

Abstract
Two questions important to the success in metalloenzyme design are how to attach or anchor metal cofactors inside protein scaffolds, and in what way such positioning affects enzymatic properties. We have previously reported a dual anchoring method to position a nonnative cofactor, MnSalen (1), inside the heme cavity of apo sperm whale myoglobin (Mb) and showed that the dual anchoring can increase both the activity and enantioselectivity over the single anchoring methods, making this artificial enzyme an ideal system to address the above questions. Here we report systematic investigations of the effect of different covalent attachment or anchoring positions on reactivity and selectivity of sulfoxidation by the MnSalen-containing Mb enzymes. We have found that changing the left anchor from Y103C to T39C has an almost identical effect of increasing rate by 1.8-fold and increasing selectivity by +14% for S, whether the right anchor is L72C or S108C. At the same time, regardless of the identity of the left anchor, changing the right anchor from S108C to L72C increases rate by 4-fold and selectivity by +66%. The right anchor site was observed to have a greater influence than the left anchor site on the reactivity and selectivity in sulfoxidation of a wide scope of other ortho-, meta- and para- substituted substrates. The 1•Mb(T39C/L72C) showed the highest reactivity (TON up to 2.31 min-1) and selectivity (ee% up to 83%) among the different anchoring positions examined. Molecular dynamic simulations indicate that these changes in reactivity and selectivity may be due to the steric effects of the linker arms inside the protein cavity. These results indicate that small differences in the anchor positions can result in significant changes in reactivity and enantioselectivity, probably through steric interactions with substrates when they enter the substrate-binding pocket, and that the effects of right and left anchor positions are independent and additive in nature. The finding that the anchoring arms can influence both the positioning of the cofactor and steric control of substrate entrance will help design better functional metalloenzymes with predicted catalytic activity and selectivity.
INTRODUCTION
Rational design of metalloenzymes has received increasing attention not only to improve our knowledge of protein structure and function but also because it can result in novel biocatalysts for industrial applications.1-14 An important branch of this field is designing artificial enzymes containing non-native metal cofactors, particularly synthetic inorganic or organometallic compounds that have been shown to be powerful catalysts.15-34 In addition to expanding enzymatic activities, such an endeavor can impart water solubility and asymmetry to these catalysts. Since these non-native metal cofactors are never found in nature, it is much more difficult to incorporate them into proteins. Therefore two questions important to the success in designing such metalloenzyme are how to attach or anchor metal cofactors inside protein scaffolds, and in what way such positioning affects enzymatic properties.
Naturally occurring metalloproteins use either a noncovalent positioning through electrostatic, hydrogen bonding and hydrophobic interactions as observed for the heme in myoglobin, or covalent attachment between amino acid side chains and substituents on the periphery of the heme, as observed in cytochrome c.35 Inspired by Nature’s way of positioning native metal cofactors, protein designers have incorporated nonnative metal cofactors into proteins through both noncovalent8,12,28-31,37-46 and covalent47-54 approaches. For example, the planar nature of the protein pocket in heme proteins such as myoglobin has been exploited for incorporation of roughly planar nonnative catalysts such as the highly active and versatile MnSalen complexes.6,28,29 Consequently, both noncovalent10,29,37,55-57 and covalent attachment strategies53 have been employed to incorporate metal salens into the myoglobin (Mb) cavity and used for asymmetric sulfoxidation. While high reaction rates and enantioselectivities similar to those reported in the mature field of small molecule asymmetric synthesis have yet to be realized, these studies represent a significant advancement in a relatively young field.
Despite the widespread recognition of the importance of metal cofactor positioning for control and fine-tuning of functional properties of metalloenzymes, especially those containing nonnative cofactors, few systematic investigations of this factor have been reported; careful systematic studies are required to obtain deeper insights into the design process and its effects on activities. We previously reported an artificial biocatalyst constructed via covalent incorporation of a N,N’-Bis(4-(2-methanesulfonylthioethoxy)salicylidene)-1,2-ethanediamino-manganese(III) bromide (MnSalen complex, 1, see Figure 1A) into the heme cavity of apo sperm whale myoglobin (Mb) and showed that covalent attachment at two points (dual anchoring) to the protein resulted in much higher incorporation yield, reaction rate and enantioselectivity than either non-covalent and single-point covalent attachment methods.53 This dual anchor artificial enzyme provides an excellent opportunity to examine the effect of anchor position on the functional properties of an enzyme such as reaction rate and selectivity. Herein we report systematic investigations of the effect of covalent attachment or anchoring position on reactivity and selectivity of these MnSalen-containing Mb enzymes in asymmetric sulfoxidation of a number of aryl methyl sulfides, and show that small differences in the anchor positions resulted in significant change in reaction rate and enantioselectivity. Most interestingly, we found for the first time that such fine-tuning is independent and additive in nature at each of the dual anchor positions. Molecular dynamic simulations were carried out to explain these observations.
Figure 1.

(A) MnSalen cofactor 1 with methyl thiosulfonate linking arms used for covalent attachment to Cys inside the Mb cavity. (B) Computer model of the Mb cavity with MnSalen (green) positioned to overlap the space occupied by the native heme cofactor. The residues selected as possible anchor positions are visible, left anchors T39 and Y103 are purple and right anchors L72 and S108 are orange.
EXPERIMENTAL
Computer Modeling and Protein Design
Computer modeling was carried out using the Molecular Operating Environment suite (MOE).58 The X-ray crystallographic coordinates of an achiral salen with an axial imidazole ligand (Cambridge structural database ID SAJLIY59) were used as a base model for 1. The linking arms (to the sulfide only) were added using the molecule builder in MOE and the atoms of the coordinating imidazole removed. After energy minimization of the arms with the Charmm22 force field, 1 was modeled into the energy minimized Mb heme pocket (oxymyoglobin, 1.6 Å PDB ID 1MBO60). Placement of the artificial cofactor was modeled to mimic the native cofactor positioning by aligning the metals and the metal coordinating atoms of the native porphyrin and the model salen complex. The heme atoms were removed and the geometry of the linking arms was adjusted by minimizing their dihedral energies. The model was then manually inspected to identify residues pointing into the Mb pocket and near the predicted location of the sulfide group of the linking arm.
Molecular dynamics were performed using MOE with an NVT ensemble at 300K with a 2 femptosecond step. Using the base model generated for selection of anchor points, point mutations were introduced using the sequence editor. Each sulfide of the Cys residue was covalently attached to the corresponding sulfide at the end of the linking arms. The position of the Mn atom, the four atoms directly coordinated to it and residues further than 10Å from the Mn were fixed and the structure minimized using the Charmm22 force field until the RMS gradient of the potential energy was less than 0.05 kJ mol-1 Å-1.
Molecular dynamic simulations of thioanisole entering the Mb cavity of the 1•Mb(T39C/L72C) and 1•apoMb(T39C/S108C) variants were evaluated from an initial state of the substrate π-π stacking with the phenyl ring of the Salen cofactor on the right side of the pocket (looking into the protein cavity with proximal histidine down). With the methyl group eclipsing the phenyl ring, the substrate was manually placed in the starting π-π stacking configuration while subjecting the system to continual energy minimization. The energy of the structure was then minimized until the RMS gradient of the potential energy fell below 1 kJ mol-1 Å-1. The simulations proceeded for 3 picoseconds at which time all models showed that the substrate molecule had reached a similar location in the back left of the protein cavity.
Construction, Expression and Purification of Myoglobin Variants and their conjugation to MnSalen
Construction, expression, and purification of myoglobin (Mb) with Y103C/S108C, T39C/S108C, Y103C/L72C and T39C/L72C mutations (as dual anchor points for MnSalen complex 1) were carried out using procedures reported previously (see supporting information).27,53,54,61,62 The heme from these proteins were then removed by extraction with butanone.35,39,41,42 To incorporate MnSalen complex (1) into the apo-protein, 10 equiv. of methanthiosulfonate-modified Mn(III)Salen complex (N,N’-bis(4-(2-methanesulfonylthioethoxy)salicylidene)-1,2-ethanediamino-manganese(III) bromide) in DMSO solution was added to 0.1 mM apo-Mb solution in 50 mM ammonia acetate buffer (pH= 5.1) at room temperature for 1-2 h, and the conjugation process was monitored by ESI mass spectrometry until the disappearance of the molecular ion peak of apo-Mb. The excess MnSalen compound and DMSO solvent was then removed from the protein by exchanging the buffer using a gel filtration column (PD-10 by Pharmacia).
Electrospray Mass spectra were obtained using a Quattro II Quadrupole Mass Spectrometer (Micromass, UK). Mass scale was calibrated using sperm whale Mb in water (25 pmoles/μl). Samples were exchanged into 50 mM ammonium acetate buffer at pH 5.1 before being injected into the mass spectrometer.
Sulfoxidation activity of the four artificial enzymes were assessed at pH 5.1 as reported previously for the 1•.Mb(Y103C/L72C) enzyme (see supporting information).53
RESULTS AND DISCUSSION
Computer modeling was used to predict the location of multiple anchor locations in sperm whale myoglobin that would position the MnSalen cofactor 1 (Figure 1) in nearly the same location as the native heme within the protein pocket. Residues selected from the model as potential anchor points are T39, L72, Y103, and S108 (Figure 1). The anchoring residues can be grouped into left (T39 or Y103) and right (L72 or S108) side anchors. Combination of all possible left and right pairs generates four unique dual anchored artificial biocatalysts, differing mainly by location of the covalent attachment.
The UV/vis spectra of the MnSalen-Mb conjugates display absorption bands at 280 nm, which is assignable to absorption by the protein, and 292 nm with a shoulder near 340 nm, which correspond to absorption by the MnSalen complex (see Supporting information). The absorption ratio between 280 and 292 nm of the purified bioconjugates is consistent with incorporation of one MnSalen per Mb based on their respective extinction coefficients at the corresponding wavelengths. To confirm the conjugation of 1 to Mb variants at the dual anchoring positions, we collected electrospray mass spectra of the variants after passing the proteins through a gel filtration column to remove excess compound 1. If no incorporation of 1 into the protein occurred, the molecular weight (MW) would be consistent with that of apo-Mb. If incorporation of 1 into the protein by a single anchor, the protein should exhibit MW of apo-Mb plus 1 containing a single thiosulfonate group, because one of the two thiosulfonate groups is removed by reaction with the anchoring Cys. Finally, if anchoring occurs at both sites the predicted MW would be that of apo-Mb plus 1 without either of the two thiosolfonate groups. Indeed the observed MW of all four variants match the latter well (see Table S1), indicating 100% conversion of the proteins into the dually anchored MnSalen biocatalysts 1•.Mb(Y103C/S108C), 1•.Mb(T39C/S108C), 1•.Mb(Y103C/L72C) and 1•.Mb(T39C/L72C).
To determine the effect of anchor position on rate and selectivity the sulfoxidation of thioanisole was carried out with each of the four artificial biocatalysts (Table 1, entries 1-4). Both the rate and selectivity of these dual anchored catalysts are higher than the MnSalen 1 alone (Table 1, entry 5), proteins without MnSalen 1 (Table 1, entry 6-7), protein with MnSalen 1 but without covalent attachment (Table 1, entry 8), or proteins with a single anchor (Table 1, entries 9-10), suggesting that the dual anchor approach is superior in positioning the metal cofactor for more efficient and selective reactions. To determine the effect of anchor position on the reactivity and selectivity, we first consider fixing the Y103C left anchor position and investigate the effect of changing the right anchor position. For the 1•Mb(Y103C/S108C) variant, a rate of 0.095 min-1 and selectivity of 15% for the R product were observed (Table 1). Interestingly, changing the right anchor from S108C to the L72C position in the 1•Mb(Y103C/L72C) variant increases the rate by over 4 fold to 0.390 min-1 and selectivity is reversed 66% to 51% selectivity for the S sulfoxide (entries 1 and 2). Based on these results, we conclude that small changes in the right anchor positions can have a significant effect on both the reaction rate and enantioselectivity.
Table 1.
Effect of Attachment and Position of Mnsalen inside Mb Scaffolds on Enantioselective Sulfoxidation of Thioanisole.a
| Entry | Catalyst (position) | Rateb (10-3 min-1) | ee%c (S) | Attachment |
|---|---|---|---|---|
| 1 | 1•Mb(Y103C/S108C) | 95 ± 16 | -15 | Double |
| 2 | 1•Mb(Y103C/ L72C) | 390 ± 30 | 51 | Double |
| 3 | 1•Mb(T39C/S108C) | 177 ± 87 | 1 | Double |
| 4 | 1•Mb(T39C/L72C) | 684 ± 74 | 66 | Double |
| 5 | MnSalenSS | 2 | 1 | Cofactor only |
| 6 | Apo Mb (T39C/L72C) | 6 | 2 | No cofactor |
| 7 | Apo-Wt Mb | 5 | 1 | No cofactor |
| 8 | Apo Mb (T39C/L72C) and MnSalen | 19 | 2 | Non-covalent |
| 9 | 1•Mb (Y103C) | 51 | 12 | single |
| 10 | 1•Mb (L72C) | 43 | 10 | single |
In 50 mM NH4OAc (pH 5.1), 130 μM catalyst, thioanisole (5 mM), H2O2 (5 mM) reacted for 10 min at 4°C.
The unit of the rate is 10-3 turnover min-1.
Reaction rates and ee% were determined by GC analysis using an ASTEC G-TA cyclodextrin column, and acetophenone was added as an internal standard. The identities of the enantiomers of sulfoxide were determined by reference to previous literature.65 Typical reaction procedures are described in supporting information.
To investigate if modulation of activity and selectivity by the left and right anchors are independent of each other, we next examine the reactivity and selectivity of Mb variants as a function of varying the left anchor positions with a common right anchor. With the L72C right anchor fixed, exchanging left anchor Y103C for T39C results in a 1.8-fold increase in rate (from 390 ± 30 × 10-3 min-1 for 1•Mb(Y103C/ L72C) to 684 ± 74 × 10-3 min-1 for 1•Mb(T39C/L72C) and +15% increase in selectivity for S (from 51% to 66%, Table 1). Interestingly, when Y103C is changed to T39C using the S108C right anchor, increases of 1.9-fold for rate (from 95 ± 16 × 10-3 min-1 for 1•Mb(Y103C/ S108C) to 187 ± 87 × 10-3 min-1 for 1•Mb(T39C/S108C) and +16 selectivity for S (15% for R to 1% for S respectively) are again observed (Table 1). It is remarkable that whether the right anchor is L72C or S108C, changing the left anchor from Y103C to T39C has an almost identical effect of increasing rate by 1.8-fold and increasing selectivity by +16% for S. At the same time, regardless of the identity of the left anchor, changing the right anchor from S108C to L72C increases rate by 4-fold and selectivity by +66%. Plotting the selectivity vs. rate data for the four variants shows well defined changes associated with altering each anchor position (see Figure 2). The parallelogram shape observed in Figure 2 highlights the fact that the effect of changing an anchor position is the same regardless of the identity of the other anchor. The selectivity and rate of catalysis also appear to correlate with each other as the faster the catalyst, the more selective it is. Therefore, we conclude that the effects of linker position in our dual anchored artificial biocatalyst are additive in nature with independent effects from right and left sides. The increases upon changing anchor positions, from 1•Mb(Y103C/S108C) to 1•Mb(T39C/L72C), result in total rate enhancements of 7 fold and an 80% increase in selectivity for the S enantiomer.
Figure 2.
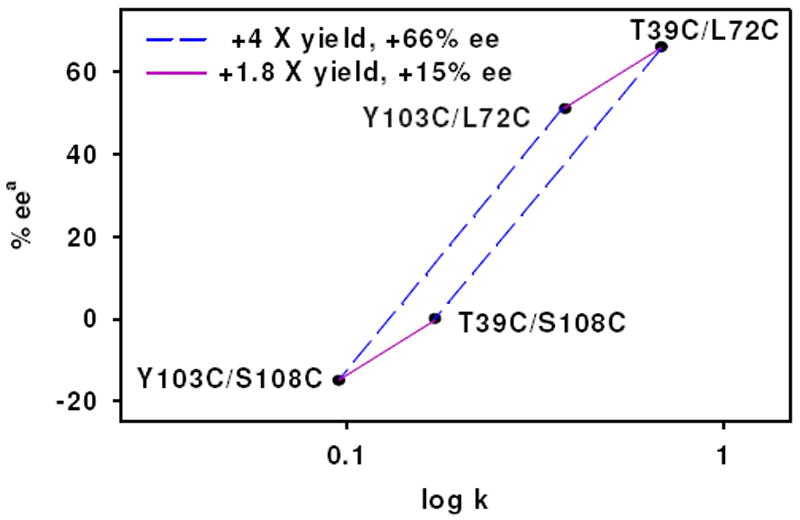
Plot of selectivity for the S sulfoxide vs. rate of sulfoxidation, plotted in log scale, demonstrating the additive nature of the anchor position effect on reactivity and selectivity. The identity of the anchors is denoted near the point. Identical changes in rate and selectivity are observed when exchanging a linking position regardless of the identity of the other anchor. Blue dashed lines indicate change of right anchor (4 fold increase in rate and 66% increased ee) and purple solid lines indicate change of left anchor (1.8 fold increase in rate and 15% increased ee). aS product is defined as positive.
To more fully understand the reason for the observed effects of anchor position on the sulfoxidation reaction we evaluated the sulfoxidation of a series of aryl methyl sulfides with a varied R groups (H, Cl, Br and Me) at different positions (ortho-, meta- and para-) on the phenyl ring under the same reaction conditions, and the results are summarized in Table 2. All of the substrates reported in Table 2 are similar in overall trend to thioanisole with regards to the influences of anchor position on reactivity and selectivity. Throughout the series of substrates, 1•Mb(T39C/L72C) is superior to 1•Mb(Y103C/S108C), 1•Mb(T39C/S108C) and 1•Mb(Y103C/L72C) regarding both reaction rate and enantioselectivity. For a given substrate, reaction rate and selectivity for the S enantiomer increase across the series of anchor positions (1•Mb(Y103C/S108C), 1•Mb(T39C/S108C), 1•Mb(Y103C/L72C) and 1•Mb(T39C/L72C)) (Table 2). The average reaction rate of all substrates by catalyst are 0.07, 0.15, 0.44 and 1.04 min-1 for 1•Mb(Y103C/S108C), 1•Mb(T39C/S108C), 1•Mb(Y103C/L72C) and 1•Mb(T39C/L72C), respectively. Interestingly, 1•Mb(T39C/L72C) showed 2.4-, 6.9-, and 14.8-folds faster reaction rates than 1•Mb(Y103C/L72C), 1•Mb(T39C/S108C) and 1•Mb(Y103C/S108C), respectively. Significant effects of the anchor position on the enantioselective sulfoxidation of these substrates were also observed. Although absolute configurations of some substrates such as 3-Cl, 4-F, 2-Br and 3-Br were not determined, 1•Mb(T39C/L72C) showed the highest ee% among the four artificial biocatalysts for all the substrates. It is noteworthy that for 1•Mb(T39C/L72C), ee% is as high as 83% was obtained using 3-chlorophenyl methyl sulfide. The 1•Mb(T39C/L72C), 1•Mb(Y103C/L72C) and 1•Mb(T39C/S108C) all have similar enantiomeric preference. However, with 1•Mb(Y103C/S108C) the preference for enantiomers is reversed for all the substrates. Clearly, the anchor positions are strongly influencing both rate of sulfoxidation and the enantioselectivity of the reaction in a systematic fashion regardless of substrate identity.
Table 2.
Effects of Anchor Sites on Asymmetric Sulfoxidation of Aryl Methyl Sulfides (R= Phenyl Substituent).a
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R | 1•Mb(Y103C/S108C) | 1•Mb(T39C/S108C) | 1•Mb(Y103C/L72C) | 1•Mb(T39C/L72C) | ||||
| Rateb,c | ee% c | Rate b,c | ee% c | Rateb,c | ee% c | Rate b,c | ee% c | ||
| 1 | H | 95 ± 16 | 15, R | 177 ± 87 | 1, S | 390 ± 30 | 51, S | 684 ± 74 | 66, S |
| 2 | 2-Cl | 114 ±12 | 21 ± 1, R | 161 ±13 | 3 ± 1,S | 464 ±41 | 37 ± 2, S | 1249±91 | 48 ± 1, S |
| 3 | 3-Cl | 108 ± 16 | 2 ± 1, (-)d | 194 ± 37 | 18 ± 1, (+)d | 525 ± 50 | 69 ± 2, (+)d | 2318 ± 119 | 83 ± 1, (+)d |
| 4 | 4-Cl | 49 ± 3 | 8 ± 4, R | 98 ± 24 | 4 ± 2, R | 439 ± 33 | 57 ± 1, S | 1459 ± 72 | 79 ± 2, S |
| 5 | 4-F | 97 ± 9 | 2 ± 2, (-)d | 158 ± 29 | 12 ± 3, (+)d | 332 ± 40 | 53 ± 0, (+)d | 1929 ± 110 | 71 ± 3, (+)d |
| 6 | 2-Br | 34 ± 9 | 7 ± 2, (-)d | 97 ±6 | 17 ± 3, (+)d | 347 ± 47 | 26 ± 1, (+)d | 698 ± 71 | 35 ± 0, (+)d |
| 7 | 3-Br | 82 ± 17 | 18 ± 3, (-)d | 178 ± 13 | 1 ± 2, (+)d | 406 ± 14 | 46 ± 2, (+)d | 971 ± 72 | 74 ± 2, (+)d |
| 8 | 4-Br | 34 ± 4 | 13 ± 2, R | 89 ± 23 | 3 ± 2, S | 325 ± 32 | 26 ± 2, S | 676 ± 76 | 37 ± 2, S |
| 9 | 3-Me | 52 ± 7 | 23 ± 1, R | 133 ± 10 | 9 ± 2, R | 516 ± 43 | 60 ± 4, S | 856 ± 63 | 67 ± 4, S |
| 10 | 4-Me | 101 ± 30 | 2 ± 1, R | 126 ± 12 | 10 ± 2, S | 302 ± 13 | 31 ± 2, S | 1260 ± 76 | 52 ± 3, S |
In 50 mM NH4OAc (pH 5.1), 130 μM catalyst, thioanisole (5 mM), H2O2 (5 mM) reacted for 10 min at 4°C.
The unit of the rate is 10-3 turnover min-1.
Reaction rates and ee% were determined by GC analysis using an ASTEC G-TA cyclodextrin column, and acetophenone was added as an internal standard. The identities of the enantiomers of sulfoxide were referred to previous literature.65
R,S order of elution not confirmed. In this work, we assigned the enantioselectivity of sulfoxides the same as that obtained by 1•Mb(T39C/L72C) as (+).
Since our goal is to understand factors useful for building the fastest and most selective biocatalysts the large influence on rate and selectivity observed for the L72 anchor position is particularly interesting. Using a noncovalently attached MnSalen, Watanabe and co-workers demonstrated dependence of selectivity on depth of the cofactor in the Mb pocket.56 Based on the results, they proposed a model that predicts increasing S selectivity with increasing depth in the pocket. Attempts to apply their model to our system were unsuccessful as the deeper S108C anchor favors R instead while the shallower L72C anchor heavily favors S. This difference between Watanabe’s noncovalent system and our dual anchored system leads us to conclude that the covalent anchors themselves must be influencing the catalyst’s reactivity.
In addition to the depth in the protein pocket, another difference in the right anchoring positions is that, while anchoring at S108C should position the right linking arm of 1 in plane with the Salen ligand, the anchoring at position L72 should draw the linking arm up into the distal protein cavity (Figure 1C). Careful X-ray crystallographic studies of the distal pocket in Mb have previously identified two small internal clefts. The first of these occurs on the left side of the pocket and has been shown to be the initial location of CO that is photo dissociated from the heme cofactor.66-69 The second cleft is located on the right side of the distal pocket and is the fourth of the Xe binding pockets (Xe4) identified by Tilton.70 X-ray crystallographic71 and kinetic studies72,73 of photolyzed CO have indicated that access to these clefts is tunable by mutagenesis and is susceptible to steric modifications of the distal pocket. Computer models of our dual anchored artificial biocatalyst predict that covalent attachment of the cofactor at L72C would position the linking arm near the entrance of the Xe4 cavity (Figure 3A). The significant effects of the position of the right anchor on reactivity and selectivity, and the presence of several bulky amino acids on the left side of the heme pocket (Phe43, Arg45 and His64) suggest that the thioanisole enters the protein cavity on the right side and should enter, at least partially, the Xe4 binding cleft. Gaining access to this slightly larger space could have important implications for catalyst selectivity by allowing the substrate more conformational freedom. The steric constraints introduced by cofactor anchoring at L72C (Figure 3A) would inhibit substrate entrance into the Xe4 binding pocket. The absence of such a barrier, as is predicted for the S108C anchor (Figure 3B), would allow the substrate to move across the phenyl ring of the cofactor and into the Xe4 pocket. Thus, by introducing steric bulk of the covalent attachment above the salen, the L72C anchor could alter the substrate access path.
Figure 3.

Computer models of A) 1•Mb(T39C/L72C) and B) 1•Mb(T39C/S108C). The residues lining the Xe4 pocket are shown in white (V68 is transparent to allow visualization of the Xe4 cavity). Cysteine anchor points are shown in red and the position of residues corresponding to anchors in the other variants are shown in orange. Atoms in the MnSalen are shown in green. Yellow arrow indicates the Xe4 cleft.
To assess the possible steric effects that the L72C anchor position may have on protein substrate interactions, we conducted molecular dynamic simulations of the substrate entering the cofactor cavity of the 1•Mb(T39C/L72C) and 1•Mb(T39C/S108C) variants. As shown in Figure 4A, the thioanisole (in blue) in 1•Mb(T39C/L72C) travels across the upper face of the salen (path indicated by white arrow). In 1•Mb(T39C/S108C), the substrate travels directly into the Xe4 binding cleft. Evidently, more room exists in the back of the cavity as multiple conformations of the substrate are observed for 1•Mb(T39C/S108C), predicting the low enantioselectivity observed experimentally. The differences in substrate access path observed in the molecular dynamic simulations support the hypothesis that the covalent link between the artificial cofactor and the L72C anchor directs the substrate away from the Xe4 binding cleft and that this interaction could alter the stereo selectivity of the artificial enzyme. Therefore, the covalent anchoring of the cofactor in our system appears to fulfill some of the roles observed for similar covalent cofactors in native protein systems. In addition to improving cofactor affinity, the covalent anchor also participate directly in positioning of the substrate during catalysis.
Figure 4.

Molecular dynamic simulations of thioanisole entering the active site of artificial biocatalysts A) 1•Mb(T39C/L72C) and B) 1•Mb(T39C/S108C). Overlaid snapshots (0.5 ps each) of thioanisole molecules are in blue with color progressing from lighter to darker with elapsed time. Residues lining the Xe4 binding cleft are shown in white (for clarity only atom positions for the first structure are shown) and V68 is transparent to allow visualization inside the Xe4 cleft, MnSalen is shown in green and histidine 64 is shown in grey. The white arrow indicates the general path taken by substrate as it enters the binding cavity.
CONCLUSIONS
We have shown that anchor positions are crucial for the dual covalent attachment of a nonnative MnSalen cofactor inside a protein pocket, even if very similar cofactor positioning is expected with different anchors. Seemingly inconsequential differences in the anchor positions can have significant effects on the reactivity and selectivity when creating artificial enzymes for asymmetric catalysis. More importantly, tuning of the reactivity of the artificial cofactor by anchor point selection was shown to be independent of the anchor position and additive in nature. Our findings that the linking arms can influence both the positioning of the cofactor and steric control of substrate entrance in a predictable way can form a strong base for future protein design.
Supplementary Material
Acknowledgments
This material is based upon work supported by National Science Foundation of China (grand no. 20971007), the US National Institute of Health (GM062211) and US National Science Foundation (CHE-10-58959).
References
- 1.Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tann C-M, Qi D, Distefano MD. Curr Opin Chem Biol. 2001;5:696. doi: 10.1016/s1367-5931(01)00268-x. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh D, Pecoraro VL. Curr Opin Chem Biol. 2005;9:97. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Reedy CJ, Gibney BR. Chem Rev (Washington, DC, U S) 2004;104:617. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 5.Harris KL, Lim S, Franklin SJ. Inorg Chem. 2006;45:10002. doi: 10.1021/ic060877k. [DOI] [PubMed] [Google Scholar]
- 6.Lu Y. Angew Chem Int Ed. 2006;45:5588. doi: 10.1002/anie.200600168. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y. Inorg Chem. 2006;45:9930. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward TR. Acc Chem Res. 2011;44:47. doi: 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- 9.Rosati F, Roelfes G. Chemcatchem. 2010;2:916. [Google Scholar]
- 10.Jing Q, Kazlauskas RJ. Chemcatchem. 2010;2:953. [Google Scholar]
- 11.Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohler V, Wilson YM, Lo C, Sardo A, Ward TR. Curr Opin Biotechnol. 2010;21:744. doi: 10.1016/j.copbio.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Ueno T, Yokoi N, Abe S, Watanabe Y. J Inorg Biochem. 2007;101:1667. doi: 10.1016/j.jinorgbio.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe Y, Hayashi T. Prog Inorg Chem. 2005;54:449. [Google Scholar]
- 15.Roth LE, Nguyen JC, Tezcan FA. J Am Chem Soc. 2010;132:13672. doi: 10.1021/ja1071866. [DOI] [PubMed] [Google Scholar]
- 16.Popp BV, Ball ZT. J Am Chem Soc. 2010;132:6660. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]
- 17.Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, DeGrado WF. J Am Chem Soc. 2010;132:15516. doi: 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivankin A, Livne L, Mor A, Caputo GA, DeGrado WF, Meron M, Lin B, Gidalevitz D. Angew Chem Int Ed. 2010;49:8462. doi: 10.1002/anie.201003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakraborty S, Touw DS, Peacock AFA, Stuckey J, Pecoraro VL. J Am Chem Soc. 2010;132:13240. doi: 10.1021/ja101812c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radford RJ, Tezcan FA. J Am Chem Soc. 2009;131:9136. doi: 10.1021/ja9000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat Chem Biol. 2009;5:882. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambasivan R, Ball ZT. J Am Chem Soc. 2010;132:9289. doi: 10.1021/ja103747h. [DOI] [PubMed] [Google Scholar]
- 23.Radford RJ, Brodin JD, Salgado EN, Tezcan FA. Coord Chem Rev. 2011;255:790. [Google Scholar]
- 24.Brustad EM, Arnold FH. Curr Opin Chem Biol. 2011;15:201. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pordea A, Ward TR. Synlett. 2009:3225. [Google Scholar]
- 26.Fruk L, Kuo CH, Torres E, Niemeyer CM. Angew Chem Int Ed. 2009;48:1550. doi: 10.1002/anie.200803098. [DOI] [PubMed] [Google Scholar]
- 27.Davies CL, Dux EL, Duhme-Klair AK. Dalton Trans. 2009:10141. doi: 10.1039/b915776j. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi T, Hitomi Y, Takimura T, Tomokuni A, Mizutani T, Hisaeda Y, Ogoshi H. Coord Chem Rev. 1999;190-192:961. [Google Scholar]
- 29.Ueno T, Abe S, Yokoi N, Watanabe Y. Coord Chem Rev. 2007 Article in Press. [Google Scholar]
- 30.Letondor C, Ward TR. ChemBioChem. 2006;7:1845. doi: 10.1002/cbic.200600264. [DOI] [PubMed] [Google Scholar]
- 31.Reetz MT, Peyralans JJP, Maichele A, Fu Y, Maywald M. Chem Commun (Cambridge, U K) 2006:4318. doi: 10.1039/b610461d. [DOI] [PubMed] [Google Scholar]
- 32.Lu Y. Curr Opin Chem Biol. 2005;9:118. doi: 10.1016/j.cbpa.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Zaykov AN, Popp BV, Ball ZT. Chem --Eur J. 2011;16:6651. doi: 10.1002/chem.200903092. [DOI] [PubMed] [Google Scholar]
- 34.Durrenberger M, Heinisch T, Wilson YM, Rossel T, Nogueira E, Knorr L, Mutschler A, Kersten K, Zimbron MJ, Pierron J, Schirmer T, Ward TR. Angew Chem Int Ed. 2011;50:3026. doi: 10.1002/anie.201007820. [DOI] [PubMed] [Google Scholar]
- 35.Stevens JM, Daltrop O, Allen JWA, Ferguson SJ. Acc Chem Res. 2004;37:999. doi: 10.1021/ar030266l. [DOI] [PubMed] [Google Scholar]
- 36.Huang L, Wojciechowski G, Ortiz De Montellano PR. J Biol Chem. 2006;281:18983. doi: 10.1074/jbc.M602307200. [DOI] [PubMed] [Google Scholar]
- 37.Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Angew Chem Int Ed. 2003;42:1005. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- 38.Marchetti M, Mangano G, Paganelli S, Botteghi C. Tetrahedron Lett. 2000;41:3717. [Google Scholar]
- 39.Hayashi T, Takimura T, Ogoshi H. J Am Chem Soc. 1995;117:11606. [Google Scholar]
- 40.Wilson ME, Whitesides GM. J Am Chem Soc. 1978;100:306. [Google Scholar]
- 41.Mahammed A, Gross Z. J Am Chem Soc. 2005;127:2883. doi: 10.1021/ja045372c. [DOI] [PubMed] [Google Scholar]
- 42.Reetz MT, Jiao N. Angew Chem Int Ed. 2006;45:2416. doi: 10.1002/anie.200504561. [DOI] [PubMed] [Google Scholar]
- 43.Collot J, Gradinaru J, Humbert N, Skander M, Zocchi A, Ward TR. J Am Chem Soc. 2003;125:9030. doi: 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]
- 44.Patel AD, Nocek JM, Hoffman BM. J Am Chem Soc. 2005;127:16766. doi: 10.1021/ja0552242. [DOI] [PubMed] [Google Scholar]
- 45.Coulter ED, Cheek J, Ledbetter AP, Chang CK, Dawson JH. Biochem Biophys Res Commun. 2000;279:1011. doi: 10.1006/bbrc.2000.4077. [DOI] [PubMed] [Google Scholar]
- 46.van de Velde F, Konemann L, van Rantwijk F, Sheldon RA. Chem Commun (Cambridge, U K) 1998:1891. [Google Scholar]
- 47.Zhang JL, Garner DK, Liang L, Chen Q, Lu Y. Chem Commun (Cambridge, U K) 2008:1665. doi: 10.1039/b718915j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies RR, Distefano MD. J Am Chem Soc. 1997;119:11643. [Google Scholar]
- 49.Reetz MT. Tetrahedron. 2002;58:6595. [Google Scholar]
- 50.Nicholas KM, Wentworth P, Harwig CW, Wentworth AD, Shafton A, Janda KD. Proc Natl Acad Sci U S A. 2002;99:2648. doi: 10.1073/pnas.052001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kruithof CA, Casado MA, Guillena G, Egmond MR, van der Kerk-van Hoof A, Heck AJR, Gebbink RJMK, van Koten G. Chem --Eur J. 2005;11:6869. doi: 10.1002/chem.200500671. [DOI] [PubMed] [Google Scholar]
- 52.Panella L, Broos J, Jin JF, Fraaije MW, Janssen DB, Jeronimus-Stratingh M, Feringa BL, Minnaard AJ, de Vries JG. Chem Commun (Cambridge, U K) 2005:5656. doi: 10.1039/b512138h. [DOI] [PubMed] [Google Scholar]
- 53.Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. J Am Chem Soc. 2004;126:10812. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 54.Zhang JL, Garner DK, Liang L, Barrios DA, Lu Y. Chem --Eur J. 2009;15:7481. doi: 10.1002/chem.200802449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ueno T, Ohashi M, Kono M, Kondo K, Suzuki A, Yamane T, Watanabe Y. Inorg Chem. 2004;43:2852. doi: 10.1021/ic0498539. [DOI] [PubMed] [Google Scholar]
- 56.Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. J Am Chem Soc. 2005;127:6556. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]
- 57.Ueno T, Koshiyama T, Abe S, Yokoi N, Ohashi M, Nakajima H, Watanabe Y. J Organomet Chem. 2007;692:142. [Google Scholar]
- 58.Molecular Operating Environment, MOE 2005.06. C.C.G., Inc., 1255 University St., Suite 1600, Montreal, Quebec, Canada, H3B 3X3; Montreal, Quebec: 2005. [Google Scholar]
- 59.Matsumoto N, Takemoto N, Ohyosia A, Okawa H. Bull Chem Soc Jpn. 1988;61:2984. [Google Scholar]
- 60.Phillips SEV. J Mol Biol. 1980;142:531. doi: 10.1016/0022-2836(80)90262-4. [DOI] [PubMed] [Google Scholar]
- 61.Springer BA, Sligar SG. Proc Natl Acad Sci U S A. 1987;84:8961. doi: 10.1073/pnas.84.24.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Natl Acad Sci U S A. 2003;100:3629. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teale FWJ. Biochim Biophys Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- 64.Fisher WR, Taniuchi H, Anfinsen CB. J Biol Chem. 1973;248:3188. [PubMed] [Google Scholar]
- 65.Anderson JL, Ding J, McCulla RD, Jenks WS, Armstrong DW. J Chromatogr A. 2002;946:197. doi: 10.1016/s0021-9673(01)01526-6. [DOI] [PubMed] [Google Scholar]
- 66.Teng T-Y, Srajer V, Moffat K. Nat Struct Biol. 1994;1:701. doi: 10.1038/nsb1094-701. [DOI] [PubMed] [Google Scholar]
- 67.Schlichting I, Berendzen J, Phillips GN, Jr, Sweet RM. Nature. 1994;371:808. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 68.Srajer V, Teng T-y, Ursby T, Pradervand C, Ren Z, Adachi S-i, Schildkamp W, Bourgeois D, Wulff M, Moffat K. Science (Washington, D C) 1996;274:1726. doi: 10.1126/science.274.5293.1726. [DOI] [PubMed] [Google Scholar]
- 69.Hartmann H, Zinser S, Komninos P, Schneider RT, Nienhaus GU, Parak F. Proc Natl Acad Sci U S A. 1996;93:7013. doi: 10.1073/pnas.93.14.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tilton RF, Jr, Kuntz ID, Jr, Petsko GA. Biochemistry. 1984;23:2849. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 71.Brunori M, Vallone B, Cutruzzola F, Travaglini-Allocatelli C, Berendzen J, Chu K, Sweet RM, Schlichting I. Proc Natl Acad Sci U S A. 2000;97:2058. doi: 10.1073/pnas.040459697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scott EE, Gibson QH. Biochemistry. 1997;36:11909. doi: 10.1021/bi970719s. [DOI] [PubMed] [Google Scholar]
- 73.Scott EE, Gibson QH, Olson JS. J Biol Chem. 2001;276:5177. doi: 10.1074/jbc.M008282200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.