

Abstract
NMR chemical shift tensors (CSTs) in proteins, as well as their orientations, represent an important new restraint class for protein structure refinement and determination. Here, we present the first determination of both CST magnitudes and orientations for 13Cα and 15N (peptide backbone) groups in a protein, the β1 IgG binding domain of protein G from Streptococcus spp., GB1. Site-specific 13Cα and 15N CSTs were measured using synchronously evolved recoupling experiments in which 13C and 15N tensors were projected onto the 1H-13C and 1H-15N vectors, respectively, and onto the 15N-13C vector in the case of 13Cα. The orientations of the 13Cα CSTs to the 1H-13C and 13C-15N vectors agreed well with the results of ab initio calculations, with an rmsd of approximately 8°. In addition, the measured 15N tensors exhibited larger reduced anisotropies in α-helical versus β-sheet regions, with very limited variation (18 ± 4°) in the orientation of the z-axis of the 15N CST with respect to the 1H-15N vector. Incorporation of the 13Cα CST restraints into structure calculations, in combination with isotropic chemical shifts, transferred echo double resonance 13C-15N distances and vector angle restraints, improved the backbone rmsd to 0.16 Å (PDB ID code 2LGI) and is consistent with existing X-ray structures (0.51 Å agreement with PDB ID code 2QMT). These results demonstrate that chemical shift tensors have considerable utility in protein structure refinement, with the best structures comparable to 1.0-Å crystal structures, based upon empirical metrics such as Ramachandran geometries and χ1/χ2 distributions, providing solid-state NMR with a powerful tool for de novo structure determination.
Keywords: magic-angle spinning, dihedral angles, cross validation, nanocrystal, quantum chemistry
The chemical shift is an exquisite and powerful probe of molecular structure, deriving from the interaction of molecular orbitals with an external magnetic field, B0. Understanding the relationships between chemical shifts and protein structure has substantial implications for modern nuclear magnetic resonance (NMR) spectroscopy, chemistry, and structural biology (1–12). The chemical shift tensor (CST) is rich with information, even when two-thirds of it is averaged to zero by molecular tumbling in solution or magic-angle spinning (MAS) of solid samples. The remaining isotopic chemical shifts remain an excellent resource for structure determination and validation, and higher-order interactions of the CST have substantial contributions to NMR relaxation (13–19). Therefore, detailed knowledge of CSTs permits a precise analysis of motion (20–22). Solid-state NMR (SSNMR) of fully aligned samples exploits amide 15N tensor information to determine the orientations of helices relative to the bilayer (23, 24). We have previously shown that use of a force field in which experimental 13Cα CSTs are compared with ab initio CSTs [generated as a function of backbone conformation (ϕ, ψ)] significantly improves the precision and accuracy of SSNMR-computed protein structures (10). In addition to determination of NMR-based structures and dynamics, CST datasets are invaluable for the continued development of quantum chemical techniques to compute isotropic and anisotropic chemical shifts, furthering our understanding of appropriate basis sets and functions for accurate MO theory of proteins (4, 6, 11, 25–27).
Over the past decade, protein structure determination by SSNMR has progressed substantially in terms of the rate of data collection and analysis, as well as in the resolution and complexity of the resulting structures (10, 28–36). In most cases, structures have been determined by using a combination of semiquantitative distance restraints (comparable to solution NOEs) together with semiempirical dihedral angle restraints, obtained from isotropic chemical shifts and chemical shift databases. More recently, we have demonstrated that tensor recoupling is also a powerful route to structure refinement since incorporating relative dipolar tensor orientations (36), CSTs (10), and precise z-filtered transferred echo double resonance (zf-TEDOR) distances (35) as restraint classes in annealing algorithms substantially enhances the precision and accuracy of the resulting protein structures. The next logical, yet technically challenging and heretofore unprecedented, step is to extend this approach by incorporating both tensor magnitudes as well as orientations into simulated annealing calculations. This approach promises further enhancement of structure quality and provides key internal controls for both backbone and side-chain conformations.
Here we present such results, first by determining the relative site-specific orientations of 1H-15N (and 1H-13C) dipolar tensors relative to the axis system of the 15N (and 13C) CST, building upon prior MAS studies of proteins (37) and static peptide samples (38–40). These NMR tensorial parameters are obtained using a set of three-dimensional (3D) synchronous recoupling pulse sequences. These datasets enable the accurate determination of CST orientations relative to the molecular frame for the majority of the backbone 15N and 13Cα sites in a crystalline protein, GB1 (41). The 13Cα tensors’ orientations are then incorporated, along with other restraint classes, into simulated annealing calculations, resulting in structures exhibiting especially high precision [defined by the backbone rmsd (bbrmsd)] and accuracy (defined by the agreement with crystal structures and structure validation metrics).
Results and Discussion
Determination of 15N and 13Cα CST Magnitudes, Orientations and Order Parameters.
We first carried out a series of 3D [Rec]-15N-13C correlation experiments each consisting of a 2D NCA plane with a tensor recoupling period in the third dimension. The resulting lineshapes depend upon the CST as well as its relative orientation to the dipole vector (Fig. 1), consistent with conventions of prior studies (37). Further details regarding the tensor notation (Fig. S1) and pulse sequence (Fig. S2) are provided in Materials and Methods and SI Text. The key elements of the data collection are ROCSA recoupling of the CST (42) and 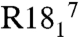 heteronuclear dipolar recoupling (43). Using a 2-13C-glycerol,U-15N GB1 sample (28, 36, 44) all 53 observable 15N and 13C resonances were resolved in the NCA isotropic 2D plane (Leu Cα is unlabeled). Four of the experiments measured tensor magnitudes for the 15N and 13C CSTs and the 1H-15N and 1H-13C vectors. The seven remaining experiments correlated the CST with these vectors using ROCSA and
heteronuclear dipolar recoupling (43). Using a 2-13C-glycerol,U-15N GB1 sample (28, 36, 44) all 53 observable 15N and 13C resonances were resolved in the NCA isotropic 2D plane (Leu Cα is unlabeled). Four of the experiments measured tensor magnitudes for the 15N and 13C CSTs and the 1H-15N and 1H-13C vectors. The seven remaining experiments correlated the CST with these vectors using ROCSA and 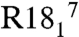 periods, evolved synchronously with different ratios of evolution time. To orient the 13Cα tensor, 1H-13C:CST ratios of 1∶1, 1∶2, and 1∶3 were utilized, with an additional 13C CST: 15N-13C ratio of 1∶1 to break mirror-plane degeneracies; to orient the 15N CST, ratios of 1H - 15N:CST of 1∶1, 2∶1, and 1∶2 were acquired. Together these experiments yielded 563 lineshapes (> 10 per residue), reporting uniquely upon each tensor magnitude and the orientation with respect to the molecular frame. Order parameters for < 100 microsecond motions, derived directly from analysis of the 1H-13Cα and 1H-15N dipolar couplings (Fig. S3), indicate a rigid backbone. Aside from G41, we anticipate minimal motional averaging (approximately 1.5%) of CST magnitudes, based upon path integral calculations of Tang et al. (26). Further examples of fitted lineshapes are provided in SI Text (Figs. S4–S6), along with the full compilation of orientations (Tables S1 and S2).
periods, evolved synchronously with different ratios of evolution time. To orient the 13Cα tensor, 1H-13C:CST ratios of 1∶1, 1∶2, and 1∶3 were utilized, with an additional 13C CST: 15N-13C ratio of 1∶1 to break mirror-plane degeneracies; to orient the 15N CST, ratios of 1H - 15N:CST of 1∶1, 2∶1, and 1∶2 were acquired. Together these experiments yielded 563 lineshapes (> 10 per residue), reporting uniquely upon each tensor magnitude and the orientation with respect to the molecular frame. Order parameters for < 100 microsecond motions, derived directly from analysis of the 1H-13Cα and 1H-15N dipolar couplings (Fig. S3), indicate a rigid backbone. Aside from G41, we anticipate minimal motional averaging (approximately 1.5%) of CST magnitudes, based upon path integral calculations of Tang et al. (26). Further examples of fitted lineshapes are provided in SI Text (Figs. S4–S6), along with the full compilation of orientations (Tables S1 and S2).
Fig. 1.

Dipolar:CST correlation spectra for both 13Cα and 15N sites. Experimental spectrum is presented in black, with simulations in red. Ratios provided are the ratio of dipolar to CST evolution. Row two of A indicates ratio of 15N-13Cα dipolar:CST evolution. (A) Fit lineshapes for [1H-13C]∶[13C CST] correlation spectra for lysines with different secondary structures are presented. K4 is located in a β-sheet, K28 in the α-helix, and K50 in a β-turn with an unusual positive value of ϕ. (b) Fit ensemble of [1H-15N]∶[15N CST] correlation spectra. Fit is representative of limited variations of 15N tensors throughout GB1.
These datasets together report upon the 13Cα CST orientation with respect to the 1H-13Cα (α1, α2, α3) and 15N-13Cα (β1, β2, β3) bond vectors. Tensor magnitudes were reported in our previous study (10); the results we report here, to restrain orientations, were acquired under identical experimental conditions. The 13Cα exhibits a broad range of total magnitude and rhombicity, depending upon residue type and secondary structure. Beyond the changes in tensor anisotropy (δ) and asymmetry (η) discussed previously (7, 10), the orientation of each element to the molecular frame varies greatly (Fig. 2 and Fig. S7). For example, in a typical β-sheet conformation the δ11 element is oriented from 0 to 25° of the 1H-13C bond vector, and closer to perpendicular to this vector in α-helical conformations. The orientation of the 13C CST to the 15N-13C bond is also a strong reporter of backbone torsion, in most cases a transposition of the β1 and β2 angles, often accompanied by a conversion to the complement of the angle. All angles fitted are provided in Table S1.
Fig. 2.

Analysis of [1H-13C] dipolar:13C CST correlation spectra. (A) Fit α angles, defining orientation of each tensor element to the HC dipole, as a function of residue number. All angles over 180° were converted to their < 90° complement for clarity. Clear trends are observed where δ11 is oriented within 20° of the dipole in β-strands but moves within 30° of the bond normal in the α-helix. δ22 and δ33 are near perpendicular to the HC bond in the β-sheet, while δ11 and δ22 reorient up to 80° in the α-helix. (B) Fit β angles defining the orientation of each tensor element to the NC bond vector. While overall variation of orientation is not as pronounced, there is a strong shift in the β2 angle between helical and sheet conformations with a concerted, smaller adjustment of β1 and β3.
We investigated the trends and consistency of the 13Cα orientations in two ways. First, we compared our measurements with ab initio chemical shielding surfaces that are available for all 20 common amino acids (http://feh.scs.uiuc.edu/amino_acid.php), (4, 6, 11) where we find excellent overall agreement. The theory-versus-experimental correlations are presented in Fig. 3 A and B, where it can be seen that all experimental values are within 30° (depicted in blue) of the predicted values. The overall agreement for each dataset is very good with an rmsd for (α1, α2, α3) of 8.0° and an R2 = 0.97 (Fig. 3A). In the case of (β1, β2, β3), the rmsd and R2 values are 8.5° and 0.95 (Fig. 3B), comparable to previously reported results for small peptides (6). Second, we computed the values for Δσ∗, defined as the difference between the shielding parallel to the 1H-13C dipole and the shielding perpendicular to the dipole; this parameter was previously measured in solution for ubiquitin and calmodulin (15).
Fig. 3.

Agreement of fit 13Cα CST orientations with ab initio predictions and reconstructed Δσ∗ values as a function of residue number. For most sites, fits are within 30° of the predicted orientation, within the maximum experimental error. (A) Experimental 13C CST orientations α1, α2, and α3 (black dots) and (B) experimental orientations β1, β2, and β3 (black dots) plotted against theoretical angles predicted by ab initio surfaces of Sun et al. assuming 2QMT crystal structure geometries. Blue lines indicate a deviation of ± 30°. (c) Reconstructed Δσ∗ magnitudes. β-sheets sites range from 20–33 ppm and α-helical from -6–8 ppm, largely consistent with values reported by Tjandra and Bax.
Observed outliers, highlighted in Fig. 3, are K10, K28, K31, and V21. In the case of K10 and V21, the agreement is actually quite good when the dihedral angles from the 2GI9 crystal structure are used, suggesting these deviations might represent a real, small difference between the microcrystal formulation used and that from the 2QMT crystal structure. In the case of the helical lysines, the measured values fit regions of the ab initio surface that are within 15° of those found in the 2QMT structure. Overall, the greatest deviations between theory and experiment are for angles near 90°, a known weak region for tensor correlation experiments, and a disproportionate number of these angles are in the α-helix. It is possible that some effects not included in the ab initio calculations, such as the helix dipole, might make a contribution to the 13Cα shielding in this region, and such effects are indeed important in computing helical 15N shifts (45). This issue might be addressed in the future by implementing even stronger 13C (CST)-(15N-13C) correlations, by using ROCSA-REDOR type correlations.
The tensor magnitudes and orientations were used to reconstruct Δσ∗ values, as measured in solution (Fig. 3C), reproducing the observed trends (14). Here, β-sheet Δσ∗ values range from 20 to 33 ppm. The largest Δσ∗ value is for K50, which has a positive value of ϕ in all available crystal structures, which is unusual for a nonglycine residue. Δσ∗ values in the α-helix (residues 23 to 36) range from -6 to 8 ppm. Turns with near α-helical conformation exhibit near-helical values of Δσ∗ but are slightly larger (by 2 to 3 ppm) as compared to those in the α-helix. Previously, it was observed that most Thr and Ser residues exhibited large negative values of Δσ∗. This is not observed in the present dataset; however, only one Thr is in the α-helix (T25), making this finding inconclusive. The chemical shift of the Cβ of T25 is nearly identical to the Cα resonance frequency, so it is possible that there is some leakage to the Cβ resonance, which is partially labeled in the 2-13C-glycerol preparation, accounting for this effect.
GB1 Structure Refined with 13C Tensor Orientations.
A series of structures were generated using 13C CST tensor magnitudes and orientations as structural constraints in an Xplor-NIH calculation, following the protocol described previously (10). These structures were solved using combinations of seven different restraint force fields, each abbreviated using a single initial for simplicity; all calculations are summarized in Table 1. They include distances from spin diffusion or 1H-1H couplings (N) (34, 36), TEDOR distances (T) (35), TALOS dihedral angles (D), CST magnitudes (C) (10), CST orientations (O), vector angles (VEAN, V) (36), and semiempirical database potentials (SE) (46–48). When compared with structures without CST or VEAN information, the tensor-refined structures possessed higher precision and better agreement with the crystal structures than those computed without these restraints. In addition to better precision and accuracy of the lowest energy structures, all 200 structures refined with CST restraints had an overall bbrmsd of 0.23–0.5 Å, and agreement with the 2QMT crystal structure of 0.51 to 1.1 Å, depending upon the distance restraints used. In all cases, the 10 lowest energy structures (of 200 total structures generated) composed the structural ensemble.
Table 1.
Structural ensemble statistical agreement internally and against the closest related crystal structure
| Restraints Used* | rmsd | |||||||||
| Structure | N | T | D | C | O | V | SE | Backbone | vs. 2QMT | All Heavy Atom |
| 1 | X | X | 0.40 ± 0.07 | 1.06 ± 0.05 | 1.07 ± 0.08 | |||||
| 2 | X | X | X | X | 0.19 ± 0.04 | 1.02 ± 0.02 | 0.98 ± 0.04 | |||
| 3 | X | X | X | 0.19 ± 0.06 | 0.69 ± 0.03 | 0.68 ± 0.04 | ||||
| 4 | X | X | X | X | 0.18 ± 0.03 | 0.60 ± 0.02 | 0.72 ± 0.03 | |||
| 5 | X | X | X | X | X | 0.15 ± 0.03 | 0.59 ± 0.02 | 0.71 ± 0.04 | ||
| 6 | X | X | X | X | X | X | 0.14 ± 0.03 | 0.57 ± 0.02 | 0.68 ± 0.03 | |
| 7 | X | X | X | X | X | 0.19 ± 0.03 | 0.55 ± 0.01 | 0.71 ± 0.04 | ||
| 8 | X | X | X | X | X | X | 0.22 ± 0.03 | 0.57 ± 0.04 | 0.75 ± 0.04 | |
| 9 | X | X | X | X | X | 0.18 ± 0.03 | 0.55 ± 0.02 | 0.71 ± 0.04 | ||
| 10 | X | X | X | X | X | X | 0.17 ± 0.06 | 0.54 ± 0.03 | 0.73 ± 0.03 | |
| 11 | X | X | X | X | X | X | X | 0.16 ± 0.03 | 0.51 ± 0.02 | 0.72 ± 0.03 |
*Abbreviations for restraints used: 1H-1H couplings/13C-13C DARR distances (N); TEDOR distances (T), TALOS dihedral angles (D), CST magnitudes (C), CST orientations (O), vector angles (V), semiempirical database potentials (SE).
To improve continuity of the spline function used to create the CST potential from the ab initio surfaces, and to eliminate any ambiguity from mirror symmetries of the CST orientations measurements, all angles were converted to the complement < 90°. This greatly improved the continuity of the constructed energy surface and thus the convergence of the annealing algorithm. In the first two calculations, distance tables from our previous CST structure refinement were used. In the first, only CST orientations and distances were used (NO) and in the second, CST magnitudes and TALOS dihedrals were added (NDCO). The bbrmsd for the NO structures was 0.4 Å, with an rmsd agreement of 1.06 Å with the 2QMT crystal structure. This is comparable in precision but a significant improvement in accuracy relative to the previously published NC structure (10). The inclusion of TALOS dihedral angles and CST magnitudes and orientations improved the resolution of the structure to 0.19 Å with comparable accuracy (1.02-Å agreement with 2QMT). If the CST magnitudes are removed (i.e., distances and CST orientations only) there was a negligible difference in the results; however, the comparable calculation lacking orientations (structure 4 of ref. 10) yields less precise structures. The major advantage of using orientations in this case is that they are independent of the tensor scaling and offset effects observed in the comparison of theoretical and experimental CST magnitudes; thus they are immune to common errors from—e.g., motional averaging and/or pulse sequence imperfections. In all cases, the structures with CST information improved upon structures solved with only distance information.
After these control structures were refined, to confirm the effectiveness of these restraints, more structures were generated using all of the restraint types defined above. These include highly precise distances from TEDOR (T) (49, 50), a technique already shown to greatly improve protein structures and described in detail elsewhere (35). A calculation including TALOS restraints and all available distances (NTD) produced a structure with a bbrmsd of 0.19 Å and an agreement with the 2QMT crystal structure of 0.69 Å. Once the CST magnitudes and orientations were added to the calculations (NTDCO), the bbrmsd improved to 0.15 Å, and the agreement with the 2QMT structure improved to 0.59 Å. Inclusion of VEAN restraints (NTDCOV) resulted in a slightly better structure (0.14-Å bbrmsd, 0.57 vs. 2QMT). A structure with all available distances and CST restraints (NTCO) showed a slight deterioration in statistical quality (0.18-Å bbrmsd, 0.60 vs. 2QMT) compared to the NTDCOV structure but still an improvement upon the NTD ensemble. We also found that allowing the calibration factors for the CST surface to vary during annealing gave slightly better results than using the previously calibrated conversion factors (though by at most 0.04 Å).
At this point, semiempirical Ramachandran database (46, 47) and hydrogen bonding energy database (SE) (48) potentials were included. This further improved the overall structural quality (0.16-Å bbrmsd, 0.51 Å vs. 2QMT) for the ensemble with all currently available restraints (NTDCOV-SE, Fig. 4). Removal of the vector angles (NTDCO-SE) or the CST restraints (NTDV-SE) yielded similar structures (0.17 Å/0.54 Å and to 0.19 Å/0.55 Å) in which the structure retaining CST information has slightly better agreement with the crystal structure, but the vector-angle structure has higher precision. Removal of TALOS and VEAN restraints (NTCO-SE) yielded a structure with 0.18-Å bbrmsd and 0.55-Å rmsd with 2QMT. Unlike TALOS (which relies upon a database of highly resolved X-ray structures), there is no explicit or implicit biasing toward favorable regions of Ramachandran space in the CST restraints presented here. Thus, it appears that employing the RAMA potential with CST restraints yields the closest accord with the X-ray results. This could of course indicate bias toward the X-ray results through the use of semiempirical database potentials; however, since the results of these calculations also exhibit the best overall agreement with all experimental restraints, this possibility seems remote. The structures (4, 8 and 9) with CST restraints, but without TALOS, exhibit improved accuracy over the structure (3) solved using only TALOS to restrain dihedral geometry. The TALOS restraints improve the convergence properties of calculations but are not required in the final stages of refinement.
Fig. 4.

GB1 structure calculated using all CST information, vector angles, TALOS dihedrals, and all distances. (A) The 10 lowest energy structures (out of 200) are presented in blue with 2QMT crystal structure represented in red; bbrmsd = 0.16 Å and agreement with 2QMT is 0.51 Å. (B) The lowest 10 energy structures are presented in CPK to illustrate the overall heavy atom order. The all heavy atom rmsd is 0.72 Å.
The quality of each structure was evaluated using several metrics. First, the internal consistency defined by the backbone and all heavy atom rmsds, followed by the agreement of each ensemble mean to the most relevant crystal structure (Table 1) were determined. Second, each structural ensemble was evaluated with PROCHECK_NMR (51). We report four metrics: regions of Ramachandran space populated by percentage, the average χ1 and χ2 deviations, and hydrogen bonding energy (Table S3). The χ1 and χ2 deviations present further evidence of overall improvements in structure quality, serving as a cross validation to illustrate that improvements in backbone geometry lead to tighter overall folds and improved geometries. Third, we determined the overall agreement of each ensemble with both CST (Table S4) and vector angle (Table S5) restraints. Finally, shifts were generated for each structure using the chemical shift prediction program SPARTA (52) and compared to the experimentally measured chemical shifts (Table S6). In all cases, these analyses indicated that all structures were highly consistent with one another having backbone resolution comparable to X-ray structures of 1.0–2.0-Å resolution. Not only do these SSNMR structures exhibit excellent structure validation metrics, the agreement of our highest-resolved structure with the 2QMT crystal structure is on the order of agreement among the four deposited crystal structures of GB1 (Table S7).
During these calculations, violations in several previously published distance, TEDOR, and TALOS dihedral restraints were identified and relaxed. In total, 20 TALOS error bars were doubled at some point during the calculations (though only 10 of the original restraints would have violated in the best final structures, within the originally published uncertainties), and the error estimates for 11 spin diffusion, 2 1H-1H, and 26 TEDOR distances were increased. The TEDOR distances that violated had predicted error bars of 0.5 Å or less, and were lengthened by 1 Å. Most often, these restraints violated in the direction of geometries in agreement with the 2QMT crystal structure. These altered restraints are given in Tables S8 and S9.
15N CST Magnitudes and Orientations.
The magnitudes of the principal elements of the 15N tensors are presented in Fig. 5A (and Table S2). Agreement between these measured values of δ11 (δ11 = δ + δiso in this instance) and our previous slow-MAS study (depicted in red in Fig. 5A) showed an rmsd between the two datasets of 1.6 ppm and an R2 of 0.95. The tensor anisotropy was larger for the α-helical residues than for the β-sheet residues, as found also in thioredoxin (53). Similarly, the measured values of η (the tensor anisotropy) were slightly smaller in α-helical (η = 0.23) than in β-sheet residues (η = 0.27). Plotting each tensor element (in the “full” representation) vs. the isotropic chemical shift reveals that, unlike C′ tensors, 15N isotropic chemical shift perturbations result from concerted shifts of all tensor elements rather than large shifts of a single element (Fig. 5B). The most deshielded 15N shift tensor element, δ11, is oriented 9–24° from the 1H-15N bond (Fig. 5C), δ22 is approximately 97° ± 12° from the 1H-15N vector and δ33 is oriented near the N-Cα bond, in or within 10° of the peptide plane (approximately 75° ± 12° from the 1H-15N bond). All CST orientations are shown in Table S2 and are in good accord with recent ab initio studies (45). Of particular interest is the observation that our tensor measurements indicate the 15N tensor deviates from ideal prolate symmetry, with η ranging from 0.15 to 0.32. Our original hypothesis was that this discrepancy between our data and other studies might result from the high covariance between relaxation and η when η < ∼ 0.5. However, recent density functional theory calculations of 15N CSTs in the α-helix of GB3 by Cai et al. (45) agree well with all of our tensor elements (Fig. S8A), (R2 = 0.99 and rmsd of 6.5 ppm) and a recent solution NMR study presents even larger tensor asymmetry (54). It should be noted that δ22 and δ33 are significantly smaller in magnitude than δ11 in the traceless representation, leading to greater uncertainty in the fitted orientation. The smaller magnitudes of δ22 and δ33 in the traceless representation, and their near perpendicular orientation to the 1H-15N vector, suggest that uncertainties in the magnitude of these elements might be inconsequential for most solution NMR relaxation studies but might be more important in the interpretation of PISEMA spectra, making a better understanding of these tensors (especially in α-helical conformations) of interest. Recent solution NMR work also reveals strong statistical agreement with our results (Fig. S8B) for the full tensor, albeit with some outliers.
Fig. 5.

Amide chemical shift tensor analysis for protein GB1. (A) Principal elements of 15N tensor in the traceless representation compared to previously published slow-MAS data. Principal elements are presented in black with error bars corresponding to one standard deviation. Tensor values from previous work are presented in red. The rmsd between δ11 elements from both datasets is 1.6 ppm, and 6 ppm for δ22 and δ33 values corresponding to a deviation of η of 0.13. (B) Chemical shift tensor elements plotted against isotropic chemical shift. The correlation reveals that the changes in the isotropic chemical shift result from largely correlated shifts of all three principal elements. R2 for each element (δ11, δ22, δ33) are 0.82, 0.75, and 0.62, respectively. (C) The angle β (a1) as a function of residue number. The angle β defines the orientation of the δ11/δzz tensor element to the HN bond dipole.
Conclusions
The chemical shift tensors of all 15N and 13C sites in a protein report upon a vast range of molecular properties, including electronic structure, backbone conformation, steric clashes, electrostatics, side-chain packing, and dynamics. Specifically, a detailed knowledge of backbone amide and Cα tensors is relevant to an increasing range of structural and dynamics work throughout NMR and structural biology in general. In this paper, we have provided a unique example of the determination of 13Cα and 15N chemical shift tensor magnitudes and orientations throughout a protein, using SSNMR tensor correlation techniques. As a first example of their utility, 13Cα CST information was used to refine the structure of a 6-kDa protein. The 13Cα shift tensor orientations are in good accord with ab initio quantum chemical prediction and provide an important parameter with which to refine SSNMR structures not currently available from experimental databases. These methods complement already established tensor refinement methods in SSNMR and further the pursuit of atomic-resolution structure determination by SSNMR. Validation of these structures reveals a quality on par with 1-Å X-ray structures. The experimental backbone amide tensors are in accordance with recent density functional theory predictions of tensor orientations in the helical residues of GB1, illustrating the importance of such measurements to macromolecular electronic structure calculations.
The methodology and information described here lays the groundwork for future CST studies determining both the structure and dynamics of a range of systems. Overall, we have established that SSNMR is a powerful tool for readily measuring important tensor quantities with site-resolution without the need for conservative mutagenesis and multiple molecular alignments. These results were achieved by using three-dimensional spectroscopy and, in principle, can be implemented on much larger systems given that spectrometers operating at twice the field used in this work are now available. Now that precise orientations and magnitudes have been determined for a range of residue types and secondary structural motifs, future structural work might include static 13Cα spectra of oriented samples as a complement to the well-established PISEMA experiment. In addition, now that more tensor orientation information is known, the chemical shift can be incorporated into an even wider array of relaxation/dynamics measurements in solution NMR.
Materials and Methods
Sample Preparation.
Samples of GB1 were prepared using 2-13C-glycerol as the 13C source in the minimal growth media. Protein was precipitated as described previously and packed wet into the central 80% of a limited-speed (thin wall) 3.2-mm Varian rotor (Varian, Inc.).
NMR Spectroscopy.
All spectra were acquired using a 500-MHz Varian InfinityPlus spectrometer and 3.2-mm T3 Balun™ 1H-13C-15N probe. Pulse widths (π/2) for 1H, 13C, and 15N were 1.9 μs, 2.5 μs, and 5.0 μs, respectively. Spinning was maintained at 11.111 ± 0.002 kHz. Periods recoupling 1H-15N/13C dipoles and 15N/13C CST line shapes were inserted into a 3D experiment using an NCA plane for site resolution, following the model used in our previous studies. Dipolar recoupling was achieved using the 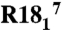 recoupling element and CST interactions were recoupled using ROCSA. Optimal resolution and sensitivity of the 15N-13C planes were achieved using SPECIFIC CP and TPPM decoupling during acquisition with approximately 75 kHz B1 on 1H. Eleven total spectra were acquired: one each for the 15N and 13C CST, the 1H-15N and 1H-13C dipoles, and seven dipole-CST correlated spectra. In these spectra ROCSA and dipolar dimensions were evolved synchronously in the same dimension, with different ratios of evolution time, in units of rotor periods. [1H-15N]∶[15N CST] ratios included 1∶1, 2∶1 and 1∶2. [1H-13C]∶[13C CST] ratios included 1∶1, 1∶2, and 1∶3. In addition a 13C CST recoupling experiment was performed where 15N was not decoupled, providing coevolution of the 13C CST and 13C-15N dipolar Hamiltonians.
recoupling element and CST interactions were recoupled using ROCSA. Optimal resolution and sensitivity of the 15N-13C planes were achieved using SPECIFIC CP and TPPM decoupling during acquisition with approximately 75 kHz B1 on 1H. Eleven total spectra were acquired: one each for the 15N and 13C CST, the 1H-15N and 1H-13C dipoles, and seven dipole-CST correlated spectra. In these spectra ROCSA and dipolar dimensions were evolved synchronously in the same dimension, with different ratios of evolution time, in units of rotor periods. [1H-15N]∶[15N CST] ratios included 1∶1, 2∶1 and 1∶2. [1H-13C]∶[13C CST] ratios included 1∶1, 1∶2, and 1∶3. In addition a 13C CST recoupling experiment was performed where 15N was not decoupled, providing coevolution of the 13C CST and 13C-15N dipolar Hamiltonians.
Data Analysis.
All eleven spectra were processed using NMRPipe. Lorentzian-to-Gaussian apodization functions were applied in the two isotropic chemical shift dimensions, with a net line broadening of 30 Hz for 13C and 15 Hz for 15N. Peak intensities in each 15N-13C (F1–F3) plane were determined and trajectories of each correlation spectrum (t2) extracted using the autoFit.tcl package in NMRPipe. All trajectories for each site were fit to exact spin simulations assuming a 2 spin basis. A subset of each experimental set were tested using a basis with all 1H within 2.5 Å; however, this only impacted the relaxation matrix and had little to no impact upon the fit angles and dipolar magnitudes. The fitting procedure was facilitated by in-house FORTRAN-77 code that called both external MINUIT minimization libraries (55), and the SPINEVOLUTION (56) simulation package.
Supplementary Material
Acknowledgments.
This research was supported by the National Institutes of Health (R01GM073770 and R01GM075937 to C.M.R. and in part by R01AI074233 to E.O.) and Molecular Biophysics Training Grant (PHS 5 T32 GM008276) and Ullyot Fellowship to L.J.S. We thank the NMR Facility at the School of Chemical Sciences, University of Illinois at Urbana-Champaign, for technical assistance, and Dr. Charles Schwieters (National Institutes of Health) for assistance with XPLOR-NIH calculations.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The 10 lowest energy protein structures have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2LGI).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103728108/-/DCSupplemental.
References
- 1.Chekmenev EY, Xu RZ, Mashuta MS, Wittebort RJ. Glycyl Cα chemical shielding in tripeptides: Measurement by solid-state NMR and correlation with X-ray structure and theory. J Am Chem Soc. 2002;124:11894–11899. doi: 10.1021/ja026700g. [DOI] [PubMed] [Google Scholar]
- 2.Chekmenev EY, Zhang Q, Waddell KW, Mashuta MS, Wittebort RJ. 15N chemical shielding in glycyl tripeptides: Measurement by solid-state NMR and correlation with X-ray structure. J Am Chem Soc. 2004;126:379–384. doi: 10.1021/ja0370342. [DOI] [PubMed] [Google Scholar]
- 3.Case DA. The use of chemical shifts and their anisotropies in biomolecular structure determination. Curr Opin Struct Biol. 1998;8:624–630. doi: 10.1016/s0959-440x(98)80155-3. [DOI] [PubMed] [Google Scholar]
- 4.Sun HH, Sanders LK, Oldfield E. Carbon-13 NMR shielding in the twenty common amino acids: Comparisons with experimental results in proteins. J Am Chem Soc. 2002;124:5486–5495. doi: 10.1021/ja011863a. [DOI] [PubMed] [Google Scholar]
- 5.Loth K, Pelupessy P, Bodenhausen G. Chemical shift anisotropy tensors of carbonyl, nitrogen, and amide proton nuclei in proteins through cross-correlated relaxation in NMR spectroscopy. J Am Chem Soc. 2005;127:6062–6068. doi: 10.1021/ja042863o. [DOI] [PubMed] [Google Scholar]
- 6.Wi S, Sun HH, Oldfield E, Hong M. Solid-state NMR and quantum chemical investigations of 13Cα shielding tensor magnitudes and orientations in peptides: Determining ϕ and ψ torsion angles. J Am Chem Soc. 2005;127:6451–6458. doi: 10.1021/ja042935b. [DOI] [PubMed] [Google Scholar]
- 7.Wylie BJ, Franks T, Graesser DT, Rienstra CM. Site-specific 13C chemical shift anisotropy measurements in a uniformly 15N,13C-labeled microcrystalline protein by 3D magic-angle spinning NMR spectroscopy. J Am Chem Soc. 2005;127:11946–11947. doi: 10.1021/ja053862e. [DOI] [PubMed] [Google Scholar]
- 8.Witter R, Sternberg U, Ulrich AS. NMR chemical shift powder pattern recoupling at high spinning speed and theoretical tensor evaluation applied to silk fibroin. J Am Chem Soc. 2006;128:2236–2243. doi: 10.1021/ja051730f. [DOI] [PubMed] [Google Scholar]
- 9.Wylie BJ, Rienstra CM. Multidimensional solid state NMR of anisotropic interactions in peptides and proteins. J Chem Phys. 2008;128:052207. doi: 10.1063/1.2834735. [DOI] [PubMed] [Google Scholar]
- 10.Wylie BJ, Schwieters CD, Oldfield E, Rienstra CM. Protein structure refinement using 13Cα chemical shift tensors. J Am Chem Soc. 2009;131:985–992. doi: 10.1021/ja804041p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Havlin RH, Le H, Laws DL, de Dios AC, Oldfield E. An ab initio quantum chemical investigation of carbon-13 NMR shielding tensors in glycine, alanine, valine, isoleucine, serine, and threonine: Comparisons between helical and sheet tensors, and the effects of χ1 on shielding. J Am Chem Soc. 1997;119:11951–11958. [Google Scholar]
- 12.Birn J, Poon A, Mao Y, Ramamoorthy A. Ab initio study of 13Cα chemical shift anisotropy tensors in peptides. J Am Chem Soc. 2004;126:8529–8534. doi: 10.1021/ja049879z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tjandra N, Szabo A, Bax A. Protein backbone dynamics and 15N chemical shift anisotropy from quantitative measurement of relaxation interference effects. J Am Chem Soc. 1996;118:6986–6991. [Google Scholar]
- 14.Tjandra N, Bax A. Solution NMR measurement of amide proton chemical shift anisotropy in 15N-enriched proteins. Correlation with hydrogen bond length. J Am Chem Soc. 1997;119:8076–8082. [Google Scholar]
- 15.Tjandra N, Bax A. Large variations in 13Cα chemical shift anisotropy in proteins correlate with secondary structure. J Am Chem Soc. 1997;119:9576–9577. [Google Scholar]
- 16.Burton RA, Tjandra N. Determination of the residue-specific 15N CSA tensor principal components using multiple alignment media. J Biomol NMR. 2006;35:249–259. doi: 10.1007/s10858-006-9037-6. [DOI] [PubMed] [Google Scholar]
- 17.Cornilescu G, Bax A. Measurement of proton, nitrogen, and carbonyl chemical shielding anisotropies in a protein dissolved in a dilute liquid crystalline phase. J Am Chem Soc. 2000;122:10143–10154. [Google Scholar]
- 18.Fushman D, Tjandra N, Cowburn D. Direct measurement of 15N chemical shift anisotropy in solution. J Am Chem Soc. 1998;120:10947–10952. [Google Scholar]
- 19.Hall JB, Fushman D. Variability of the 15N chemical shielding tensors in the B3 domain of protein G from 15N relaxation measurements at several fields. Implications for backbone order parameters. J Am Chem Soc. 2006;128:7855–7870. doi: 10.1021/ja060406x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer AG. NMR probes of molecular dynamics: Overview and comparison with other techniques. Annu Rev Biophys Biomol Struct. 2001;30:129–155. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 21.Palmer AG, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 22.Palmer AG. NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 23.Opella SJ, Zeri AC, Park SH. Structure, dynamics, and assembly of filamentous bacteriophages by nuclear magnetic resonance spectroscopy. Annu Rev Phys Chem. 2008;59:635–657. doi: 10.1146/annurev.physchem.58.032806.104640. [DOI] [PubMed] [Google Scholar]
- 24.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sitkoff D, Case DA. Theories of chemical shift anisotropies in proteins and nucleic acids. Prog Nucl Magn Reson Spectrosc. 1998;32:165–190. [Google Scholar]
- 26.Tang S, Case DA. Vibrational averaging of chemical shift anisotropies in model peptides. J Biomol NMR. 2007;38:255–266. doi: 10.1007/s10858-007-9164-8. [DOI] [PubMed] [Google Scholar]
- 27.Cai L, Fushman D, Kosov DS. Density functional calculations of 15N chemical shifts in solvated dipeptides. J Biomol NMR. 2008;41:77–88. doi: 10.1007/s10858-008-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellani F, et al. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 29.Castellani F, van Rossum BJ, Diehl A, Rehbein K, Oschkinat H. Determination of solid-state NMR structures of proteins by means of three-dimensional 15N-13C-13C dipolar correlation spectroscopy and chemical shift analysis. Biochemistry. 2003;42:11476–11483. doi: 10.1021/bi034903r. [DOI] [PubMed] [Google Scholar]
- 30.Loquet A, et al. 3D structure determination of the Crh protein from highly ambiguous solid-state NMR restraints. J Am Chem Soc. 2008;130:3579–3589. doi: 10.1021/ja078014t. [DOI] [PubMed] [Google Scholar]
- 31.Lange A, et al. A concept for rapid protein-structure determination by solid-state NMR spectroscopy. Angew Chem Int Ed Engl. 2005;44:2089–2092. doi: 10.1002/anie.200462516. [DOI] [PubMed] [Google Scholar]
- 32.Zech SG, Wand AJ, McDermott AE. Protein structure determination by high-resolution solid-state NMR spectroscopy: Application to microcrystalline ubiquitin. J Am Chem Soc. 2005;127:8618–8626. doi: 10.1021/ja0503128. [DOI] [PubMed] [Google Scholar]
- 33.Manolikas T, Herrmann T, Meier BH. Protein structure determination from 13C spin-diffusion solid-state NMR spectroscopy. J Am Chem Soc. 2008;130(12):3959–3966. doi: 10.1021/ja078039s. [DOI] [PubMed] [Google Scholar]
- 34.Zhou DH, et al. Solid-rate protein-structure determination with proton-detected triple-resonance 3D magic-angle-spinning NMR spectroscopy. Angew Chem Int Ed Engl. 2007;46:8380–8383. doi: 10.1002/anie.200702905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nieuwkoop AJ, Wylie BJ, Franks WT, Shah GJ, Rienstra CM. Atomic resolution protein structure determination by three-dimensional transferred echo double resonance solid-state nuclear magnetic resonance spectroscopy. J Chem Phys. 2009;131:095101. doi: 10.1063/1.3211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franks WT, et al. Dipole tensor-based atomic-resolution structure determination of a nanocrystalline protein by solid-state NMR. Proc Natl Acad Sci USA. 2008;105:4621–4626. doi: 10.1073/pnas.0712393105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou G, Paramasivam S, Byeon IJ, Gronenborn AM, Polenova T. Determination of relative tensor orientations by gamma-encoded chemical shift anisotropy/heteronuclear dipolar coupling 3D NMR spectroscopy in biological solids. Phys Chem Chem Phys. 2010;12:14873–14883. doi: 10.1039/c0cp00795a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartzell CJ, Whitfield M, Oas TG, Drobny GP. Determination of the nitrogen-15 and carbon-13 chemical-shift tensors of L-[13C]Alanyl-L-[15N]Alanine from the dipole-coupled powder patterns. J Am Chem Soc. 1987;109:5966–5969. [Google Scholar]
- 39.Oas TG, Hartzell CJ, Dahlquist FW, Drobny G. 15N Amide chemical-shift tensors of several dipeptides. Biophys J. 1986;49:A328. [Google Scholar]
- 40.Oas TG, Hartzell CJ, Dahlquist FW, Drobny GP. The amide 15N chemical-shift tensors of 4 peptides determined from 13C dipole-coupled chemical-shift powder patterns. J Am Chem Soc. 1987;109:5962–5966. [Google Scholar]
- 41.Gronenborn AM, et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein-G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 42.Chan JCC, Tycko R. Recoupling of chemical shift anisotropies in solid-state NMR under high-speed magic-angle spinning and in uniformly 13C-labeled systems. J Chem Phys. 2003;118:8378–8389. [Google Scholar]
- 43.Zhao X, Eden M, Levitt MH. Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chem Phys Lett. 2001;342:353–361. [Google Scholar]
- 44.LeMaster DM, Kushlan DM. Dynamical mapping of E-coli thioredoxin via 13C NMR relaxation analysis. J Am Chem Soc. 1996;118:9255–9264. [Google Scholar]
- 45.Cai L, Fushman D, Kosov DS. Density functional calculations of chemical shielding of backbone 15N in helical residues of protein G. J Biomol NMR. 2009;45:245–253. doi: 10.1007/s10858-009-9358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clore GM, Kuszewski J. χ1 Rotamer populations and angles of mobile surface side chains are accurately predicted by a torsion angle database potential of mean force. J Am Chem Soc. 2002;124:2866–2867. doi: 10.1021/ja017712p. [DOI] [PubMed] [Google Scholar]
- 47.Kuszewski J, Gronenborn AM, Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grishaev A, Bax A. An empirical backbone-backbone hydrogen-bonding potential in proteins and its applications to NMR structure refinement and validation. J Am Chem Soc. 2004;126:7281–7292. doi: 10.1021/ja0319994. [DOI] [PubMed] [Google Scholar]
- 49.Hing AW, Vega S, Schaefer J. Transferred-echo double-resonance NMR. J Magn Reson. 1992;96:205–209. [Google Scholar]
- 50.Jaroniec CP, Filip C, Griffin RG. 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly 13C, 15N-labeled solids. J Am Chem Soc. 2002;124:10728–10742. doi: 10.1021/ja026385y. [DOI] [PubMed] [Google Scholar]
- 51.Doreleijers JF, Rullmann JAC, Kaptein R. Quality assessment of NMR structures: A statistical survey. J Mol Biol. 1998;281:149–164. doi: 10.1006/jmbi.1998.1808. [DOI] [PubMed] [Google Scholar]
- 52.Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR. 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Tasayco ML, Polenova T. Dynamics of reassembled thioredoxin studied by magic angle spinning NMR: Snapshots from different time scales. J Am Chem Soc. 2009;131:13690–13702. doi: 10.1021/ja9037802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao LS, Grishaev A, Cornilescu G, Bax A. Site-specific backbone amide 15N chemical shift anisotropy tensors in a small protein from liquid crystal and cross-correlated relaxation measurements. J Am Chem Soc. 2010;132:4295–4309. doi: 10.1021/ja910186u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.James F, Roos M. Minuit—system for function minimization and analysis of parameter errors and correlations. Comput Phys Commun. 1975;10:343–367. [Google Scholar]
- 56.Veshtort M, Griffin RG. SPINEVOLUTION: A powerful tool for the simulation of solid and liquid state NMR experiments. J Magn Reson. 2006;178:248–282. doi: 10.1016/j.jmr.2005.07.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.