

In this report, we confirm that the RhoA-binding protein citron kinase (CIT-K) is required for midbody abscission in late cytokinesis, while it has little or no role in early cytokinesis. Moreover, we show that CIT-K, despite being commonly considered a RhoA effector, promotes midbody stability through RhoA and anillin during late cytokinesis.
Abstract
The small GTPase RhoA plays a crucial role in the different stages of cytokinesis, including contractile ring formation, cleavage furrow ingression, and midbody abscission. Citron kinase (CIT-K), a protein required for cytokinesis and conserved from insects to mammals, is currently considered a cytokinesis-specific effector of active RhoA. In agreement with previous observations, we show here that, as in Drosophila cells, CIT-K is specifically required for abscission in mammalian cells. However, in contrast with the current view, we provide evidence that CIT-K is an upstream regulator rather than a downstream effector of RhoA during late cytokinesis. In addition, we show that CIT-K is capable of physically and functionally interacting with the actin-binding protein anillin. Active RhoA and anillin are displaced from the midbody in CIT-K-depleted cells, while only anillin, but not CIT-K, is affected if RhoA is inactivated in late cytokinesis. The overexpression of CIT-K and of anillin leads to abscission delay. However, the delay produced by CIT-K overexpression can be reversed by RhoA inactivation, while the delay produced by anillin overexpression is RhoA-independent. Altogether, these results indicate that CIT-K is a crucial abscission regulator that may promote midbody stability through active RhoA and anillin.
INTRODUCTION
Cytokinesis is the final stage of the cell division cycle that physically separates the two daughter cells at the end of mitosis (Glotzer, 2005; Barr and Gruneberg, 2007). During anaphase, positional cues emanating from the central spindle and/or from astral microtubules drive the assembly of an equatorial ring of actin and myosin that constricts and furrows the plasma membrane (Barr and Gruneberg, 2007; Pollard, 2010). When the cleavage furrow meets the central spindle, the two daughter cells remain connected by a cytoplasmic bridge that contains the midbody, a specialized structure formed at the interdigitating plus ends of central spindle microtubules (Glotzer, 2005; Barr and Gruneberg, 2007). Finally, the midbody is resolved by an active process known as abscission, which requires the dismantling of actin filaments and specific membrane trafficking events (Barr and Gruneberg, 2007; Schiel and Prekeris, 2010). The tetraploid state that results from cytokinesis failure can lead to chromosomal instability and tumorigenesis (Fujiwara et al., 2005; Caldwell et al., 2007; Li et al., 2010; Sagona and Stenmark, 2010). The small GTPase RhoA is a master component of cytokinesis control mechanisms and plays essential roles in cytokinesis throughout eukaryotes (Piekny et al., 2005). In animal cells, active RhoA is absolutely required for the initial stages of cytokinesis, promoting formation and stabilization of the cleavage furrow through the control of actin polymerization (Piekny et al., 2005). In addition, it is also essential for driving contraction of the actin ring, through the stimulation of myosin light chain (MLC) phosphorylation (Werner and Glotzer, 2008). In contrast, once the midbody has formed, RhoA must be inactivated, since an excess of active RhoA may inhibit abscission (Morin et al., 2009). Thus, to ensure the orderly progression of cytokinesis until the final cut, animal cells must tightly control in time and space the cortical localization of RhoA and its activation state, which impinges on the activity of several effector proteins (Piekny et al., 2005).
The scaffold protein anillin (Field and Alberts, 1995; Oegema et al., 2000) is one of the most crucial partners of RhoA during cytokinesis and plays a fundamental role in the assembly and stabilization of the contractile ring by interacting with RhoA, septins, F-actin, myosin II, and mDia2 (Kinoshita et al., 2002; Straight et al., 2005; Hickson and O’Farrell, 2008; Piekny and Glotzer, 2008; Watanabe et al., 2010). During anaphase, anillin is recruited to the equatorial cortex (D’Avino et al., 2008) by at least two RhoA-dependent mechanisms (Hickson and O’Farrell, 2008) involving an interaction with F-actin or with the plus ends of microtubules, respectively. Anillin depletion results in cleavage furrow instability, leading to characteristic cytoplasmic oscillations and preventing midbody formation (Straight et al., 2005; Zhao and Fang, 2005). In addition to this early function, a late role for anillin in cytokinesis has been proposed. Indeed, in Drosophila S2 cells, anillin depletion leads to a late phenotype characterized by excessive membrane blebbing around the midbody and abscission failure (Somma et al., 2002). However, this possibility has not been confirmed so far in mammalian cells, due to the strong early phenotype caused by anillin inactivation in this context (Hickson and O’Farrell, 2008; Piekny and Glotzer, 2008). Interestingly, the anillin phenotype observed in anillin-depleted Drosophila cells is very similar to the phenotype caused by depletion of citron kinase (CIT-K; Naim et al., 2004), a conserved cytokinesis protein that binds active-RhoA (Di Cunto et al., 1998; Madaule et al., 1998). In mammalian cells, CIT-K localizes at the cleavage furrow through a RhoA-dependent mechanism (Eda et al., 2001) and concentrates in the midbody at later stages. Studies in CIT-K knockout mice (Di Cunto et al., 2000) and rats (Liu et al., 2003) would indicate that, in vivo, the protein is absolutely required for cytokinesis only in specific cell types. However, studies performed in HeLa cells by overexpressing CIT-K mutants (Madaule et al., 1998) or by RNA interference (RNAi)-based knockdown (Gruneberg et al., 2006) are consistent with a more general role for CIT-K in mammalian cytokinesis. It was initially proposed that mammalian CIT-K may act during cleavage furrow ingression, regulating the levels of di-phosphorylated MLC downstream of RhoA (Eda et al., 2001; Yamashiro et al., 2003; Matsumura, 2005). However, more recent observations suggest that, even in mammals, CIT-K could be specifically involved in the latest stages of cytokinesis (Gruneberg et al., 2006; Neumann et al., 2010). The similarity of their RNAi phenotypes in Drosophila cells suggested that CIT-K and anillin could functionally interact (Naim et al., 2004), but this possibility has not yet been investigated. In addition, although it is still commonly believed that CIT-K is a RhoA effector, studies performed in both Drosophila (Dean and Spudich, 2006) and mammalian cells (Matsumura, 2005) indicate that it has little or no role in regulating MLC phosphorylation in vivo.
In this report, we address in more detail the dynamics of cytokinesis failure in CIT-K-depleted or overexpressing mammalian cells. Moreover, we set out to analyze the functional relationships among CIT-K, RhoA, and anillin in late cytokinesis of HeLa cells.
RESULTS
Appropriate levels of CIT-K are necessary for abscission
CIT-K has been shown to be involved in cytokinesis control on the basis of both in vitro (Madaule et al., 1998; Naim et al., 2004; Gruneberg et al., 2006) and in vivo (Di Cunto et al., 2000; D’Avino et al., 2004; Naim et al., 2004) evidence. However, the previous studies did not directly clarify when CIT-K is required during mammalian cell cytokinesis and by which mechanisms it may act. To start resolving these issues, we analyzed cell division in HeLa cells depleted of endogenous CIT-K, using a previously described small interfering RNA (siRNA) sequence (Gruneberg et al., 2006). CIT-K is efficiently depleted 48 h after transfection (Figure 1A). As expected, CIT-K depletion significantly increased the formation of multinucleated cells (37.12% ± 1.51%) compared with cells transfected with control siRNA (5.51% ± 0.79%). To assess which stages of cytokinesis are affected by CIT-K depletion, we observed cells 30 h posttransfection using time-lapse video microscopy. In all CIT-K-depleted cells, nuclear division, cleavage furrow formation, cleavage furrow ingression, and midbody formation were indistinguishable from controls (Figure 1A and Supplemental Movies S1–S3). However, the majority of CIT-K-depleted cells were unable to complete cytokinesis. In particular, we observed two distinct abscission phenotypes, which will be referred to as “early fusion” and “late fusion. In early fusion (Figure 1A and Movie S2), soon after cleavage furrow ingression and midbody formation, the two daughter cells exhibit excessive cortical blebbing, after which the midbody regresses, leading to binucleated cells. In late fusion (Figure 1A and Movie S3), after midbody formation, cells do not exhibit membrane instabilities, reattach normally, and remain interconnected by a cytoplasmic bridge for an interval comparable to the interval observed in control cells. However, cells subsequently fail to undergo abscission and fuse back together, resulting in the formation of binucleated cells. Similar results (Supplemental Figure S1A and Movies S4 and S5) were obtained by using, instead of siRNAs, a previously published small hairpin RNA (shRNA)-expressing construct (Camera et al., 2008). To further confirm the specificity of the phenotype detected in HeLa cells, and to address whether it is relevant to the in vivo phenotype that we have previously described (Di Cunto et al., 2000), we analyzed the mitosis of cerebellar granule precursor cells (GPC) in primary culture using time-lapse video microscopy. We chose these cells because they are extremely sensitive to the absence of CIT-K (Di Cunto et al., 2000). GPC prepared from control postnatal day 5 mouse cerebella divided normally under our culture conditions (Figure 1B and Movie S6). In contrast, most of the GPC prepared from littermate CIT-K−/−mice failed cytokinesis, with kinetics and morphology very similar to those observed in CIT-K-depleted HeLa cells (Figure 1B and Movie S7).
FIGURE 1:

CIT-K is require for abscission both in vitro and in vivo. (A) Selected frames from time-lapse series of HeLa cells undergoing cytokinesis. HeLa cells were transfected with control or CIT-K siRNA and observed 30 h posttransfection by time-lapse video microscopy. Top, A control cytokinesis. CIT-K-depleted cells displayed an “early fusion” (middle) and a “late fusion” (bottom) phenotype. At least 50 cells in cytokinesis were analyzed for each condition. For full movies, see Movies S1–S3. The level of endogenous CIT-K and RhoA expression in whole-cell lysates was determined by immunoblotting using the corresponding antibodies. Tubulin was the internal loading control (right). (B) Selected frames from time-lapse series of GPCs undergoing cytokinesis. GPCs were isolated from P5 wild-type (WT) and CIT-K knockout (CIT-K−/−) mice and observed by time-lapse video microscopy 3 h after plating. At least 40 cells in cytokinesis were analyzed for CIT-K−/−mice. For full movies, see Movies S6 and S7.
These results indicate that, even in mammalian cells, CIT-K is required for midbody abscission, but not for the earlier cytokinesis events.
To further understand the function of CIT-K during cytokinesis, we analyzed the effects of CIT-K overexpression on cell division. HeLa cells were transfected with a plasmid encoding a fusion between CIT-K and Cherry fluorescent protein (CIT-K-Cherry), or with Cherry alone. As previously described for the other tagged version of CIT-K (Eda et al., 2001), the overexpressed CIT-K-Cherry protein accumulated during interphase in characteristic cytoplasmic dots, which were dispersed during metaphase. Afterward, CIT-K-Cherry daccumulated at the cleavage furrow and at the midbody (Figure S2 and Movie S8). Interestingly, CIT-K-Cherry overexpression led to a fourfold increase in the frequency of multinucleated cells citron kinase active (CKA)-Cherry 38.08% ± 10.15%; Cherry 10.85% ± 6.38%; p = 0017). As seen by time-lapse video microscopy, ∼35% of the overexpressing cells that initiate mitosis fail to complete cytokinesis. In these cells, anaphase and cytokinesis initiation was normal. However, after cleavage furrow ingression, the cytosol and the DNA of the two daughter cells underwent rapid oscillatory movements, followed by cleavage furrow regression and formation of binucleated cells (Figure 2 and Movie S9). It must be noted that this phenotype, which was previously detected after the expression of mutant forms of CIT-K (Madaule et al., 1998), is strongly reminiscent of the phenotype displayed by anillin-depleted cells (Straight et al., 2005; Zhao and Fang, 2005). In the remaining CIT-K-overexpressing cells, push–pull movements were not detectable, and cytokinesis progressed normally to the midbody stage. However, these cells took a very long time to divide and, in some cases, did not separate at all within the period of observation (10–12 h; Figure 2 and Movie S10). We measured the time from anaphase to abscission of each cell division: the abscission time of CIT-K-overexpressing cells is almost double the time observed in control cells (Figure 2).
FIGURE 2:

Appropriate levels of CIT-K are necessary for abscission. (A) Selected frames from time-lapse series of HeLa cells transfected with either Cherry or CIT-K-Cherry, using the same conditions as in Figure 1. Insets in the first frames represent red fluorescence of the imaged cells. During cytokinesis, CIT-K overexpression leads to both oscillatory movements (top) and delayed abscission (bottom). For full movies, see Movies S9 and S10. (B) Quantification of abscission time of HeLa cells expressing either Cherry or CIT-K-Cherry and not undergoing oscillatory movements during the time-lapse analysis. A total of 30 CIT-K-Cherry and 42 control cells were scored. Error bars represent SEM. Asterisks indicate that the difference is statistically significant using Student’s t test (p < 0.01).
Altogether, our knockdown and overexpression experiments confirm that, even in mammalian cells, the main role of CIT-K is to regulate abscission.
CIT-K is required to maintain anillin and RhoA at the midbody
To establish how CIT-K regulates abscission, we hypothesized that it may affect the localization of critical cytokinesis components. Therefore, we studied the localization of central spindle proteins (RACGAP1, ECT2, Aurora B) and of cleavage furrow proteins (actin, myosin IIB, anillin, and RhoA) in CIT-K-depleted cells 30 h after transfection. In particular, we concentrated on two cytokinesis stages: early midbody, when the midbody is formed but cells are round and have condensed DNA, and late midbody, when the nuclei are re-formed and the two daughter cells have already spread, but are still linked by the cytoplasmic bridge. Actin, myosin IIB, ECT2, RACGAP1, and Aurora B display the expected localizations in both control and CIT-K-depleted cells (Figure S3A). Interestingly, in the cases of RACGAP1 and Aurora B, we did not observe significant differences, even with phosphospecific antibodies recognizing, respectively, S387 and T232 (Figure S3A), two Aurora B target sites that play a major role in cytokinesis (Minoshima et al., 2003; Yasui et al., 2004). In contrast, the localization of anillin was strongly affected. In control cells, anillin concentrated at the cleavage furrow upon anaphase onset and localized prominently to the midbody-surrounding cortex in cytokinesis. At later stages, anillin localized in clusters along the cytoplasmic bridge (Figure 3A). Conversely, in CIT-K-depleted cells, after accumulating normally at the cleavage furrow, anillin displayed an abnormal distribution. Indeed, at the early midbody stage, CIT-K depletion led to a dramatic increase of cells showing an extended anillin signal in the midbody (68% ± 6.75%), compared with the control cells (18.64% ± 2.72%; Figure 3A). Even more strikingly, at the late midbody stage, anillin was completely lost from the cytoplasmic bridge in 67.18% of CIT-K-depleted cells (Figure 3A). Quite unexpectedly, we even observed a similar delocalization phenotype for the active pool of RhoA, revealed by a trichloroacetic acid (TCA)-fixation protocol (Yonemura et al., 2004). In control cells, active RhoA localized exclusively at the midbody (Figure 3B). In contrast, in ∼50% of early midbody cells depleted of CIT-K, although active RhoA was clearly accumulated at the midzone, it formed an asymmetrical and much-less-focused band, and was clearly detectable at the cell cortex, outside the midzone (Figure 3B). At later stages, RhoA became completely undetectable in ∼85% of CIT-K-depleted cells (Figure 3B). Similar results were obtained using a previously published shRNA-expressing construct instead of siRNAs (Camera et al., 2008; Figure S1, B and C).
FIGURE 3:

CIT-K is required to maintain anillin and RhoA at the midbody, whereas RhoA activity is required for anillin but not for CIT-K localization. (A and B) Asynchronous HeLa cells were transfected with control or CIT-K siRNA and analyzed by immunofluorescence 30 h posttransfection. The localization of (A) anillin (ANLN) and (B) active RhoA was determined in cells in early and in late cytokinesis. Anti–α-tubulin antibodies (Tub) and DAPI were used to reveal midbodies and nuclei, respectively. At least 100 cells in cytokinesis in three independent experiments were scored. (C) HeLa cells, blocked at prometaphase with thymidine and nocodazole, were released into fresh medium for 90 min to allow them to progress through mitosis and cytokinesis. Cells were then treated with solvent alone (CTRL) or with a cell-permeable C3 transferase (2 μg/ml) for 1 h and processed for immunofluorescence. Immunofluorescence of late cytokinesis figures revealed the midbody localization of anillin and CIT-K. Cells were counterstained for α-tubulin (Tub) and DAPI to reveal midbodies and nuclei, respectively. At least 150 cells in cytokinesis in four independent experiments were scored.
These results suggest that, during late cytokinesis, CIT-K may stabilize the midbody by promoting the fixation of anillin and of active RhoA.
RhoA activity is required in late cytokinesis for anillin localization but not for CIT-K localization
The delocalization of RhoA and anillin in CIT-K-depleted cells may indicate that CIT-K retains RhoA and anillin at the midbody, or that these proteins can be interdependent during late cytokinesis. To discriminate between these possibilities, we specifically inactivated RhoA in late cytokinesis, treating synchronized HeLa cells with a cell-permeable Clostridium botulinum C3-toxin (Winton et al., 2002). In particular, HeLa cells were synchronized with the thymidine/nocodazole method and were treated with C3 90 min after nocodazole release (Figure 3C). Parallel immunofluorescence analysis indicated that, after this treatment, the majority of cells had passed through mitosis and were in cytokinesis (unpublished data). As expected, C3 treatment strongly decreased the number of late midbody cells in which RhoA could be detected at the midbody (39.25% ± 0.12% vs. 86.99% ± 0.06% in the control cells, unpublished data). Moreover, C3 treatment strongly affected the localization of anillin (Figure 3C). In particular, anillin could not be detected in the majority of C3-treated late cytokinesis cells (54.22% ± 0.18% vs. 16.94% ± 0.13% in control cells).
Strikingly, a CIT-K signal indistinguishable from the signal observed in control cells was detected in all the late cytokinesis we analyzed (Figure 3C), thus indicating that the disappearance of active RhoA from the midbody has no effect on CIT-K localization at this stage. These data indicate that, in late cytokinesis, CIT-K is necessary to retain at the midbody both RhoA and anillin, while active RhoA is necessary only to retain anillin. Due to the penetrant early phenotype elicited by anillin depletion, we could not evaluate whether anillin might be required for CIT-K and RhoA localization in late cytokinesis.
CIT-K forms a physical complex with anillin
The results presented in the preceding section, together with the previous suggestion of a possible functional interaction between CIT-K and anillin (Naim et al., 2004), prompted us to ask whether the two proteins may physically interact in dividing cells. Moreover, we wanted to address whether an eventual interaction could be modulated during the cell cycle. To this end, HeLa cells were synchronized at the G1/S boundary by a thymidine block, released into fresh media, and harvested hourly. The time window for mitosis was identified by the levels of cyclin B. As expected (Liu et al., 2003; Zhao and Fang, 2005), Western blot analysis indicated that levels of CIT-K and anillin are regulated throughout the cell cycle, peaking in mitosis and dropping at G1. A reciprocal coimmunoprecipitation of the two endogenous proteins was detected throughout mitosis. The association was particularly obvious 11 h after release, that is, when levels of cyclin B decreased and cells exited mitosis (Figure 4A).
FIGURE 4:

CIT-K forms a physical complex with anillin. (A) HeLa cells were synchronized at the G1/S boundary by a thymidine block, released into fresh media, and harvested every hour. Left, Total cell extracts from asynchronous cells (asy) or from the indicated time points were analyzed by Western blotting to monitor the amount of CIT-K and anillin around the time of mitosis exit (corresponding to 11 h, as shown by cyclincyclin B decrease). Vinculin was used as internal loading control. Right, Total extracts from the indicated time points were immunoprecipitated with anti-CIT-K or anti-anillin antibodies. Western blotting with the indicated antibodies revealed reciprocal coimmunoprecipitation of the endogenous proteins. (B) The indicated GFP-anillin fusion constructs were cotransfected with Myc-tagged CITK-expression plasmid in HEK293T for 48 h. Immunoprecipitations were then performed from total cell lysates using anti-GFP antibody, and Western blotting was used to reveal the immunoprecipitated proteins for the GFP and Myc epitopes. Conserved regions and previously described domains are indicated: *, 2 × NLS sequences; My, myosin-binding domain (aa 146–258); Ac, actin-binding and -bundling region (aa 258–371); AHD, anillin homology domain (aa 608–943); PH domain (aa 943–1087). (C) The indicated Myc-CIT-K fusion constructs were cotransfected with GFP-anillin, as in (B). Immunoprecipitations and Western blotting were performed as in (B). Conserved regions and previously described domains are indicated: KD, Ser/Thr kinase domain; Coiled-coil, coiled-coil region; RhoBD, Rho-binding domain; ZF domain; PH domain; CMG; SH3B domain; PDZB domain. In every case, the results shown are representative examples of at least three different experiments.
To determine which anillin domains are involved in the interaction with CIT-K, we tested several green fluorescent protein (GFP)–anillin constructs for their ability to bind cotransfected CIT-K. Interestingly, we found that the interaction was strongly decreased by deletion of the first 100 amino acids of anillin (Figure 4B), which contain the nuclear localization signal (NLS). Moreover, the interaction was completely lost with a construct in which the whole anillin N-terminal region, including not only the NLS but also the myosin- and the actin-binding domains, was deleted. Conversely, we found that the N-terminal region is the minimal portion of anillin sufficient for binding to CIT-K (Figure 4B). Similar experiments, performed with wild-type anillin and CIT-K mutants, revealed that the N-terminal half of CIT-K (containing the kinase domain and the coiled-coil region) is not capable of interacting with anillin. Instead, we found a strong association between anillin and the C-terminal portion of CIT-K, which contains several protein–protein interaction domains. However, constructs lacking either the Rho-binding domain, the Citron, MRCK, Genegis-Khan homology domain (CMG) domain (CMG), the proline-rich, putative SH3-binding domain (SH3B), and the PDZ-binding domain (PDZB), or the zinc finger (ZF) and pleckstrin homology (PH) domains, were still able to bind to anillin (Figure 4C). Therefore an extensive C-terminal region of CIT-K and the N-terminal region of anillin are responsible for their interaction.
CIT-K overexpression affects the localization of anillin and of active RhoA
Considering that the knockdown of CIT-K impairs the retention of anillin and of active RhoA at the midbody and that CIT-K may form a physical complex with both proteins, a simple explanation for the overexpression phenotypes we observed could be that high levels of CIT-K may dominantly interfere with localization of anillin and RhoA. To address this possibility, we first studied the localization of endogenous anillin and of active RhoA in CIT-K-overexpressing cells. Interestingly, in interphase cells, both molecules were strongly recruited to the characteristic CIT-K clusters (Figure 5A). In particular, while anillin showed a diffuse nuclear localization in control cells, in CIT-K-overexpressing cells, the nucleus was mostly negative and the anillin signal was detected in the CIT-K dots, which in the majority of cases resided in the cytoplasm (Figure 5A). On the other hand, not only was the acid-fixable pool of RhoA concentrated in the CIT-K clusters, but the overall intensity of its signal was strongly increased in the CIT-K-overexpressing cells (Figure 5A). These results indicate CIT-K can efficiently recruit both anillin and RhoA, and it can stabilize RhoA in an acid-fixable form. To address whether CIT-K overexpression may alter the localization of anillin and RhoA during cytokinesis, we used dynamic fluorescence microscopy to analyze HeLa cells cotransfected with CIT-K-Cherry and with either GFP-anillin or Caenorhabditis elegans (ce) GFP-RhoA. The localization of the latter protein in dividing mammalian cells faithfully represents the localization of endogenous active RhoA (Yuce et al., 2005). As in the case of the endogenous proteins, overexpressed GFP-anillin and GFP-RhoA accumulated in the CIT-K clusters during interphase. Moreover, when the cells started to round up at the beginning of mitosis, the clusters of overexpressed proteins were almost completely disassembled (Figures 5B and S4 and Movies S11 and S12). In cells that underwent oscillatory movements, CIT-K-Cherry displayed an asymmetrical distribution from the beginning of mitosis, with prominent crescents already being detectable at the metaphase stage (Figure 6B and Movie S11). Interestingly, in these cells, GFP-anillin displayed essentially the same localization of CIT-K-Cherry throughout mitosis (Figure S4 and Movie S12), while GFP-RhoA behaved differently at the earliest stages. Indeed, GFP-RhoA showed a diffuse localization during metaphase and started to enrich at the midzone during anaphase. However, when cells started to oscillate, even GFP-RhoA became colocalized with the CIT-K asymmetrical enrichments (Figure 6B and Movie S11).
FIGURE 5:

CIT-K overexpression affects the localization of anillin and of active RhoA. (A) HeLa cells were transfected with either Cherry or Cherry-CIT-K. Interphase cells were analyzed by immunofluorescence for localization of the endogenous anillin or active RhoA. (B) Selected frames from time-lapse series of HeLa cells undergoing cytokinesis. HeLa cells were cotransfected with GFP-ceRhoA and CIT-K-Cherry and observed 30 h posttransfection by time-lapse video microscopy. For full movie, see Movie S11. (C) HeLa cells in late cytokinesis, expressing either Cherry or CIT-K-Cherry, were analyzed for the localization of active RhoA by immunofluorescence 30 h posttransfection. Nuclei were counterstained with DAPI.
FIGURE 6:
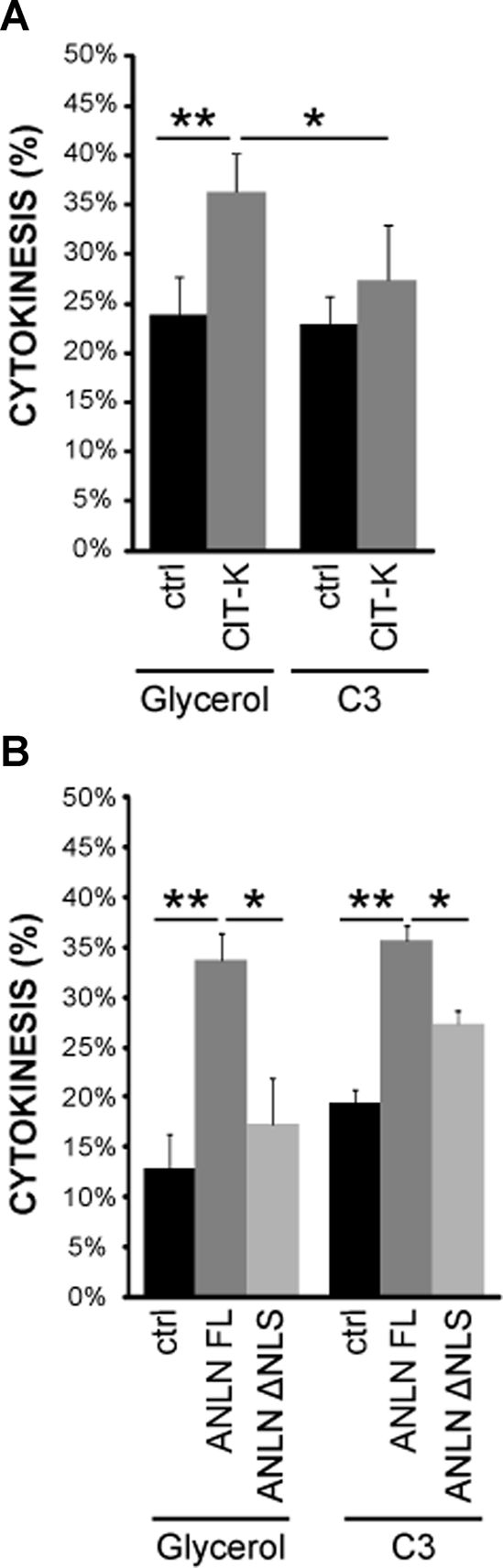
CIT-K or anillin overexpression delays abscission. (A) HeLa cells expressing either Cherry or CKA-Cherry were synchronized with the thymidine/nocodazole method, treated with vehicle or with C3 transferase protein 2 h after release, and analyzed after one additional hour. Slides were then scored for the fraction of telophase cells, as determined by DAPI and α-tubulin staining and fluorescence microscopy. In every case, error bars represent SEM. Asterisks indicate that the difference is statistically significant using Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001). (B) HeLa cells were transfected with GFP, anillin (ANLN)-FL, or anillin-ΔNLS and analyzed as in (A).
On the other hand, in the cotransfected cells that formed a midbody, CIT-K overexpression did not dramatically change the distribution of GFP-anillin and of GFP-RhoA throughout mitosis and cytokinesis. Nevertheless, in analyzing the localization of the endogenous proteins during late cytokinesis, we observed that CIT-K overexpression induced a stronger and broader accumulation of RhoA-GTP at the midbody (Figure 5C).
These data show that CIT-K overexpression is able to perturb anillin and RhoA localization not only during cytokinesis but also during interphase. Moreover, they suggest that CIT-K overexpression could cause abscission delay through a prolonged accumulation of active RhoA at the midbody.
Functional interaction between CIT-K, anillin, and RhoA in late cytokinesis
The data presented in the preceding section strongly suggest that CIT-K, RhoA, and anillin stabilize the midbody in late cytokinesis, with CIT-K and anillin working upstream and downstream of RhoA, respectively. To test this model, we treated late cytokinesis cells overexpressing CIT-K or anillin with a cell-permeable C3 toxin and evaluated their kinetic of cytokinesis exit. As expected, under basal conditions, the percentage of cytokinesis was significantly increased by CIT-K-Cherry overexpression 180 min after nocodazole release (Figure 6A). Interestingly, the carboxy-terminal region of CIT-K, which localizes to the midbody (Figure S5A), was even more effective than the wild-type protein in this assay (Figure S5B). Importantly, the overexpression of GFP-anillin results in a similar abscission delay (Figure 6B), thus confirming that anillin may play a role in late cytokinesis. However, C3-toxin treatment revealed that the delay induced by CIT-K and by its C-terminal region are sensitive to RhoA inactivation (Figures 6A and S5B), while the anillin-induced delay is independent from RhoA activity (Figure 6B). To address whether the interaction between CIT-K and anillin is functionally relevant, we investigated the role of the N-terminal domain of anillin. Overexpression of this domain fused to GFP had no dominant effects on cytokinesis, presumably because the protein is rapidly concentrated into the nucleus and does not localize to the midbody after anaphase (Figure S5C). Conversely, compared with wild-type anillin, the Δ-NLS anillin mutant, which has a strongly reduced CIT-K-binding ability (Figure 4C), still localized to the midbody, although it also showed a diffuse cytoplasmic localization (Figure S5C). Interestingly, although this mutant can rescue the early phenotype induced by anillin depletion (Piekny and Glotzer, 2008), its overexpression had no effect on cytokinesis exit, either under basal conditions or upon C3 treatment (Figure 6B). Altogether, these results strongly suggest that CIT-K could stabilize anillin at the midbody both through RhoA and through a physical interaction with the anillin N-terminal region.
CIT-K regulates the activation state of RhoA in late cytokinesis
Although CIT-K is commonly considered a downstream effector of RhoA, the phenotypes we have observed with CIT-K depletion and overexpression would be rather consistent with the idea that it may regulate RhoA activity at the midbody. To address this hypothesis, we measured the levels of GTP-RhoA in synchronized HeLa cells in which CIT-K was either down-regulated or overexpressed. More precisely, we measured the levels of active RhoA 180 min after nocodazole release, because at this time most cells were in late cytokinesis. Importantly, as shown in Figure 7A, active RhoA was significantly reduced in cells depleted of CIT-K. Conversely, overexpressed wild-type CIT-K and CIT-K C-terminal region significantly increased the levels of active RhoA (Figure 7B). Interestingly, the RhoA-binding domain was necessary for RhoA stimulation in the latter assay; neither the N-terminal region of CIT-K nor a mutant deleted of the Rho-binding domain elicited significant changes. Finally, we addressed whether CIT-K could modulate the levels of GTP-RhoA through the stimulation of a guanine nucleotide exchange factor (GEF) activity or through the inhibition of a GAP activity. To this end, we measured the total Rho-GEF and the total Rho-GAP activities of lysates obtained from cells in late cytokinesis after treatment with control or with CIT-K siRNAs. As shown in Figure 7C, CIT-K depletion dproduced a significant decrease of the total Rho-GEF activity. Moreover, Figure 7D shows that CIT-K RNAi even produced a significant decrease in the total RhoGAP activity. Despite these differences, we did not detect significant changes in the total levels of the GEFs and GAPs known to play a role in cytokinesis, such as ECT2 (Yuce et al., 2005), GEF-H1 and its activated form (pS885; Birkenfeld et al., 2007), p190RhoGAP (Mikawa et al., 2008), and RACGAP1 and its activated form (pS387; Minoshima et al., 2003; Figure S3B). Moreover, in the cases we could evaluate with the reagents available, we did not detect significant changes in their localization to the midbody (Figure S3A). The implications of these results are discussed in the following section.
FIGURE 7:

CIT-K overexpression affects RhoA activation state. (A) HeLa cells were transfected with control or with CIT-K siRNA and synchronized with the thymidine/nocodazole method. Three hours after nocodazole release, active RhoA was measured by G-LISA assay (Cytoskeleton). RhoA activation was expressed as percent activation relative to control siRNA (set to 100%). (B) HeLa cells were transfected with Myc, CIT- K-Myc, or the indicated Myc-CIT-K fusion constructs and were processed as in (A). (C) HeLa cells were transfected with control or CIT-K siRNA and synchronized with the thymidine/nocodazole method. Cells were lysed 3 h after nocodazole release. Cell extracts were incubated with [3H]GDP-loaded RhoA in the presence of free GTP. After 15 min of incubation, aliquots were taken in triplicate, and the amount of bound [3H]GDP was measured. This parameter is inversely related to the RhoA-GEF activity. (D) Cell extracts prepared as in (C) were incubated with [3H]GTP-loaded RhoA in the presence of free GTP. After 15 min of incubation, aliquots were taken in triplicate, and the amount of bound [3H]GTP was measured. This parameter is inversely related to the RhoA-GAP activity. In both (C) and (D), the level of nucleotide-binding in the presence of lysis buffer was set to 100%. In every case, error bars represent SEM. Asterisks indicate that the difference is statistically significant using Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001).
DISCUSSION
In this report, we have provided conclusive evidence that, even in mammalian cells, the main role of CIT-K in cytokinesis is to regulate abscission. Indeed, our dynamic microscopy analysis has shown that CIT-K knockdown has no detectable effects on cleavage furrow formation, on cleavage furrow ingression, and on midbody formation, but results in midbody instability (Figure 1A). Remarkably, the same phenotype was observed with a dramatically increased penetrance in proliferating cerebellar GPC cultured from CIT-K−/− mice, thus confirming its in vivo relevance (Figure 1B). Conversely, the overexpression of CIT-K may lead to the formation of an overstable midbody, which may result in abscission delay (Figure 2).
Our conclusions are fully consistent with previous loss-of-function experiments performed both in HeLa cells (Neumann et al., 2010) and in Drosophila (Naim et al., 2004).
On the basis of the available knowledge, we cannot completely exclude the possibility that CIT-K may act at even earlier stages, but we consider it quite unlikely that this may be of great physiological relevance. First of all, the early anillin-like cytokinesis phenotype obtained by overexpressing wild-type CIT-K (the present study; Figure 2) or some mutant variants of it (Madaule et al., 1998) is not a proof that CIT-K acts early in cytokinesis, but indicates only that it may dominantly interfere with early cytokinesis events. Secondly, although it has been suggested that CIT-K might di-phosphorylate MLC during furrow ingression (Yamashiro et al., 2003; Matsumura, 2005), this concept was only based on in vitro evidence. In contrast, the in vivo analysis of myosin phosphorylation in CIT-K knockout mice revealed that, if anything, MLC phosphorylation is increased, rather than decreased, in the neuronal precursors of mutant mice (Matsumura, 2005). The accumulation of myosin at the cell equator during cytokinesis and its functional modulation through MLC phosphorylation are very robust processes, probably controlled through both RhoA-dependent and -independent mechanisms operating in parallel (Uehara et al., 2010). Accordingly, it has been reported that primary hepatocytes obtained from CIT-K knockout rats display remarkably normal furrow ingression, even when Rho kinases (ROCKs) are inactivated by means of a specific chemical inhibitor (Liu et al., 2003), thus implying that other RhoA-dependent and -independent kinases may act in parallel with ROCKs and CIT-K during early cytokinesis.
How does CIT-K regulate abscission? We here provide evidence indicating CIT-K may stabilize the midbody, principally by preventing anillin loss through the maintenance of active RhoA. The specific interaction of these molecules and the “master” role of CIT-K were supported first of all by localization studies, indicating that anillin and RhoA are specifically lost from the midbody when CIT-K levels are reduced (Figure 3) and that they are prominently recruited by overexpressed CIT-K (Figure 5). Moreover, in addition to the direct physical interactions previously reported for CIT-K and RhoA (Madaule et al., 1995) and for RhoA and anillin (Piekny and Glotzer, 2008), we found that CIT-K and anillin form a physiological complex during mitosis exit that is detectable by reciprocal coimmunoprecipitation (Figure 4). The idea that CIT-K works upstream of RhoA in late cytokinesis is in apparent contrast with the studies performed on early cytokinesis (Eda et al., 2001; Yamashiro et al., 2003), but it is well supported by our data. Indeed, while CIT-K loss strongly affects active RhoA localization and levels (Figures 3B and 7, A and B), the inactivation of RhoA does not affect CIT-K localization (Figure 3C). Moreover, in cells that undergo early midbody failure with oscillatory movements due to CIT-K overexpression, the first abnormality is the formation of a CIT-K crescent, which is followed by the displacement of active RhoA from the midzone (Figure 5B and Movie S11). Finally, the inactivation of RhoA is capable of rescuing the cytokinesis delay elicited by CIT-K overexpression (Figures 6A and S5B).
On the other hand, our data indicate that anillin works downstream of CIT-K and RhoA. Indeed, its normal localization to the midbody is affected both by CIT-K loss and by RhoA inactivation (Figure 3, A–C). In addition, anillin overexpression is capable of delaying abscission as well as CIT-K, but this property is insensitive to RhoA inactivation (Figure 6B). Although the binding to RhoA is clearly important, the full stabilization of anillin to the midbody may also require an additional interaction with CIT-K. Indeed, the overexpression of a mutant lacking the amino-terminal nuclear localization domain, not involved in RhoA binding and fully functional in early cytokinesis (Piekny and Glotzer, 2008), but strongly contributing to the interaction with CIT-K (Figure 4B), is not capable of delaying abscission (Figure 6B).
The simplest model to reconcile our data with the previous literature is that, during anaphase, CIT-K and anillin are recruited to the midzone by active RhoA, independently or as a preassembled complex. In this phase, CIT-K may weakly contribute to stabilize the cleavage furrow, in concert with ROCKs and other kinases, by phosphorylating MLC and other substrates. However, after being concentrated at the midbody, CIT-K becomes essential for its stabilization, through the stabilization of RhoA and anillin. Finally, a change in CIT-K activity or in its scaffolding capabilities, determined by the eventual fulfilling of checkpoint mechanisms, may lead to RhoA inactivation, anillin loss, and abscission. With respect to this model, we find it particularly suggestive that the yeast homologues of anillin have been implicated in the so-called “no-cut pathway,” in which they act downstream of Aurora B (Norden et al., 2006). Since a similar mechanism linking Aurora B to actin persistence has also been recently demonstrated in mammalian cells (Steigemann and Gerlich, 2009), and since Aurora B is not affected by CIT-K inactivation, it is conceivable that the CIT-K/RhoA/anillin pathway that we have here described may play a role in preventing premature abscission.
Our knockdown and the overexpression studies indicate CIT-K can regulate, locally and globally, the activation state of RhoA. However, the precise mechanisms by which it may do so remain to be fully characterized. An obvious possibility would be that CIT-K may regulate the localization and/or the activity of RhoA-GEF and/or RhoA-GAP proteins. However, the data shown in Figure S3 allow us to exclude most of the well-characterized mechanisms. Indeed, the total levels and localization of ECT2 (Yuce et al., 2005), the total levels of GEF-H1 and its activation state (revealed by the phosphoporylation of the S885 residue; Birkenfeld et al., 2007), the total levels of p190RhoGAP (Mikawa et al., 2008), and the total levels and localization of RACGAP1 and its activated form (phosphorylated on S387; Minoshima et al., 2003) are not significantly affected by CIT-K depletion. On the basis of a crude measurement of RhoA-GEF and RhoA-GAP activities, we consider it unlikely that CIT-K may repress a GAP protein, since the total GAP activity of late cytokinesis cells is decreased, rather than increased, by CIT-K depletion. On the other hand, the decrease of total Rho-GEF activity produced by CIT-K inactivation would be consistent with the involvement of a strong Rho-GEF, but its identity remains to be established and its role should be evaluated in light of the increased GAP activity. An alternative mechanism by which CIT-K could increase active RhoA is through a scaffolding function, which could be mediated, for instance, by its RhoA-binding domain. At first glance, this mechanism may seem less intuitive, because it can be argued that an interaction with CIT-K could prevent active RhoA from interacting with anillin and/or with other effector proteins responsible for midbody stability. Nevertheless, the fact that the RhoA-binding domain is necessary for the increase of active RhoA produced by CIT-K overexpression is consistent with this scenario. Since it has been shown that RhoA binds CIT-K and Rhotekin using two distinct regions, while both regions bind ROCKs (Fujisawa et al., 1998), it is conceivable that RhoA and CIT-K may form higher-order complexes with other RhoA effector molecules. This could simultaneously prevent the interaction of RhoA with GAP proteins and make possible the deployment of RhoA function. It will be very interesting to investigate these possibilities in future work.
MATERIALS AND METHODS
Molecular biology
The mammalian expression construct coding for full-length mouse Myc-CIT-K has been previously described (Di Cunto et al., 2003). The Myc-CIT-K mutant constructs were generated from full-length Myc-CIT-K by deleting via restriction digestions the sequences encoding the following amino acids: Ct (0–955), Nt (955–2055), ΔC2 (1746–2055), ΔPHZF (1388–1588). The expression construct coding for CIT-K-Cherry was generated by inserting the Myc-CIT-K cDNA into the pm-Cherry-N1 plasmid (Clontech, Mountain View, CA). The GFP-ceRhoA and GFP-anillin constructs were kindly provided by A. Piekny (Piekny and Glotzer, 2008).
Antibodies and inhibitors
The following antibodies were used: rabbit polyclonal anti-GFP (Abcam, Cambridge, MA), mouse monoclonal anti–Myc 9E10 (Sigma-Aldrich, St. Louis, MO), rabbit polyclonal anti–CIT-K (Di Cunto et al., 2000), mouse monoclonal anti–CIT-K (Transduction Laboratories, BD Biosciences, Franklin Lakes, NJ), rabbit polyclonal anti-anillin for immunofluorescence (Oegema et al., 2000), rabbit polyclonal anti-anillin for immunoprecipitation and Western blot (Bethyl Laboratories, Montgomery, TX), mouse monoclonal anti–cyclin B (Abcam), mouse monoclonal anti-RhoA (Santa Cruz, Santa Cruz, CA), goat polyclonal anti-RACGAP1 (Abcam), rabbit polyclonal anti–p387S-RACGAP1 (Abcam), rabbit polyclonal anti-ECT2 (Santa Cruz), rabbit polyclonal anti–myosin IIB (Sigma), mouse monoclonal anti-GEF-H1 (Abcam), rabbit polyclonal anti-p885S-GEF-H1 (Abcam), mouse monoclonal anti–Aurora B (BD Biosciences), rabbit polyclonal anti–p232T-Aurora B (Rockland Immunochemicals, Gilbertsville, PA), and mouse monoclonal anti-p190RhoGAP (Becton Dickinson). C3 transferase protein was obtained from Cytoskeleton (Denver, CO) and used in accordance with the manufacturer’s instructions.
Cell culture and synchronization
HeLa cells were cultured in Roswell Park Memorial Institute medium (RPMI) supplemented with 10% fetal bovine serum and penicillin/streptomycin at 37°C in 5% CO2. For synchronization, asynchronous cultures were supplemented with 2 mM thymidine (Sigma) and maintained under these conditions for 16 h, then released for 4–6 h in fresh complete medium supplemented with 30 μM deoxycytidine. After release, cells were cultured for a further 16 h in fresh complete medium in the presence of 50 ng/ml nocodazole (Sigma), to block cells at prometaphase. Finally, cells were washed twice with fresh medium and allowed to progress through mitosis/cytokinesis for the times indicated in the different figures.
For immunoprecipitation experiments, HeLa cells were synchronized at the G1/S boundary with 2 mM thymidine (Sigma) for 16 h, released in fresh complete medium supplemented with 30 μM deoxycytidine, and harvested at the times indicated in the different figures.
Cerebellar cell isolation and fractionation
Cerebellar cells were purified using a modification of the procedures described by Wechsler-Reya and Scott (1999).
Transfection and RNAi
In this study, we used the previously described anillin and CIT-K double-stranded RNAs (dsRNAs; Gruneberg et al., 2006; Piekny and Glotzer, 2008); as a control for potential off-target effects, a nontargeting siRNA control was used (Dharmacon, Thermo Scientific, Lafayette, CO). Moreover, we used a pSuper-based shRNA vector, which has been previously described, together with the appropriate control (Camera et al., 2008). HeLa cells plated on six-well plate were transfected using 1 μg of the required plasmid DNA and 3 μl Trans-IT-LT1 transfection reagent (Mirus Bio, Madison, WI), or 6.25 μl of the required siRNA (20 μM) and 2.5 μl Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturers’ instructions.
Immunofluorescence
For the detection of RhoA, cells were fixed in 10% cold TCA for 15 min, as previously described (Yonemura et al., 2004). Methanol fixation at −20°C for 2 min was used for staining RACGAP1, ECT2, and myosin IIB. For the detection of CIT-K, anillin, and actin, cells were fixed in 10% paraformaldehyde. Cells were then permeabilized with 0.1% (vol/vol) Triton X-100 for 10 min, blocked for 30 min at room temperature in blocking solution (phosphate-buffered saline, 5% bovine serum albumin [BSA]), and incubated for 1 h with the primary antibodies. Primary antibodies were detected with anti–rabbit Alexa Fluor 488 or 568 (Molecular Probes, Invitrogen), anti–mouse Alexa Fluor 488 or 568 antisera (Molecular Probes), or anti–goat 594 (Molecular Probes) used at 1:1000 dilution. Polymeric F-actin was detected with phalloidin tetramethylrhodamine isothiocyanate (TRITC; Sigma). To visualize DNA, fixed cells were stained with 4’,6-diamidino-2-dhenylindole (DAPI; Sigma). Images were acquired using an ApoTome system (Zeiss, Germany).
Immunoprecipitation and Western blotting
For all immunoprecipitations, cells were extracted with lysis buffer containing 1% Triton X-100, 120 mM NaCl, 50 mM Tris-HCl (pH 7.5), protease inhibitors (Roche, Mannheim, Germany), and 1 mM phenylmethylsulfonyl fluoride (PMSF). The extracts were then clarified by centrifugation. Antibodies and protein-G–Sepharose beads (Amersham Biosciences, Uppsala, Sweden) were added to lysates and incubated overnight at 4°C. Pellets were washed four times with lysis buffer and analyzed by SDS–PAGE.
For immunoblots, immunoprecipitates or equal amounts of proteins from whole-cell lysates were resolved by reduction with SDS–PAGE and blotted to nitrocellulose filters, which were then incubated with the indicated antibodies and developed using the ECL System (Amersham Biosciences).
Rho activation assay
To detect GTP-RhoA during late stages of cytokinesis, we used a Rho G-LISA assay (Cytoskeleton), according to the manufacturer’s specifications.
GEF activity assay
GDP-loaded RhoA was prepared by incubating 0.5 μg affinity-purified GST-tagged Rac1 in loading buffer (25 mM Tris/HCl, pH 7.5, 1.0 mM dithiothreitol [DTT], 100 mM NaCl, 5 mM EDTA) and 0.5 μCi of [3H]GDP at 30°C for 10 min; this was followed by the addition of 20 mM MgCl2 and incubation at 30°C for 10 min.
For GEF activity measurements, cells were lysed in 100 μl of a buffer containing 25 mmol/l Tris-HCl (pH 7.5), 1% NP40, 100 mmol/l NaCl, 1% glycerol, 10 mmol/l MgCl2, 1 mmol/l PMSF, 1 mmol/l Na3VO4, and protease inhibitors (Roche). Lysates were clarified by centrifugation (12,500 × g, 10 min), and 600 μg of protein extracts were incubated in 200 μl of Rac1GEF buffer (25 mmol/l Tris-HCl, pH 7.5, 1 mmol/l DTT, 1 mg/l ml BSA, 2 mmol/l GTP, 100 mmol/l NaCl, 1 mmol/l MgCl2). To start the reaction, 100 μl of [3H]GDP-loaded recombinant RhoA was added. Samples were analyzed 15 min after incubation at 24°C under shaking. The reaction was stopped by adding 5 ml of ice-cold wash buffer (20 mmol/l Tris-HCl, pH7.5, 100 mmol/l NaCl, 20 mmol/l MgCl2). Samples were filtered through a nitrocellulose membrane (0.22-mm pore size; Millipore, Bedford, MA) under vacuum. The filter was washed twice with 5 ml of wash buffer, air-dried, and placed in plastic vials with 5 ml of scintillation mixture (Ultima Gold; PerkinElmer, Waltham, MA); bound residual radioactivity was measured using a scintillation counter.
Relative GEF activity was represented as the reciprocal function of the residual radioactivity normalized after background subtraction.
GAP activity assay
For GAP activity, GTP-loaded RhoA was prepared by incubating 1 μg of affinity-purified GST-tagged RhoA in loading buffer (25 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.1 mM DTT, 1 mg/ml BSA, 5 μCi of [γ-32P]GTP) at room temperature for 10 min. Thereafter, to block further loading, the MgCl2 concentration was brought to 25 mM and the sample was placed on ice. An aliquot was filtered through a nitrocellulose membrane (0.22-mm pore size; Millipore) under vacuum. The filter was washed twice with 5 ml of wash buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 20 mM MgCl2, 1 mM DTT), air-dried, and placed in plastic vials with 5 ml of scintillation mixture (Ultima Gold; PerkinElmer). Bound radioactivity was measured by using a scintillation counter and considered as total GTP-loaded RhoA (100%). For GAP-activity measurements, HeLa cells were lysed in 100 μl of a buffer containing 25 mM Tris-HCl (pH 7.5), 1% Nonidet P-40, 100 mM NaCl, 1% glycerol, 10 mM MgCl2, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitors (Roche). Lysates were clarified by centrifugation (12,500 × g, 10 min), and 700 μg of protein extract were incubated in 100 μl of GAP buffer (25 mM Tris-HCl, pH 7.5, 1 mM DTT, 1 mg/ml BSA, 2 mM GTP, 100 mM NaCl, 11 mM MgCl2) for 5 min at room temperature. To start the reaction, 130 μl of [γ-32P]GTP-loaded recombinant RhoA was added, and samples were incubated for 5 min at 24°C under shaking. The reaction was stopped by adding 5 ml of ice-cold wash buffer. Samples were filtered through nitrocellulose membranes, washed, and counted as described above; remaining RhoA-GTP was expressed as a percentage of total GTP-loaded RhoA.
Supplementary Material
Acknowledgments
We are grateful to Alisa Piekny (Concordia University, Montreal, QC) and Michael Glotzer (University of Chicago, Chicago, IL) for the anillin expression constructs and to Christine Field (Harvard Medical School, Boston, MA) for providing anti-anillin antibodies. This work was funded by the Italian Ministry of University and Research through the Progetti di Rilevante Interesse Nazionale (PRIN) program and by the Regione Piemonte through the “Converging Technologies” program, Modeling Oncogenic Pathways: from Bioinformatics to Diagnosis and Therapy (BIOTHER) project.
Abbreviations used:
- BSA
bovine serum albumin
- ce
Caenorhabditis elegans
- CIT-K
citron kinase
- CKA
citron kinase active
- CGM
Citron, MRCK, Genegis-Khan homology domain
- DAPI
4′,6-diamidino-2-phenylindole
- dsRNA
double-stranded RNA
- DTT
dithiothreitol
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- GPC
granule precursor cells
- MLC
myosin light chain
- NLS
nuclear localization signal
- PDZB
PDZ-binding
- PH
pleckstrin homology
- PMSF
phenylmethylsulfonyl fluoride
- RNAi
RNA interference
- ROCK
Rho kinase
- SH3B
proline-rich, putative SH3-binding domain
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- TCA
trichloroacetic acid
- ZF
type-2 zinc finger
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-12-0952) on August 17, 2011.
REFERENCES
- Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell. 2007;131:847–860. doi: 10.1016/j.cell.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell CM, Green RA, Kaplan KB. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol. 2007;178:1109–1120. doi: 10.1083/jcb.200703186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camera P, Schubert V, Pellegrino M, Berto G, Vercelli A, Muzzi P, Hirsch E, Altruda F, Dotti CG, Di Cunto F. The RhoA-associated protein Citron-N controls dendritic spine maintenance by interacting with spine-associated Golgi compartments. EMBO Rep. 2008;9:384–392. doi: 10.1038/embor.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avino PP, Savoian MS, Glover DM. Mutations in sticky lead to defective organization of the contractile ring during cytokinesis and are enhanced by Rho and suppressed by Rac. J Cell Biol. 2004;166:61–71. doi: 10.1083/jcb.200402157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Avino PP, Takeda T, Capalbo L, Zhang W, Lilley KS, Laue ED, Glover DM. Interaction between anillin and RacGAP50C connects the actomyosin contractile ring with spindle microtubules at the cell division site. J Cell Sci. 2008;121:1151–1158. doi: 10.1242/jcs.026716. [DOI] [PubMed] [Google Scholar]
- Dean SO, Spudich JA. Rho kinase’s role in myosin recruitment to the equatorial cortex of mitotic Drosophila S2 cells is for myosin regulatory light chain phosphorylation. PLoS One. 2006;1:e131. doi: 10.1371/journal.pone.0000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cunto F, Calautti E, Hsiao J, Ong L, Topley G, Turco E, Dotto GP. Citron rho-interacting kinase, a novel tissue-specific ser/thr kinase encompassing the Rho-Rac-binding protein Citron. J Biol Chem. 1998;273:29706–29711. doi: 10.1074/jbc.273.45.29706. [DOI] [PubMed] [Google Scholar]
- Di Cunto F, Ferrara L, Curtetti R, Imarisio S, Guazzone S, Broccoli V, Bulfone A, Altruda F, Vercelli A, Silengo L. Role of citron kinase in dendritic morphogenesis of cortical neurons. Brain Res Bull. 2003;60:319–327. doi: 10.1016/s0361-9230(03)00058-3. [DOI] [PubMed] [Google Scholar]
- Di Cunto F, et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;28:115–127. doi: 10.1016/s0896-6273(00)00090-8. [DOI] [PubMed] [Google Scholar]
- Eda M, Yonemura S, Kato T, Watanabe N, Ishizaki T, Madaule P, Narumiya S. Rho-dependent transfer of Citron-kinase to the cleavage furrow of dividing cells. J Cell Sci. 2001;114:3273–3284. doi: 10.1242/jcs.114.18.3273. [DOI] [PubMed] [Google Scholar]
- Field CM, Alberts BM. Anillin, a contractile ring protein that cycles from the nucleus to the cell cortex. J Cell Biol. 1995;131:165–178. doi: 10.1083/jcb.131.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa K, Madaule P, Ishizaki T, Watanabe G, Bito H, Saito Y, Hall A, Narumiya S. Different regions of Rho determine Rho-selective binding of different classes of Rho target molecules. J Biol Chem. 1998;273:18943–18949. doi: 10.1074/jbc.273.30.18943. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307:1735–1739. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- Gruneberg U, Neef R, Li X, Chan EH, Chalamalasetty RB, Nigg EA, Barr FA. KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol. 2006;172:363–372. doi: 10.1083/jcb.200511061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson GR, O’Farrell PH. Rho-dependent control of anillin behavior during cytokinesis. J Cell Biol. 2008;180:285–294. doi: 10.1083/jcb.200709005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ. Self- and actin-templated assembly of mammalian septins. Dev Cell. 2002;3:791–802. doi: 10.1016/s1534-5807(02)00366-0. [DOI] [PubMed] [Google Scholar]
- Li J, Wang J, Jiao H, Liao J, Xu X. Cytokinesis and cancer: Polo loves ROCK‘n’ Rho(A) J Genet Genomics. 2010;37:159–172. doi: 10.1016/S1673-8527(09)60034-5. [DOI] [PubMed] [Google Scholar]
- Liu H, Di Cunto F, Imarisio S, Reid LM. Citron kinase is a cell cycle-dependent, nuclear protein required for G2/M transition of hepatocytes. J Biol Chem. 2003;278:2541–2548. doi: 10.1074/jbc.M210391200. [DOI] [PubMed] [Google Scholar]
- Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S. Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature. 1998;394:491–494. doi: 10.1038/28873. [DOI] [PubMed] [Google Scholar]
- Madaule P, Furuyashiki T, Reid T, Ishizaki T, Watanabe G, Morii N, Narumiya S. A novel partner for the GTP-bound forms of rho and rac. FEBS Lett. 1995;377:243–248. doi: 10.1016/0014-5793(95)01351-2. [DOI] [PubMed] [Google Scholar]
- Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005;15:371–377. doi: 10.1016/j.tcb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Mikawa M, Su L, Parsons SJ. Opposing roles of p190RhoGAP and Ect2 RhoGEF in regulating cytokinesis. Cell Cycle. 2008;7:2003–2012. doi: 10.4161/cc.7.13.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoshima Y, et al. Phosphorylation by Aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev Cell. 2003;4:549–560. doi: 10.1016/s1534-5807(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Morin P, Flors C, Olson MF. Constitutively active RhoA inhibits proliferation by retarding G(1) to S phase cell cycle progression and impairing cytokinesis. Eur J Cell Biol. 2009;88:495–507. doi: 10.1016/j.ejcb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim V, Imarisio S, Di Cunto F, Gatti M, Bonaccorsi S. Drosophila citron kinase is required for the final steps of cytokinesis. Mol Biol Cell. 2004;15:5053–5063. doi: 10.1091/mbc.E04-06-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell. 2006;125:85–98. doi: 10.1016/j.cell.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Oegema K, Savoian MS, Mitchison TJ, Field CM. Functional analysis of a human homologue of the Drosophila actin binding protein anillin suggests a role in cytokinesis. J Cell Biol. 2000;150:539–552. doi: 10.1083/jcb.150.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekny A, Werner M, Glotzer M. Cytokinesis: welcome to the Rho zone. Trends Cell Biol. 2005;15:651–658. doi: 10.1016/j.tcb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Piekny AJ, Glotzer M. Anillin is a scaffold protein that links RhoA, actin, and myosin during cytokinesis. Curr Biol. 2008;18:30–36. doi: 10.1016/j.cub.2007.11.068. [DOI] [PubMed] [Google Scholar]
- Pollard TD. Mechanics of cytokinesis in eukaryotes. Curr Opin Cell Biol. 2010;22:50–56. doi: 10.1016/j.ceb.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584:2652–2661. doi: 10.1016/j.febslet.2010.03.044. [DOI] [PubMed] [Google Scholar]
- Schiel JA, Prekeris R. Making the final cut—mechanisms mediating the abscission step of cytokinesis. ScientificWorldJournal. 2010;10:1424–1434. doi: 10.1100/tsw.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somma MP, Fasulo B, Cenci G, Cundari E, Gatti M. Molecular dissection of cytokinesis by RNA interference in Drosophila cultured cells. Mol Biol Cell. 2002;13:2448–2460. doi: 10.1091/mbc.01-12-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigemann P, Gerlich DW. An evolutionary conserved checkpoint controls abscission timing. Cell Cycle. 2009;8:1814–1815. [PubMed] [Google Scholar]
- Straight AF, Field CM, Mitchison TJ. Anillin binds nonmuscle myosin II and regulates the contractile ring. Mol Biol Cell. 2005;16:193–201. doi: 10.1091/mbc.E04-08-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara R, Goshima G, Mabuchi I, Vale RD, Spudich JA, Griffis ER. Determinants of myosin II cortical localization during cytokinesis. Curr Biol. 2010;20:1080–1085. doi: 10.1016/j.cub.2010.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Okawa K, Miki T, Sakamoto S, Morinaga T, Segawa K, Arakawa T, Kinoshita M, Ishizaki T, Narumiya S. Rho and anillin-dependent control of mDia2 localization and function in cytokinesis. Mol Biol Cell. 2010;21:3193–3204. doi: 10.1091/mbc.E10-04-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Werner M, Glotzer M. Control of cortical contractility during cytokinesis. Biochem Soc Trans. 2008;36:371–377. doi: 10.1042/BST0360371. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Dubreuil CI, Lasko D, Leclerc N, McKerracher L. Characterization of new cell permeable C3-like proteins that inactivate Rho and stimulate neurite outgrowth on inhibitory substrates. J Biol Chem. 2002;277:32820–32829. doi: 10.1074/jbc.M201195200. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Totsukawa G, Yamakita Y, Sasaki Y, Madaule P, Ishizaki T, Narumiya S, Matsumura F. Citron kinase, a Rho-dependent kinase, induces di-phosphorylation of regulatory light chain of myosin II. Mol Biol Cell. 2003;14:1745–1756. doi: 10.1091/mbc.E02-07-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Urano T, Kawajiri A, Nagata K, Tatsuka M, Saya H, Furukawa K, Takahashi T, Izawa I, Inagaki M. Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J Biol Chem. 2004;279:12997–13003. doi: 10.1074/jbc.M311128200. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Hirao-Minakuchi K, Nishimura Y. Rho localization in cells and tissues. Exp Cell Res. 2004;295:300–314. doi: 10.1016/j.yexcr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Yuce O, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol. 2005;170:571–582. doi: 10.1083/jcb.200501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WM, Fang G. Anillin is a substrate of anaphase-promoting complex/cyclosome (APC/C) that controls spatial contractility of myosin during late cytokinesis. J Biol Chem. 2005;280:33516–33524. doi: 10.1074/jbc.M504657200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.