

Abstract
Peptide nucleic acid (PNA) is a synthetic analogue of DNA that commonly has an N-aminoethlyl-glycine backbone. The crystal structure of two PNA duplexes, one containing eight standard nucleobase pairs (GGCATCGG)2 (pdb: 3MBS), and the other containing the same nucleobase pairs and a central pair of bipyridine ligands (pdb: 3MBU), has been solved with a resolution of 1.2 Å and 1.05 Å, respectively. The non-modified PNA duplex adopts a P-type helical structure s i m i l a r t o that of previously characterized PNAs. The atomic-level resolution of the structures allowed us to observe for the first time specific modes of interaction between the terminal lysines of the PNA and the backbone and nucleobases situated in the vicinity of the lysines, which are considered an important factor in the induction of a preferred handedness in PNA duplexes. These results support the notion that while PNA typically adopts a P-type helical structure, its flexibility is relatively high. For example, the base pair rise in the bipyridine-containing PNA is the largest measured to date in a PNA homoduplex. The two bipyridines are bulged out of the duplex and are aligned parallel to the minor groove of the PNA. In the case of the bipyridine-containing PNA, two bipyridines from adjacent PNA duplexes form a π-stacked pair that relates the duplexes within the crystal. The bulging out of the bipyridines causes bending of the PNA duplex, which is in contrast to the structure previously reported for biphenyl-modified DNA duplexes in solution, where the biphenyls are π-stacking with adjacent nucleobase pairs and adopt an intrahelical geometry [Johar et al., Chem. Eur. J., 2008, 14, 2080]. This difference shows that relatively small perturbations can significantly impact the relative position of nucleobase analogues in nucleic acid duplexes.
Keywords: PNA structure, X-ray crystallography, nucleic acids, bipyridine, nucleic acid bending
Introduction
Peptide nucleic acid (PNA) is a synthetic analogue of DNA that has a pseudo-peptide backbone instead of the sugar diphosphate backbone of the DNA (Figure 1).[1–3] PNA forms homoduplexes by Watson Crick hybridization. To date crystal structures have been reported for a palindromic 6-base pair (bp) PNA duplex,[4] for another 6-bp PNA duplex with the same sequence but which contained lysine at the C-terminus,[5] and for a partly self-complementary single stranded (ss) PNA that formed within the crystal short duplex and triplex motifs.[6] These studies showed that PNA homoduplexes adopt a P-type helix structure, which is more unwound and has larger diameter than DNA duplexes.
Figure 1.

The four single strand PNAs in the asymmetric unit of Bipy PNA crystal structure.
Our research focuses on the incorporation of metal ions at specific locations within PNA duplexes to create hybrid inorganic-nucleic acid nanostructures, which have potential applications in nanotechnology and biology. The substitution of pairs of nucleobases with ligands enables incorporation of metal ions into PNA or other nucleic acid duplexes and it allows control of the number of metal ions and of their position in the duplexes. Numerous DNA and PNA duplexes incorporating one or multiple metal complexes have been studied in the last decade.[7–11] In most of these studies, the ligands incorporated in the nucleic acid duplex were aromatic and the metal complexes were square planar to (1) make possible π stacking between the complex and adjacent base pairs, and (2) minimize structural distortions of the duplex. While the thermal stability of the ligand- and metal-containing nucleic acids has been routinely studied, structural information about the duplexes before and after the incorporation of ligands and metal ions has been obtained only in a few cases. Such information is important for the rational design of metal-containing PNA nanostructures.
The crystal structure of a modified DNA duplex that had an alternating purine-pyrimidine sequence of natural nucleobases and contained two [Dipic•Cu2+•Py] complexes, where Dipic and Py stand for pyridine-2,6-dicarboxylate and pyridine, respectively, revealed a Z-type conformation for the duplex.[12] Each of the two Cu2+ ions situated in the duplex had a distorted octahedral coordination with four equatorial coordination sites occupied by donor atoms from the pyridine-2,6-dicarboxylate and the pyridine ligands, and two axial sites occupied by monodentate nucleobases from the base pairs adjacent to the metal complex. The very recent crystal structure of a DNA duplex that contained three central, adjacent [Imidazole·Ag+·Imidazole] complexes showed that the duplex adopts a B-like structure in which the Ag+ ions are aligned on the helical axis at Ag-Ag distances of 3.9 Å.[13] Based on molecular modeling, Brostchi et al. proposed a “zipper-like” model for the stacking of 2,2′-bipyridine and biphenyl in DNA duplexes.[14] In this model, the distal pyridyl rings of bipyridine ligands situated in complementary positions in the DNA are alternatively stacked, with the DNA backbone being axially stretched to accommodate the pair of stacked bipyridines. A recent NMR spectroscopy and molecular dynamics study of a 10-basepair DNA duplex containing a central pair of asymmetric biphenyls showed that the biphenyl groups are intercalated in the duplex, each of the two biphenyls being π stacked head to tail between the other biphenyl and a neighboring nucleobase pair.[15] The overall structure of the DNA duplex containing the biphenyl displayed only minor differences when compared with B-DNA.
Although the crystal structures of several PNA duplexes have been published,[4–6] no crystal structure of a ligand-modified PNA has been reported to date. In this paper, we describe the crystal structure of an 8-base pair PNA duplex with a palindromic sequence, 8-bp PNA, and of a PNA duplex with the same sequence of nucleobases but which contains a pair of bipyridine (Bipy) ligands situated in the middle of the duplex, Bipy PNA (Scheme 2). The high resolution of these structures, 1.20 Å and 1.05 Å, respectively, allows us to identify atomic details of the structures which had not been observed in previously reported crystal structures of PNA homoduplexes, which had resolutions of 1.70 – 2.60 Å.[4–6]
Scheme 2.

Sequences of non-modified and bipyridine-modified PNA duplexes
Results
With more than 950 non-H-atoms in the asymmetric unit, including more than 200 non H-atoms from ordered solvent and water molecules, the Bipy PNA structure represents the second largest novel equal-atom structure solved by ab intio methods, with the largest being the antibiotic feglymycin, which has 1033 non H-atoms.[16] Although the 8-bp PNA structure is smaller than that of Bipy PNA, with more than 500 atoms in the asymmetric unit it is still one of the largest all-equal atom structures solved to date by direct methods.
The crystal structures of the 8-bp PNA and Bipy PNA are at 1.20 and 1.05 Å resolution, respectively. This high resolution made possible the observation of extensive geometric and conformational details. Four PNA strands comprise the asymmetric unit in both the 8-bp PNA and Bipy PNA structures (Figures 1 and S1). These strands form two antiparallel duplexes, one of which has a left(L)-configuration and the other a right(R)-configuration. The duplexes are stacked head to tail within the crystal leading to a continuous, curvilinear network of alternating right- and left-handed helices (Figure S1b). The only disordered region is at the C-terminal lysines of the 8-bp PNA, which cannot be traced beyond the Cβ position in any of the four strands. However, in the Bipy PNA, two of the four lysines within the unit cell are well-ordered, with low thermal factors (23–28 Å2) and strong electron densities (Figures 2 and S2).
Figure 2.

Structural details of the hydrogen bonds that involve two of the four terminal lysines in the unit cell of Bipy PNA and atoms from the G1 nucleobase and the PNA backbone. Hydrogen bonds are identified by dotted lines.
In the crystal structures of both 8-bp PNA and Bipy PNA, the nucleobases and the PNA backbone are very well-resolved in the electron density maps. All nucleobases participate in Watson-Crick hydrogen bonding. The carbonyl groups of the linker that connects the nucleobases to the PNA backbone point towards the C-end of the PNA (Figure 3). The amide groups of the backbone are in trans configuration and the majority of the backbone carbonyl groups are directed towards the C-end of the PNA and the solution (Figures 3 and S3). In 8-bp PNA, two backbone carbonyl groups, specifically those of the terminal cytosine C8 and of the thymidine T5 have opposite orientation, namely towards the N-end of the PNA and the minor groove of the duplex (Figure S3). For both nucleobases, a well-ordered water molecule is located at the position that would be occupied by the oxygen atom of the backbone carbonyl group if oriented away from the duplex (Figure 4b). The same properties are observed for the backbone carbonyls of Bipy PNA, with only those of the nucleobases A4 and T6 being oriented towards the duplex and with a water molecule found at the position that would be occupied by the oxygen atom of the carbonyl group in the “away from the duplex” configuration.
Figure 3.

Structural features of the Bipy PNA duplex. Note the out-of-duplex position of the central bipyridines. The figure identifies the N- and C-end of the two PNA strands at one of the ends of the PBA duplex and the central base pairs of the PNA duplex.
Figure 4.

(a) Pair of π-stacked bipyridine ligands in the crystal structure of Bipy PNA. The view of the backbone of Bipy PNA shows the orientation of backbone carbonyl of T6 towards the N-end of the PNA and the minor groove of the duplex in contrast to that of the carbonyl groups in A4 and G7, which are oriented towards the C end and away from the duplex. (b) PNA backbone for the central base pairs in Bipy PNA and water molecules (red crosses) that are situated closest to the PNA backbone; (c) Relative orientation of bipyridines with respect to the Bipy PNA duplex from which they are extruded.
Within the asymmetric unit of the Bipy PNA, the two lysines situated in the R-configuration oligomers are well-resolved (Figures 2 and S2). They have an extended conformation stabilized by several hydrogen bonds formed between atoms in the lysine monomer and atoms of nucleobases or backbone of adjacent duplexes. One of these bonds is between the peptide carbonyl group of the lysine and the H8 proton of a terminal G, and two other hydrogen bonds are between the C=O and NH2 groups of the CONH2 end of the lysine and the terminal NH2 and the last peptide C=O groups, respectively, of the same duplex to which G belongs (Figure 2). Two more hydrogen bonds anchor the side chain amino group of the lysine to C=O groups of two backbone peptide groups of another PNA duplex.
The central Bipy ligands of the Bipy PNA are situated outside the duplex and form inter-duplex, π-stacked Bipy-Bipy pairs that bridge adjacent duplexes with identical handedness in the unit cell. The orientation of the Bipy rings is almost parallel to the major grooves (Figure 4). The backbone of the protruding Bipy is compressed so that the A-T and T-A base pairs flanking the Bipy are π stacked and form a base pair step with geometry similar to that of the rest of the base pairs in the duplex (Figure 4b and Table S1).
Discussion
The crystal structures of PNA homoduplexes reported to date had resolution of 1.70 to 2.60 Å.[4–6] The two structures of non-modified and bipyridine-modified PNAs reported in this paper have significantly higher resolution, namely 1.20 and 1.05 Å, respectively. Consequently, we have obtained additional details of the structures, specifically in the area of the terminal lysines and the solvent situated in closest proximity of the PNA.
The major features of the non-modified 8-bp PNA helix and of the two halves of the Bipy PNA duplex that flank the bipyridines coincide with those observed for other PNAs. For example, helical parameters and the torsion angles of the 8-bp PNA duplexes are similar to those characteristic for a P-form helix (Tables 1 and S3), with each of the torsion angles α – χ3 of the non-terminal bases, i.e. G2-C9, having a relatively narrow distribution similar to that observed in other PNA duplexes (Figure S4). The 8-bp PNA duplexes have large base pair displacement from the helical axis and small twist angles (Table 1 and S3). Consequently, they are wider and more unwounded than B-DNA duplexes. As it was the case in previously reported PNA structures, the carbonyl group of the linker that connects the nucleobase to the backbone is oriented towards the C-end of the PNA oligomer, and the majority of backbone carbonyls are oriented towards the C-end of each strand and the solution. Only the backbone carbonyl groups of the terminal cytosine and central thymine monomers point towards the N-end and the duplex. It is interesting to note that in the crystal structure of a 6-bp PNA duplex, it was also observed that the backbone carbonyls of the terminal cytosines and the middle thymines were oriented towards the N-end and the duplex in 50% of the PNA molecules.[4] The flipping of the carbonyl in a minority of the PNA monomers is unlikely to be a consequence of lattice-packing interactions because it occurs in PNAs that crystallize in different space groups. Furthermore, analysis of the interactions between symmetry-related molecules in crystals of the 8-bp PNA showed that the region surrounding both types of carbonyls have similar steric constraints.
Table 1.
Helical Parameters of PNA and Modified DNA Duplexes
| Duplex | Helical sense | Disp. (Å) | Rise* (Å) | Inclination (°) | Tilt (°) | Twist * (°) | Slide (°) | Roll (°) | BP/turn | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H-GGCATGCC-Lys-NH2[b] | X-ray | R | −8.2 | 3.3 | 1.0 | −0.3 | 18.8 | −0.3 | −0.2 | 19 | this paper |
| L | 8.4 | 3.2 | 1.8 | 0.3 | −18.8 | −0.2 | 0.4 | 19 | |||
| H-GGCATGCC-Lys-NH2[b] | NMR | L | 7.9 | 3.7 | −5.0 | −0.2 | −17.3 | −0.22 | −0.60 | 21 | [46] |
| H-CGTACG-NH2 | X-ray | R | 8.3 | 3.2 | 0.3 | 1.0 | 19.8 | 0.06 | 1.81 | 18 | [4] |
| H-CGTACG-L-Lys-NH2 | X-ray | L | 6.6 | 3.4 | −5.02 | 0 | −19 | −0.74 | −5.12 | 18 | [5] |
| R | 7.7 | 3.2 | −5.47 | 0 | 20.5 | −0.41 | 5.17 | 18 | |||
| H-GTAGATCACT-L-Lys-NH2 | X-ray | L | 7.9 | 3.3 | −8.9 | 0 | −18 | / | / | 20 | [6] |
| R | 3.5 | 3.4 | −26.7 | 0 | 19.1 | / | / | 19 | |||
| 3′-CGHATHCG-2′[c] | X-ray | R | −7.0 | 3.8 | / | 0 | 22.9 | −3.4 | −2.8 | 16 | [47] |
| 4′-dd(CGAATTCG)-6′[d] | R | / | 3.8 | / | 1.9 | 14.3 | 4.4 | 4.9 | 25 | [48] | |
| 5′-h(GTGTACAC)-3′[e] | Hex, R | −6.2 | 2.2 | 24.3 | / | 33.2 | −2.5 | 11.5 | 11 | [49] | |
| Trig, R | −6.5 | 3.3 | 3.4 | / | 24.2 | −3.3 | 4.1 | 15 |
Helical parameters calculated using Curves and assuming a linear helical axis.
(S)-glycol nucleic acid with an acyclic, three-carbon propylene glycol phosphodiester backbone. H represents hydroxypyridone ligand instead of a nucleobase.
Homo-DNA with 2′,3′-dideoxy-β-D-glucopyranose sugar-phosphodiester backbone.
Hexitol nucleic acid with an extra methylene group between C1′ and O4′ of the backbone β-D-2′-deoxyribose.
In previous crystal structures of PNA, the conformation of the terminal lysines could not be determined due to the flexibility of the side chain of this amino acid. For example, in the crystal structure of a 6-bp PNA duplex containing a C-terminal lysine in each oligomer, the lysines had B-factors higher than the rest of the PNA.[5] Also, the side chains of the lysines pointed into solvent channels within the crystal and there were no contacts between the lysine and the PNA. On the other hand, molecular mechanics calculations showed low energy conformations containing hydrogen bonds between the ammonium group of the L-lysine side chain and the linker carbonyl oxygen atom or the backbone amide NH, besides cation-π interactions between RNH3+ and guanine.[5] In the current structure of Bipy PNA, both the backbone and the side chain of two terminal L-Lysines within the asymmetric unit are involved in a relatively large hydrogen bonding network with atoms of other PNA duplexes and consequently are well ordered (Figure 2). While it is possible that the hydrogen bonds in which the lysines are involved are due to packing interactions, they do reveal the ability of lysines to participate in hydrogen bonds similar to those identified in molecular mechanics calculations and consequently to exert their chiral induction effect on the PNA duplexes.
Based on molecular dynamics modelling, Brotschi and collaborators suggested that two or more bipyridines situated in complementary positions in a DNA duplex form a zipper-like motif in which the distal pyridine rings of bipyridines from opposite strands participate in π-stacking interactions.[14] Recent NMR studies demonstrated the validity of this model for a pair of biphenyl groups situated in a DNA duplex. We find that Bipy ligands of the Bipy PNA are bulged out of the PNA duplex and form π-stacked pairs that bridge PNA duplexes that are adjacent within the crystal (Figure 4). The backbone of the protruding Bipy is compressed so that the A-T and T-A base pairs flanking the Bipy are π stacked and the rise between these two base pairs is ~3 Å, which is comparable to that of the other base pair steps (Table S1). The torsion angles of the central Bipy5 monomer as well as some of the torsion angles of the PNA monomers adjacent to Bipy5 have values distinct from those of the other PNA monomers of typical PNA duplexes (Table S4). The difference in the Bipy arrangement in the PNA and that of biphenyl in DNA duplexes may be due to the differences between the two nucleic acids or to packing effects manifested in the crystal.
The two duplexes 8-bp PNA and Bipy PNA have the same melting temperature, namely 63°C. This result suggests that the two bipyridines are outside the Bipy PNA duplex in solution too, and thus do not have an effect on the thermal stability of the Bipy PNA duplex. In previous studies, we have found that in a 10-bp PNA duplex formed from the PNA complementary oligomers GTAGATCACT-Lys-NH2 and AGTGATCTAC-Lys-NH2 the substitution of an AT base pair with a pair of bipyridine ligands produces a strong destabilization of the PNA duplex, for which the melting temperature changes from 66°C to 47°C. The melting temperature of a 9-bp PNA duplex with the same sequence as the 10-bp PNA but lacking the central AT base pair was 63°C. The difference between the effect of the bipyridines on the thermal stability of PNA duplexes with different sequences indicates that the ligand effect is modulated by the duplex sequence and suggests that the two bipyridines can adopt different orientations with respect to the duplex depending on the PNA sequence.
The two half-duplexes situated on each side of the pair of bipyridines have helical parameters identical to each other except for the tilt angles, which have the same absolute value but opposite sign (Table 2). The displacement of the base pairs from the helical axis is smaller and the tilt is larger than in most P-type helices, but the values of these parameters are similar to those observed for the short duplex motif formed within a crystal of a single stranded PNA.[6] The base pair rise in each half duplex of the Bipy PNA is the largest observed to date in a PNA structure. The half duplexes have relatively small twist angles and are thus unwound, as it is typical for P-type duplexes.
Table 2.
Helical Parameters[a] for Half-Duplexes Situated on Each Side of the Bipyridines in Bipy PNA.
| Duplex | X-Disp. (Å) | Inclination (°) | Rise (Å) | Tilt (°) | Twist (°) | Slide (°) | Roll (°) |
|---|---|---|---|---|---|---|---|
| R-AB | −3.04 | −24.67 | 4.08 | 5.70 | 20.20 | −0.01 | 8.88 |
| −3.04 | −24.67 | 4.08 | −5.70 | 20.20 | −0.01 | 8.88 | |
| R-CD | −2.82 | −24.70 | 4.07 | 4.89 | 20.82 | −0.07 | 9.01 |
| −2.82 | −24.70 | 4.07 | −4.89 | 20.82 | −0.07 | 9.01 | |
| L-EF | 3.80 | −21.75 | 3.99 | 6.16 | −19.54 | −0.03 | −7.80 |
| 3.80 | −21.75 | 3.99 | −6.16 | −19.54 | −0.03 | −7.80 | |
| L-GH | 3.75 | −21.41 | 3.97 | 5.78 | −19.53 | −0.10 | −7.35 |
| 3.74 | −21.42 | 3.97 | −5.78 | −19.53 | −0.10 | −7.35 |
Calculated with respect to a linear helical axis determined for each half.
The apparent persistence length of a DNA duplex, which is the distance over which a segment of the duplex is linear, is estimated to be ~ 50 nm (~150 bp). DNA bending is important for the packaging of genetic material, the regulation of gene expression and the interaction of nucleic acids with proteins.[17,18] Short tracts of adenines (An-tracts) cause macroscopic curvature of the DNA molecule by additive contributions of the helically-phased A-tracts leading to overall curvature angles of up to 110°.[19, 20] On the other hand, repair of damaged DNA, including DNA containing modified bases, abasic sites or mismatches, involves recognition of the damage site by DNA repair proteins. Crystal structures and spectroscopic studies of complexes formed between the DNA and the repair proteins have shown that DNA is bent and that the bases to be repaired or the abasic sites can be extrahelical.[21–26] Molecular dynamics studies of DNA showed that kinks of 20–30° may occur at abasic sites contained in the DNA and that the abasic site is particularly flexible.[27, 28] A recent study by NMR spectroscopy of the effect of nucleotides with a conformationally-locked, bicyclo[3.1.0]hexane pseudosugar backbone on the structure of DNA duplexes showed that such a modification leads to duplex bending towards the major groove.[29] The comparative study of the crystal structure of Bipy PNA and 8-bp PNA shows that the effect of bulging out of the PNA duplex of the two bipyridines causes a ~53o bending angle for the duplex, which is significantly larger than the corresponding angle for 8-bp PNA (~25°) (Table S5) and is similar to that observed for a DNA duplex containing a synthetic CPD-like lesion in its complex with DNA photolyase after in situ repair.[30]
Conclusion
The crystal structure of non-modified and bipyridine-modified PNAs reported in this paper have the highest resolution reported to date for a PNA crystal structure. This resolution allowed us to identify for the first time specific modes of interaction between the terminal lysines of the PNA and the backbone and nucleobases situated in the vicinity of the lysines, which are considered an important factor in the induction of a preferred handedness in PNA duplexes. The results described in this paper support the notion that while PNA typically adopts a P-type helical structure, its flexibility is relatively high. For example, the base pair rise in Bipy PNA is the largest measured to date in a PNA homoduplex (Tables 1 and 2). The significant bending of the PNA duplex that contains a pair of bipyridines situated in extrahelical positions is in contrast to the situation in DNA duplexes in solution where these ligands are πstacking with adjacent nucleobase pairs and adopt an intrahelical geometry shows that relatively small perturbations, such as packing forces, can significantly impact the relative position of the ligands in the nucleic acid duplex. It will be interesting to examine the effect of the PNA sequence and metal coordination to the bipyridine ligands on the ligands’ relative orientation with respect to the PNA duplex. Studies aimed at addressing these questions are in progress in our laboratories.
Experimental Section
PNA Monomer and Oligomer Synthesis
The bipyridine-containing PNA monomer has been synthesized according to a previously published method.[31] Briefly, PNA monomers A, T, C and G were purchased from Applied Biosystems and used without further purification. PNA oligomers have been synthesized on lysine preloaded p-methylbenzhydrylamine resin using the Boc-protection strategy.[32] After cleavage using TFMSA/TFA, the PNA was precipitated using ethyl ether and was purified by reverse-phase HPLC using a C18 silica column on a Waters 600 System (Waters, City, State). Absorbance was measured with a Waters 2996 Photodiode Array Detector. Oligomers were characterized by MALDI-ToF mass spectrometry, using an Applied Biosystems Voyager Biospectrometry Workstation with delayed extraction using a R-cyano-4-hydroxycinnamic acid matrix (10 mg/mL in 1:1 water:acetonitrile, 0.1% TFA). MALDI-ToF calcd/found for (PNA+H)+ m/z: 8-bp PNA: 2315.24/2314.90; Bipy PNA: 2611.57/2610.73. After HPLC purification, the PNA was lyophilized and then stored at −20°C. PNA stock solutions were prepared by dissolving solid PNA in 10 mM pH 7.0 HEPES buffer. The concentration of the stock solution was determined by UV-vis spectroscopy using ε values for nucleobases at 260 nm of 13,700 M−1 for A; 8,600 M−1 for T; 11,700 M−1 for G; 6,600 M−1 for C;[32] and 9,770M−1 for Bipy. The concentration of the stock solutions was 2.16 mM ss PNA for 8-bp PNA and 1.91 mM for Bipy PNA, which corresponds to 5 mg PNA/ml. To form the PNA duplexes, the solutions were annealed by heating to 95oC for 5 min, followed by slow cooling to room temperature. Crystallization
8-bp PNA
Single crystals of the non-modified 8-bp PNA duplex were grown from a crystallization (reservoir) solution comprised of 40% (v/v) ethylene glycol and pH 6.2 0.1 M Na/K phosphate buffer. The 5 mg/ml stock solution of the 8-bp PNA duplex in pH 7.0 10 mM HEPES buffer was used for screening and final optimization. All crystallization setups were vapor-diffusion, in which 1 μl of PNA stock solution was added to 1 μl of reservoir solution on a cover slip, then suspended over 0.7 ml of reservoir solution. All trays were maintained at 20 °C. Nucleation was observed after five days and crystals continued to grow over two weeks to yield plate-like crystals, the largest with final dimensions of up to 0.08 × 0.08 × 0.04 mm. The ethylene glycol in the crystallization solution served dual roles of precipitant and cryoprotectant, which permitted the crystals to be mounted in a rayon loop and flash-cooled to 100K for data collection.[33]
Bipy PNA
Single crystals of the bipyridine-containing PNA duplex were grown from a crystallization (reservoir) solution comprised of 40% (v/v) 1,2-propanediol, 0.1 M citrate pH 5.5, and 0.2 M NaCl. A stock solution of Bipy PNA at 5mg/mL and crystallization setups were prepared as described above for the non-modified PNA Nucleation was observed after about one week, with continued crystal growth over three weeks before reaching final crystal dimensions of approximately 0.1 × 0.05 × 0.05 mm with rod-like morphology. The 1,2-propanediol in the crystallization solution served dual roles of precipitant and cryoprotectant, which permitted the crystals to be flash-cooled to 100 K for data collection.
Structure Determination
A combination of direct methods in conjunction with maximum likelihood residual refinement and/or density modification was used to solve and iteratively build and refine the crystal structures of 8-bp PNA and Bipy PNA.
Bipy PNA
Complete data sets were collected from two crystals on the undulator beam line X06SA at the Swiss Light Source equipped with a mar165 CCD detector with the parameters shown in Table 3. Since each data set alone could not easily be solved by direct methods, a more complete data set (overall completeness 97%, 90.7 in the last shell) was obtained by merging the two data sets using XPREP. The merged data set was solved by SHELXD[34] using the dual space strategy.[35] Density modification as implemented in SHELXE[36] significantly improved the resulting electron density and allowed us to build the first model comprising of the PNA backbone and all bases with COOT.[37] The structure determination was completed by iterative cycles of model building and refinement with SHELXL.[38] Local NCS symmetry restraints were only used in the initial phases of refinement in order to improved convergence.[39] The course of the refinement was carefully monitored using 5% of the reflections to calculated free R-factors,[40] and completed by restrained anisotropic refinement against all data using a conjugate gradient method as implemented in SHELXL.[38] All non-Hydrogen atoms were refined anisotropically using standard bond lengths and angles and applying suitable rigid-bond and similarity restraints. Additional isotropic restraints were applied to solvent molecules and parts of the molecule that showed unreasonable ADP ellipsoids. Additional crystallographic data are summarized in Table 4.
Table 3.
Data Collection Phasing and Refinement Statistics for Bipy PNA Crystals
| Data set | P9-I | P9-II |
|---|---|---|
| Beam line | X10SA, SLS | X10SA, SLS |
| Temperature [K] | 100 | 100 |
| Wavelength [Å] | 0.8 | 0.8 |
| Oscillation range [°] | 0.5 | 0.5 |
| Detector distance [mm] | 75 | 75 |
| a,b,c [Å] | 38.20, 25.73, 53.05 | 38. 1, 25. 7, 52.90 |
| α,β,γ [°] | 90, 105.3,90 | 90, 105.3, 90 |
| Space group | P2 | P2 |
| resolution range [Å] | 50–1.05 | 50–1.05 |
| No, of reflections | 277897 | 249854 |
| No. of unique | 42731 | 43541 |
| Redundancy | 6.5 | 5.7 |
| Completeness[a] | 92.1 (71.9) | 93.4(66.7) |
| Rmerge | 0.06 (0.43) | 0.05 (0.35) |
| I/σ(I) | 23.3 (1.7) | 29.0 (2.4) |
– last shell 1.05–1.09
Table 4.
Structure Refinement Parameters for 8-bp PNA and Bipy PNA
| P9-merged | P8-III | |
|---|---|---|
| resolution [Å] | 50–1.1 | 50–1.2 |
| No. of data | 38601 | 29399 |
| No. of restraints | 11775 | 9536 |
| No of parameter | 8683 | 7484 |
| No of non-H-atoms | 963 | 831 |
| No of solvent molecules* | 8 | 5 |
| No of carbonate | 2 | 0 |
| No of water | 171 | 189 |
| wR2 (all data) | 0.356 | 0.454 |
| R1[F>4σ] | 0.127 | 0.179 |
| R1free [I>4σ] | 0.169 | 0.220 |
ethylene glycol, glycerol or propanediol
The final model contains four PNA strands, two carbonate ions, and several solvent molecules, including ethylene glycol, glycerol or propanediol from the crystallization solution. Water molecules were added using COOT selected from peaks in the Fo-Fc map with a minimum height of 3.5 sigma and good hydrogen-bond geometry. 184 water molecules were included in the refinement. The four PNA strands display a pseudo-inversion centre. However, the terminal lysine residues that were only partially visible in the electron density and the central bipyridines break this symmetry. Moreover, the backbone shows significant deviations from the pseudo-inversion centre. As a consequence, the rmsd’s for the superposition of an inverted strand with its pseudo-symmetry mate are approximately 0.5 Å, which clearly justifies the refinement in the non-centrosymmetric space group P2. The refinement converged at R-factors of R= 0.128 and Rfree = 0.168.
8-bp PNA
Solving the structure of 8-bp PNA proved to be challenging, which is most likely due to the fact that the crystals were (at least partially) non-merohedrally twinned. The diffraction patterns clearly showed two (or more) components, which hampered the solution and refinement as described below. Two data sets, P8-I and P8-II in Table 5, were collected from two crystals on beam line X10SA at the Swiss Light Source (SLS) using a marmosaic 225 CCD detector.[41] These two data sets were integrated with HKL2000.[42] Since the individual data proved difficult to solve, the two SLS data sets (P8-I and P8-II) were merged using XPREP,[43] which did not affect the completeness significantly. However, this merged data set was finally solved with SHELXD.[43] The best solution resulted in a correlation coefficient of only 65%. Consequently, while the electron density map showed clearly defined solvent regions and some helical features, it was not sufficient for model building. Density modification with SHELXE increased the correlation coefficient to 78% and the resulting map was easily interpretable.[36]
Table 5.
Data Collection Parameters for 8-bp PNA
| Data set | P8-I | P8-II | P8-III |
|---|---|---|---|
| Beam line | X10SA, SLS | X10SA, SLS | ID23B, APS |
| Temperature [K] | 100 | 100 | 100 |
| Wavelength [Å] | 0.8266 | 0.8266 | 1.0 |
| Oscillation range [°] | 0.5 | 0.5 | 1.0 |
| a, b, c [Å] | 18.87, 28.86, 54.94 | 18.91, 28.93, 55.07 | 18.88, 28.91, 54.98 |
| α, β, γ [°] | 87.9, 85.8, 79.5 | 88.0, 86.1, 79.4 | 88. 1, 85.8, 79.6 |
| Space group | P1 | P1 | P1 |
| resolution range [Å] | 50–1.1 | 50–1.1 | 50–1.27 |
| No. reflections | 156663 | 67947 | 81240 |
| No. unique reflections | 43264 | 43541 | 30089 |
| Redundancy | 3.6 | 1.7 | 2.3 |
| Completeness | 94.6 (91.4)[a] | 88.6 (78.4) | 91.7 (76.5)[b] |
| Rmerge | 0.11 (0.47) | 0.06 (0.25) | 0.06, (0.19) |
| I/σ(I) | 11.8 (2.4) | 12.4(2.7) | 21.2 (2.2) |
last resolution shell: 1.1–1.4 [Å]
last resolution shell: 1.27–1.47 [Å]
The course of the refinement was again monitored using 5% of the reflections to calculated free R-factors,[40] and completed by restrained anisotropic refinement against all data using a conjugate gradient method as implemented in SHELXL.[36] All non-Hydrogen atoms were refined anisotropically using standard bond lengths and angles and applying suitable rigid-bond and similarity restraints. Additional isotropic restraints were applied to solvent molecules and parts of the molecule that showed unreasonable ADP ellipsoids. Nevertheless, the refinement was stuck at R-factors around 28%, presumably due to the non-merohedral twinning observed during data collection. In order to resolve this issue, a third data set for a crystal obtained from a slightly modified crystallization condition which contained an additional 10% ethanol in the crystallization solution was collected at Argonne National Laboratory, beam line ID23B (P8-III in Table 5). As the crystals suffered from severe radiation damage, a 10 micrometer collimator was used and data were collected with 1 degree oscillation range per frame until the average intensity dropped per image to about 60% of the initial value, which on average related to approximately 20 degrees oscillation. Data collection was resumed after the crystal was translated by 50 micrometer until the entire crystal has been utilized. This data set was independently solved by using SHELXS[44] to identify the 50 strongest peaks as starting coordinates for the less flexible PNA peptide backbone atoms to be used in ACORN.[45]
The 8-bp PNA structure was refined following the same protocols as described above for the Bipy PNA. The model contains four PNA strands again related by a pseudo-inversion centre. Since the lysine residues are for the most part disordered, the remaining PNA strands almost fulfil the pseudo-inversion centre. The rmsd’s for the superposition of an inverted strand with its counterpart is only 0.3 Å. However, this value is still larger than the estimated uncertainties and hence the structure was refined in the space group P1. The final model comprises 4 PNA strands, 4 ethylene glycol molecules and 197 well-defined water molecules. The refinement converged at R=0.18 (Rfree=0.22). These relatively high R-factors are presumably due to the fact that even these crystals suffered from partial non-merohedral twinning described above.
The structures have been deposited in the Protein Structure Database and have numbers 3MBS and 3MBU.
Supplementary Material
Scheme 1.
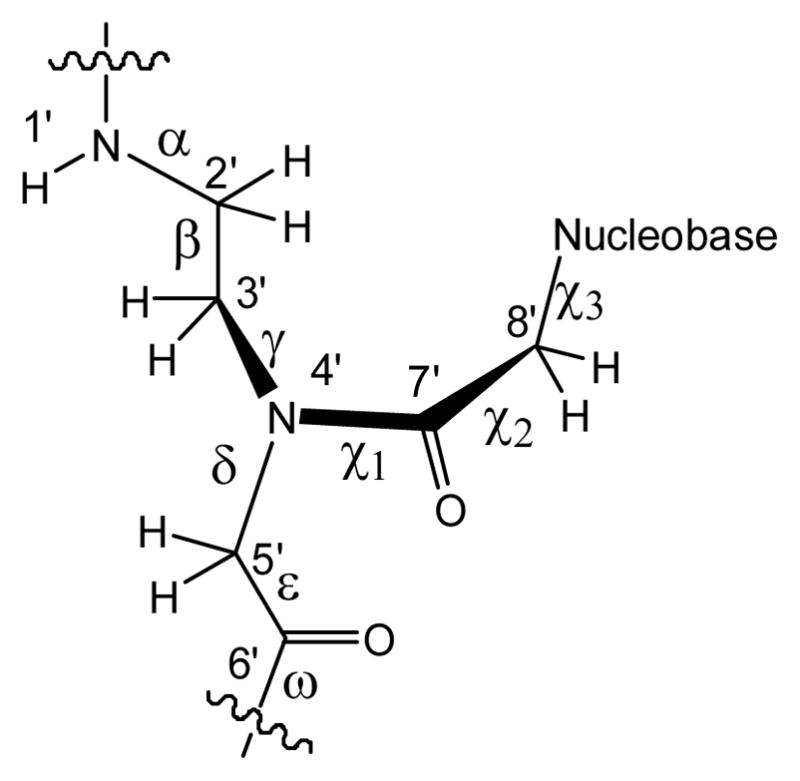
Chemical structure of PNA and definitions of torsion angles: α = C6′-N1′-C2′-C3′ ; β = N1′-C2′-C3′-N4′ ; γ = C2′-C3′-N4′-C5′ ; δ= C3′-N4′-C5′-C6′; ε = N4′-C5′-C6′-O1′; ω= C5′-C6′-N1′-C2′; χ1 = C3′-N4′-C7′-C8′; χ2 = N4′-C7′-C8′-N1(py)/N9(pu); χ3 = C7′-C8′-N1(py)/N9(pu)-C2(py)/C4(pu). The angle ε defines the orientation of the backbone carbonyl group. An alternative definition of ε as N4′-C5′-C6′-N1′ has been used in literature,[4] which corresponds to the definition of the dihedral angle of ε in DNA.
Acknowledgments
This work was supported by the National Science Foundation [CHE-0347140 to C.A. and in part by CHE-9808188 to the Center for Molecular Analysis at Carnegie Mellon], Deutsche Forschungsgemeinschaft (SH14/5 to GMS), The National Institutes of Health [GM066466 to JIY] The Pennsylvania Department of Health [to JIY] W. H. acknowledges support from an Astrid and Bruce McWilliams Fellowship. We thank the staff members at the General Medicine and Cancer Institutes Collaborative Access Team (GM/CA-CAT) and the Southeast Regional Collaborative Access Team, both at the Advanced Photon Source, Argonne National Laboratory, and the Paul Scherrer Institute (X10SA) at the Swiss Light Source for access and technical assistance. GM/CA CAT has been funded in whole or in part by the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). Use of the Advanced Photon Source is supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Nielsen PE, Egholm M. Current Issues in Molecular Biology. 1999;1:89. [PubMed] [Google Scholar]
- 2.Nielsen PE, Egholm M, Berg RH, Buchardt O. Science. 1991;254:1497. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 3.Achim C, Armitage BA, Ly DH, Schneider JW. Wiley Encyclopedia of Chemical Biology. John Wiley & Sons; 2008. [Google Scholar]
- 4.Rasmussen H, Kastrup JS, Nielsen JN, Nielsen JM, Nielsen PE. Nat Struct Biol. 1997;4:98. doi: 10.1038/nsb0297-98. [DOI] [PubMed] [Google Scholar]
- 5.Rasmussen H, Liljefors T, Petersson B, Nielsen PE, Kastrup JS. J Biomol Struct Dyn. 2004;21:495. doi: 10.1080/07391102.2004.10506943. [DOI] [PubMed] [Google Scholar]
- 6.Petersson B, Nielsen BB, Rasmussen H, Larsen IK, Gajhede M, Nielsen PE, Kastrup JS. Journal of the American Chemical Society. 2005;127:1424. doi: 10.1021/ja0458726. [DOI] [PubMed] [Google Scholar]
- 7.He W, Franzini RM, Achim C. Progress in Inorganic Chemistry. 2007;55:545. [Google Scholar]
- 8.Mueller J. European Journal of Inorganic Chemistry. 2008:3749. [Google Scholar]
- 9.Tanaka K, Shionoya M. Coordination Chemistry Reviews. 2007;251:2732. [Google Scholar]
- 10.Clever GH, Kaul C, Carell T. Angewandte Chemie, International Edition. 2007;46:6226. doi: 10.1002/anie.200701185. [DOI] [PubMed] [Google Scholar]
- 11.Shionoya M, Tanaka K. Current Opinion in Chemical Biology. 2004;8:592. doi: 10.1016/j.cbpa.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Atwell S, Meggers E, Spraggon G, Schultz PG. Journal of the American Chemical Society. 2001;123:12364. doi: 10.1021/ja011822e. [DOI] [PubMed] [Google Scholar]
- 13.Johannsen S, Megger N, Böhme D, Siegel RKO, Müller J. Nature Chemistry. 2010;2:229. doi: 10.1038/nchem.512. [DOI] [PubMed] [Google Scholar]
- 14.Brotschi C, Haberli A, Leumann CJ. Angewandte Chemie, International Edition. 2001;40:3012. doi: 10.1002/1521-3773(20010817)40:16<3012::AID-ANIE3012>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.Johar Z, Zahn A, Leumann CJ, Jaun B. Chemistry--A European Journal. Vol. 14. 2008. p. 1080. [DOI] [PubMed] [Google Scholar]
- 16.Bunkoczi G, Vertesy L, Sheldrick George M. ngewandte Chemie, International Edition. 2005;44:1340. doi: 10.1002/anie.200461933. [DOI] [PubMed] [Google Scholar]
- 17.Maher LJ., III Current Opinion in Chemical Biology. 1998;2:688. doi: 10.1016/s1367-5931(98)80104-x. [DOI] [PubMed] [Google Scholar]
- 18.Swinger KK, Rice PA. Current Opinion in Structural Biology. 2004;14:28. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Strahs D, Schlick T. Journal of Molecular Biology. 2000;301:643. doi: 10.1006/jmbi.2000.3863. [DOI] [PubMed] [Google Scholar]
- 20.Gohlke C, Murchie AIH, Lilley DMJ, Clegg RM. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:11660. doi: 10.1073/pnas.91.24.11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horton NC, Perona JJ. Journal of Molecular Biology. 1998;277:779. doi: 10.1006/jmbi.1998.1655. [DOI] [PubMed] [Google Scholar]
- 22.Kim JL, Nikolov DB, Burley SK. Nature. 1993;365:520. doi: 10.1038/365520a0. [DOI] [PubMed] [Google Scholar]
- 23.Kugel JF. Biochemistry and Molecular Biology Education. 2008;36:341. doi: 10.1002/bmb.20202. [DOI] [PubMed] [Google Scholar]
- 24.Lorenz M, Hillisch A, Goodman SD, Diekmann S. Nucleic Acids Research. 1999;27:4619. doi: 10.1093/nar/27.23.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorenz M, Hillisch A, Payet D, Buttinelli M, Travers A, Diekmann S. Biochemistry. 1999;38:12150. doi: 10.1021/bi990459+. [DOI] [PubMed] [Google Scholar]
- 26.Parkinson G, Wilson C, Gunasekera A, Ebright YW, Ebright RE, Berman HM. Journal of Molecular Biology. 1996;260:395. doi: 10.1006/jmbi.1996.0409. [DOI] [PubMed] [Google Scholar]
- 27.Barsky D, Foloppe N, Ahmadia S, Wilson DM, III, MacKerell AD., Jr Nucleic Acids Research. 2000;28:2613. doi: 10.1093/nar/28.13.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curuksu J, Zakrzewska K, Zacharias M. Nucleic Acids Res. 2008;36:2268. doi: 10.1093/nar/gkm1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu Z, Maderia M, Barchi JJ, Jr, Marquez VE, Bax A. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:24. doi: 10.1073/pnas.0408498102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mees A, Klar T, Gnau P, Hennecke U, Eker APM, Carell T, Essen L-O. Science. 2004;306:1789. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]
- 31.Franzini R, Watson RM, Popescu D-L, Patra GK, Achim C. Polymer Preprints. 2004;45:337. [Google Scholar]
- 32.Nielsen PE. Peptide Nucleic Acids: Protocols and Applications. 2. 2004. [Google Scholar]
- 33.Ten TY. J Appl Cryst. 1990;23:387. [Google Scholar]
- 34.Uson I, Sheldrick GM. Curr Opin Struct Biol. 1999;9:643. doi: 10.1016/s0959-440x(99)00020-2. [DOI] [PubMed] [Google Scholar]
- 35.Miller R, DeTitta GT, Jones R, Langs DA, Weeks CM, Hauptman HA. Science. 1993;259:1430. doi: 10.1126/science.8451639. [DOI] [PubMed] [Google Scholar]
- 36.Sheldrick GM. Zeitschrift fuer Kristallographie. 2002;217:644. [Google Scholar]
- 37.Emsley P, Cowtan K. Acta Crystallographica, Section D: Biological Crystallography. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Sheldrick GM, Schneider TR. Methods in Enzymology. 1997;277:319. [PubMed] [Google Scholar]
- 39.Uson I, Pohl E, Schneider TR, Dauter Z, Schmidt A, Fritz H-J, Sheldrick GM. Acta Crystallographica, Section D: Biological Crystallography. 1999;D55:1158. doi: 10.1107/s0907444999003972. [DOI] [PubMed] [Google Scholar]
- 40.Brünger AT. Nature. 1992;355:472. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 41.Pohl E, Pradervand C, Schneider R, Tomizaki T, Pauluhn A, Chen Q, Ingold G, Zimoch Z, Schulze-Briese C. Synch Rad News. 2006;19:24. [Google Scholar]
- 42.Otwinowski Z, Minor W. Methods in Enzymology. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Sheldrick GM. Bruker AXS. 2007 [Google Scholar]
- 44.Sheldrick GM. Acta Crystallographica, Section A: Foundations of Crystallography. 1990;A46:467. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 45.Yao JX, Dodson EJ, Wilson KS, Woolfson MM. Acta Crystallographica, Section D: Biological Crystallography. 2006;D62:901. doi: 10.1107/S0907444906008122. [DOI] [PubMed] [Google Scholar]
- 46.He W, Hatcher E, Balaeff A, Beratan DN, Gil RR, Madrid M, Achim C. Journal of the American Chemical Society. 2008;130:13264. doi: 10.1021/ja800652h. [DOI] [PubMed] [Google Scholar]
- 47.Schlegel MK, Essen LO, Meggers E. Journal of the American Chemical Society. 2008;130:8158. doi: 10.1021/ja802788g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egli M, Pallan PS, Pattanayek R, Wilds CJ, Lubini P, Minasov G, Dobler M, Leumann CJ, Eschenmoser A. Journal of the American Chemical Society. 2006;128:10847. doi: 10.1021/ja062548x. [DOI] [PubMed] [Google Scholar]
- 49.Declercq R, Van Aerschot A, Read RJ, Herdewijn P, Van ervelt L. Journal of the American Chemical Society. 2002;124:928. doi: 10.1021/ja016570w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.