

Abstract
More than 50 new inhibitors of the oncogenic Stat3 protein were identified through a structure–activity relationship (SAR) study based on the previously identified inhibitor S3I-201 (IC50 = 86 µm, Ki > 300 µm). A key structural feature of these inhibitors is a salicylic acid moiety, which, by acting as a phosphotyrosine mimetic, is believed to facilitate binding to the Stat3 SH2 domain. Several of the analogues exhibit higher potency than the lead compound in inhibiting Stat3 DNA binding activity, with an in vitro IC50 range of 18.7–51.9 µm, and disruption of Stat3–pTyr peptide interactions with Ki values in the 15.5–41 µm range. One agent in particular exhibited potent inhibition of Stat3 phosphorylation in both breast and multiple myeloma tumor cells, suppressed the expression of Stat3 target genes, and induced antitumor effects in tumor cells harboring activated Stat3 protein.
Keywords: antitumor agents, molecular recognition, molecular therapeutics, protein–protein interactions, stat3
Introduction
The signal transducer and activator of transcription 3 (Stat3) protein mediates the relay of extracellular cytokine or growth factor stimulation to the nucleus, where it initiates the expression of gene profiles that promote cell proliferation, differentiation, and cell survival.[1] In normal cells, Stat3 transcriptional activity is transient and responsive to physiological cues. However, numerous human cancer cell lines, including breast,[2] prostate,[3] ovarian,[4] brain,[5] and lung[6] have been found to harbor persistently activated Stat3 protein. Aberrant Stat3 activity is widely acknowledged to be a master regulator of the cancer phenotype and to play a critical role in malignant transformation and tumorigenesis.[1] Moreover, dysregulated Stat3 transcriptional function has been implicated in the induction of tumor immune tolerance.[7] Overactivation of Stat3 promotes tumorigenesis by the up-regulation of cell survival proteins, cell cycle regulators and induction of angiogenesis.[8] Inhibition of Stat3 signaling correlates with suppression of cell transformation, motility, growth, and the induction of apoptosis in malignant cells.[9] Cell lines that lack aberrant Stat3 activation are more tolerant to Stat3 inhibitors, possibly identifying an irreversible dependence on persistent Stat3 activation for survival in vulnerable cell lines.[9] Of clinical and therapeutic significance, earlier studies from our research group and others have shown that in vivo administration of inhibitors of Stat3–Stat3 dimerization induce tumor regression in xenograft models.[10,11] In summary, Stat3 protein is considered an exciting and high-value target for cancer therapeutics.
The canonical view of Stat3 signaling describes latent Stat3 protein (monomeric[1] or dimeric[12]) residing predominantly in the cytoplasm. Ligand binding to the extracellular domain of transmembrane receptors induces intracellular activation of tyrosine kinases such as Janus kinases (JAKs). Receptors are phosphorylated on critical tyrosine residues of their cytoplasmic domain, creating docking sites for the recruitment of monomeric unphosphorylated Stat3 protein via its SH2 domain. Stat3 is phosphorylated on a key tyrosine residue, Tyr 705, which leads to receptor dissociation and the formation of activated Stat3–Stat3 dimers through reciprocal SH2–pTyr705 interactions. After translocation to the nucleus, dimeric Stat3 complexes bind to DNA response elements and promote gene transcription.[13, 14]
Inhibition of constitutive Stat3–Stat3 complexes by disruption of binding interfaces offers significant value as a molecular targeted therapy for cancer treatment.[10] Disruption of Stat3 complexes has been achieved through SH2 domain binders that compete with phosphorylated Stat3 monomers for the phosphotyrosine (pTyr) binding module. Numerous research groups, including our own, have shown that disruption of Stat3 transcriptional activity through dimer disruption leads to suppression of Stat3 gene expression profiles and induction of apoptosis. Stat3 dimers have been effectively disrupted by peptides,[15] peptidomimetics,[16] small molecules,[17] and metal complexes (Figure 1).[18] Peptidic inhibitors have been derived from the cognate binding sequence of Stat3 (pYLKTK) and from the Stat3-binding gp130 receptor (GpYLPQTV).[10] Inspired by these proof-of-principle peptidic probes, our groups[15a, 16a, d, 17d–g] and many others have synthesized optimized, more drug-like second-generation peptidomimetic inhibitors.[16b–c, e–g] Most notably, these include ISS610 (4-CN-Ph-pTyr-Leu (1))[16a] derived from pYLKTK, pCin-Leu-Pro-Glu-NHBn (2)[16b] derived from GpYLPQTV, and, most recently, the cell-permeable macrocyclic compound CJ-1383 (3).[16c] In addition to peptidomimetics, small-molecule inhibitors such as Stattic (4),[17a] LLL12 (5),[17b] STA-21 (6),[17c] and S3I-M2001 (7)[17d, e] have been identified through a combination of in silico and in vitro screening of chemical libraries as well as de novo rational design.
Figure 1.

Structures of Stat3 inhibitors 1–8.
By conducting an in silico structure-based virtual screen of the National Cancer Institute (NCI) chemical libraries, our research groups recently identified the potent Stat3 inhibitor S3I-201 (Figure 1, compound 8: IC50 = 86 µm as determined by an electrophoretic mobility shift assay (EMSA)).[17f] We identified that S3I-201 offers several opportunities for structural diversification, and embarked on a medicinal chemistry program to identify more potent analogues of S3I-201. Broadly speaking, the Stat3 SH2 domain is composed of three subpockets: a hydrophilic domain bounded by Lys 591, Arg 609, Ser 611, and Ser 613, and two hydrophobic domains; the first comprises Ile634 and the hydrocarbon portions of the side chains of Lys591 and Arg 595, and the second comprises Trp623, Val 637, Ile 659, and Phe716. The structural core of S3I-201 is glycolic acid, the carboxylic acid of which has been condensed with 4-aminosalicylic acid to furnish the amide bond, and the hydroxy group of which has been tosylated. Because S3I-201 carries only two appendages off the main scaffold, GOLD[19] docking unsurprisingly demonstrated that this small molecule can simultaneously occupy only two of these three subpockets (Figure 2). The salicylic acid moiety of S3I-201 is a known pTyr mimetic,[20] and low-energy GOLD docking studies consistently placed it in the pTyr binding site. The potential for hydrogen bonds and salt bridges here suggests that this component is responsible for a considerable portion of the binding energy with the Stat3 SH2 domain. GOLD docking studies suggested that the O-toluenesulfonyl (tosyl) group binds in the Arg 595/Ile 634 subpocket, leaving the Trp623/Phe716 subpocket unoccupied; the secondary amide NH of S3I-201 offers an excellent opportunity to gain access to this third subpocket. Thus, we were confident that a rational, synthetic program, facilitated by the inherent modular design of S3I-201, would allow the optimization of contacts between small molecule and the Stat3 SH2 domain to furnish more potent analogues of S3I-201.
Figure 2.

Low-energy GOLD[19] docking conformation of S3I-201 (8); hydrophobic residues are indicated in light grey, hydrophilic residues are dark grey.
The tosylate moiety in S3I-201 is an excellent leaving group, allowing nucleophilic attack at the carbon atom to which it is attached. Moreover, in this case, S3I-201 is especially prone to nucleophilic attack, due to the interaction of the σ* orbital (the LUMO) of the C–OTs bond with the π* orbital of the adjacent carbonyl group. Whilst we have no conclusive proof that S3I-201 functions as an irreversible inhibitor, there are several nucleophilic residues on the Stat3 SH2 domain surface, including Cys 418 and Cys 712, that may form a covalent bond to S3I-201. Such an event might compromise an SAR study because the majority of the inhibitory activity would be derived from the irreversible conjugation to the protein surface, which would be common to all S3I-201 analogues. Moreover, it is probable that irreversible inhibitors would exhibit poorer protein selectivity profiles than their reversible inhibitor counterparts. Therefore, we decided to replace the scaffold oxygen atom with a nitrogen atom to convert the labile tosylate into a non-labile tosylamide. The resulting secondary sulfonamide possesses a polar NH group, but despite this, GOLD docking studies consistently placed the tosylamide in the same hydrophobic subpocket (Arg 595/Ile 634) as the parent tosylate in S3I-201 (compound 9 or SF-1-082,[16g] Figure 3 A). Nevertheless, to further encourage occupancy of this hydrophobic subpocket, we elected to convert the NH group of the tosylamide to the more hydrophobic NCH3 group. In addition, as alluded to previously, a key aspect of this work was to functionalize the secondary amide NH, as it was anticipated that doing so would allow access to the third, as-yet-unexplored hydrophobic subpocket (Trp 623, Val 637, Ile659, and Phe716). Indeed, several low-energy GOLD docked poses of the N-benzyl derivative 14 (previously reported as SF-1-062)[16g] revealed that, as well as the salicylic acid and the N(CH3)-tosyl components binding the same subpockets as the corresponding components in the parent S3I-201, the N-benzyl group is projected into the third subpocket (Trp 623/Phe 716) as predicted (Figure 3 B).
Figure 3.

Low-energy GOLD[19] docking conformations of A) compound 9 and B) compound 14; hydrophobic residues are indicated in light grey, hydrophilic residues are dark grey.
The N(CH3)-tosylamide analogue of S3I-201 offers four potential optimization sites: 1) the salicylic acid component, 2) the secondary amide NH, 3) the tosyl moiety, and 4) the N(CH3) unit of the tosylamide. The salicylic acid moiety is a known phosphotyrosine mimetic, and because its modification would add considerably to the synthetic effort required for this research, we chose to keep this component constant. The remaining three sites would be subjected to SAR studies. Herein we elaborate on our previous communication[16g] by expanding on the SAR work of our initial lead compound S3I-201 and by providing additional biological characterizations in vitro and in whole cells.
Results and Discussion
If the considerable inhibitory activity of S3I-201 is due to its ability to covalently modify the Stat3 target, then conversion of the labile O-tosyl group to the non-labile N(CH3)-tosyl group would be expected to cause a significant decrease in the inhibition of Stat3. To investigate this, we first prepared a focused set of non-labile analogues of S3I-201 (shown in Table 1), the syntheses of which are described in full in the Supporting Information. Unfortunately, replacement of the scaffold oxygen atom with NH, NCH3, or NBoc led to a decrease in activity in all cases, from an IC50 value of 86 µm (by EMSA) to > 300 µm for all non-labile analogues, suggesting that S3I-201 might indeed operate, at least in part, as an irreversible inhibitor. On the other hand, benzylation of the amide NH of S3I-201 also led to a loss in inhibitory activity (compound 12, SF-1-120:[16g] IC50 > 300 µm) despite the alkylating potential of this analogue remaining intact. Nevertheless, within this series (R1 = benzyl), we observed a slight recovery in activity if the scaffold oxygen atom is replaced with the NCH3 unit (14: IC50 = 292 µm), and thus we elected to constrain the X heteroatom/group as NCH3 for most of this research project. Because 14 demonstrated some activity against Stat3, and we believed the R1 = benzyl group of that inhibitor makes favorable interactions with the Trp623/Phe716 hydrophobic subpocket, as predicted by GOLD docking experiments (Figure 3 B), we decided to investigate the effects of modifying the benzyl group, in particular at the para and meta positions, where deeper access to the subpocket might be realized.
Table 1.
EMSA inhibition data for disruption of the Stat3–Stat3:DNA complex in vitro by a focused set of S3I-201 (8) analogues.
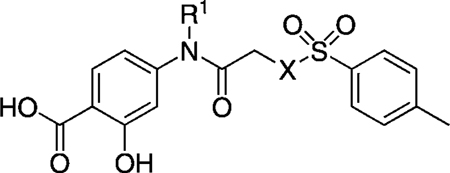 | |||
|---|---|---|---|
| Compd | R1 | X | IC50 [µm] |
| 8 (S3l-201) | H | O | 86 ± 33 |
| 9 | H | NH | > 300 |
| 10 | H | NCH3 | > 300 |
| 11 | H | NBoc | > 300 |
| 12 | O | > 300 | |
| 13 | NH | > 300 | |
| 14 | NCH3 | 292 ± 35 | |
| 15 | NBoc | > 300 | |
Probing the Trp623, Val 637, Ile 659, and Phe716 hydrophobic subpocket: SAR of the R1 group
The series of S3I-201 analogues listed in Table 2, where X = NCH3, were furnished by following the synthetic steps outlines in Scheme 1. After the one-pot and stepwise benzylations of the carboxylic acid and hydroxy functionalities of 4-aminosalicylic acid (16), which proceeded in moderate yield (54 %), several hydrophobic aldehydes (RCHO) were reductively aminated with the resultant aniline 17 to afford the series of secondary anilines 18 and 19a–i in very good to excellent yields. Meanwhile, sulfonylation of glycine methyl ester (20) with para-toluenesulfonyl chloride (p-TsCl) furnished secondary sulfonamide 21, which was subsequently N-methylated with methyl iodide, and then saponified with lithium hydroxide to generate carboxylic acid 23 (75 % yield over three steps). Condensation of the primary aniline 17 and the secondary anilines 18 and 19a–i with acid 23 to deliver the secondary amide 24 and the tertiary amides 25 and 26a–i, respectively, was achieved with the highly reactive peptide coupling agent dichlorotriphenylphosphorane (PPh3Cl2), which is believed to generate the corresponding acid chloride of 23 in situ. Finally, a global debenzylation of compounds 24, 25, and 26 a–i with hydrogen gas over 10 % palladium on carbon yielded the series of S3I-201 analogues 10, 14, and 27 a–i. Importantly, the aryl nitrile moieties were essentially untouched in the debenzylation reactions, with the reducing conditions proving chemoselective for removal of the benzyl protecting groups. These phenomena are likely due to a combination of rapid reaction times (both aryl nitrile-containing intermediates 26 c and 26 d were doubly debenzylated in ~ 1 h), which limited the exposure of the nitrile functional group to the reducing conditions, and the fortuitous limited solubilities of the compounds in neat methanol, requiring the use of THF as co-solvent, which is known to suppress hydrogenation of nitriles.[21] Conversely, because the reduction of aryl–bromide bonds with H2 and Pd/C catalyst is known to be a relatively facile reaction,[22] hydrogenolysis of the benzyl protecting groups in intermediates 26 a and 26 b was not attempted. Instead, we employed a high-yielding, non-reducing, two-step protocol. First, the benzyl ester was selectively hydrolyzed with lithium hydroxide (the tertiary amide was slowly hydrolyzed under these conditions), and then the benzyl ether was cleaved under acidic conditions with trifluoroacetic acid (TFA). Removal of the benzyl ether under these conditions is believed to be facilitated by chelation of a proton between the carboxylic acid and ether functionalities.[23]
Table 2.
EMSA inhibition data for disruption of the Stat3–Stat3:DNA complex in vitro by a series of R1-functionalized analogues of compound 10.
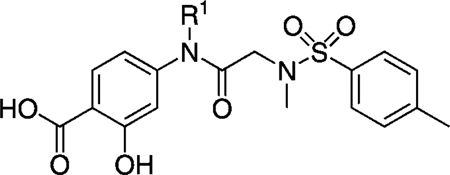 | |||||
|---|---|---|---|---|---|
| Compd | R1 | IC50 [µm] | Compd | R1 | IC50 [µm] |
| 10 | H | > 300 | 27 e | > 300 | |
| 14 | 295 ± 35 | 27 f | 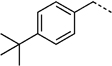 |
194 ± 47 | |
| 27 a | 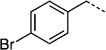 |
290 | 27 g | 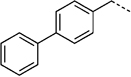 |
115 ± 60 |
| 27 b | 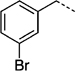 |
> 300 | 27 h | 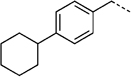 |
35 ± 9 |
| 27 c | 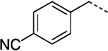 |
260 ± 47 | 27 i | 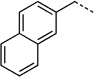 |
280 ± 15 |
| 27 d | 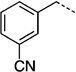 |
298 ± 11 | |||
Scheme 1.

a) 1. BnBr, KOtBu, DMF, 0 °C →RT, 5 h; 2. BnBr, KOtBu, DMF, 0 °C →RT, 16 h, 54 %; b) 1. RCHO, AcOH, 4 Å MS, MeOH, 45 °C, 3 h; 2. NaCNBH3, RT, 12 h,75–96 %; c) p-TsCl, DIPEA, CH3CN, 0 °C →RT, 1 h, 93 %; d) MeI, Cs2CO3, DMF, RT, 16 h, 85 %; e) LiOH·H2O, THF/MeOH/H2O (3:1:1), RT, 1 h, 95 %; f) PPh3Cl2, CHCl3, 60 °C, 12 h, 89–95 %; g) H2, 10 % Pd/C, MeOH/THF (1:1), RT, 1–16 h, 85–100 %; or for 26 a and 26 b: h) LiOH·H2O, THF/H2O (3:1), RT, 24 h, 76–86 %; i) TFA/toluene (1:2), RT, 16 h, 85–93 %
Replacement of the R1 benzyl group in 14 with 4-cyanobenzyl (27 c, SF-1-073)[16g] led to an improvement in activity (IC50 = 260 µm for 27 c; cf. IC50 = 292 µm for 14). This enhancement in Stat3 inhibition may be due to improved hydrophobic interactions with the larger and more electron-poor aromatic system, and/or from a hydrogen bond between the nitrile group and the SH2 domain. More interesting is the observation that Stat3 inhibition improved with increasing size of the hydrophobic R1 group. Specifically, 4-(tert-butyl)benzylated agent 27 f (SF-1-068)[16g] showed marked improvement in activity over both 14 (R1 = benzyl) and 27 c (R1 = 4-cyanobenzyl), whilst replacement of the tert-butyl group with a phenyl ring to give the large biphenyl-based inhibitor 27 g (SF-1-070, R1 = 4-phenylbenzyl) led to a further approximate twofold increase in potency (27 g, IC50 = 115 µm; cf. IC50 = 194 µm for 27 f). Furthermore, the inclusion of the especially hydrophobic 4-cyclohexylbenzyl group at the R1 position furnished an inhibitor that exhibited Stat3 inhibitory activity with more than double the potency of our lead agent: IC50 = 35 µm for 27 h (SF-1-066);[16g] cf. IC50 = 86 µm for S3I-201 (8).
N-Substituted piperidinylmethyl derivatives and N-substituted 4-(piperidinyl)benzyl derivatives
Because greater Stat3 inhibitory activity was furnished by substitution at the para position of the R1 benzyl group in 14, we were keen to functionalize this position further still. However, owing to a simpler synthetic demand, it was decided to determine whether substitution at the 4-position of the cyclohexyl group (a good match for benzyl) would also enhance inhibitor activity. Replacement of the cyclohexylmethyl moiety in 27 e with 4-piperidinylmethyl would allow facile elaboration of the inhibitor through functionalization of the piperidine nitrogen to probe deeper into the proposed subpocket. To this end, compound 26 j (Scheme 2) was accessed by following the steps in Scheme 1, where the RCHO aldehyde was N-Boc-piperidinylformaldehye (the Boc group was inadvertently removed during the peptide coupling step with PPh3Cl2; full details for the synthesis of 26 j are given in the Supporting Information). Because the piperidinylmethyl group was proposed to bind in a hydrophobic subpocket, we appreciated that conjugation of groups to the piperidine nitrogen that would considerably decrease its basicity would be required. Thus, the transformations conducted on the piperidine nitrogen (Scheme 2) included re-tert-butoxycarbonylation and arylation with 4-fluorobenzonitrile or 2-chloropyrimidine to afford, after benzyl deprotections, inhibitors 27 jb, 27 jc, and 27 jd, respectively. Unfortunately, as shown in Table 3, none of the inhibitors were active; all exhibited EMSA IC50 values > 300 µm.
Scheme 2.

a) R3 = Boc: Boc2O, cat. DMAP, CH2Cl2, RT, 1 h, 95 %; R3 = aryl: R3F or R3Cl, DIPEA, DMSO, 120 °C, 16 h, 76–96 %; b) H2, 10 % Pd/C, MeOH/THF (1:1), RT, 1–16 h, 85–100 %.
Table 3.
EMSA inhibition data for disruption of the Stat3–Stat3:DNA complex in vitro by a series of R1=N-(4-piperidinyl)methyl-based analogues of compound 10.
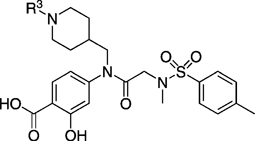 | ||
|---|---|---|
| Compd | R3 | IC50 [µm] |
| 27 ja | H | > 300 |
| 27 jb | > 300 | |
| 27 jc | 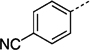 |
> 300 |
| 27 jd | > 300 | |
Next, we tackled functionalization of the 4-position of the cyclohexyl component of inhibitor 27 h in a similar manner. This time, preparation of the requisite aldehyde 4-[N-trifluoroacetyl(piperidin-4-yl)]benzaldehyde (32) was slightly more complicated, and its synthesis is illustrated in Scheme 3. Briefly, protection of the piperidine nitrogen of 4-phenylpiperidine (29) was accomplished as its acid-stable trifluoroacetamide 30. Subsequently, regioselective para-chlorocarbonylation of 30 was effected under Friedel–Crafts conditions,[24] and then the crude acid chloride 31 was reduced to the target aldehyde 32 in a modification of the Rosenmund reaction. Employing 32 as the RCHO aldehyde, the corresponding compound 26 k was then furnished by following the appropriate steps in Scheme 1. Next, as shown in Scheme 4, the trifluoroacetyl group of 26 k was cleaved in excellent yield by brief treatment with lithium hydroxide to reveal the piperidine nitrogen atom in 33. Subsequent functionalization of this nitrogen was accomplished with a variety of reagents to furnish, after the standard benzyl deprotections, the series of compounds 27 ka–kg depicted in Table 4. As in the case of the N-piperidinylmethyl series of inhibitors 27 ja–jd, we elected to substitute the piperidine nitrogen atom in 33 with functionalities that would decrease its basicity through withdrawal of its lone pair of electrons into aryl systems, and acyl and sulfonyl groups. Inhibitors 27 kh and 27 ki were prepared as shown in Scheme 5. Specifically, deprotection of the tert-butyl ester of 34 h with TFA also led to the concomitant removal of the benzyl ether, as reported by us previously, to deliver monobenzyl-protected compound 35. Facile condensation of the carboxylic acid of 35 with ammonium chloride, employing O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HBTU) as the coupling agent, generated carboxamide 36 in excellent yield. Deprotection of the benzyl esters of 35 and 36 under the usual hydrogenolytic conditions furnished the corresponding inhibitors 27 kh and 27 ki. As the N-(piperidin-4-yl)benzyl moiety was predicted to bind in a hydrophobic subpocket, we anticipated that the polar acid and carboxamide-containing inhibitors might demonstrate poor activity against Stat3. In fact, as Table 4 illustrates, among the entire series 27 ka–ki, only 4-cyanophenyl-based 27 kd and 4-cyanobenzenesulfonyl-based 27 kg exhibited Stat3 inhibitory activity (< 300 µm), with IC50 values of 45 and 50 µm, respectively. Both 27 kd and 27 kg share a 4-benzonitrile moiety, so it may seem curious that the related inhibitor 4-cyanobenzamide 27 kf demonstrated no inhibition of Stat3. This result could be due to the nature of the EMSA, which is conducted on nuclear extracts that contain various other members of the STAT protein family. It may be the case that 27 kf is a potent inhibitor of a different STAT isoform, decreasing the concentration of free compound available to inhibit Stat3, leading to an apparent IC50 lower than the actual value.
Scheme 3.

a) (CF3CO)2O, DIPEA, CH2Cl2, 0 °C →RT, 3 h, 93 %; b) (COCl)2, AlCl3, CH2Cl2, 0 °C, 1 h; c) H2, 10 % Pd/C, DIPEA, EtOAc, RT, 2 h, 63 % (two steps).
Scheme 4.

a) LiOH·H2O, THF/H2O (3:1), RT, 10 min, 98 %; b) R3 = Boc: Boc2O, cat. DMAP, CH2Cl2, RT, 1 h, 95 %; R3 = aryl: R3F or R3Cl, DIPEA, DMSO, 120 °C, 16 h, 80–99 %; R3 = p-CNC6H4SO2: p-CNC6H4SO2Cl, DIPEA, RT, 16 h, 99 %; R3 = p-CNC6H4CO2: p-CNC6H4CO2H, HBTU, DIPEA, DMF, RT, 16 h, 89 %; c) H2, 10 % Pd/C, MeOH/THF (1:1), RT, 1–16 h, 85–100 %.
Table 4.
EMSA inhibition data for disruption of the Stat3–Stat3:DNA complex in vitro by a series of R1=N-(4-piperidinyl)benzyl-based analogues of compound 10.
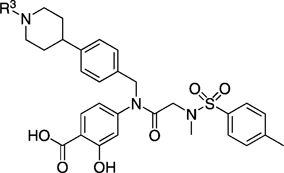 | |||||
|---|---|---|---|---|---|
| Compd | R3 | IC50 [µm] | Compd | R3 | IC50 [µm] |
| 27 ka | > 300 | 27 kf | > 300 | ||
| 27 kb | H | > 300 | 27 kg | 50 ± 15 | |
| 27 kc | > 300 | 27 kah | > 300 | ||
| 27 kd | 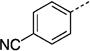 |
45 ± 12 | 27 ki | > 300 | |
| 27 ke | 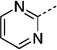 |
> 300 | |||
Scheme 5.

a) TFA/toluene (1:1), RT, 4 h, 95 %; b) NH4Cl, DIPEA, HBTU, DMF, RT, 16 h, 99 %; c) H2, 10 % Pd/C, MeOH/THF (1:1), RT, 1–16 h, 85–100 %.
Biphenyl and terphenyl derivatives
After elaborating our inhibitors through functionalization of the 4-position of the cyclohexyl moieties in 27 e (IC50 > 300 µm) and 27 h (IC50 = 35 µm), the next logical approach to probe deeper into the Trp623/Phe 716 hydrophobic subpocket was to modify the biphenyl unit of 27 g (IC50 = 115 µm). The aryl bromide moiety in 26 a provides an excellent handle for facile substitution reactions via Suzuki chemistry, facilitating access to the desired biphenyl analogues of 27 g. To this end, and as described in Scheme 6, 26 a was treated with a variety of aryl boronic acids in the presence of catalytic Pd(PPh3)4 to furnish, after the standard benzyl group deprotections, the series of meta- and para-substituted biphenyl-based inhibitors 27 la–lh shown in Table 5. Likewise, the corresponding 4-(4-bromophenyl)benzyl derivative 26 m furnished the terphenyl-based inhibitors 27 na–nh (Table 5).
Scheme 6.

a) R4B(OH)2, Pd(PPh3)4, K2CO3, DMF, 100 °C, 24 h, 16–73 %; b) LiOH·H2O, THF/H2O (3:1), RT, 24 h, 76–99 %; c) TFA/toluene (1:2), RT, 16 h, 85 %; d) H2, 10 % Pd/C, MeOH/THF (1:1), RT, 1–16 h, 85–100 %.
Table 5.
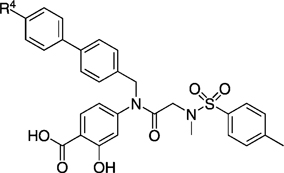
EMSA inhibition data for disruption of the Stat3–Stat3:DNA complex in vitro by a series of biphenyl- and terphenyl-based derivatives of inhibitor 27 a.
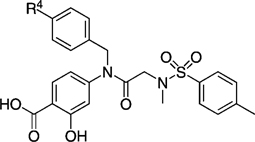 |
 |
||||
|---|---|---|---|---|---|
| Compd | R4 | IC50 [µm] | Compd | R4 | IC50 [µm] |
| 27 g | 115 ± 760 | 27 na | 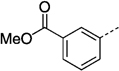 |
63 ± 2 | |
| 27 la | 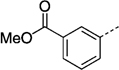 |
191 ± 8 | 27 nb | 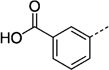 |
> 300 |
| 27 lb | 250 ± 23 | 27 nc | 82 ± 1 | ||
| 27 lc | 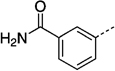 |
> 300 | 27 nd | 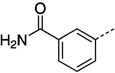 |
90 |
| 27 gld | 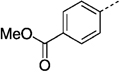 |
141 ± 10 | 27 ne | 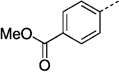 |
97 ± 17 |
| 27 le | 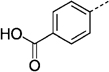 |
> 300 | 27 nf | 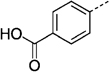 |
> 300 |
| 27 lf | 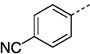 |
274 ± 10 | 27 ng | 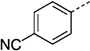 |
74 ± 9 |
| 27 lg | 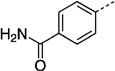 |
> 300 | 27 nh | 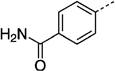 |
43 ± 1.3 |
As shown in Table 5, none of the biphenyl-based inhibitors 27 la–27lh offered any improvement in Stat3 inhibitory activity over the parent biphenyl inhibitor 27 g (IC50 = 115 µm). However, excluding the carboxylic acid-substituted compounds 27 nb and 27 nf, the terphenyl-based inhibitors 27 na–nh proved more potent that the parent inhibitor 27 g, with the most active compound 27 nh disrupting the Stat3–Stat3:DNA ternary complex with an IC50 value of 43 µm. The improved activity of the terphenyl-based inhibitors over their biphenyl-based counterparts is likely due, at least in part, to enhanced van der Waals contacts between the larger terphenyl moieties and the protein surface, possibly in the proposed Trp623/Phe 716 subpocket. The observation that the 4-carboxamide terphenyl 27 nh was the most potent of the series is probably due to a hydrogen bond between the carboxamide functional group and the protein surface.
Probing the Arg595/Ile634 hydrophobic subpocket: SAR of the sulfonamide X group
To complete our research program, we modified the X = NCH3 component (X = O in S3I-201) whilst invoking optimized R1 and R2 groups to help identify even more potent Stat3 inhibitors. Once more, R1 was constrained as the 4-cyclohexylbenzyl group. Thus, a focused variation of the NCH3 group in 27 h was executed. The substitutes chosen were the more hydrophobic NBoc group, the more polar NH group, and oxygen, affording, in the latter case, a potentially irreversible inhibitor. The syntheses of these target molecules are depicted in Scheme 7. Briefly, secondary aniline 19 h was coupled to TsN-(Boc)CH2CO2H (39) using PPh3Cl2, which, due to the generation of HCl in situ, led to the inadvertent loss of the Boc group to furnish 40. Standard hydrogenolytic debenzylation of 40 gave 41 (SF-1-083),[16g] or, alternatively, the NH of 40 was re-tert-butoxycarbonylated and then debenzylated as usual to deliver 43 (SF-1-087).[16g] To synthesize the labile O-tosyl analogue of 41, compound 19 h was first coupled to 2-acetoxyacetyl chloride to produce 44. Simple hydrolysis of the acetate group in the presence of the aryl benzyl ester proceeded smoothly. Tosylation of the resultant primary alcohol to give 45 was nontrivial and required the use of 20 equiv p-TsCl in order to suppress symmetrical ether formation through the reaction of the starting alcohol with the product tosylate. Debenzylation of 45 was closely monitored, and fresh Pd catalyst (10 mol %) was added every 2 h to minimize reaction time and the likelihood of loss of the O-tosyl group through nucleophilic attack by the methanol co-solvent. S3I-201 analogue 46 (SF-1-121)[16g] was thus furnished in very good yield (85 %).
Scheme 7.

a) TsN(Boc)CH2CO2H (46), PPh3Cl2, CHCl3, 60 °C, 12 h, 48 %; b) AcOCH2COCl, DIPEA, CH2Cl2, RT, 4 h, 64 %; c) (Boc)2O, cat. DMAP, THF, 12 h, 81 %; d) H2, 10 % Pd/C, MeOH/THF (1:1), RT, 1–16 h, 85–94 %; e) LiOH·H2O, THF/MeOH/H2O (3:1:1); 89 % f) p-TsCl, DIPEA, CH2Cl2, RT, 3 h, 85 %.
The EMSA data for compounds 41 and 43 in Table 6 indicate that changing the X = NCH3 group in compound 27 h to NH or NBoc, respectively, had a detrimental effect on Stat3 inhibitory activity, increasing the IC50 value from 35 to ~ 100 µm. However, more interestingly, the O-tosyl analogue 46 was equipotent with the parent inhibitor 27 h, within experimental error. Compound 46, carrying the labile O-tosyl group, has the capacity to function as an irreversible inhibitor, whilst 27 h, with the non-labile N(CH3)-tosyl moiety, possesses no such potential. These data thus suggest that the inhibitory activity of 46 likely arises chiefly from noncovalent interactions with the Stat3 SH2 domain. Interestingly, however, the similar activities of 46 and 27 h are in stark contrast to the very different activities of the analogous R1 = H derivatives S3I-201 (8) and 10, respectively, for which replacement of the X = O atom with NCH3 abolished Stat3 inhibitory activity (> 300 µm). Taken together, these results suggest that the R1 = 4-(cyclohexyl)benzyl moiety in 27 h and 46 contributes significantly to the inhibition of Stat3. Furthermore, it is evident that the nature of the X group in the S3I-201 scaffold plays a considerable role in the subsequent Stat3 inhibitory activity, and for this reason our current research efforts are focused toward a more extensive SAR study of this group. This work shall be reported in due course.
Table 6.
EMSA inhibition data for disruption of the Stat3–Stat3:DNA complex in vitro by a series of X-substituted para-toluenesulfonyl analogues of inhibitor 27 h.
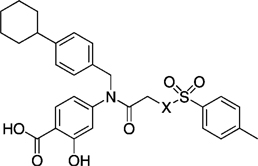 | ||
|---|---|---|
| Compd | X | IC50 [µm] |
| 27 h | NCH3 | 35 ± 9 |
| 41 | NH | 95 ± 9 |
| 43 | NBoc | 115 ± 35 |
| 46 | O | 43 ± 13 |
We selected several of our analogues of S3I-201 (8) for more thorough biophysical characterization by evaluating their inhibition of the Stat3 protein in isolation by using an in vitro fluorescence polarization (FP) assay.[25] The principle of this assay works on the decrease in FP that occurs upon displacement of the 5-carboxyfluorescein (F*)-labeled Stat3 SH2 domain inhibitor F*-GpYLPQTV from the Stat3 protein by the small molecule of interest. Generally, the FP Ki data for the selected inhibitors (Table 7) corroborate those data observed in the EMSA, with potent activity in one assay reflected by potent activity in the other. These data support our hypothesis that the S3I-201 analogues disrupt the ternary Stat3–Stat3:DNA complex, as quantified in the EMSA, through direct inhibition of the Stat3 protein. It is reasonable to expect that disruption of the Stat3–Stat3 dimer (full-length protein) bound to DNA in the EMSA may be more difficult than disrupting the Stat3–phosphopeptide interaction in the FP assay, and this is probably the reason why, in many cases, the FP-determined Ki values were lower than the corresponding EMSA-determined IC50 values.
Table 7.
EMSA and FP inhibition data of selected inhibitors for disruption of the Stat3–Stat3:DNA and Stat3–gp130 sequence complexes in vitro.
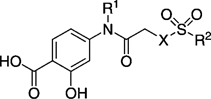 | |||||
|---|---|---|---|---|---|
| Compd | R1 | X | R2 | EMSA IC50 [µm] | FP Ki [µm] |
| 8 (S3I-201) | H | O | 86 ± 33 | > 50 | |
| 27 h | 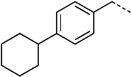 |
NCH3 | 35 ± 9 | 15 ± 4.9 | |
| 27 kd | 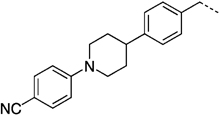 |
NCH3 | 45 ± 12 | 18.7 ± 1.4 | |
| 27 kd | 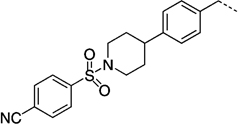 |
NCH3 | 50 ± 15 | 21.5 ± 2.2 | |
| 27 kd | 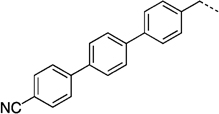 |
NCH3 | 74.3 ± 9.3 | 21.6 ± 0.9 | |
| 27 kd | 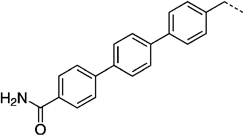 |
NCH3 | 43 ± 1.3 | 16.7 ± 1.7 | |
STAT isoform selectivities
Using a similar Stat1 SH2 domain FP-based binding assay,[26] we also investigated the isoform selectivity of some of our most potent Stat3 inhibitors by evaluating their inhibitory activities against Stat1, which exhibits 78 % sequence identity to Stat3.[27] The results of our findings are disclosed in Table 8 and in the Supporting Information. Compound 27 h exhibited a greater than threefold selectivity for Stat3 (Stat3: Ki = 15 µm, Stat1: Ki ≧ 50 µm). In contrast to 27 h, the 4-cyanobenzenesulfonyl-based compound 27 kg showed only limited isoform specificity (Stat3: Ki = 21 µm, Stat1: Ki = 28 µm), which, given the structural similarities of these two compounds, suggests that the 4-cyclohexylbenzyl group at the R1 position is also a source of Stat3 isoform specificity.
Table 8.
Comparative Stat isoform selectivity as assessed by a Stat3 and Stat1 FP assay.
| Compd | Ki [µm] | |
|---|---|---|
| Stat3 | Stat1 | |
| 27 h | 15 ± 5 | > 50 |
| 27 kd | 8.4 ± 1 | > 50 |
| 27 kg | 21 ± 2 | 28 ± 2 |
| 27 nh | 8.4 ± 2 | 9.5 ± 2 |
Whole-cell cytotoxicity
Agents 27 h, 27 kd, 27 kg, 27 nh, and 46 all show significantly improved in vitro inhibitory activity against Stat3 DNA binding activity (as determined by EMSA), with IC50 values of 18.7–51.9 µm (Tables 5–8) and Stat3–pTyr peptide interaction in the FP assay, with Ki values of 13–26.5 µm (Tables 7 and 8 and Supporting Information). For select active compounds, whole-cell activities were investigated by screening inhibitors at a concentration of up to 100 µm across a range of human tumor cell lines, namely breast cancer (MDA-MB-468),[28] prostate cancer (DU145),[29] acute myeloid leukemia (OCI-AML-2),[30] and human multiple myeloma (JJN-3), all of which harbor constitutively active Stat3 (data not shown). The inhibitory activities (IC50 values) for select compounds are listed in Table 9.
Table 9.
IC50 values for selected R1-substituted analogues in whole-cell viability studies.
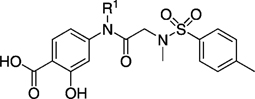 | ||||
|---|---|---|---|---|
| Compd | R1 | IC50 [µm] | ||
| MDA-MB-468[a] | DU145[b] | OCI-AML-2[c] | ||
| 27 h | 17 ± 4 | 37 ± 12 | 36 ± 13 | |
| 27 kd | > 100 | > 100 | > 100 | |
| 27 kg | 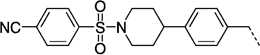 |
95 ± 9 | > 100 | > 100 |
| 27 ng | 33 ± 16 | 94 ± 7 | 58 ± 14 | |
| 27 nh | > 100 | > 100 | > 100 | |
MDA-MB-468: breast cancer.
DU145: prostate cancer.
OCI-AML-2: acute myeloid leukemia.
Indeed, there was good correlation between the whole-cell effects and the inhibition of Stat3 in nuclear extracts. Treatment of cells with compounds 10, 14, 27 a–e, 27 i, 27 ja–jd, 27 ld, 27 lh, and 27 nd had no effect on cell growth at inhibitor concentrations < 100 µm, reflecting their poor IC50 values in the EMSA (data not shown), whereas the active Stat3 inhibitors in the in vitro EMSA inhibited the growth of cells dependent on constitutively active Stat3. Accordingly, the whole-cell activities observed with 27 h, 27 kg, and 46 mirror their inhibitory activities in the Stat3 DNA binding activity/EMSA (Table 2) and the Stat3–pTyr peptide interactions in the FP assay (Tables 7 and 8). Agent 27 h inhibited the growth of MDA-MB-468 breast cancer cells with an IC50 value of 17 µm, of DU145 with IC50 = 37.2 µm, and of OCI-AML-2 with IC50 = 35.9 µm, consistent with its EMSA results or the Ki value for the inhibition of Stat3–pTyr peptide interactions (Tables 7 and 8). Notably, although 27 h inhibited Stat3 activity in EMSA with an IC50 value of 35 µm, it inhibited Stat3–pTyr peptide interactions with an IC50 value of 15 µm (Table 8), suggesting that in cells, it might be more effective to disrupt Stat3 binding to pTyr peptide motifs of receptors, as has been previously reported for dimerization disruptors by our group.[15, 17g] Consistent with the findings from the EMSA (Tables 3 and 4), the N-(4-piperidinyl)methyl- (27 ja–27d) and N-(4-piperidinyl)benzyl-based (27 ka–kf) inhibitors proved ineffective toward the growth of Stat3-dependent tumor cell lines.
Generally, a good correlation was observed between the EMSA data for the biphenyl- and terphenyl-based compounds (Table 5) and the whole-cell data for the Stat3-dependent cell lines, particularly with the MDA-MB-468 cell line. It is probable that the polar carboxamide functional groups in 27 nd and 27 nh hindered cellular entry of these compounds, hence their poorer whole-cell activities than might have otherwise been anticipated based on their activities in the EMSA. Generally speaking, these series of compounds were equipotent at inhibiting the growth of breast cancer MDA-MB-468 cells and acute myeloid leukemia OCI-AML-2 cells, and were around half as active or worse in the prostate cancer DU145 cell line. Compound 27 h shows strong cellular effects (Table 10), consistent with the inhibition of Stat3 activity in cells (Figure 4).
Table 10.
IC50 values for a series of para-toluenesulfonyl X analogues of inhibitor 27 h in whole-cell viability studies.
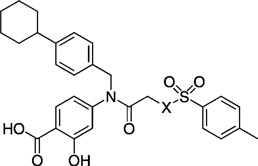 | ||||
|---|---|---|---|---|
| Compd | X | IC50 [µm] | ||
| MDA-MB-468[a] | OCI-AML-2[b] | DU145[c] | ||
| 27 h | NCH3 | 17 ± 4 | 37 ± 12 | 36 ± 13 |
| 41 | NH | 21 ± 6 | 38 ± 14 | 33 ± 8 |
| 43 | NBoc | 10 ± 9 | 24 ± 16 | 18 ± 9 |
| 46 | O | 43 ± 6 | 52 ± 13 | 62 ± 12 |
MDA-MB-468: breast cancer.
OCI-AML-2: acute myeloid leukemia.
DU145: prostate cancer.
Figure 4.

Western blot analysis showing A) inhibition of Stat3 phosphorylation (pYStat3) and B) repression of Stat3-regulated gene products, Bcl-xL and Survivin, in human breast (MDA-MB-468) and multiple myeloma (JJN-3) cells as a function of treatment with 27 h (100 µm, 24 h). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a lane loading control.
As detailed in Table 10, analogues of S3I-201 (8) carrying the optimized R1 = 4-cyclohexylbenzyl and R2 = p-tolyl groups all exhibited sub-100 µm activities in the three Stat3-dependent tumor cell lines. Compounds 27 h (X = NCH3) and 41 (X = H) were roughly equipotent. The most potent compound of this series in the Stat3-dependent cell lines was compound 43, the X = NBoc analogue of 27 h, inhibiting MDA-MB-468 cell growth with an IC50 value of 10.5 µm. The improved whole-cell activities of 43 relative to 27 h is possibly due to the greater hydrophobicity of the NBoc group over the NCH3 group, which might facilitate more efficient cellular entry. Encouragingly, when assessed by FP for Stat3 binding potency, compound 43 was shown to have Ki = 15 ± 0.2 µm (Supporting Information). The O-tosyl derivative 46, a potentially irreversible inhibitor, demonstrated activities in the Stat3-dependent cell lines that were around half that exhibited by the parent 27 h. These findings may, in part, be a consequence of the possible hydrolysis of 46 to primary alcohol 47, which was synthesized and found to show no inhibition of Stat3 in vitro (IC50 > 300 µm), nor appreciable whole-cell activity (MDA-MB-468: IC50 > 100 µm).
Inhibition of intracellular aberrant Stat3 phosphorylation and the induction of known Stat3-regulated genes
Consistent with the effects on viability, 27 h strongly inhibited constitutively active Stat3 in tumor cells, including the human breast cancer (MDA-MB-468) and multiple myeloma (JJN-3) lines, as measured by Western blot analysis (Figure 4 A), confirming that select agents inhibit aberrant Stat3 activation in tumor cells. Furthermore, treatment with 27 h inhibited the expression of Bcl-xL and Survivin, the genes for which are known to be regulated by Stat3 (Figure 4 B). These findings suggest that the modulation of aberrant Stat3 in MDA-MB-468 and JJN-3 cells leads to suppression of Stat3-mediated gene regulation. These events contribute to the loss of viability observed following the treatment of malignant cells that harbor aberrant Stat3 activity by the newly identified small-molecule inhibitors.
Conclusions
In summary, we have conducted an extensive SAR study centered on the previously identified Stat3 inhibitor S3I-201 (8) to derive analogues with improved Stat3 inhibitory activity. These studies have led to the identification of several diverse classes of agents equipped with an additional appendage that promotes interaction with the hitherto unexplored pocket on the Stat3 protein surface, thereby intensifying the binding to Stat3 and enhancing the Stat3 inhibitory activity. Specifically, compounds 27 h, 27 nh, 27 kd, 27 kg, and 46 all show significantly improved in vitro inhibitory activity against Stat3, with IC50 values of 18.7–51.9 µm. Moreover, at these concentrations, select compounds inhibit constitutively active Stat3 and Stat3 tyrosine phosphorylation in malignant cells and promote anti-tumor cell effects consistent with the inhibition of aberrant Stat3 activity. The improved inhibitory activity against Stat3 activation is derived in part from the successful occupation of the third subdomain of the Stat3 SH2 domain, as supported by computational modeling; S3I-201 can simultaneously occupy only two of these subdomains. Importantly, with a labile O-tosyl group a to a carbonyl group, S3I-201 has the capacity to operate, at least in part, as an irreversible inhibitor, which would be anticipated to lead to poor protein selectivity profiles.
Experimental Section
Anhydrous solvents MeOH, DMSO, CH2Cl2, THF, and DMF were purchased from Sigma–Aldrich and were used directly from Sure-Seal bottles. Molecular sieves were activated by heating at 300 °C under vacuum overnight. All reactions were performed under an atmosphere of dry N2 in oven-dried glassware and were monitored for completeness by thin-layer chromatography (TLC) using silica gel (visualized by UV light, or developed by treatment with KMnO4 stain or phosphomolybdic acid stain). 1H and 13C NMR spectra were recorded on Bruker 400 MHz and Varian 500 MHz spectrometers in either CDCl3, CD3OD, or [D6]DMSO. Chemical shifts (d) are reported in ppm after calibration to residual isotopic solvent. Coupling constants (J) are reported in Hz. Before biological testing, inhibitor purity was evaluated by reversed-phase HPLC (RP-HPLC). Analysis by RP-HPLC was performed by using a Microsorb-MV 300A C18 250 × 4.6 mm column run at 1 mL min−1, and using gradient mixtures of A) H2O with 0.1 m CH3COONH4 and B) MeOH. Ligand purity was confirmed by using linear gradients from 75 % A and 25 % B to 100 % B after an initial 2 min period of 100 % A. The linear gradient consisted of a changing solvent composition of either I) 4.7 % per minute and UV detection at λ 254 nm, or II) 1.4 % per minute and detection at λ 214 nm, each ending with 100 % B for 5 min. For reporting HPLC data, percentage purity is given in parentheses after the retention time for each condition. All biologically evaluated compounds are of > 95 % chemical purity as measured by HPLC. Full characterization for all final compounds and intermediate compounds are provided in the Supporting Information.
Supplementary Material
Acknowledgements
We thank Jeffrey L. Wrana and Alessandro Datti (Mt. Sinai Hospital) for access to the SMART high-throughput screening facility. This work was supported by the Leukemia and Lymphoma Society of Canada (P.T.G.), NSERC (P.T.G.), the University of Toronto (P.T.G.), and by National Cancer Institute grants CA106439 (J.T.) and CA128865 (J.T.). A.S. is a Leukemia and Lymphoma Society Scholar in Clinical Research.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201100194: full synthetic protocols, characterization of intermediate compounds, whole-cell tumor data, fluorescence polarization binding data, and EMSA results.
References
- 1.a) Buettner R, Mora LB, Jove R. Clin. Cancer Res. 2002;8:945–954. [PubMed] [Google Scholar]; b) Darnell JE., Jr Nat. Med. 2005;11:595–596. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]; c) Bowman T, Garcia R, Turkson J, Jove R. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 2.Berishaj M, Gao SP, Leslie K, Al-Ahmadie H, Gerald WL, Bornmann W, Bromberg JF. Breast Cancer Res. 2007;9:R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, Zellweger T, Alanen K, Mirtti T, Visakorpi T, Bubendorf L, Nevalainen MT. Am. J. Pathol. 2008;172:1717–1728. doi: 10.2353/ajpath.2008.071054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke WM, Jin X, Lin H-J, Huang M, Liu R, Reynolds KR, Lin J. Oncogene. 2001;20:7925–7934. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 5.Carro M, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akca H, Tani M, Hishida T, Matsumoto S, Yokota J. Lung Cancer. 2006;54:25–33. doi: 10.1016/j.lungcan.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 7.a) Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. Cell Cycle. 2005;4:1131–1133. doi: 10.4161/cc.4.9.1985. [DOI] [PubMed] [Google Scholar]; b) Schlessinger K, Levy DE. Cancer Res. 2005;65:5282–5834. doi: 10.1158/0008-5472.CAN-05-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bromberg J, Darnell JE., Jr Oncogene. 2000;19:2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 9.Bromberg JF, Wrzeszczynska MH, Devagan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 10.a) Fletcher S, Turkson J, Gunning PT. ChemMedChem. 2008;3:1159–1168. doi: 10.1002/cmdc.200800123. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fletcher S, Drewry J, Shahani V, Page BDG, Gunning PT. Biochem. Cell Biol. 2009;87:825–833. doi: 10.1139/o09-044. [DOI] [PubMed] [Google Scholar]; c) Page BDG, Ball D, Gunning PT. Expert Opin. Ther. Pat. 2011;21:1–19. [Google Scholar]; d) Haftchenary S, Avadisian M, Gunning PT. Anti-Cancer Drugs. 2011;22:115–127. doi: 10.1097/CAD.0b013e328341185b. [DOI] [PubMed] [Google Scholar]
- 11.Siddiquee KAZ, Gunning PT, Glenn M, Katt WP, Zhang S, Schroeck C, Sebti SM, Jove R, Hamilton AD, Turkson J. ACS Chem. Biol. 2007;12:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 12.a) Schröder M, Kroeger K, Volk HD, Eidne KA, Grütz G. J. Leukoc. Biol. 2004;75:792–797. doi: 10.1189/jlb.1003496. [DOI] [PubMed] [Google Scholar]; b) Sehgal PB. Semin. Cell Dev. Biol. 2008;19:329–340. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paillard V, Kaptein A, Saunders M. J. Biol. Chem. 1996;271:5961–5964. doi: 10.1074/jbc.271.11.5961. [DOI] [PubMed] [Google Scholar]
- 14.Faruqi T, Gomez D, Bustelo X, Bar-Sagi D, Reich N. Proc. Natl. Acad. Sci. USA. 2001;98:9014–9019. doi: 10.1073/pnas.161281298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti SM, Hamilton AD, Jove R. J. Biol. Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]; b) Ren Z, Cabell LA, Schaefer TS, McMurray JS. Bioorg. Med. Chem. Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- 16.a) Turkson J, Kim JS, Zhang S, Yaun J, Haung Glenn M, Haura MPE, Sebti SM, Turkson J, Hamilton AD. Mol. Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]; b) Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Bioorg. Med. Chem. Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. ACS Med. Chem. Lett. 2010;1:85–89. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gunning PT, Katt WP, Glenn MP, Siddiquee KAZ, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Bioorg. Med. Chem. Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]; e) Coleman DR, IV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Int. J. Pept. Res. Ther. 2008;14:1–9. doi: 10.1007/s10989-012-9313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Dourlat J, Valentin B, Liu W-C, Garbay C. Bioorg. Med. Chem. Lett. 2007;17:3943–3946. doi: 10.1016/j.bmcl.2007.04.107. [DOI] [PubMed] [Google Scholar]; g) Chen J, Nikolovska-Coleska Z, Yang C-Y, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Bioorg. Med. Chem. Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]; h) Shahani VM, Yue P, Fletcher S, Sharmeen S, Sukhai MA, Luu DP, Zhang X, Sun H, Zhao W, Schimmer AD, Turkson J, Gunning PT. Bioorg. Med. Chem. 2011;19:1823–1838. doi: 10.1016/j.bmc.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Chem. Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]; b) Lin L, Hutzen B, Li P-K, Ball S, Zuo M, DeAngelis S, Foust E, Sobo M, Friedman L, Bhasin D, Cen L, Li C, Lin J. Neoplasia. 2010;12:39–50. doi: 10.1593/neo.91196. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Song H, Wang R, Wang S. J. Lin, Proc. Natl. Acad. Sci. USA. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gunning PT, Glenn M, Siddiquee KAZ, Katt WP, Masson E, Sebti SM, Turkson J, Hamilton AD. Chem-BioChem. 2008;9:2800–2803. doi: 10.1002/cbic.200800291. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Siddiquee KAZ, Gunning PT, Glenn M, Glenn WP, Sebti SM, Jove R, Hamilton AD, Turkson J. ACS Chem. Biol. 2007;12:787–796. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]; f) Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, Laughlin MM, Lawrence NJ, Sebti SM, Turkson J. Proc. Natl. Acad. Sci. USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Fletcher S, Singh J, Zhang X, Yue P, Page BDG, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunning PT. ChemBioChem. 2009;10:1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Shahani VM, Yue P, Haftchenary S, Zhao W, Lukkarila J, Zhang X, Ball D, Nona C, Gunning PT, Turkson J. ACS Med. Chem. Lett. 2011;2:79–81. doi: 10.1021/ml100224d. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.a) Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, Sebti S, Yu H, Jove R. Mol. Cancer Ther. 2004;3:1533–1542. [PubMed] [Google Scholar]; b) Turkson J, Zhang S, Mor LB, Sebti S, Jove R. J. Biol. Chem. 2005;280:32979–32988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- 19.Jones G, Willett P, Glen RC, Leach AC, Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 20.Zhang S, Zhang ZY. Drug Discovery Today. 2007;12:373–381. doi: 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Maegawa T, Fujita Y, Sakurai A, Akashi A, Sato M, Oono K, Sajiki H. Chem. Pharm. Bull. 2007;55:837–839. doi: 10.1248/cpb.55.837. [DOI] [PubMed] [Google Scholar]
- 22.Pandey PN, Purkayastha ML. Synthesis. 1982:876–878. [Google Scholar]
- 23.Fletcher S, Gunning PT. Tetrahedron Lett. 2008;49:4817–4819. [Google Scholar]
- 24.Valerio C, Moulines F, Ruiz J, Blais J-C, Astruc D. J. Org. Chem. 2000;65:1996–2002. doi: 10.1021/jo9914622. [DOI] [PubMed] [Google Scholar]
- 25.Schust J, Berg T. Anal. Biochem. 2004;330:114–118. doi: 10.1016/j.ab.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 26.Wu P, Brasseur M, Schindler U. Anal. Biochem. 1997;249:29–36. doi: 10.1006/abio.1997.2158. [DOI] [PubMed] [Google Scholar]
- 27.Zhao W, Jaganathan S, Turkson J. J. Biol. Chem. 2010;285:35855–35865. doi: 10.1074/jbc.M110.154088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alimirah F, Chen J, Basrawala Z, Xin H, Choubey D. FEBS Lett. 2006;580:2294–2300. doi: 10.1016/j.febslet.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Curtis JE, Minden MD, McCulloch EA. Leukemia. 1989;3:264–269. [PubMed] [Google Scholar]
- 30.Carlesso DAN, Frank, Griffin JD. J. Exp. Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.