

Abstract
An accurate scoring function is a key component for successful protein structure prediction. To address this important unsolved problem, we develop a generalized orientation and distance-dependent all-atom statistical potential. The new statistical potential, generalized orientation-dependent all-atom potential (GOAP), depends on the relative orientation of the planes associated with each heavy atom in interacting pairs. GOAP is a generalization of previous orientation-dependent potentials that consider only representative atoms or blocks of side-chain or polar atoms. GOAP is decomposed into distance- and angle-dependent contributions. The DFIRE distance-scaled finite ideal gas reference state is employed for the distance-dependent component of GOAP. GOAP was tested on 11 commonly used decoy sets containing 278 targets, and recognized 226 native structures as best from the decoys, whereas DFIRE recognized 127 targets. The major improvement comes from decoy sets that have homology-modeled structures that are close to native (all within ∼4.0 Å) or from the ROSETTA ab initio decoy set. For these two kinds of decoys, orientation-independent DFIRE or only side-chain orientation-dependent RWplus performed poorly. Although the OPUS-PSP block-based orientation-dependent, side-chain atom contact potential performs much better (recognizing 196 targets) than DFIRE, RWplus, and dDFIRE, it is still ∼15% worse than GOAP. Thus, GOAP is a promising advance in knowledge-based, all-atom statistical potentials. GOAP is available for download at http://cssb.biology.gatech.edu/GOAP.
Introduction
One key to the solution of the protein folding and structure prediction problems is an accurate energy function. A perfect energy function should have its global minimum free energy in the native state of a protein. In principle, such an energy function can be obtained from quantum mechanics (1). This is only feasible for small molecules and in general is not yet possible for large systems such as a protein in a solvent. Thus, by necessity, current physics-based approaches approximate the energy function using empirical molecular mechanics force fields (2–5) that contain terms associated with bond lengths, angles, torsional angles, van der Waals, and electrostatic interactions (2,3). The parameters associated with these terms are typically obtained by fitting data from quantum mechanical calculations of small peptide fragments and data from experiment (2–4,6). The resulting physics-based potentials often ignore the contribution of multibody interactions beyond pairs.
In practice, physics based potentials are currently less successful than knowledge-based potentials (7). Knowledge-based potentials make use of the growing number of experimental protein structures and can be categorized into contact potentials (8–10) and distance-dependent potentials (11–17) and describe interactions at the residue- or atomic level (8,9,12–19). Whereas most potentials are pairwise-additive, some multibody potentials have been developed (20–23); these are often residue-based (24–30). On the atomic level, orientation dependencies for subsets of atoms have also been investigated (31–33). For example, in dDFIRE, Yang and Zhou (31,34) introduced into DFIRE the orientation dependence of polar atom interactions (treated as dipoles), which includes hydrogen-bonding interactions, and some improvement over DFIRE (14) in refolding the protein terminal regions with secondary structures was observed.
Lu et al. (32) developed an all-atom, orientation-dependent side-chain contact potential. The orientations are defined for blocks of atoms bonded rigidly to the same residue that lie in the same plane. Because the interaction centers are on the block, rather than on individual atoms, this requires that the orientation angles be defined at high resolution to accurately determine the atomic positions within the block. Zhang and Zhang (33) added a side-chain orientation-dependence to their all-atom, distance-dependent potential that uses a reference state generated by random walk theory and showed some improvement over potentials lacking such an orientation dependence. Kortemme et al. (35) developed an orientation-dependent potential specifically for hydrogen bonding.
In this work, because an all-atom distance-dependent potential is likely needed for atomic resolution modeling and refinement, we focus on developing a more accurate knowledge-based, all-atom distance-dependent potential. We generalize the treatment of the orientation-dependence of polar atoms, blocks of atoms, or side chains to all 167 residue-specific, heavy atom types. This generalization is based on the observation that the environment around each atom is anisotropic. This effect is more pronounced for polar atoms and cannot be fully captured by introducing a vectorlike dipole (31) that still requires rotational symmetry around the dipole vector. When residues are hydrogen-bonded, this rotational symmetry might be broken (35). To better characterize their anisotropic environment, a planelike object is introduced for each atom using two of its bonded neighboring atoms and itself. When there is only one bonded neighboring atom (e.g., a backbone oxygen), a next neighboring atom is used (e.g., the Cα atom for the backbone oxygen). The introduction of a plane associated with each heavy atom requires five angle parameters in addition to the distance between interaction centers to describe a pair interaction.
We decompose the potential, named “generalized orientation-dependent all-atom potential” (GOAP), into a distance-dependent and a conditional (dependent on the given distance) angle (orientation)-dependent part. The distance dependence is treated identically as in DFIRE (14), a potential that has performed well across various applications (31,36–40). The angle-dependent part is denoted as GOAP AnGular (GOAP_AG). GOAP naturally integrates orientation-dependent polar atom interactions (34), hydrogen-bonding (35), and side-chain interactions (33). It also captures the geometry of the Cysteine disulfide bond. The only free parameter needed to derive GOAP is the sequence separation cutoff for the orientation-dependent part GOAP_AG that ignores the angular dependence between heavy atoms in residues that are close in sequence. This cutoff does not require any training and is determined from simulating the angle distributions with steric interactions and chain-connectivity (i.e., background distributions when specific pairwise interactions are switched off). GOAP was tested on 11 commonly used decoy sets for native structure recognition (33,41–44,46 and (R. Samudrala, E. Huang, and M. Levitt, unpublished)). We describe the results of this evaluation below.
Method
Definition of the relative orientation of interacting heavy atom planes
In this method, for each heavy (nonhydrogen) atom, we define an associated plane defined by it and the neighboring bonded heavy atoms; see Fig. 1. When an atom has two or more bonded heavy atoms (atom A in Fig. 1, left), any two of the bonded heavy atoms can be used (of course, a consistent selection is always made in deriving and evaluating the energy score). When there is only one bonded heavy atom (atom A in Fig. 1, right, e.g., main-chain oxygen), the next-neighbor, bonded atom is used (e.g., the Cα atom for the main-chain oxygen).
Figure 1.
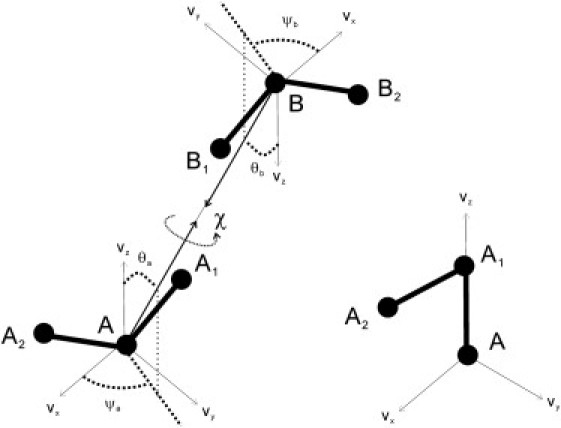
Definition of the plane associated with a given heavy atom and a description of the relative orientation of the planes.
For each plane defined by these three atoms (e.g., A, A1, A2 in Fig. 1, left), we define a local coordinate system using the following unit vectors,
| (1) |
where are the relative vectors from atom A to atoms A1 and A2, respectively; and lie within the plane, and is the normal vector to the plane. When there is only one bonded heavy atom (Fig. 1, right), we change the definition of to . The values and do not change.
To specify the relative position of the two planes associated with the interacting atoms, we require the distance among A and B, rab, and five angles as defined in Fig. 1: The polar angles (θa, ψa) of vector in the local coordinate system of atom A, the polar angles (θb, ψb) of vector in the local coordinate system of atom B, and the torsional angle χ between and around the axis or .
The GOAP potential
The GOAP potential is extracted from known protein structures based on the inverse Boltzmann equation,
| (2) |
where a and b are the atom types of the two interacting atoms, pobs is the probability of the property (rab,θa,ψa,θb,ψb,χ) observed in known protein structures, and pexp is the expected probability of the same property in a reference state without specific interactions (i.e., when E(rab,θa,ψa,θb,ψb,χ) = 0). R is the universal gas constant and T is the absolute temperature at which all the observed states equilibrate. T is usually assumed to be room temperature (∼300 K). In this work, as in others (14,17,19), we consider 167 heavy atom types. Equation 2 can be decomposed into two terms, as
| (3) |
where the first term depends only on the distance rab, and the second term depends on the conditional probabilities pobs(θa,ψa,θb,ψb,χ|rab) and pexp(θa,ψa,θb,ψb,χ|rab). We deal with the two types of terms separately.
The DFIRE potential
For the term that depends only on distance in Eq. 3, we employ the DFIRE (14) reference state for extracting the energy score. The DFIRE reference state is a uniformly distributed set of ideal gas (or equivalently an ideal solution of) points in a finite space. In an unbounded system comprised of an ideal gas (noninteracting point particles), the number of pairwise counts at a given distance is density × 4πr2abΔrab. Here, Δrab is the bin size at the given distance. Proteins are of course finite in size. The finite size effect is taken into account by introducing a scaling factor α < 2; then, the dependence on distance in the reference state becomes density × 4πrαabΔrab. Another important feature of the DFIRE reference state is the assumption that at a large distance cutoff (rcut), the distribution in the reference state equals the observed distribution in real protein structures: Nobs(rcut) = density × 4πrαcutΔrcut.
This assumption not only eliminates the problem of an unphysical nonzero energy at the cutoff distance found in other statistical potentials (17,19), but also determines the unknown density parameter. Therefore, the pairwise counts in the reference state can be written as
| (4) |
By substituting the probabilities in the first term in Eq. 3 with the number of pairwise observations and expected values, we obtain the DFIRE energy function:
| (5) |
The cutoff rcut is set to 15 Å, and α = 1.61 as determined by the best fit of rα to the actual distance-dependent number of ideal gas points in the 1011 finite protein-size spheres that have sizes corresponding to the 1011 nonredundant high-resolution protein structures used for deriving DFIRE (14). Beyond the cutoff distance rcut (i.e., for rab > rcut), EDFIRE(rab) is set to zero.
The GOAP_AG potential
To overcome the problem of insufficient statistics if the angle is treated as nonseparable, we make the assumption for GOAP_AG that the dependence of the potential on the angles θa, ψa, θb, ψb, and χ are independent of each other at the given distance. This gives for the angular contribution
| (6) |
where E(θi|rab) = –RTlog(pobs(θi|rab)/pexp(θi|rab)); E(ψi|rab) = −RTlog(pobs(ψi|rab)/pexp(ψi|rab)), i = a,b; and E(χ|rab) = –RTlog(pobs(χ|rab)/pexp(χ|rab)). Here, pobs(angle|rab) and pexp(angle|rab) with angle = θa,ψa,θb,ψb,χ, are the conditional probabilities of the observed and expected angles at the given distance rab. This assumption of independence of angular distributions has also been made in treatments of H-bonding (36) and in dDFIRE (31,35). In deriving EGOAP_AG, we bin the cos(θa,b), ψa,b, χ-values into Nbin = 12 equally sized bins and assume that the expected probabilities are constant for all bins,
However, this assumption is only good when a suitable sequence separation cutoff is applied for EGOAP_AG. To avoid a zero count, the initial count values for each angle bin are set to 0.1.
The sequence separation cutoff for GOAP_AG
The rationale of applying a sequence separation cutoff s (i.e., two interacting atoms a and b must reside in separate residues i and j satisfying |i − j| ≥ s) for the angle-dependent energy term EGOAP_AG is based on the observation that at small s, the angle distributions are mainly determined by steric interactions and direct chain connectivity rather than by nonbonded, nonsteric interactions. Therefore, the expected distributions (when nonbonded, pairwise interactions are switched off) will not be constant (i.e., independent of angle). It is not trivial to obtain accurate expected angle distributions when they are not constant, which is what happens for small cutoff values of s. Therefore, we introduce the cutoff parameter s into GOAP and ignore the orientation dependence when |i – j|< s (because it cannot be accurately obtained). Then, GOAP can be written as
| (7) |
where i and j are the residue numbers on which the two interacting atoms reside.
To determine the value of s for which the expected angle distribution is essentially constant, we employed a Monte Carlo simulation to simulate the angular distributions allowing only steric interactions (we exclude any nonbonded heavy atom pair atoms within a distance of 3.3 Å from each other) in a 50-residue Alanine peptide with ideal bond lengths and bond angles taken from CHARMM (3). The standard deviations of the binned distributions are then examined. The standard deviation is defined as
where the average 〈〉 is over all bins at given distance, in which Nbin = 12 is the number of bins for each angle parameter. The value σ measures the uniformity of the distribution. The value s should be large enough so that the background distribution is close to uniform, i.e., σ = 0. It should also be small enough so that the contribution of GOAP_AG to the total potential will not be neglected too much (note that σ = 0 when s = ∞). Therefore, a suitable value of s is a compromise of the two effects. In practice, we look at the change of the background standard deviation at each s (the slope of σ(s) versus s curve). A reasonable choice of s is when the change in slope is close to zero (then, an increase of s will result in a negligible change of the standard deviation).
Fig. 2 shows the average standard deviations from the simulation of expected angle distributions on a 50-residue Alanine peptide when only steric interactions are applied. The main-chain dihedral angles of the peptide were randomly sampled for 5,000,000 steps. At each step, all the dihedral angles are set to random values. When no steric clashes are present, the conformation will be used for counting the angle distributions. The angle distributions (normalized summation over bins to be 1 for each angle) at each distance bin (20 bins span from 0 to 15 Å) were obtained. The plot shows the standard deviations (σ) of the distributions averaged over all-atom type pairs and all distance bins. The steric interactions and chain-connectivity most affect the θ-angle and least the χ-dihedral angle. At a small cutoff of s = 3, the distributions deviate the most from uniform. Beyond s = 7, the curves are almost flat with little change. Defining the slope at s as σ(s+1)–σ(s), we find the slope at s = 7 for θ-angle distribution to be −0.013. The slopes at s = 5, s = 6, and s = 8 are −0.045, −0.020, and −0.010, respectively. Therefore, at ∼s = 7, σ starts to change very slowly. In this work, s = 7 is applied to GOAP_AG. For the distance-dependent part, DFIRE potential, we set s = 1, so as to exclude interactions within the same residue.
Figure 2.
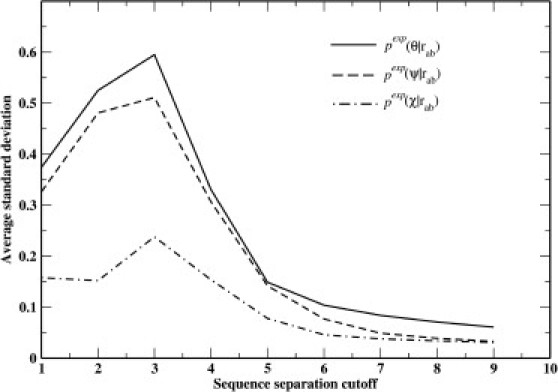
Dependence of standard deviations of the expected distributions on the sequence separation cutoff from a Monte Carlo simulation on a 50-residue Alanine peptide where only steric interactions are allowed.
URL for Protein Structure Library and GOAP
GOAP is obtained using the same 1011 protein structures as in DFIRE (14); the list of structures along with the GOAP potential is available at http://cssb.biology.gatech.edu/GOAP. Using more structures to obtain the potential does not significantly change the performance of GOAP.
Decoy sets and potentials evaluated
Tested decoy sets include the multiple decoy sets from the ‘R’ Us s decoy set at http://dd.compbio.washington.edu/. These sets are the 4state_reduced (42), fisa (41), fisa_casp3 (41), lmds (43), lattice_ssfit (44,47), hg_structal and ig_structal (R. Samudrala, E. Huang, and M. Levitt, unpublished), and ig_structal_hires (45). The MOULDER decoy set (46) is downloaded from http://salilab.org/decoys/. The ROSETTA all-atom decoy set is obtained from http://depts.washington.edu/bakerpg/decoys/, and the I-TASSER set (33) from the Zhang lab is obtained from http://zhanglab.ccmb.med.umich.edu/.
We compare our GOAP potential with the following all-atom potentials: the DFIRE potential (14) that is part of GOAP; the RWplus potential (33) that uses random walk theory for the reference state and includes side-chain orientations; the dDFIRE potential (31,34) that includes polar-polar, polar-nonpolar atom orientations described with vectorlike dipoles; and the OPUS-PSP potential (32) that defines orientations of blocks of side-chain atoms. OPUS-PSP is a contact potential, whereas the others are distance-dependent. The programs dDFIRE, RWplus, and OPUS-PSP are downloaded from the corresponding author's websites. A perfect potential should rank the native structure as the lowest energy structure. The significance of the native structure energy (Enative) is given by its Z-score defined as Z-score = (Enative–Eave)/σE where Eave is the average energy of all decoys and σE is the energy standard deviation of all decoys.
Results
Native structure recognition from decoys
The performance of various potentials on the 11 decoy sets for native structure recognition is compared in Table 1. GOAP achieves the best success rate with 226 out of 278 targets having their native energy as the lowest and the best average Z-score per target. Compared to DFIRE (at 128), RWplus (at 135), and dDFIRE (at 164), GOAP provides for a significant improvement in both success rate and Z-score. Only the performance of OPUS-PSP (at 196) is comparable. Still, our method shows a 15% better success rate compared to OPUS-PSP. All the improvements of GOAP are from the three homology modeling sets (hg_structal, ig_structal, and ig_structal_hires (45)) and the ab initio ROSETTA set.
Table 1.
Performance of different potentials in native structure recognition
| Decoy sets | DFIRE | RWplus | dDFIRE | OPUS-PSP | GOAP | No. of targets |
|---|---|---|---|---|---|---|
| 4state_reduced | 6(−3.48) | 6(−3.51) | 7(−4.15) | 7(−4.49) | 7(−4.38) | 7 |
| fisa | 3(−4.87) | 3(−4.79) | 3(−3.80) | 3(−4.24) | 3(−3.97) | 4 |
| fisa_casp3 | 4(−4.80) | 4(−5.17) | 4(−4.83) | 5(−6.33) | 5(−5.27) | 5 |
| lmds | 7(−0.88) | 7(−1.03) | 6(−2.44) | 8(−5.63) | 7(−4.07) | 10 |
| lattice_ssfit | 8(−9.44) | 8(−8.85) | 8(−10.12) | 8(−6.75) | 8(−8.38) | 8 |
| hg_structal | 12(−1.97) | 12(−1.74) | 16(−1.33) | 18(1.87) | 22(−2.73) | 29 |
| ig_structal | 0(0.92) | 0(1.11) | 26(−1.02) | 20(0.69) | 47(−1.62) | 61 |
| ig_structal_hires | 0(0.17) | 0(0.32) | 16(−2.05) | 14(−0.77) | 18(−2.35) | 20 |
| MOULDER | 19(−2.97) | 19(−2.84) | 18(−2.74) | 19(−4.84) | 19(−3.58) | 20 |
| ROSETTA | 20(−1.82) | 20(−1.47) | 12(−0.83) | 39(−3.00) | 45(−3.70) | 58 |
| I-TASSER | 49(−4.02) | 56(−5.77) | 48(−5.03) | 55(−7.43) | 45(−5.36) | 56 |
| No. total (Z-score) | 128(−1.94) | 135(−2.13) | 164(−2.52) | 196(−2.86) | 226(−3.57) | 278 |
Numbers in parentheses are the average Z-scores of the native structures. More negative is better. Highlighted entries are the best ones in the respective set.
These sets have the common feature that their decoys have more realistic bond lengths and angles than decoys in most other sets. They are relatively hard for conventional methods such as DFIRE and RWplus without fully incorporating orientation dependence. dDFIRE's success rate on the homology modeling sets is comparable to OPUS-PSP, but performs poorest on the ROSETTA set. For the five traditional Decoy ‘R’ Us sets (4stat_reduced, fisa, fisa_casp3, lmds, and lattice_ssfit) that mostly used in the literature (14,17,19,21,33), GOAP recognizes the native energy as lowest for 30 out of 34 targets, whereas DFIRE, RWplus, and dDFIRE all recognize 28, and OPUS-PSP recognizes 31 targets whose native energy is the lowest. It should be noted that OPUS-PSP has a free parameter (the weight of the repulsive Lennard-Jones term) trained on the 4stat_reduced set.
Correlation of energy score with model quality and model selection
Although the ability to assign the native structure as being lowest in energy is the most important characteristic of a good potential, for an energy function to be useful for guiding conformation sampling, it should also have a good correlation with model quality. In Table 2, we compare the performance of different potentials as assessed by both their Pearson correlation coefficient of energy and TM-score (48) and the TM-score of the lowest energy structure. The 112-protein CASP9 (49) target set (models were generated by all CASP9 servers and downloaded from the CASP9 website http://predictioncenter.org/casp9/; most are homology modeling structures) is also included. Here, we use the TM-score (48) instead of the root mean-square deviation of the model to native, because if the majority of the structure is of good quality, the TM-score is insensitive to local substructures that differ significantly from native, whereas root mean-square deviation is quite sensitive to such effects.
Table 2.
Average Pearson's correlation coefficient of energy score with TM-score and average TM-score of selected models
| Decoy sets | DFIRE | RWplus | dDFIRE | OPUS-PSP | GOAP | No. of targets |
|---|---|---|---|---|---|---|
| 4state_reduced | −0.635 0.659 | −0.606 0.667 | −0.693 0.732 | −0.589 0.755 | −0.694 0.818 | 7 |
| fisa | −0.446 0.449 | −0.462 0.434 | −0.461 0.454 | −0.282 0.405 | −0.347 0.475 | 4 |
| fisa_casp3 | −0.243 0.288 | −0.240 0.277 | −0.149 0.309 | −0.095 0.270 | −0.221 0.300 | 5 |
| lmds | −0.118 0.333 | −0.147 0.346 | −0.248 0.364 | −0.091 0.339 | −0.146 0.339 | 10 |
| lattice_ssfit | −0.094 0.247 | −0.097 0.251 | −0.070 0.266 | −0.051 0.248 | −0.058 0.248 | 8 |
| hg_structal | −0.817 0.890 | −0.806 0.891 | −0.796 0.891 | −0.752 0.891 | −0.825 0.889 | 29 |
| ig_structal | −0.785 0.945 | −0.782 0.948 | −0.766 0.948 | −0.779 0.953 | −0.865 0.946 | 61 |
| ig_structal_hires | −0.876 0.947 | −0.879 0.950 | −0.844 0.946 | −0.832 0.946 | −0.885 0.944 | 20 |
| MOULDER | −0.859 0.734 | −0.840 0.745 | −0.881 0.748 | −0.802 0.738 | −0.886 0.771 | 20 |
| ROSETTA | −0.441 0.507 | −0.444 0.505 | −0.393 0.480 | −0.343 0.506 | −0.476 0.511 | 58 |
| I-TASSER | −0.519 0.571 | −0.488 0.577 | −0.525 0.578 | −0.284 0.547 | −0.477 0.567 | 56 |
| CASP9 | −0.604 0.618 | −0.585 0.609 | −0.481 0.593 | −0.448 0.624 | −0.611 0.627 | 112 |
| All average | −0.610 0.669 | −0.599 0.668 | −0.566 0.663 | −0.500 0.670 | −0.626 0.677 | 390 |
| P value∗ | 0.010 | 5.0 × 10−5 | 1.8 × 10−10 | 1.8 × 10−45 | — | |
| P value† | 0.042 | 0.036 | 9.2 × 10−4 | 0.065 | — | |
| No. of targets‡ | 274 | 273 | 269 | 268 | 274 |
Native structures are excluded from all sets. Highlighted entries are the best ones in the respective set. The first number in each cell is the Pearson correlation coefficient; the second number is the TM-score of lowest energy selected model.
Two-sided P value of Student's t-test of the difference of the Pearson's correlation coefficient between GOAP and the given method.
Two-sided P value of Student's t-test of the difference of TM-score of top-ranked model between GOAP and the given method.
Number of targets whose top-rank model has a TM-score to native >0.5.
We find that GOAP gives the best Pearson coefficient of the energy score with TM-score (48) and has the best average TM-scores of the selected models. OPUS-PSP does much worse as assessed by the correlation coefficient, but comes in second in model selection. DFIRE is very close to OPUS-PSP in model selection but does much better than OPUS-PSP in its correlation with TM-score. Because DFIRE is part of GOAP, its good performance in correlation and model selection is passed on to GOAP. Because of the inclusion of the orientation-dependent part GOAP_AG, GOAP performs better than DFIRE; e.g., for the three homology modeling decoy sets, GOAP is >5% better, on average, than the other methods in terms of its correlation with TM-score.
Fig. 3 shows some examples of the correlation of TM-score and GOAP energy. It is noteworthy that for target T0581 in the CASP9 set, a template-free modeling target, only GOAP identifies the single good model with a TM-score = 0.66 (BAKER-ROSETTASERVER_TS4; the next best has a TM-score of 0.36) in the first position (see Fig. 3 d). The ranking of this model by other methods are: fourth in DFIRE and RWplus, third in dDFIRE, and fifth in OPUS-PSP. These results show the advantage of GOAP in selecting the best models.
Figure 3.
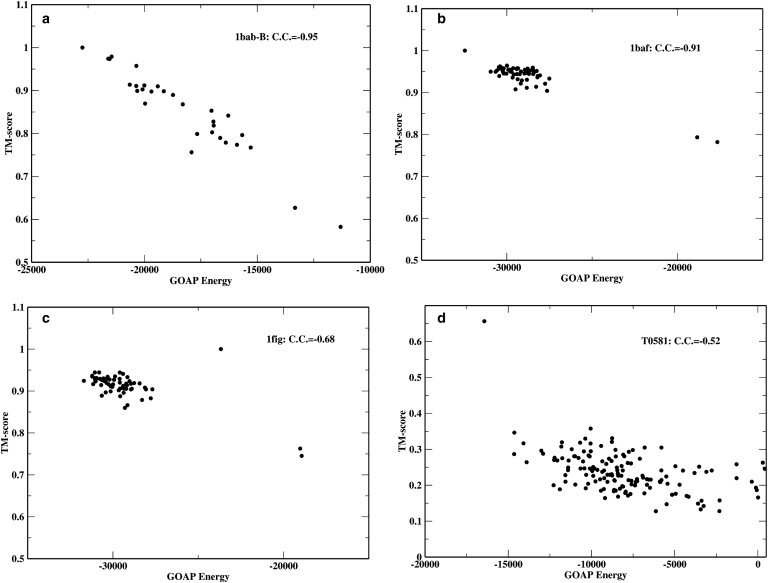
Examples of correlations between GOAP score and TM-score. (a–c) Native structures are included to show their positions in the energy landscapes.
To establish the statistical significance of the small difference between GOAP and other methods, two-sided P values of the Student's t-test of the differences between GOAP and other methods for the Pearson's correlation coefficients and the TM-scores of the lowest energy models are also shown in Table 2. Except for the TM-score difference between GOAP and OPUS-PSP (P value = 0.065), GOAP gives statistically significant (P value < 0.05) better results than all other methods for both TM-score and Pearson correlation. The number of targets whose top-ranked models have a TM-score to native >0.5 are also given; clearly, there is very little difference between methods.
We have shown that GOAP performs much better than other all-atom statistical potentials in native structure recognition and consistently better than those potentials in the correlation of the energy with TM-score of the models and in selecting the best models. In what follows, we shall examine the factors that could contribute to the better performance of GOAP as well as the validity of its approximations.
Effects of sequence separation cutoff and main-chain atoms
The angle-dependent GOAP_AG potential depends on the sequence separation cutoff s. We have suggested that s should not be too small and have chosen a reasonable value s = 7. To show that this choice indeed results in better potential than a smaller, or a larger s (more orientation-dependent energy will be neglected), in Table 3, we give the performance of GOAP with s = 2 and s = 10. Because two of the compared methods RWplus (33) and OPUS-PSP (32) considered orientations using only side-chain atoms, we also give in Table 3 the performance of GOAP with s = 7 (the default value) and evaluated using GOAP_AG for main-chain, side-chain atoms, respectively. Clearly, with a smaller s = 2, or larger s = 10, GOAP's performance is worse than with the default choice s = 7 in native recognition success rate and Z-score (compare Table 3 with Table 1).
Table 3.
Performance of GOAP using a sequence cutoff s = 2, 10, and the default s = 7 and GOAP_AG evaluated only for main-chain or side-chain atoms, respectively
| Decoy sets | s = 2 | s = 10 | Default s = 7 and main-chain atoms only for GOAP_AG | Default s = 7 and side-chain atoms only for GOAP_AG | GOAP | No. of targets |
|---|---|---|---|---|---|---|
| 4state_reduced | 7(−4.34) | 7(−4.15) | 7(−3.95) | 6(−4.25) | 7(−4.38) | 7 |
| fisa | 2(−2.31) | 3(−3.89) | 3(−4.28) | 3(−4.58) | 3(−3.97) | 4 |
| fisa_casp3 | 4(−4.11) | 4(−4.94) | 5(−5.55) | 4(−5.47) | 5(−5.27) | 5 |
| lmds | 7(−4.03) | 6(−3.42) | 7(−3.20) | 6(−2.68) | 7(−4.07) | 10 |
| lattice_ssfit | 8(−10.23) | 8(−7.82) | 8(−9.19) | 8(−9.29) | 8(−8.38) | 8 |
| hg_structal | 21(−2.20) | 22(−2.85) | 18(−2.08) | 18(−2.68) | 22(−2.73) | 29 |
| ig_structal | 38(−1.49) | 45(−1.50) | 45(−1.80) | 8(−0.31) | 47(−1.62) | 61 |
| ig_structal_hires | 18(−2.38) | 18(−2.16) | 19(−2.72) | 6(−0.75) | 18(−2.35) | 20 |
| MOULDER | 19(−3.36) | 19(−3.52) | 19(−2.88) | 19(−3.88) | 19(−3.58) | 20 |
| ROSETTA | 20(−1.28) | 43(−3.81) | 37(−2.76) | 35(−2.95) | 45(−3.70) | 58 |
| I-TASSER | 47(−7.50) | 45(−5.11) | 47(−4.81) | 46(−4.23) | 45(−5.36) | 56 |
| No. total (Z-score) | 191(−3.40) | 220(−3.46) | 215(−3.20) | 159(−2.78) | 226(−3.57) | 278 |
Numbers in parentheses are the average Z-scores of the native structures.
Even worse performance is seen when main-chain atoms are not included in the GOAP_AG energy. Therefore, the contribution to GOAP_AG from main-chain atoms is more important than that from side-chain atoms. However, the sensitivities of decoy sets to the s cutoff and main-chain atom inclusion are different. The most sensitive sets are the homology modeling sets hg_structal, ig_structal, ig_structal_hires, and ROSETTA ab initio set. Thus, proper choice of sequence separation cutoff and inclusion of all atoms in the orientation-dependent energy term are crucial for our method to improve over other orientation-dependent/independent, all-atom potentials.
Examples of orientation dependence
Here, we examine some examples of angle distributions involving polar-polar, polar-nonpolar, and nonpolar-nonpolar atom pairs. To show that it is necessary to consider the orientations of all, not just polar, atoms, and at what distance the effects of orientations are most important, in Fig. S1 a–c (see Supporting Material), we present the average standard deviation of the angle-dependent energy terms (see Eq. 6) of the GOAP potential over all polar-polar, polar-nonpolar, and nonpolar-nonpolar pairs, respectively for 1), E(θ|rab), 2), E(ψ|rab), and 3), E(χ|rab). Polar atoms are nitrogen, oxygen, and sulfur in Cysteine; all other atoms are nonpolar. The standard deviation for the energy term of a given pair at given distance is defined as
| (8) |
where the average 〈〉 is over Nbin = 12 of angle bins. From Fig. S1, we see that all three angles (θ, ψ, and χ) for all three kinds of pairs (polar-polar, polar-nonpolar, and nonpolar-nonpolar) deviate most from uniform at ∼4 Å, but the differences between different kinds of pairs become obvious at ∼6 Å. It is understandable that the differences between different kinds of pairs are larger at distances <4 Å. These results demonstrate that even for nonpolar-nonpolar atom pairs, their full orientation dependence is required. When GOAP is used to calculate the energy scores on the 1011 native protein structures that are used for deriving the GOAP potential, the average DFIRE score per protein is −21,565, whereas the average GOAP_AG score is −19,769. This means that the energy contribution of orientation-dependent part is almost the same as that of the distance-dependent part for a typical protein.
In Fig. S2, we show some specific examples of the angular dependence of polar-polar, polar-nonpolar, and nonpolar-nonpolar pairs. We shall focus mainly on the ψ-dependence because the θ, χ dependences for polar atoms have been investigated in the dDFIRE potential (31,34). Fig. S2 a shows the nonuniform ψ-dependence of the disulfide bond Cys SG-Cys SG at 2.25 Å. The energy has two favorable positions of ±75°. Fig. S2 b shows the ψ-dependence of a typical hydrogen bond (H-bond) between Ala N and Ala O at 2.75 Å. The dip at −105° shows that the ψ-degree of freedom is necessary for accurately describing a H-bond. In the dDFIRE potential (31), polar atoms are represented by a dipole and only θ is defined for each atom. Fig. S2 c shows an example of a polar-nonpolar interaction, Ala O-Ala CB at 3.25 Å. The ψ-dependence of the nonpolar atom Ala CB is shown. The interaction is favored when ψ > 75°. Fig. S2, d–f, shows the θ-, ψ-, and χ-dependences of the Ala CB-Ala CB interactions at 3.75 Å, respectively. Even though this interaction involves only nonpolar atoms, its dependence on all three angles is not uniform. Thus, they are required to describe this typical nonpolar atom interaction accurately.
Effects of orientation dependence on GOAP's performance
The above analysis presented with some observations regarding the orientation dependence of atomic pair interactions. Here, we analyze the contributions of different orientational terms and the overall orientational contribution of the nonpolar-nonpolar interactions. In Table 4, we show the performance of GOAP when the contribution of each of the three types of angle (θ, ψ, and χ) terms is not included and when all angle-dependent terms are not considered for nonpolar-nonpolar pairs. Table 4 shows that the contribution from θ-angle is the most important and from ψ the least. It also shows that, consistent with previous analysis, the orientational dependence from nonpolar-nonpolar interactions contributes somewhat positively to GOAP's performance.
Table 4.
Performance of GOAP when different angular components are turned off
| Decoy sets | Angle θ | Angle ψ | Angle χ | All angle terms for nonpolar-nonpolar | GOAP | No. of targets |
|---|---|---|---|---|---|---|
| 4state_reduced | 7(−4.33) | 7(−4.34) | 7(−4.37) | 7(−4.33) | 7(−4.38) | 7 |
| fisa | 3(−3.86) | 3(−4.57) | 3(−4.00) | 3(−4.21) | 3(−3.97) | 4 |
| fisa_casp3 | 4(−4.94) | 5(−6.01) | 5(−5.20) | 5(−5.33) | 5(−5.27) | 5 |
| lmds | 6(−3.40) | 7(−4.27) | 7(−3.85) | 7(−3.79) | 7(−4.07) | 10 |
| lattice_ssfit | 8(−9.04) | 8(−8.42) | 8(−8.40) | 8(−9.00) | 8(−8.38) | 8 |
| hg_structal | 20(−2.59) | 22(−2.66) | 22(−2.71) | 20(−2.53) | 22(−2.73) | 29 |
| ig_structal | 35(−1.26) | 43(−1.66) | 44(−1.46) | 46(−1.69) | 47(−1.62) | 61 |
| ig_structal_hires | 17(−1.96) | 18(−2.52) | 18(−2.15) | 18(−2.56) | 18(−2.35) | 20 |
| MOULDER | 19(−3.58) | 19(−3.51) | 19(−3.55) | 19(−3.30) | 19(−3.58) | 20 |
| ROSETTA | 41(−3.30) | 43(−3.80) | 40(−3.46) | 40(−3.42) | 45(−3.70) | 58 |
| I-TASSER | 45(−4.95) | 46(−4.94) | 45(−5.07) | 47(−5.28) | 45(−5.36) | 56 |
| No. total (Z-score) | 205(−3.27) | 221(−3.56) | 218(−3.40) | 220(−3.50) | 226(−3.57) | 278 |
Numbers in parentheses are the average Z-scores of the native structures.
The independence of angular distributions
The assumption made in Eq. 6 that all angle distributions are independent is intended to overcome the problem of too few cases that satisfy the joint distribution of five angles at each distance. How well this assumption holds is not yet known. We examine here a typical polar-polar interaction of main-chain N-O pairs using amino-acid nonspecific atom types to increase the statistics of the joint distribution and derive the joint distribution with a larger dataset of 3506 proteins downloaded from http://dunbrack.fccc.edu/PISCES.php (50). The covariance of all angle pairs at different distances is given in Fig. S3, a–c. From these figures, we observe that θN has a relatively stronger covariance with θo at ∼6 Å and 8 Å, whereas, with the other three angles, it shows weaker covariation for all distances (see Fig. S3 a). The covariance of ψN with ψo and χ has a dip or peak at ∼2 Å. Beyond 4 Å, the covariance is weak (see Fig. S3 b). These results indicate that except for a somewhat narrow range of distances, the assumption of independent angular distributions holds reasonably well.
Discussion
In this article, we have improved the description of pairwise atomic interactions by introducing the orientation dependence of all individual heavy atoms. However, to obtain the orientational contribution to the potential, a sequence separation cutoff is needed. The cutoff is the only free parameter and is obtained by Monte Carlo simulation of a noninteracting peptide. We find that inclusion of main-chain atoms has a greater effect on GOAP's performance than the cutoff (see Table 3). This is consistent with the findings in the OPUS-PSP article (32), where the authors reported the results for the decoy sets ig_structal and ig_structal_hires when main-chain block types were included.
The results in Wu et al. (26) (46(−2.79) for ig_structal and 19(−3.03) for ig_structal_hires) are much better than the ones (20(0.693) and 14(−0.768)) we obtained using the downloaded OPUS-PSP program that ignores such main-chain interactions. The main-chain blocks include the main-chain amide and carbonyl groups, and therefore, they take into account the hydrogen-bond interactions. However, when these blocks are included in OPUS-PSP, it only recognizes 24 of 34 native structures in the five Decoys ‘R’ Us sets. Thus, inclusion of main-chain interactions does not necessarily improve the overall performance of OPUS-PSP. The authors of OPUS-PSP(32) suggest that their rigid-body description is not suited for optimizing main-chain hydrogen-bond interactions. Another reason could be that OPUS-PSP defines main-chain blocks that do not depend on specific amino-acid types, whereas in real proteins, there are different preferences of different amino acids for different secondary structures; this feature is included in GOAP.
In testing of the GOAP potential on commonly used decoy sets, we find that GOAP performs better than other all-atom potentials in native structure recognition and is consistently better in terms of the correlation of energy score with model quality as assessed by the correlation of the TM-score to native and in good model selection. The close homology modeling decoys and the ROSETTA ab initio decoys are particularly sensitive to the performance of all-atom potentials. Here, GOAP performs consistently better than other potentials on these decoy sets. Thus, GOAP might prove to be useful in high accuracy protein structure refinement and in ab initio structure prediction, but this remains to be demonstrated. Its application to side-chain modeling and protein design might give better results than the OPUS-PSP potential, because it has atomic resolution and a distance dependence, and it includes main-chain atoms compared to the block resolution, contact nature, and side-chain atom restrictions of the OPUS-PSP potential (51). Applications of GOAP to these areas are under investigation.
GOAP can also be included in possible composite knowledge-based scores like the QMEAN score (52) and that employed by Eramian et al. (53) to develop a more accurate score function for model rank and selection, and for absolute model quality prediction (54,55). These methods integrate different kinds of scores using a machine learning approach or a linear combination with trained weights. Because GOAP does not include short-range (<7 sequence separation) angle correlations, some kind of backbone torsional, angle-dependent, knowledge-based scores as in the QMEAN approach might further enhance its performance in native structure recognition and model selection.
The physical source of the orientation-dependence is the anisotropic nature of the atomic electronic environment that also depends on the position and identity of the interacting partner. Our potential demonstrates that such anisotropy is found in all kinds of atoms (polar and nonpolar). The improved performance of our GOAP potential and other orientation-dependent potentials over orientation-independent ones (such as the DFIRE) has implications for the development of more accurate physics-based, all-atom potentials. Traditional physics-based all-atom force fields (2,3) represent atoms as hard spheres and take into account orientation dependencies only for bonded atoms in the angle and dihedral angle terms.
Nonbonded interactions are described by short-range van der Waals and long-range electrostatic terms and lack any angular orientation dependence. In recent developments of molecular-mechanics force fields, the electronic polarization of the atomic environment has been taken into account by calculating induced charges during the simulation (56). However, this is too computationally expensive for protein simulations even though a dipole description of electronic polarization is still inadequate for protein atoms. In contrast, GOAP naturally takes into account the orientation-dependence of H-bonds, disulfide bonds, salt-bridges, and other possible pair interactions at all distances.
However, due to the introduction of a sequence separation cutoff s = 7 because of the inaccurate estimation of expected angle distributions at shorter cutoffs, only nonlocal H-bonds (e.g., those in β-sheets and long separated side chains) are included. Although the knowledge-based potential can be directly used in Monte Carlo simulations and in model selection, its application to molecular dynamics simulations requires differentiable functions. This could be done using splines. Although GOAP might be useful for sampling conformations using molecular dynamics, the resulting thermodynamic properties might be unrealistic because GOAP is a potential of mean force derived from the statistics of solved protein structures. However, as a means of generating good quality models, our ranking results here are suggestive; but it remains to be demonstrated whether the good performance of GOAP will be retained when it is used to drive the conformational search rather than to select among extrinsically generated decoys. This is a promising avenue that is currently being pursued.
Acknowledgments
The authors thank Dr. Bartosz Ilkowski for managing the cluster on which this work was conducted.
This work is supported by National Institutes of Health grant GM-48835.
Supporting Material
References
- 1.Senn H.M., Thiel W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. Engl. 2009;48:1198–1229. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 2.Weiner S., Kollman P., Case D. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986;7:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 3.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 4.Ponder J.W., Case D.A. Force fields for protein simulations. Adv. Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 5.Jagielska A., Wroblewska L., Skolnick J. Protein model refinement using an optimized physics-based all-atom force field. Proc. Natl. Acad. Sci. USA. 2008;105:8268–8273. doi: 10.1073/pnas.0800054105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnautova Y.A., Jagielska A., Scheraga H.A. A new force field (ECEPP-05) for peptides, proteins, and organic molecules. J. Phys. Chem. B. 2006;110:5025–5044. doi: 10.1021/jp054994x. [DOI] [PubMed] [Google Scholar]
- 7.Skolnick J. In quest of an empirical potential for protein structure prediction. Curr. Opin. Struct. Biol. 2006;16:166–171. doi: 10.1016/j.sbi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Miyazawa S., Jernigan R.L. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- 9.DeBolt S.E., Skolnick J. Evaluation of atomic level mean force potentials via inverse folding and inverse refinement of protein structures: atomic burial position and pairwise non-bonded interactions. Protein Eng. 1996;9:637–655. doi: 10.1093/protein/9.8.637. [DOI] [PubMed] [Google Scholar]
- 10.Zhang C., Vasmatzis G., DeLisi C. Determination of atomic desolvation energies from the structures of crystallized proteins. J. Mol. Biol. 1997;267:707–726. doi: 10.1006/jmbi.1996.0859. [DOI] [PubMed] [Google Scholar]
- 11.Hendlich M., Lackner P., Sippl M.J. Identification of native protein folds amongst a large number of incorrect models. The calculation of low energy conformations from potentials of mean force. J. Mol. Biol. 1990;216:167–180. doi: 10.1016/S0022-2836(05)80068-3. [DOI] [PubMed] [Google Scholar]
- 12.Sippl M.J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 13.Jones D.T., Taylor W.R., Thornton J.M. A new approach to protein fold recognition. Nature. 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H., Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen M.Y., Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tobi D., Elber R. Distance-dependent, pair potential for protein folding: results from linear optimization. Proteins. 2000;41:40–46. [PubMed] [Google Scholar]
- 17.Lu H., Skolnick J. A distance-dependent atomic knowledge-based potential for improved protein structure selection. Proteins. 2001;44:223–232. doi: 10.1002/prot.1087. [DOI] [PubMed] [Google Scholar]
- 18.Rykunov D., Fiser A. New statistical potential for quality assessment of protein models and a survey of energy functions. BMC Bioinformatics. 2010;11:128. doi: 10.1186/1471-2105-11-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samudrala R., Moult J. An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J. Mol. Biol. 1998;275:895–916. doi: 10.1006/jmbi.1997.1479. [DOI] [PubMed] [Google Scholar]
- 20.Munson P.J., Singh R.K. Statistical significance of hierarchical multi-body potentials based on Delaunay tessellation and their application in sequence-structure alignment. Protein Sci. 1997;6:1467–1481. doi: 10.1002/pro.5560060711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Y., Kloczkowski A., Jernigan R.L. Four-body contact potentials derived from two protein datasets to discriminate native structures from decoys. Proteins. 2007;68:57–66. doi: 10.1002/prot.21362. [DOI] [PubMed] [Google Scholar]
- 22.Krishnamoorthy B., Tropsha A. Development of a four-body statistical pseudo-potential to discriminate native from non-native protein conformations. Bioinformatics. 2003;19:1540–1548. doi: 10.1093/bioinformatics/btg186. [DOI] [PubMed] [Google Scholar]
- 23.Li X., Liang J. Geometric cooperativity and anticooperativity of three-body interactions in native proteins. Proteins. 2005;60:46–65. doi: 10.1002/prot.20438. [DOI] [PubMed] [Google Scholar]
- 24.Gilis D., Biot C., Rooman M. Development of novel statistical potentials describing cation-π interactions in proteins and comparison with semiempirical and quantum chemistry approaches. J. Chem. Inf. Model. 2006;46:884–893. doi: 10.1021/ci050395b. [DOI] [PubMed] [Google Scholar]
- 25.Miyazawa S., Jernigan R.L. How effective for fold recognition is a potential of mean force that includes relative orientations between contacting residues in proteins? J. Chem. Phys. 2005;122:024901. doi: 10.1063/1.1824012. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y., Lu M., Ma J. OPUS-Ca: a knowledge-based potential function requiring only Cα positions. Protein Sci. 2007;16:1449–1463. doi: 10.1110/ps.072796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoppe C., Schomburg D. Prediction of protein thermostability with a direction- and distance-dependent knowledge-based potential. Protein Sci. 2005;14:2682–2692. doi: 10.1110/ps.04940705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchete N.V., Straub J.E., Thirumalai D. Development of novel statistical potentials for protein fold recognition. Curr. Opin. Struct. Biol. 2004;14:225–232. doi: 10.1016/j.sbi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y., Kolinski A., Skolnick J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 2003;85:1145–1164. doi: 10.1016/S0006-3495(03)74551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koliński A., Bujnicki J.M. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins. 2005;61(Suppl 7) doi: 10.1002/prot.20723. 84–90. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y., Zhou Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins. 2008;72:793–803. doi: 10.1002/prot.21968. [DOI] [PubMed] [Google Scholar]
- 32.Lu M., Dousis A.D., Ma J. OPUS-PSP: an orientation-dependent statistical all-atom potential derived from side-chain packing. J. Mol. Biol. 2008;376:288–301. doi: 10.1016/j.jmb.2007.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J., Zhang Y. A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction. PLoS ONE. 2010;5:e15386. doi: 10.1371/journal.pone.0015386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y., Zhou Y. Ab initio folding of terminal segments with secondary structures reveals the fine difference between two closely related all-atom statistical energy functions. Protein Sci. 2008;17:1212–1219. doi: 10.1110/ps.033480.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortemme T., Morozov A.V., Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C., Liu S., Zhou Y. The dependence of all-atom statistical potentials on structural training database. Biophys. J. 2004;86:3349–3358. doi: 10.1529/biophysj.103.035998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C., Liu S., Zhou Y. Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci. 2004;13:391–399. doi: 10.1110/ps.03411904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S., Zhang C., Zhou Y. A physical reference state unifies the structure-derived potential of mean force for protein folding and binding. Proteins. 2004;56:93–101. doi: 10.1002/prot.20019. [DOI] [PubMed] [Google Scholar]
- 39.Zhou H., Zhou Y. Quantifying the effect of burial of amino acid residues on protein stability. Proteins. 2004;54:315–322. doi: 10.1002/prot.10584. [DOI] [PubMed] [Google Scholar]
- 40.Zhu J., Xie L., Honig B. Structural refinement of protein segments containing secondary structure elements: local sampling, knowledge-based potentials, and clustering. Proteins. 2006;65:463–479. doi: 10.1002/prot.21085. [DOI] [PubMed] [Google Scholar]
- 41.Simons K.T., Kooperberg C., Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J. Mol. Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 42.Park B., Levitt M. Energy functions that discriminate x-ray and near native folds from well-constructed decoys. J. Mol. Biol. 1996;258:367–392. doi: 10.1006/jmbi.1996.0256. [DOI] [PubMed] [Google Scholar]
- 43.Keasar C., Levitt M. A novel approach to decoy set generation: designing a physical energy function having local minima with native structure characteristics. J. Mol. Biol. 2003;329:159–174. doi: 10.1016/S0022-2836(03)00323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samudrala R., Xia Y., Huang E. A combined approach for ab initio construction of low resolution protein tertiary structures from sequence. Proc. Pac. Symp. Biocomput. 1999:505–516. doi: 10.1142/9789814447300_0050. [DOI] [PubMed] [Google Scholar]
- 45.Reference deleted in proof.
- 46.John B., Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucl. Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia Y., Huang E.S., Samudrala R. Ab initio construction of protein tertiary structures using a hierarchical approach. J. Mol. Biol. 2000;300:171–185. doi: 10.1006/jmbi.2000.3835. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y., Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 49.Moult J., Fidelis K., Tramontano A. Critical assessment of methods of protein structure prediction (CASP)—round IX. Proteins Struct. Funct. Bioinformat. 2011 doi: 10.1002/prot.23200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G., Dunbrack R.L., Jr. PISCES: a protein sequence culling server. Bioinformatics. 2003;19:1589–1591. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 51.Ma J. Explicit orientation dependence in empirical potentials and its significance to side-chain modeling. Acc. Chem. Res. 2009;42:1087–1096. doi: 10.1021/ar900009e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benkert P., Tosatto S.C., Schomburg D. QMEAN: a comprehensive scoring function for model quality assessment. Proteins. 2008;71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 53.Eramian D., Shen M.Y., Marti-Renom M.A. A composite score for predicting errors in protein structure models. Protein Sci. 2006;15:1653–1666. doi: 10.1110/ps.062095806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Z., Tegge A.N., Cheng J. Evaluating the absolute quality of a single protein model using structural features and support vector machines. Proteins. 2009;75:638–647. doi: 10.1002/prot.22275. [DOI] [PubMed] [Google Scholar]
- 55.Benkert P., Biasini M., Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamoureux G., Roux B. Modeling induced polarization with classical Drude oscillators: theory and molecular dynamics simulation algorithm. J. Chem. Phys. 2003;119:3025–3039. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.