen SP, Lin CC, Tsai NM, Cheng YL, et al. Activation of nonsteroidal anti-inflammatory drug-activated gene-1 via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase revealed a isochaihulactone-triggered apoptotic pathway in human lung cancer A549 cells. J Pharmacol Exp Ther. 2007;323:746–56. doi: 10.1124/jpet.107.126193. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi K, Lee SH, Eling TE, Baek SJ. Identification of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) as a novel downstream target of phosphatidylinositol 3-kinase/AKT/GSK-3beta pathway. J Biol Chem. 2004;279:49617–23. doi: 10.1074/jbc.M408796200. [DOI] [PubMed] [Google Scholar]
- 29.Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21:255–62. doi: 10.1093/annonc/mdp304. [DOI] [PubMed] [Google Scholar]